

Abstract
As global fisheries decline, microbial single-cell protein (SCP) produced from brewery process water has been highlighted as a potential source of protein for sustainable animal feed. However, biotechnological investigation of SCP is difficult because of the natural variation and complexity of microbial ecology in wastewater bioreactors. In this study, we investigate microbial response across a full-scale brewery wastewater treatment plant and a parallel pilot bioreactor modified to produce an SCP product. A pyrosequencing survey of the brewery treatment plant showed that each unit process selected for a unique microbial community. Notably, flow equalization basins were dominated by Prevotella, methanogenesis effluent had the highest levels of diversity, and clarifier wet-well samples were sources of sequences for the candidate bacterial phyla of TM7 and BD1-5. Next, the microbial response of a pilot bioreactor producing SCP was tracked over 1 year, showing that two different production trials produced two different communities originating from the same starting influent. However, SCP production resulted generally in enrichment of several clades of rhizospheric diazotrophs of Alphaproteobacteria and Betaproteobacteria in the bioreactor and even more so in the final product. These diazotrophs are potentially useful as the basis of a SCP product for commercial feed production.
Introduction
Already half of all global fish stocks have been deemed fully exploited (Cressey, 2009), which has led to the collapse of several fisheries and the potential collapse of others over the next several decades (Worm et al., 2006). Concomitantly, aquaculture (the farm rearing of fish) has grown at an annual rate of 14% since 1970 (FAO Fisheries Department, 2003). Because aquaculture feed production relies on significant amounts of non-sustainable fish meal protein harvested from ocean fisheries, further aquaculture growth will result in more fish meal shortages and further depletion of ocean fisheries. Therefore, there has been renewed interest in the development of less expensive and more sustainable fish meal replacements.
In the brewing industry, solid byproducts of various forms (spent grains, hops, yeasts, etc.), once a costly landfill waste, have become a livestock feed source. Even after this removal of solids, a large amount of dissolved carbon still remains in the typical brewery wastewater (Hough, 1985). This brewery waste can be aerobically and microbiologically treated in a process-wastewater treatment facility and the carbon-degrading microbiota harvested as dried microbial biomass, called single-cell protein (SCP). Researchers have recognized SCP's potential as an animal feed for decades, although SCP has never fully replaced fish meal at production scale (El-Sayed, 1999). A major concern has been the negative performance and connotations associated with wastewater and, in particular, reuse of raw sewage. Initial studies in SCP production from domestic wastewater biomass sources were often plagued by heavy metal contamination and faecal pathogens as part of their process stream (Tacon, 1979; Lovell, 1989). However, food-processing wastes have minor or no exposure to domestic sewage (Vriens et al., 1989) and are known to have a much higher chemical oxygen demand (COD) and lower total nitrogen profile than domestic sewage (Gray, 2004). Specifically, brewery process water possesses distinct characteristics that make the technology more feasible than most food-processing process water types, such as a continuous global year-round production of dissolved carbon process water (Huige, 2006), and an amino acid profile rich in lysine and methionine (two essential amino acids absent from many plant and fungal sources) that is comparable with fish meal (Vriens et al., 1989; Tacon et al., 2009). Several major challenges to bring SCP to market are to reliably maintain a high crude protein and essential amino acid content, and to continue to produce a treated process-wastewater that meets local water regulations. An understanding of the underlying microbial community responsible for SCP product formation is needed to help achieve these goals.
Fortuitously, knowledge of microbial communities is enabled by the rapid increase in DNA sequencing throughput with decreased cost, which has greatly expanded the detection coverage of microbial diversity in environmental samples and has the potential to identify the functioning and turnover of species in environmental engineering applications such as wastewater technology. Several recent studies have established that high-throughput sequencing can track how wastewater community consortia fluctuate in real-world systems (Werner et al., 2011) and how wastewater treatment diversity can have notable biogeographical trends between plants (Werner et al., 2011; Zhang et al., 2012). The production of novel bioproducts from wastewater such as fuel and chemicals requires a closer look at how these microbial systems function in order to identify relevant communities responsible for mixed community biotechnologies [e.g. microbial fuel cells (Lee et al., 2010), and lignin-cellulose degradation (Hollister et al., 2011)]. In this study, the microbial turnover and diversity of an entire brewery wastewater treatment works was analysed by pyrotag sequencing technology in order to identify metabolisms of biotechnological interest and to develop possible strategies to improve quality or economic competitiveness. Specifically, this study attempted to identify what common microbial community responses were seen in an SCP production pilot bioreactor running under a non-conventional aerobic treatment regime and the relationship of this community compared with the microbial consortia of the larger treatment plant facility.
Results
Physical characteristics and operational conditions of the wastewater treatment facility
Table 1 shows the operational conditions of unit processes in this study. For the wastewater treatment facility, operational parameters were tracked for each stage by plant operators. This data indicated high COD wastewaters and a sizable suspended solids fraction in both the influent and in the acidogenic basins. Organic acid and volatile fatty acids (VFA) monitoring of the basin indicated production of organic acids and (in conjunction with free ammonia measurements) also indicated a relatively low ratio of free nitrogen to carbon available for microbes. In the methanogenic upflow anaerobic sludge blanket (UASB) basin, further acid production was noted, and the majority of this was consumed in the UASB with a notable sludge blanket observable as total suspended solids (TSS). These constituents were then aerobically treated to discharge standards in the aerobic basin. In contrast, the pilot bioreactor was marked by a higher mixed liquor suspended solids (MLSS) content than most conventional domestic wastewater treatment regimes, as well as much lower dissolved oxygen (DO) levels and higher VFA levels due to influent from the acidogenic basin.
Table 1.
Operating parameters of each unit process
| Sample location | Operating conditions | Average (SD) | |
|---|---|---|---|
| Plant influent sample | Flow rate | 402 (174) | m3 d−1 |
| pH | 10.85 (1.3) | ||
| COD total | 10 147 (3848) | mg l−1 | |
| COD soluble | 7859 (2573) | mg l−1 | |
| TSS | 1989 (1011) | mg l−1 | |
| Temperature | 35 | °C | |
| Acidogenic basin mixed liquor | Volume | 600 | m3 |
| pH | 5.68 (0.43) | ||
| VFA | 1654 (284) | mg l−1 acetic acid | |
| Total organic acids | 4489 (1439) | mg l−1 | |
| COD total | 10 554 (1854) | mg l−1 | |
| COD soluble | 8005 (1714) | mg l−1 | |
| TSS | 1275 (352) | mg l−1 | |
| Total N | 164 (18) | mg l−1 | |
| P-PO4 | 152 (21) | mg l−1 | |
| NH3-N | 8.76 (9.48) | mg l−1 | |
| Methanogenesis UASB outfall | Volume | 800 | m3 |
| VFA | 4418 (2567) | mg l−1 acetic acid | |
| TSS | 4705 (2785) | mg l−1 | |
| Aerobic basin mixed liquor | Volume | 3820 | m3 |
| SRT | 16 | d | |
| Clarifier outfall | COD total | 131 (22) | mg l−1 |
| COD soluble | 123 (22) | mg l−1 | |
| TSS | 12 (5) | mg l−1 | |
| Pilot bioreactor sample port | Volume | 38 | m3 |
| Flow rate | 4.75 | m3 d−1 | |
| pH | 7.15 (0.36) | ||
| MLSS | 2600 (461) | mg l−1 | |
| Total organic acids | 3666 (1301) | mg l−1 | |
| DO | 0.22 (0.05) | mg l−1 | |
| Excess N, P added as urea and phosphoric acid | |||
SD, standard deviation; SRT, solids retention time.
