

Abstract
The fitness of sensitive and resistant Pseudomonas aeruginosa in different aquatic environments depends on genetic capacities and transcriptional regulation. Therefore, an antibiotic-sensitive isolate PA30 and a multi-resistant isolate PA49 originating from waste waters were compared via whole genome and transcriptome Illumina sequencing after exposure to municipal waste water and tap water. A number of different genomic islands (e.g. PAGIs, PAPIs) were identified in the two environmental isolates beside the highly conserved core genome. Exposure to tap water and waste water exhibited similar transcriptional impacts on several gene clusters (antibiotic and metal resistance, genetic mobile elements, efflux pumps) in both environmental P. aeruginosa isolates. The MexCD-OprJ efflux pump was overexpressed in PA49 in response to waste water. The expression of resistance genes, genetic mobile elements in PA49 was independent from the water matrix. Consistently, the antibiotic sensitive strain PA30 did not show any difference in expression of the intrinsic resistance determinants and genetic mobile elements. Thus, the exposure of both isolates to polluted waste water and oligotrophic tap water resulted in similar expression profiles of mentioned genes. However, changes in environmental milieus resulted in rather unspecific transcriptional responses than selected and stimuli-specific gene regulation.
Introduction
The increasing numbers of infections by multi-resistant bacteria turn out to be a great threat to our daily life. Bacteria develop resistance against antibiotics used in human health care, agriculture and animal husbandry or against pollutants from industry by accumulating genetic adaptations or acquisition of mobile genetic elements via horizontal gene transfer. To overcome multi-drug resistance, we have to undergo a thorough study to unravel how bacteria adapt to different habitats, to finally discover novel strategies to handle such infections.
One of the most prominent bacterial pathogens that is infamous for its high potential to develop multi-drug resistance is Pseudomonas aeruginosa, a Gram-negative, ubiquitous opportunistic bacterium that can cause acute and chronic infections especially in patients in intensive care or suffering from predisposing conditions like cystic fibrosis. The rate of infections in human body differs according to the site of infection as 2% on skins, 3.3% on nasal mucosa, 6.6% for the throat, 24% for fecal samples (Morrison and Wenzel, 1984). Pseudomonas aeruginosa is found in hospital waste water, respiratory equipment, solutions, medicines, disinfectants, sinks, mops, food mixtures and vegetables (Trautmann et al., 2005). An important characteristic of P. aeruginosa is its ability to form biofilms as an adaptation to adverse environmental conditions. The microbes attach to the surface and embed themselves in extracellular polymeric substances such as proteins (e.g. extracellular enzymes), lipids and nucleic acids (Flemming and Wingender, 2001), usually leading to increased resistance towards harsh conditions such as temperature changes, pH fluctuations, presence of antibiotics (Kwon and Lu, 2006) and immune cells of humans (Donlan and Costerton, 2002).
Sequencing of several P. aeruginosa strains genomes revealed that a large fraction (around 10%) of the genome is dedicated to gene regulation, which is consistent with its high versatility (Stover et al., 2012; Mathee et al., 2008). This high versatility enables evolutionary adaptations and facilitates the bacterium to colonize vigorous and diverse ecological niches. The core genome is usually highly conserved between different Pseudomonas strains (Mathee et al., 2008; Klockgether et al., 2011).