Pyrotag microbial diversity across the wastewater treatment plant
Table 2 shows alpha diversity information and sequencing depth for each sample. A total of 808 near full-length Sanger and a total of ∼ 54 000 pyrotag 16S amplicon sequences were completed. Microbial diversity coverage estimates based on alpha diversity were bracketed on the low end by the Chao1 estimator and on the high end by the CatchAll estimate. Chao1 and abundance-based coverage estimator (ACE) metrics of samples rarified to the same sequencing depth showed that the influent and acidogenic basin had the lowest levels of diversity, while the majority of operational taxonomic units (OTUs) in this study were found from both the UASB bioreactor and the aerobic basin of the brewery wastewater treatment plant (WWTP). Figure 2 describes phylum-level diversity from all samples arranged by unweighted pair group method with arithmetic mean (UPGMA) cladogram based on the unweighted UniFrac distance, which shows that samples clustered primarily because of unit process type (jackknife sensitivity analysis of sequencing depth in Supporting Information Fig. S1). Samples tended to primarily cluster by treatment regime, except the pilot reactor mixed liquor samples clustered closely to the corresponding time point of the final dried SCP product. The four main dominant phyla found from pyrotags in this study were the Bacteroidetes, Firmicutes, Actinobacteria and Proteobacteria (from the alpha, beta and gamma classes), and were consistent with previous research on wastewater aerobic treatment (Seviour and Nielsen, 2010) and process wastewater specifically (Manz et al., 1994). The candidate divisions of SR1 and RF3, seen previously in methanogenesis bioreactor surveys (Chouari et al., 2005; Riviere et al., 2009), comprised 2.8% and 2.3% of sequences respectively from the methanogenesis UASB process. TM7 (Hugenholtz et al., 2001) and BD1-5 (Li et al., 1999) accounted for 5.2% and 5.1% of sequences detected in the treatment plant wet well. Krona hierarchical pie charts (Supplemental Information, http://inside.mines.edu/~jspear/resources.html) showed the relative abundance of OTUs across lower taxonomic levels. The influent was dominated by several groups of Firmicutes from the Lactobacillus and Enterococcus families. Acidogenic basin pyrotag sequences were dominated by the genus Prevotella, a saccharolytic fermenter. The remaining stages showed no highly dominant clades across time, although several clades of closely related enriched OTUs (from Bacteroidetes, Firmicutes and Proteobacteria) appear to dominate in the pilot bioreactor and SCP product. In addition, approximately 14.3% of all OTUs (representing 4% of total sequences) remained unclassified beyond the domain level (5.9% of pilot plant OTUs, 5.2% of methanogenesis OTUs and 4.0% of aerobic basin OTUs).
Table 2.
Sampling schedule, sequencing depth and alpha diversity of each unit process
| Alpha diversity estimator | |||||||
|---|---|---|---|---|---|---|---|
| Sample location | Date, day number | Number of Sanger reads | Number of pyrotags (V1-V2) | Observed OTUs (97%) | % coverage | Chao1 Average (SD)a | ACE Average (SD)a |
| Plant influent sample port | 1/30/08, 0 | – | 459 | 52 | 46–76 | 68.2 | 76.0 |
| 3/19/08, 49 | – | 816 | 38 | 54–66 | 48.4 (11.8) | 54.0 (12.2) | |
| 4/30/09, 456 | – | 674 | 8 | 56–84 | 8.8 (2.0) | 10.2 (2.6) | |
| Acidogenic basin mixed liquor | 1/30/08, 0 | – | 1127 | 38 | 70–73 | 39.4 (11.0) | 43.4 (10.8) |
| 3/19/08, 49 | 92 | 3162 | 85 | 45–58 | 66.1 (16.5) | 71.2 (15.2) | |
| 3/19/09, 414 | 52 | 3450 | 66 | 67–93 | 51.4 (12.1) | 52.5 (7.3) | |
| 4/30/09, 456 | 43 | 884 | 41 | 58–76 | 48.4 (12.9) | 50.1 (10.5) | |
| Methanogenesis UASB outfall | 1/30/08, 0 | – | 975 | 128 | 51–60 | 159.8 (23.2) | 172.4 (21.4) |
| 3/19/08, 49 | 14 | 2373 | 220 | 59–71 | 193.5 (31.5) | 191.8 (27.0) | |
| 4/30/09, 456 | 72 | 805 | 131 | 32–58 | 192.1 (25.1) | 192.2 (18.3) | |
| Aerobic basin mixed liquor | 4/30/09, 456 | 34 | 1097 | 194 | 45–48 | 255.4 (38.7) | 267.5 (30.2) |
| 4/30/09, 456 | – | 722 | 155 | 43–65 | 222.8 (29.9) | 239.7 (22.4) | |
| 4/30/09, 456 | – | 1226 | 211 | 46–63 | 266.2 (42.4) | 275.8 (37.4) | |
| Clarifier outfall | 1/30/08, 0 | – | 1176 | 138 | 40–59 | 174.9 (46.7) | 193.8 (32.3) |
| 3/19/08, 49 | 52 | 2439 | 315 | 53–61 | 276.2 (41.7) | 305.6 (45.0) | |
| 4/30/09, 456 | 38 | 960 | 176 | 45–66 | 220.3 (25.9) | 223.8 (21.8) | |
| Pilot bioreactor sample port | 03/19/08, 49 | 48 | 4090 | 88 | 52–71 | 68.6 (30.7) | 71.0 (16.7) |
| 04/08/08, 69 | – | 1217 | 127 | 39–63 | 167.8 (34.2) | 177.3 (27.7) | |
| 09/10/08, 224 | 24 | 4874 | 197 | 59–71 | 127.8 (31.1) | 136.2 (26.6) | |
| 01/06/09, 342 | 29 | 2840 | 193 | 50–70 | 163.2 (25.4) | 164.1 (21.9) | |
| 02/16/09, 383 | 56 | 3005 | 163 | 53–75 | 130.9 (32.1) | 133.7 (25.9) | |
| 03/19/09, 414 | 51 | 1711 | 151 | 63–70 | 159.2 (26.2) | 165.2 (19.1) | |
| 04/30/09, 456 | 25 | 1231 | 115 | 33–59 | 137.5 (28.1) | 143.9 (25.8) | |
| Dried SCP product | 05/07/08, 98 | 46 | 3266 | 92 | 63–79 | 72.4 (13.4) | 78.9 (12.8) |
| 09/10/08, 224 | 19 | 3580 | 123 | 55–70 | 90.3 (19.1) | 102.3 (19.8) | |
| 01/06/09, 342 | 41 | 2122 | 171 | 64–74 | 160.5 (25.9) | 171.1 (24.3) | |
| 02/16/09, 383 | – | 1309 | 103 | 52–71 | 119.2 (22.4) | 128.3 (23.8) | |
| 03/19/09, 414 | 37 | 2262 | 147 | 56–68 | 142.2 (32.0) | 152.9 (28.7) | |
| 04/30/09, 456 | 35 | 1104 | 94 | 46–60 | 118.8 (22.9) | 123.9 (18.6) | |
Jackknifed to 500 sequences sample–1 for all samples except first.
SD, standard deviation.
Fig 2.
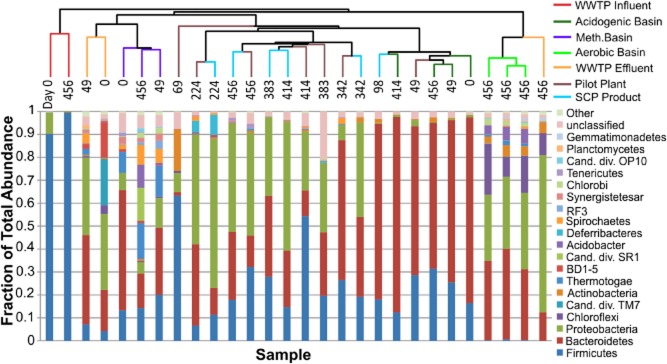
Phylum-level distribution shown sorted by UPGMA clustering of UniFrac unweighted distance. Clades common to a unit process colored based on Fig. 1. Stacked bar graphs of phyla distributions are sorted by total dataset rank abundance starting with the most common from the bottom. Leaves are labeled with the day of sampling from initial date.
Tracking microbial response in the pilot bioreactor by principal component analysis
When tracked over time, each trial run within the pilot plant produced the same pattern of shifts with the three beta diversity metrics studied (Fig. 3A–C). In trial 1, the pilot reactor community was initially similar to the influent environment and then shifted to a new community composition. Before trial 2, the reactor was drained and refilled but not re-inoculated or sterilized. In trial 2, a second microbial community developed that was different from the influent and trial 1. For principal component analyses (PCoAs) of the pilot bioreactor and influent, results did not change with jackknifing to 800 sequences per sample (Supporting Information Fig. S2). No significant time–decay relationship (representing steady succession or turnover) was seen in the study, and most comparisons between time points showed the same level of dissimilarity distance (Fig. 3D). Bi-plots of the most common family-level classifications of OTUs found in this study were graphed in conjunction with PCoA data (Fig. 3A–C) and showed a correlation of Rhodospirillales with trial 1, several family types from Proteobacteria with trial 2 and Prevotellaceae with the influent.
Fig 3.
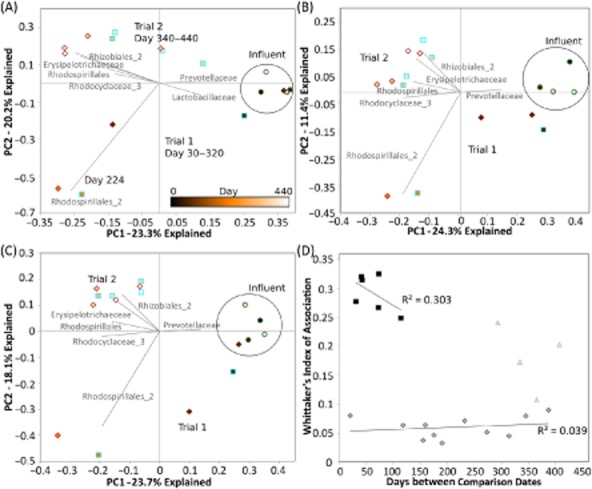
Microbial turnover across a bioreactor over two production trials. PCoA plots of (A) Whittaker's index of association, (B) unweighted UniFrac, (C) Sørensen index and (D) distance–decay of Whittaker's index of association. (A, B, C) PCoA plots include influent samples as a baseline (green circle) and pilot plant mixed liquor (brown diamond) and SCP (blue square) time courses (fill color indicates time progression). PCoA bi-plots of the level 4 SILVA 104 non-redundant database taxonomy of the most abundant OTUs are denoted. (D) Distance–decay pairwise comparisons are graphed according to the difference in days of time points. Distance–decay trends are seen only for sample comparisons for trial 2 (square) but not trial 1 (triangle), and all other comparisons (diamond).
Co-abundance, metastats and microbial lifestyle analysis of pilot bioreactor and SCP product OTUs
To address the question of which OTUs might be biotechnologically relevant, the variations of OTU abundances in the pilot reactor and SCP product samples over time were used to generate Bray–Curtis distances (a generalized distance metric) between repeatedly occurring OTUs. These data were clustered using UPGMA (Supporting Information Fig. S3) and identified that a small number of recurrent OTUs with high abundance (6.2% of OTUs and 67.6% of sequences) contributed to a deep clade within the clustergram. For these OTUs to be responsible for SCP formation, they should have a distribution that was enriched within the pilot bioreactor and SCP product when compared with the community of the acidogenic basin (which served as the influent community to the pilot bioreactor). When the MetaStats statistic was used to compare these OTUs with the pilot bioreactor influent distribution of OTUs, about 44.7% of the sequences (from the combined set of SCP and pilot bioreactor OTUs) were significantly enriched over the influent, and 29.6% of the sequences from the combined set were from OTUs that were significantly depleted compared with the influent (Supporting Information Fig. S4). When a phylogenetic tree was constructed from representative pyrotags of these OTUs, a phylogenetically coherent trend was observed. As shown in Fig. 4, when sequences from enriched or depleted OTUs were cross-referenced with the known microbial lifestyle of the taxonomic classification of the sequence, the depleted OTUs tended to be more from saccharolytic fermenters, primarily Prevotellaceae. Enriched OTUs tended to be from genera consisting of rhizospheric diazotrophs such as Azospirillum, Azonexus and Telmatospirillum, and a smaller fraction of saccharolytic fermenters. OTUs identified as having a rhizospheric diazotroph lifestyle were absent from both the pilot bioreactor influent and from depleted OTUs, but accounted for 39.4% of sequences from enriched OTUs in the pilot bioreactor and 73.0% of sequences from enriched OTUs in the final product.
Fig 4.