It has a disparate variety of metabolism; it can degrade very distinct compounds such as alcohols, fatty acids, sugars, di- and tri-carboxylic acids, aromatics, amines and amino acids, which can be used up as sources of carbon. Pseudomonas aeruginosa has both aerobic and anaerobic metabolism. It is capable of anaerobic metabolism by converting nitrate to nitrite (Schreiber et al., 2007). Additionally, the genome harbours a huge repertoire of enzymes and efflux pumps that contribute to a high intrinsic resistance towards different classes of antibiotics. Additional resistance can easily develop by mutation or horizontal gene transfer, rendering P. aeruginosa a common cause of multi-drug resistant infections (Breidenstein et al., 2011). Regular use of high amounts of antibiotics in hospitals and other practices were assumed to be the sources of origin of antibiotics in the waste water systems and responsible for supporting emergence of multi-resistant bacteria (Rizzo et al., 2013). These resistances may not only develop from chromosomally encoded genes but also from mobile genetic elements like plasmids or integrons (Merlin et al., 2011). Not only waste water systems contribute to the development of resistance in bacteria, but also pollutants from industries and agricultural activities where the antibiotics and pollutants are directly released into the environmental water like rivers and lakes, creating selective pressure on these bacteria and making them evolve as resistance strains. The sensitive strains accept the resistant genes from these resistant donors and propagate as resistant strains. The concentrations of antibiotics in waste water might not be high enough to stimulate inhibitory effects but stimulate stress response mechanisms, which contribute to horizontal gene transfer and relevant transcriptional activities. It has also been proven by mutant investigations that sub-inhibitory concentrations of antibiotics can drive the evolution of antimicrobial resistance (Pedró et al., 1996). It all depends on the substance, concentration and strain present in the waste water systems. There is no final suggestion about long terms effects of sub-inhibitory concentration antibiotics and other micro-pollutants. It is commonly accepted that beside the linkage between antibiotics and antibiotic resistance, co-selection and the presence of heavy metal ions in the environments contributes to increasing resistance mechanisms due to the localization of resistance genes in close neighborhood on genetic mobile elements (Seiler and Berendonk, 2012).
Beside antibiotic and heavy metal stress, starvation is another widespread adverse stimulus present in many aquatic environments where P. aeruginosa is found in nature (Bernier et al., 2013). Tap water represents an oligotrophic matrix with very low organic matter, and P. aeruginosa has recently been shown to persist and proliferate as biofilms in municipal drinking water distribution systems (Wang et al., 2012). The molecular responses of P. aeruginosa strains to starvation stress in tap water are so far unknown. In this study, we compared the transcriptional response of an antibiotic sensitive and a multi-resistant P. aeruginosa waste water isolate cultivated in municipal waste water and tap water focusing on regulatory mechanisms that could promote the development of antibiotic resistance.
Results and discussion
Bacteria have developed highly orchestrated processes to respond to environmental stresses, which when elicited alter the cellular physiology in a manner that enhances the organism's survival and its ability to cause disease. This study focused on the behaviour of two natural isolates of P. aeruginosa as a Gram-negative bacterium exposed to municipal waste water containing complex mixtures of xenobiotics and, as a second scenario, exposed to tap water simulating nutrient limitation (starvation). Since bacteria have to deal with unfavourable growth conditions in addition to diverse stresses in nature, bacteria that reached the stationary growth phase were used to imitate this environment and then exposed to stress. During transition from exponential growth to stationary phase, growth becomes unbalanced especially in laboratory systems, i.e. the synthesis of different macromolecules and cell constituents do not slow down synchronically (Nyström, 2004). Thus, stationary phase is an operational definition and does not describe a specific and fixed physiological state or response of the bacteria. It is more or less a change in physiology due to, e.g. phosphate limitation or accumulation of toxic waste products. Beside the changes in morphologies of bacteria, the gene expression pattern could be altered in stationary phase. In consequence, transcriptome analyses were run with ribonucleic acid (RNA) extracted from early stationary growth phase. In the present study, two different P. aeruginosa isolates were exposed to water matrices containing quite different compositions. Waste water from the influent of a municipal waste water treatment plants (WWTP) is composed of complex mixtures of xenobiotics like antibiotics, other pharmaceuticals, biocide etc., whereas tap water, in opposite, contains very low level of organic matter (including xenobiotics) as a result of the intensive drinking water conditioning processes at waterworks. We analysed the transcriptional responses from two P. aeruginosa isolates: the antibiotic sensitive strain PA30 and the multi-resistant strain PA49. Both P. aeruginosa strains did neither show any differences in growth in diluted brain heart infusion (BHI) or BM2 broth nor in yields of extracted total RNA after exposure in tap water or waste water.
Genome analyses
Large fractions of the P. aeruginosa genome belong to the highly conserved core genome containing only few highly variable genes (Dötsch et al., 2010), while most of the genetic variation between species is restricted to the so-called accessory genome organized in various regions of genomic plasticity (RGPs) (Mathee et al., 2008). Most of these RGPs represent mobile elements originating from horizontal gene transfer and include transposons, phages, plasmids and genomic islands, which are a major source of resistance genes (Battle et al., 2009; Kung et al., 2010; Klockgether et al., 2011). The large amount of homology between the core regions of different P. aeruginosa strains enabled us to employ the genomic sequences of strain PAO1 chromosome and a selection of genomic islands as a blueprint for de novo assembly. The resulting draft genomes consist of 207 contigs with a total length of 6.77 Mb for the strain PA30 and 269 contigs with 7.01 Mb for strain PA49 respectively (Table S1).