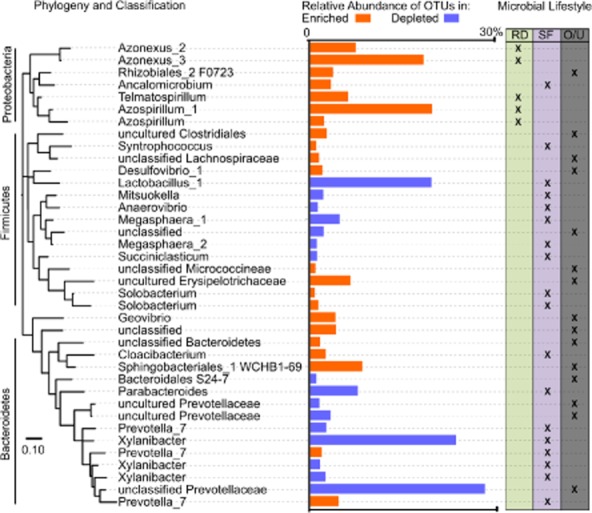
Phylogenetic tree and sequence abundance of depleted and enriched OTUs. Enriched OTUs (orange) and depleted OTUs (blue) show clustering by phylogeny and have differential metabolic roles when classified by rhizospheric diazotroph (RD, pea green), saccharolytic fermenter (SF, light purple) and other/unclassified (O/U, grey). References for metabolic assignment and phylogenetic tree bootstrap values in Supporting Information Fig. S6.
Full-length 16S sequences of rhizospheric diazotrophs from the pilot bioreactor
Phylogenetic trees of full-length Sanger 16S sequences from rhizospheric diazotrophs in Alphaproteobacteria and Betaproteobacteria are shown in Fig. 5 (full phylogenetic tree in Supporting Information Fig. S5). In the Betaproteobacteria (Fig. 5A), near full-length sequences were related to known isolates of Azospira sp., Azovibrio restrictus and Azonexus caeni (but not genera Azoarcus). In the Alphaproteobacteria (Fig. 5B and C), near full-length clone sequences were not related to any of the known rhizospheric strains for Magnetospirillum and were related to one strain of Azospirillum [Azospirillum fermentarium CC-LY743 isolated from a fermentation tank (Lin et al., 2013)]. Closest basic local assignment search tool (BLAST) matches were commonly from strains isolated in large part from either wastewater and microbial fuel cell sources (Kaksonen et al., 2004; Quan et al., 2006; Borole et al., 2009; de Cárcer et al., 2011; Croese et al., 2011; Sun et al., 2011), as well as rhizosphere studies (Ashida et al., 2010; Knief et al., 2012) for which rhizospheric diazotrophs are primarily associated.
Fig 5.
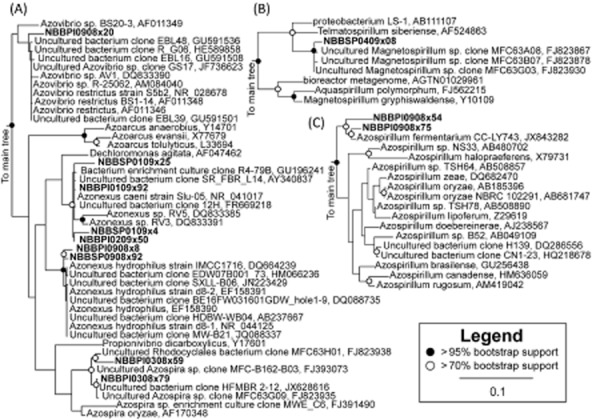
Phylogenetic diversity of full-length Sanger sequences related to rhizospheric diazotrophs detected from the pilot bioreactor and final product for sequences from (A) Azoarcus, Azonexus and Azospira clades, (B) Magnetospirillum and Telmatospirillum clades, (C) and Azospirillum clades. Symbols denoting bootstrap support values are for maximum likelihood analyses. Representative sequences for OTUs identified from this study are in bold.
Discussion
Insights into microbial diversity in WWTPs
Prior to this work, it was unclear if large segments of the microbial community would pass through the entire treatment plant or if communities would shift entirely from one treatment regime to another. This study reveals that the influent waste beer from brewery process water was limited in bacterial diversity and had little impact on the colonization of subsequent unit processes in a brewery wastewater treatment works, and that the microbial community of each unit process remained largely self-selective. In concurrence with previous work on wastewater microbial variation (Fernandez et al., 2000; Wells et al., 2011; Werner et al., 2011), constant turnover of even the most common species was a normal occurrence in samples from this study. In terms of novel diversity discovery, unclassifiable OTUs were found in all environments downstream of the acidogenic basin. Particularly surprising was the number of unclassified OTUs found in the methanogenesis UASB and the clarifier wet-well outfall (purple and orange squares, Fig. 1). The design of the UASB may be one contributor to the diversity, as the sludge granules are known to be highly complex physical structures with differing chemical and microbial composition based on depth from the surface of the granule (Sekiguchi et al., 1999; Liu et al., 2002; Diaz et al., 2006). The clarifier wet well receives low settling COD effluent from clarifier operations prior to discharge from the plant and may be an overlooked environment for sampling, likely harbouring organisms related to wastewater treatment washout.
Fig 1.
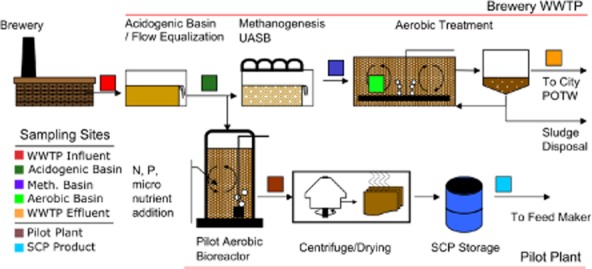
Overview of the study site showing the brewery treatment works (top) and the pilot-scale reactor (bottom). Coloured squares indicate sampling location and are used to illustrate figures throughout this paper.
Organisms responsible for SCP cell growth
The significantly depleted OTUs were found to be primarily saccharolytic fermentative anaerobes such as Prevotella (Fig. 4), whereas several separate types of diazotrophs (as well as some fermentative anaerobes) comprised much of the enriched sequences in the pilot bioreactor and SCP product. The presence of saccharolytic bacteria in aerobic conditions was unusual as Prevotella are strict anaerobes. In the final SCP product, even higher abundances of rhizospheric diazotroph sequences were seen than in the pilot bioreactor. We interpret the reduction of Prevotella and an increase in rhizospheric diazotrophs in SCP as indicating that the primary treatment effect responsible for SCP production was from rhizospheric diazotrophs and that the saccharolytic fermenter sequences, although common, may be from inactive cells washed in from the acidogenic basin. While diazotrophy exists across numerous phyla in nature, in this pilot bioreactor only diazotrophs from the Proteobacteria commonly associated with wastewater consortia were detected. The large relative abundance of diazotrophs seen in this study has a parallel in the treatment of certain high-strength wastewaters such as olive oil wastes and paper and pulp mill wastes where Proteobacteria rhizospheric diazotrophs were isolated and nifH genes and transcripts and nitrogenase activity detected (Papadelli et al., 1996; Clark et al., 1997; Gauthier et al., 2000; Bowers et al., 2008). These environments are often replete with simple carbon sources, yet are relatively limited in nitrogen and oxygen and require a diverse suite of microorganisms to generate fixed nitrogen to support the broader microbial consortia. In the pilot reactor of our studied system, aeration estimates produced DO levels 0.1–0.5 mg l−1 in the pilot reactor. There may be similarities with this environment and the rhizospheric habitat where such organisms autochthonously exist, particularly the abundance of simple organic acids from acidogenesis, which in the rhizosphere are released from plant roots to diazotrophs (Christiansen-Weniger et al., 1992).