An alignment of the contigs with P. aeruginosa reference strain PAO1 showed a huge overlap of 95.8% for PA30 and 96.4% for PA49 (Fig. 1; Table S2), reflecting the highly conserved character of the P. aeruginosa core genome. Comparing the contigs with the genome islands that were used in the alignment process revealed a distinct pattern of accessory genomic elements for the two strains covering large fractions of the various genomic islands (Fig. 1; Table S2). Strain PA30 contains full length or near-full length sequences of PAGI-5 to PAGI-11, larger fractions of PAGI-1 and PAGI-2 and several regions of PAGI-3, whereas only insignificant fractions of the remaining genomic elements occurred. In case of the multi-resistant strain PA49, all genomic islands except the smaller PAGI-9 to PAGI-11 were covered at varying percentages (Table S2). The scattered distribution of regions within the genomic islands that actually showed homology with PA49 contigs may be partially explained by incomplete sequence assembly. However, the fact that both the contigs and the genomic island reference sequences contained a large amount of non-overlapping regions (data not shown) suggests that at least in some cases, the accessory elements found in PA30 and PA49 only partially contain sequences that are homologous to the genomic islands and also include a substantial amount of new and previously uncharacterized sequences.
Figure 1.
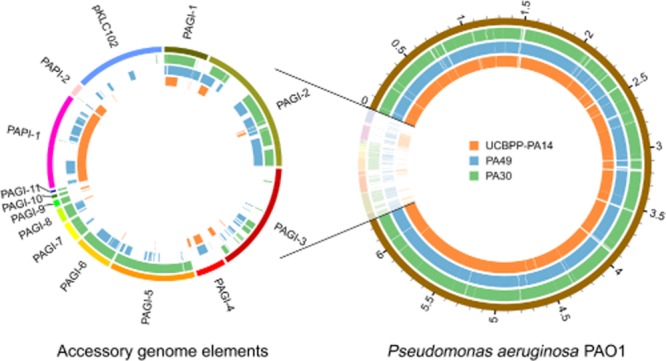
Coverage of genomic reference sequences by the newly assembled genomes. The reference sequences that were used for the hybrid de novo assembly are displayed on the outer circles (diagram to the right) with an additional display of the accessory elements alone (missing the PAO1 chromosome, diagram to the left). Regions that are covered by the contigs of strains PA30 and PA49 or overlap with the chromosome of another reference strain PA14 are highlighted by coloured areas in the concentric inner circles as specified by the color legend.
Since the genomes of PA30 and PA49 were nearly completely covered, the sequence types according to the multi-locus sequence typing (MLST) scheme by Curran and colleagues (2004) could be determined, enabling a phylogenetic classification of the two strains. As demonstrated by the phylogenetic tree (Fig. 2), PA30 and PA49 are members of the lineage that includes the type strain PAO1 and some recently sequenced strains. The question about their origin is open, since the sampling sites were influenced by hospital and housing waste waters. Selective pressures like the presence of antibiotics and other environmental criteria are a general concept that refers to many factors that create an evolutionary landscape and allow organisms with novel mutations or newly acquired characteristics to survive and proliferate (Kümmerer, 2009). There is evidence that even in sub-inhibitory concentration, antibiotics or other xenobiotics may still exert their impact on microbial communities (Goh et al., 2002; Davies et al., 2006). The direct link between antibiotics or heavy metal ions and development/selection of resistance mechanisms is obvious and manifold described (Seiler and Berendonk, 2012; Rizzo et al., 2013). The impact of other harsh environmental conditions on resistance activities, recombination and horizontal gene transfer remains to be determined. Long-term effects of environmental exposure to low levels of antibiotics like these present in surface waters or in the outflow of sewage plants are also still unknown.
Figure 2.

Multi-locus sequence typing phylogenetic tree. Phylogenetic relations of the two newly sequenced strains PA30 and PA49 (bold) with eight previously published genomes of P. aeruginosa. The phylogenetic tree is based on seven genes that are commonly used for MLST scheme by Curran and colleagues (2004).
The prediction of protein coding sequences (CDS) yielded for both strains a comparatively large number of genes, about 99% of which were successfully annotated according to their best-hit BLAST alignments (Table S1). The vast majority of the predicted genes were found in both strains and also in the PAO1 reference genome (5262 genes), representing the conserved core genome of P. aeruginosa. Regarding the development of multi-drug resistance, P. aeruginosa is known for its high intrinsic resistance that is caused by a combination of low membrane permeability, efflux pumps and resistance genes encoded in the core genome (Nikaido, 2001; Schweizer, 2003), together with the potential to develop high-level resistance by accumulation of small mutations (Fajardo et al., 2008; Dötsch et al., 2009; Martinez et al., 2009; Alvarez-Ortega et al., 2010; Breidenstein et al., 2011; Bruchmann et al., 2013). However, the most obvious cause of multi-drug resistance is the acquisition of resistance genes by horizontal gene transfer (Davies and Davies, 2010). Therefore, we performed a blast search of the predicted genes of the two strains in the Comprehensive Antibiotic Resistance Database (CARD) (McArthur et al., 2013) and scanned both genomes for genetic variations of intrinsic resistance determinants. In a previous work, strain PA49 was found to be resistant towards the antibiotics gentamicin (GM), amikacin (AN), azlocillin (AZ), ceftazidime (CAZ), piperacillin/tazobactam (PT), ciprofloxacin (CIP) and imipenem (IPM) (Schwartz et al., 2006). Searching its genome for resistance determinants revealed the presence of one aminoglycoside acetyltransferase of the AAC(6’)-type, two aminoglycoside adenylyltransferases of type ANT(2'’) and ANT(3'’) and one VIM metallo-beta-lactamase (Table 1). Two additional genes were annotated as beta-lactamases in PA49 only by the BLAST search in the National Center for Biotechnology Information (NCBI) non-redundant (nr) protein database but not found in the CARD database (Fig. 3A). Taken together, these genes confer resistance towards a wide range of aminoglycosides and beta-lactam antibiotics, explaining the resistance towards GM, AN, AZ, CAZ and PT. Fluoroquinolones like CIP target the DNA gyrase and Topoisomerase IV enzyme complexes, and high-level resistance towards these antibiotics is often caused by sequence variations of the two subunits GyrA (gyrase) and ParC (topoisomerase) (Ruiz, 2003) and indeed, both proteins contained a single amino acid exchange in the resistance determining region (Table 1). These two mutations represent the most common type of variations found in fluoroquinolone resistant isolates of P. aeruginosa and have recently been shown to be sufficient for the development of high-level resistance towards CIP (Bruchmann et al., 2013). Finally, a frameshift mutation in the outer membrane porin OprD was found that is likely to cause misfolding or decreased functionality of the protein. Defective mutations of OprD are known to cause resistance towards carbapenems including IPM in combination with intrinsic beta-lactamases and efflux pumps (Pirnay et al., 2002). Of note, the strain PA30 that is sensitive towards all these antibiotics did not contain any known horizontally acquired resistance genes and harboured wild-type alleles of the target genes gyrA, parC and oprD (Table 1). In summary, these results provide a comprehensive explanation for the resistance phenotype covering all the antibiotics that were tested, since all resistance determining genes and alleles (besides the ones intrinsic to P. aeruginosa) were exclusively found in PA49 (Fig. 3A; Table 2).
Table 1.