However, we must caution that without further investigation, nitrogen fixation cannot conclusively be attributed as the primary biological advantage, especially since unlike conventional brewery wastewater treatment, nitrogen (as urea) was added to excess, and nitrogen fixation would be rapidly suppressed by the presence of fixed nitrogen. A major challenge in this study was monitoring the entire mixed liquor of such large bioreactors; therefore, it is possible that urea was not optimally dissolved, mixed or sorbed to microbial flocs. Another possible association is that the saccharolytic fermenting anaerobic bacteria in the acidogenic upstream treatment produced easily accessible organic acids that become the influent of the pilot bioreactor, where microbes adapted to have affinity for organic acids, and other pilot bioreactor conditions are then enriched. Under both scenarios, the wide presence of wastewater-specific diazotrophs found in this study indicates a potential target for future metabolic biotechnology exploitation. For example, because of the variable flow nature of batch fermentation in beer production, breweries have large flow equalization basins to regulate the supply of wastewater to continuous flow treatment stages. The conversion of such basins to an intentional acidogenic pretreatment stage upstream of the treatment bioreactors is one possibility to produce a constant supply of organic acids for uptake and conversion to SCP by aerobic or microaerophilic organic acid consuming microbes. Another potential avenue of research centres on the use of methanogenic UASBs as an inoculant source for these types of diazotrophs to seed their growth.
In summary, this study highlights the overall distinctiveness of each treatment stage within a single process WWTP, especially the occurrence of Prevotella and related saccharolytic fermenters in flow equalization basins and the role of wastewater-associated rhizospheric diazotrophs in high-strength wastewaters. This research indicated that rather than growing a single culture of these organisms, endogenous enrichment of rhizospheric diazotrophic bacteria in high-strength wastewater treatment systems can be used to produce SCP and should be studied as a way to deliberately manipulate microbial ecology for the production of a high protein content ingredient for aquaculture feed. However, before large-scale implementation of this technology can occur, several challenges remain to bring such a product to market such as regulatory and feed safety approval, scale up of production and large-scale animal feeding trials.
Experimental procedures
Project site description and sample handling
Figure 1 shows the brewery WWTP and pilot reactor research site used in this study (New Belgium Brewing, Fort Collins, CO, USA). The brewery treatment works itself consists of an unaerated influent flow equalization basin with acidogenic conditions, followed in series by a methanogenesis basin consisting of an UASB bioreactor. Next follows an aerobic treatment basin and clarifier for activated sludge growth and settling prior to discharge to the city publicly owned treatment works.
The pilot reactor received wastewater from the acidogenic basin to aerobically treat the wastewater at low mean cell residence time (< 8 days) with nutrient addition (N as urea, P as phosphoric acid and a customized micronutrient cocktail) to increase the protein concentration via growth of cell mass. The pilot bioreactor was initially fed and seeded from the methanogenesis UASB for about 1 month prior to the start of trial 1 before being switched to the acidogenic basin.
Two separate production trials using the pilot bioreactor were completed over a 450 day study period (trial 1: day 30–320, trial 2: day 340–440), and enough SCP was consistently produced at > 55% crude protein content for use in commercial feeding trials (Logan et al., 2011). Sampling locations and reactor conditions are shown in Fig. 1 (colored squares) and Table 1. Treatment plant influent samples (Fig. 1, red square) were taken from the waste influent receiving line upstream of the acidogenic basin. Acidogenic basin (dark green square) samples were taken from basin mixed liquor. Methanogenesis UASB (purple square) samples were taken from the basin outfall stream. Aerobic basin (light green square) samples were taken from basins directly, and clarifier wet well (orange square) was taken from the wet-well outfall channel. Pilot bioreactor samples (brown square) were taken from an effluent sampling port leading to the drying and centrifuge assembly. Finally, dried SCP product (blue square) was sampled directly from storage containers containing the most recent batch of manufacture. Liquid grab samples from basins and outfalls were collected over the study period in autoclaved 1-L serum bottles (rinsed with sample first) and returned to the lab and spun down at 10 000 relative centrifugal force (RCF) for 5 min. Grab samples and dry SCP product samples were stored at −20°C until DNA extraction with a Mo-Bio PowerSoil Kit (Carlsbad, CA, USA) per the manufacture's protocol with the exception of a 1 min bead beating step in a Biospec (Bartlesville, OK, USA) MiniBeadbeater-8 instead of vortex bead beating.
Wastewater chemical analysis
WWTP characterization (N, P, COD and VFA) analysis was conducted using commercial Hach TNT kits (Loveland, CO, USA). TSS was measured using filter paper and drying at 105°C (APHA, 1985) collected daily from January to March 2009. Pilot bioreactor MLSS (Royce 711, Australia) measurements and pH were collected daily from October 2008 to May 2009. Grab samples collected at the same time as nucleic acid samples were returned to the lab for organic acid analysis. An ion exclusion organic acid column Animex HPX-87H (Bio-Rad, Hercules, CA, USA) with a pre-column filter (Bio-Rad) was used with an Agilent 1100 HPLC. Samples were spun (10 000 RCF, 5 min) and filtered (0.45 μm) before use. Standards for formic, acetic, lactic, propionic, succinic, butyric, isobutyric, valeric, isovaleric, 2-methylbutyric and citric acid were run at three intervals in replicates of three with an injection volume of 50 um. Running buffer consisted of 0.04 N phosphoric acid, 0.60 ml min−1, 40°C, 35 min. Diode array detector signal frequency was 210 nm, reference 360 nm. Concentrations of all detected organic acids were summed to determine total organic acid content in samples.
Sampling and Sanger sequencing
Samples were collected over a period of 1 year from throughout the brewery treatment works as well as from the pilot plant and final dried product to track changes in community composition across treatment stages and in time. Sample DNA extraction, Sanger sequencing and full-length 16S small subunit (SSU) ribosomal RNA (rRNA) gene bioinformatic methods were adapted from Sahl and colleagues (2010) for 8F and 1492R bacterial primers and with details listed in the Supplemental Methods. Briefly, DNA was extracted from samples and amplified by polymerase chain reaction (PCR) followed by vector cloning of amplicons and Sanger sequencing of the T3/T7/515F reads, and finally contig formation to determine full-length 16S SSU rRNA gene reads. Reads were clustered at the 97% OTU level and unique representative sequences extracted. Representative sequences derived from Sanger sequencing OTUs of full-length 16S sequence clones from Proteobacteria were used with 64 full-length 16S sequences of BLAST closest matches and 37 SILVA reference sequences for phylogenetic reconstruction. These sequences were trimmed to a contiguous aligned [Nearest Alignment Space Termination (NAST)-based aligner of mothur (Schloss, 2009)] 1142 bp length and used with the pos_var_bac ‘0’ filter in phyml (Guindon et al., 2010) with default parameters and 100 bootstraps for detailed phylogenetic comparison.