Comparison of antibiotic resistance determinants found in the genomes of PA30 and PA49. Identifiers state PAO1 gene IDs or RefSeq Accession where applicable. Genotypes refer to presence or absence or genes or specific alleles with ‘wt’ indicating the genotype found in the reference strains PAO1
| Gene ID/accession | Gene name | Resistance type | PA30 genotype | PA49 genotype | Affected antibioticsa |
|---|---|---|---|---|---|
| gi|32470063 | aac(6’)-Ib | AAC(6’) | – | Present | Aminoglycosides (GM, AN) |
| gi|378773997 | aadB | ANT(2'’) | – | Present | Aminoglycosides (GM, AN) |
| gi|489251134 | blaVIM-2 | VIM | – | Present | Beta-lactams (AZ, CAZ, PT) |
| gi|88853419 | aadA10 | ANT(3'’) | – | Present | Aminoglycosides |
| gi|489211498 | – | AmpC | – | Present | Beta-lactams (AZ, CAZ, PT) |
| gi|489217979 | – | Metallo-beta-lactamase | – | Present | Beta-lactams (AZ, CAZ, PT) |
| gi|407937916 | – | Metallo-beta-lactamase | Present | – | Beta-lactams (AZ, CAZ, PT) |
| PA3168 | gyrA | Target modification | wt | T83I | Fluoroquinolones (CIP) |
| PA4964 | parC | Target modification | wt | S87L | Fluoroquinolones (CIP) |
| PA0958 | oprD | Decreased permeability | Multiple SNPs | Multiple SNPs, frameshift | Carbapenems (IPM) |
| insertion CC at position 279 |
Abbreviations indicate specific antibiotics. AN, amikacin; PT, Piperacillin + Tazobactam.
AZ, azlocillin; CAZ, ceftazidime; CIP, ciprofloxacin; GM, gentamicin; IPM, imipenem.
Figure 3.
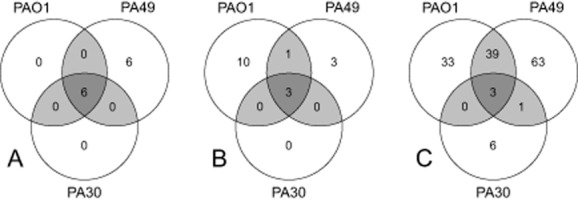
Comparison of specific gene classes found in the genomes of PA30 and PA49 with type strain PAO1. The circles of this Venn Diagram contain the numbers of genes that were predicted from the genome sequence of the two newly sequenced strains, in comparison with the known genes of the PAO1 reference genome. PAO1 genome annotation was taken from www.pseudomonas.com (Winsor et al., 2011).A. Genes involved in antibiotic resistance (excluding efflux pumps).B. Genes involved in metal ion resistance.C. Gene involved in genetic mobility – transposases, integrases, recombinases and conjugation-related proteins.
Table 2.
Expression of genes related to antibiotic resistance (excluding efflux pumps)
| PA30 | PA49 | ||||
|---|---|---|---|---|---|
| Gene ID/accession | Product | T | W | T | W |
| gi|489182848 | Acriflavin resistance protein | n.p.a | n.p. | 524 | 416 |
| gi|496684660 | Bleomycin resistance protein | n.p. | n.p. | 1808 | 1737 |
| gi|489211498 | Beta-lactamase | n.p. | n.p. | 1028 | 435 |
| gi|489251134 | VIM-1 protein | n.p. | n.p. | 35 957 | 39 477 |
| gi|32470063 | Hypothetical protein | n.p. | n.p. | 50 184 | 48 460 |
| gi|88853419 | Aminoglycoside-modifying enzyme | n.p. | n.p. | 4358 | 3280 |
| gi|378773997 | 2.-aminoglycoside nucleotidyltransferase | n.p. | n.p. | 7275 | 5761 |
| gi|496684693 | Glyoxalase/bleomycin resistance protein/dioxygenase | n.p. | n.p. | 362 | 1547 |
| gi|160901172 | Acriflavine resistance protein B | n.p. | n.p. | 468 | 408 |
| gi|489217979 | Beta-lactamase | n.p. | n.p. | 109 | 64 |
| PA0706 | Chloramphenicol acetyltransferase | 399 | 304 | 48 | 73 |
| gi|489208693 | Fusaric acid resistance protein | 25 | 18 | 30 | 18 |
| gi|116049746 | Beta-lactamase | 107 | 64 | 272 | 131 |
| PA1129 | Probable fosfomycin resistance protein | 82 | 54 | 33 | 11 |
| PA5514 | Probable beta-lactamase | 258 | 285 | 191 | 422 |
| PA1959 | Bacitracin resistance protein | 385 | 925 | 110 | 219 |
| PA4110 | Beta-lactamase precursor AmpC | 694 | 382 | 101 | 74 |
| PA4119 | Aminoglycoside 3'-phosphotransferase type IIb | 113 | 177 | 37 | 40 |
| PA5159 | Multi-drug resistance protein | 79 | 66 | 65 | 120 |
| PA1858 | Streptomycin 3'’-phosphotransferase | 145 | 131 | 31 | 52 |
| gi|407937916 | beta-lactamase domain-containing protein | 233 | 201 | n.p. | n.p. |
n.p. – gene sequence is not present in this strain.