Pyrosequencing and bioinformatics pipeline
Pyrosequencing PCR of the 16S SSU rRNA gene using the bacterial 27F and 338R primers with sequence barcoding (Hamady et al., 2008) adapted from Sahl and colleagues (2010) and processed using qiime (Caporaso et al., 2010), with details listed in the Supplemental Methods. Briefly, sequences were denoised filtered, and clustered using qiime, then aligned and classified using the NAST-based aligner and classifier of mothur (Schloss, 2009) trained on the customized SILVA 104 non-redundant database (80% confidence level cut-off). Note that taxonomic assignments in figures and text in this paper with underscores or number assignments at the end (e.g. firmicutes_bacilli or azonexus_2) indicate paraphyletic classification in the SILVA reference tree system.
Alpha and beta diversity analysis
qiime was used to compute alpha diversity estimates of the Chao1 and ACE metrics (Colwell, 2009). The CatchAll statistic (Bunge, 2011) was also used with default parameters, and the best model total species observed values were used. All alpha diversity estimators were computed with samples rarified up to the same sequencing depth with 50 replicates each. Coverage estimates were based on the percentage of observed OTUs to the Chao1 estimator as a low estimate and to the CatchAll as the high estimate using all sequences per sample for computation. Several metrics of beta diversity originally derived from classical macroecology [particularly plant communities (Sørensen, 1948; Whittaker, 1972)], such as the Sørensen (a qualitative sharing metric) and Whittaker's Index of Association (quantitative relative abundance sharing metric), were chosen to study the pilot plant system. These have been applied for the study of variations in bacterioplankton communities across distance in the North Pacific (Hewson et al., 2006) and could indicate temporal-spatial distance–decay relationships. Additionally, based on recent work on wastewater variation (Zhang et al., 2012), the unweighted UniFrac metric (Lozupone et al., 2011) was used to compare wastewater samples. qiime was used to calculate the beta diversity metrics of unweighted UniFrac (using a whole tree of the entire dataset), the complement of Whittaker's index of association (Legendre and Legendre, 1998) (using a custom python script rather than the qualitative Whittaker's found in qiime), and the qualitative Sørensen index of similarity. UPGMA clusters and principal coordinate analysis were determined using qiime scripts. Distance matrices from each metric were also jackknife subsampled to the smallest sequence count to examine the sensitivity of sequencing depth on clustering and principal coordinate analysis. Bi-plots of only the top 10 most commonly found taxonomic families were displayed.
Determining biotechnologically relevant OTUs
While it is useful to compare environments over time in a holistic manner with beta diversity metric analysis, we also wanted to identify the distribution of OTUs responsible for the community shift, especially the fraction that indicates SCP formation. To determine organisms that might be of biotechnological interest, two tests were used. The first was to test if OTUs are co-abundant (i.e. co-incident in time and relative abundance). This can highlight patterns of variation of consortia in response to natural wastewater source variations. Additionally, a significance test was used to determine OTUs that were differentially enriched in the mixed liquor as compared with the reactor influent.
To understand the co-abundance of OTUs in both the pilot plant and final SCP product, OTUs that were present in these two environments were used. OTUs were included if an OTU was present in these two environments (pilot bioreactor and final product) more than once and had more than two sequences associated with it. 319 out of 1604 OTUs passed this criteria. The Bray–Curtis pairwise distance metric (Legendre and Legendre, 1998) was computed for each OTU based on the abundances of each OTU across SCP and pilot bioreactor samples (n = 13).
Each OTU was then grouped into a functional category based on the metabolic profile of the nearest taxonomic representative found in Bergey's Index (Brenner et al., 2011) (characteristic of the entire known genus or family level) or related primary literature (of the genus level only). A conservative approach was taken where clades with significant known metabolic variation or poor classification (at the genus level) were labeled as ‘unknown or unclassified’.
OTUs were additionally compared by online metastats (http://metastats.cbcb.umd.edu/detection.html) (White et al., 2009), a program developed to examine differential abundance of elements associated between patients from a control and treatment category for medical microbiome studies. Abundance information from the series of acidogenic basin samples (representing the influent to the pilot plant) and the pilot plant mixed liquor samples were used as the pre- and post-treatment cases with a threshold p-value < 0.05 and a difference in average relative abundance of ± 1% absolute magnitude as being significantly ‘enriched’ or ‘depleted’ respectively.
A major concern in this work was the role of PCR and extraction biases in calculating co-abundance dissimilarity distances. This work did not seek and does not serve to capture the true compositional nature of the sample, but rather relative abundance data is used to understand patterns in OTU distributions undergoing the same DNA extraction, amplification and analysis conditions. Another concern was misidentification or the inclusion of spurious sequences leading to poor taxonomic identification. Therefore, no conclusions are made on any single OTU, and full-length 16S SSU Sanger-based gene sequencing was used to verify the findings of short sequencing reads.
Nucleotide accession numbers and online resources
A total of 809 Sanger sequences were deposited in GenBank under accession numbers JQ072092–JQ072899. Sanger sequences were named according to the location and date in which they were sampled [i.e. NBBEQMMYY_44; where NBB = New Belgium Brewing, EQ = flow equalization/acidogenic basin, ME = methanogenic UASB, PI = pilot plant, SP = SCP product, AB = aerobic basin, OT = wet well outfall, MMYY = (month/year) 44 = sequence number]. 454 pyrotags taken from quality score processing were submitted in FASTQ format to mg-rast (ID 4477674.3). Custom python scripts and interactive Krona pie charts (Ondov et al., 2011) used in this study can be obtained from http://inside.mines.edu/~jspear/resources.html.
Acknowledgments
Authors wish to thank Brandon Weaver and New Belgium Brewing, Fort Collins, CO, for facilities, space and process wastewater treatment support (http://www.newbelgium.com). Thanks to the Pace Lab at the University of Colorado, Boulder for assistance with Sanger sequencing. The authors would like to acknowledge J. M. R., R. C. E. and members of the GEM Lab for useful comments on the manuscript.
Conflict of interest
Andrew Logan and Seth Terry are both Vice-Presidents of Research of Nutrinsic, Corp.
Supporting Information
Fig. S1. Jackknife UPGMA node sensitivity analysis for Figure 2.
Fig. S2. Jackknife PCOA ellipsoid plots, subsampled to 800 sequences per sample with 100 jackknife replicates.
Fig. S3. OTUs in a pilot reactor and SCP product clustered over time.
Fig. S4. Breakdown of sequences in co-abundance and lifestyle analysis showing the distribution of enriched and depleted sequences across pilot bioreactor mixed liquor and final SCP product.
Fig. S5. Phylogenetic diversity of full-length Sanger sequences related to rhizospheric diazotrophs detected from the pilot bioreactor and final product.
Fig. S6. Phylogenetic tree and sequence abundance of depleted and enriched OTUs.
References
- APHA. Standard Methods for the Examination of Water and Wastewater. 16th edn. Washington, DC, USA: American Public Health Association; 1985. [Google Scholar]
- Ashida N, Ishii S, Hayano S, Tago K, Tsuji T, Yoshimura Y, et al. Isolation of functional single cells from environments using a micromanipulator: application to study denitrifying bacteria. Appl Microbiol Biotechnol. 2010;85:1211–1217. doi: 10.1007/s00253-009-2330-z. [DOI] [PubMed] [Google Scholar]
- Borole AP, Mielenz JR, Vishnivetskaya TA. Hamilton CY. Controlling accumulation of fermentation inhibitors in biorefinery recycle water using microbial fuel cells. Biotechnol Biofuels. 2009;2:7. doi: 10.1186/1754-6834-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers TH, Reid NM. Lloyd-Jones G. Composition of nifH in a wastewater treatment system reliant on N2 fixation. Appl Microbiol Biotechnol. 2008;79:811–818. doi: 10.1007/s00253-008-1486-2. [DOI] [PubMed] [Google Scholar]
- Brenner DJ, Krieg NR, Garrity GM, Staley JT, Boone DR, Vos P, et al. Bergey's Manual of Systematic Bacteriology. New York, USA: Springer; 2011. [Google Scholar]
- Bunge J. Estimating the number of species with catchall. Pac Symp Biocomput. 2005;2011:121–130. doi: 10.1142/9789814335058_0014. [DOI] [PubMed] [Google Scholar]
- de Cárcer DA, Ha PT, Jang JK. Chang IS. Microbial community differences between propionate-fed microbial fuel cell systems under open and closed circuit conditions. Appl Microbiol Biotechnol. 2011;89:605–612. doi: 10.1007/s00253-010-2903-x. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouari R, Le Paslier D, Dauga C, Daegelen P, Weissenbach J. Sghir A. Novel major bacterial candidate division within a municipal anaerobic sludge digester. Appl Environ Microbiol. 2005;71:2145–2153. doi: 10.1128/AEM.71.4.2145-2153.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen-Weniger C, Groneman AF. Veen JA. Associative N2 fixation and root exudation of organic acids from wheat cultivars of different aluminium tolerance. Plant Soil. 1992;139:167–174. [Google Scholar]
- Clark TA, Dare PH. Bruce ME. Nitrogen fixation in an aerated stabilization basin treating bleached kraft mill wastewater. Water Environ Res. 1997;69:1039–1046. [Google Scholar]
- Colwell RK. 2009. EstimateS: statistical estimation of species richness and shared species from samples. Version 8.2 User's Guide.
- Cressey D. Aquaculture: future fish. Nature. 2009;458:398–400. doi: 10.1038/458398a. [DOI] [PubMed] [Google Scholar]
- Croese E, Pereira MA, Euverink G-JW, Stams AJM. Geelhoed JS. Analysis of the microbial community of the biocathode of a hydrogen-producing microbial electrolysis cell. Appl Microbiol Biotechnol. 2011;92:1083–1093. doi: 10.1007/s00253-011-3583-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz EE, Stams AJM, Amils R. Sanz JL. Phenotypic properties and microbial diversity of methanogenic granules from a full-scale upflow anaerobic sludge bed reactor treating brewery wastewater. Appl Environ Microbiol. 2006;72:4942–4949. doi: 10.1128/AEM.02985-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sayed A-FM. Alternative dietary protein sources for farmed tilapia, Oreochromis spp. Aquaculture. 1999;179:149–168. [Google Scholar]
- FAO Fisheries Department. 2003. Review of the state of world aquaculture. FAO Fisheries Circular No. 886. Food and Agriculture Organization of the United Nations, Rome.
- Fernandez AS, Hashsham SA, Dollhopf SL, Raskin L, Glagoleva O, Dazzo FB, et al. Flexible community structure correlates with stable community function in methanogenic bioreactor communities perturbed by glucose. Appl Environ Microbiol. 2000;66:4058–4067. doi: 10.1128/aem.66.9.4058-4067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier F, Neufeld JD, Driscoll BT. Archibald FS. Coliform bacteria and nitrogen fixation in pulp and paper mill effluent treatment systems. Appl Environ Microbiol. 2000;66:5155–5160. doi: 10.1128/aem.66.12.5155-5160.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray N. Biology of Wastewater Treatment. 2nd edn. London, UK: Imperial College Press; 2004. [Google Scholar]
- Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W. Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Hamady M, Walker JJ, Harris JK, Gold NJ. Knight R. Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat Methods. 2008;5:235–237. doi: 10.1038/nmeth.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewson I, Steele JA, Capone DG. Fuhrman JA. Remarkable heterogeneity in meso- and bathypelagic bacterioplankton assemblage composition. Limnol Oceanogr. 2006;51:1274–1283. [Google Scholar]
- Hollister EB, Hammett AM, Holtzapple MT, Gentry TJ. Wilkinson HH. Microbial community composition and dynamics in a semi-industrial-scale facility operating under the MixAlco™ bioconversion platform. J Appl Microbiol. 2011;110:587–596. doi: 10.1111/j.1365-2672.2010.04919.x. [DOI] [PubMed] [Google Scholar]
- Hough J. The Biotechnology of Malting and Brewing. New York, USA: Cambridge University Press; 1985. [Google Scholar]
- Hugenholtz P, Tyson GW, Webb RI, Wagner AM. Blackall LL. Investigation of candidate division TM7, a recently recognized major lineage of the domain bacteria with no known pure-culture representatives. Appl Environ Microbiol. 2001;67:411–419. doi: 10.1128/AEM.67.1.411-419.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huige N. Brewery by-products and effluents. In: Priest F, Stewart G, et al., editors. Handbook of Brewing. Boca Raton, USA: CRC/Taylor & Francis; 2006. pp. 655–714. [Google Scholar]
- Kaksonen AH, Plumb JJ, Franzmann PD. Puhakka JA. Simple organic electron donors support diverse sulfate-reducing communities in fluidized-bed reactors treating acidic metal- and sulfate-containing wastewater. FEMS Microbiol Ecol. 2004;47:279–289. doi: 10.1016/S0168-6496(03)00284-8. [DOI] [PubMed] [Google Scholar]
- Knief C, Delmotte N, Chaffron S, Stark M, Innerebner G, Wassmann R, et al. Metaproteogenomic analysis of microbial communities in the phyllosphere and rhizosphere of rice. ISME J. 2012;6:1378–1390. doi: 10.1038/ismej.2011.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TK, Van Doan T, Yoo K, Choi S, Kim C. Park J. Discovery of commonly existing anode biofilm microbes in two different wastewater treatment MFCs using FLX titanium pyrosequencing. Appl Microbiol Biotechnol. 2010;87:2335–2343. doi: 10.1007/s00253-010-2680-6. [DOI] [PubMed] [Google Scholar]
- Legendre P. Legendre L. Numerical Ecology 2nd English Ed. New York, USA: Elsevier; 1998. [Google Scholar]
- Li L, Kato C. Horikoshi K. Bacterial diversity in deep-sea sediments from different depths. Biodivers Conserv. 1999;8:659–677. [Google Scholar]
- Lin S-Y, Liu Y-C, Hameed A, Hsu Y-H, Lai W-A, Shen F-T. Young C-C. Azospirillum fermentarium sp. nov., a nitrogen-fixing species isolated from a fermenter. Int J Syst Evol Microbiol. 2013;63:3762–3768. doi: 10.1099/ijs.0.050872-0. [DOI] [PubMed] [Google Scholar]
- Liu W-T, Chan O-C. Fang HHP. Characterization of microbial community in granular sludge treating brewery wastewater. Water Res. 2002;36:1767–1775. doi: 10.1016/s0043-1354(01)00377-3. [DOI] [PubMed] [Google Scholar]
- Logan A, Lawrence A, Dominy W. Tacon A. Bacterial single cell protein produced from brewery wastes as a protein source in white shrimp diets. Aquafeed Spring. 2011;2011:7–12. [Google Scholar]
- Lovell T. Nutrition and Feeding of Fish. New York, USA: Chapman & Hall; 1989. [Google Scholar]
- Lozupone C, Lladser ME, Knights D, Stombaugh J. Knight R. UniFrac: an effective distance metric for microbial community comparison. ISME J. 2011;5:169–172. doi: 10.1038/ismej.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manz W, Wagner M, Amann R. Schleifer K-H. In situ characterization of the microbial consortia active in two wastewater treatment plants. Water Res. 1994;28:1715–1723. [Google Scholar]
- Ondov BD, Bergman NH. Phillippy AM. Interactive metagenomic visualization in a Web browser. BMC Bioinformatics. 2011;12:385. doi: 10.1186/1471-2105-12-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadelli M, Roussis A, Papadopoulou K, Venieraki A, Chatzipavlidis I, Katinakis P. Ballis K. Biochemical and molecular characterization of an Azotobacter vinelandii strain with respect to its ability to grow and fix nitrogen in olive mill wastewater. Int Biodeterior Biodegradation. 1996;38:179–181. [Google Scholar]
- Quan Z-X, Im W-T. Lee S-T. Azonexus caeni sp. nov., a denitrifying bacterium isolated from sludge of a wastewater treatment plant. Int J Syst Evol Microbiol. 2006;56:1043–1046. doi: 10.1099/ijs.0.64019-0. [DOI] [PubMed] [Google Scholar]
- Riviere D, Desvignes V, Pelletier E, Chaussonnerie S, Guermazi S, Weissenbach J, et al. Towards the definition of a core of microorganisms involved in anaerobic digestion of sludge. ISME J. 2009;3:700–714. doi: 10.1038/ismej.2009.2. [DOI] [PubMed] [Google Scholar]
- Sahl JW, Fairfield N, Harris JK, Wettergreen D, Stone WC. Spear JR. Novel microbial diversity retrieved by autonomous robotic exploration of the world's deepest vertical phreatic sinkhole. Astrobiology. 2010;10:201–213. doi: 10.1089/ast.2009.0378. [DOI] [PubMed] [Google Scholar]
- Schloss PD. A high-throughput DNA sequence aligner for microbial ecology studies. PLoS ONE. 2009;4:e8230. doi: 10.1371/journal.pone.0008230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi Y, Kamagata Y, Nakamura K, Ohashi A. Harada H. Fluorescence in situ hybridization using 16S rRNA-targeted oligonucleotides reveals localization of methanogens and selected uncultured bacteria in mesophilic and thermophilic sludge granules. Appl Environ Microbiol. 1999;65:1280–1288. doi: 10.1128/aem.65.3.1280-1288.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seviour R. Nielsen PH. Microbial Ecology of Activated Sludge [New ed.] London, UK: IWA Publishing; 2010. [Google Scholar]
- Sørensen T. A method of establishing groups of equal amplitude in plant sociology based on similarity of species and its application to analyses of the vegetation on Danish commons. Biol Skr. 1948;5:1–34. [Google Scholar]
- Sun Y, Wei J, Liang P. Huang X. Electricity generation and microbial community changes in microbial fuel cells packed with different anodic materials. Bioresour Technol. 2011;102:10886–10891. doi: 10.1016/j.biortech.2011.09.038. [DOI] [PubMed] [Google Scholar]
- Tacon A, Metian M. Hasan M. 2009. , and Feed ingredients and fertilizers for farmed aquatic animals: sources and composition. UN FAO, Technical Paper 540.
- Tacon AGJ. Activated sewage sludge, a potential animal foodstuff II. Nutritional characteristics. Agric Environ. 1979;4:271–279. [Google Scholar]
- Vriens L, Nihoul R. Verachtert H. Activated sludges as animal feed: a review. Biol Waste. 1989;27:161–207. [Google Scholar]
- Wells GF, Park H-D, Eggleston B, Francis CA. Criddle CS. Fine-scale bacterial community dynamics and the taxa–time relationship within a full-scale activated sludge bioreactor. Water Res. 2011;45:5476–5488. doi: 10.1016/j.watres.2011.08.006. [DOI] [PubMed] [Google Scholar]
- Werner JJ, Knights D, Garcia ML, Scalfone NB, Smith S, Yarasheski K, et al. Bacterial community structures are unique and resilient in full-scale bioenergy systems. PNAS. 2011;108:4158–4163. doi: 10.1073/pnas.1015676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JR, Nagarajan N. Pop M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput Biol. 2009;5:e1000352. doi: 10.1371/journal.pcbi.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker RH. Evolution and measurement of species diversity. Taxon. 1972;21:213–251. [Google Scholar]
- Worm B, Barbier EB, Beaumont N, Duffy JE, Folke C, Halpern BS, et al. Impacts of biodiversity loss on ocean ecosystem services. Science. 2006;314:787–790. doi: 10.1126/science.1132294. [DOI] [PubMed] [Google Scholar]
- Zhang T, Shao M-F. Ye L. 454 pyrosequencing reveals bacterial diversity of activated sludge from 14 sewage treatment plants. ISME J. 2012;6:1137–1147. doi: 10.1038/ismej.2011.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Jackknife UPGMA node sensitivity analysis for Figure 2.
Fig. S2. Jackknife PCOA ellipsoid plots, subsampled to 800 sequences per sample with 100 jackknife replicates.
Fig. S3. OTUs in a pilot reactor and SCP product clustered over time.
Fig. S4. Breakdown of sequences in co-abundance and lifestyle analysis showing the distribution of enriched and depleted sequences across pilot bioreactor mixed liquor and final SCP product.
Fig. S5. Phylogenetic diversity of full-length Sanger sequences related to rhizospheric diazotrophs detected from the pilot bioreactor and final product.
Fig. S6. Phylogenetic tree and sequence abundance of depleted and enriched OTUs.