Identifiers state PAO1 gene IDs or RefSeq accession where applicable. Transcriptional activity in tap water (T) and waste water (W) was normalized for the two strains independently and is shown as normalized pseudocounts.
Both PA30 and PA49 harbour a set of genes involved in metal ion resistance that are not found in the P. aeruginosa core genome. Strain PA30 contains several genes encoding resistance genes related to copper, mercury and arsenic/arsenate, while most of the genes could not be found in PA49 (Fig. 3B; Table 3).
Table 3.
Expression of genes related to metal tolerance
| PA30 | PA49 | ||||
|---|---|---|---|---|---|
| Gene ID/accession | Product | T | W | T | W |
| gi|489181230 | Mercuric reductase | n.p.a | n.p. | 695 | 892 |
| gi|134047226 | MerP | n.p. | n.p. | 190 | 376 |
| gi|410691713 | Hg(II)-responsive transcriptional regulator MerR | n.p. | n.p. | 626 | 2339 |
| gi|498493737 | Mercuric reductase | 515 | 520 | n.p. | n.p. |
| gi|134047116 | MerT | 65 | 53 | 132 | 339 |
| gi|152989484 | Mercuric resistance operon regulatory protein | 239 | 310 | n.p. | n.p. |
| PA2065 | Copper resistance protein A precursor | 253 | 95 | 654 | 20 |
| PA2064 | Copper resistance protein B precursor | 114 | 59 | 238 | 12 |
| PA0950 | Probable arsenate reductase | 472 | 615 | 268 | 451 |
| gi|498490899 | Arsenical resistance protein ArsH | 102 | 139 | n.p. | n.p. |
| gi|493249841 | Arsenate reductase | 75 | 97 | n.p. | n.p. |
| gi|493264869 | Arsenic transporter | 119 | 196 | n.p. | n.p. |
| gi|493249844 | ArsR family transcriptional regulator | 486 | 235 | n.p. | n.p. |
| gi|495544062 | Copper oxidase | 1420 | 203 | n.p. | n.p. |
| gi|493265806 | Copper resistance protein B | 612 | 100 | n.p. | n.p. |
| gi|386717894 | Copper resistance protein C | 358 | 141 | n.p. | n.p. |
| gi|489188126 | Copper resistance protein CopD | 717 | 392 | n.p. | n.p. |
n.p. – gene sequence is not present in this strain.
Identifiers state PAO1 gene IDs or RefSeq accession where applicable. Transcriptional activity in tap water (T) and waste water (W) was normalized for the two strains independently and is shown as normalized pseudocounts.
The extent of the accessory genomes found in PA30 and PA49 point towards a high incidence of horizontal gene transfer in the evolutionary history of these strains. Therefore, we also searched the annotated genomes for genes associated with genomic mobility, mostly classified as recombinases, transposases, integrases or conjugative elements. Since mobile genetic elements per definition belong to the accessory genome, it is not surprising that nearly all genes associated with genetic mobility that were found in PA30 and PA49 are not present in the genome of PAO1 (Fig. 3C). Both strains contain a large number of mobility genes (75 in PA30, 103 in PA49) (Table 4).
Table 4.
Expression of genes related to genetic mobility and horizontal gene transfer
| PA30 | PA49 | ||||
|---|---|---|---|---|---|
| Gene ID/accession | Product | T | W | T | W |
| gi|386063806 | Conjugal transfer protein TrbJ | n.p.a | n.p. | 4 | 7 |
| gi|446855879 | Conjugal transfer protein TrbE | n.p. | n.p. | 58 | 33 |
| gi|446855879 | Conjugal transfer protein TrbE | n.p. | n.p. | 26 | 21 |
| gi|386063808 | Conjugal transfer protein TrbC | n.p. | n.p. | 10 | 16 |
| gi|51492563 | TnpA | n.p. | n.p. | 481 | 609 |
| gi|66045904 | TnpR resolvase | n.p. | n.p. | 2370 | 3179 |
| gi|19352419 | Putative transposase | n.p. | n.p. | 3570 | 2090 |
| gi|498491672 | Excisionase family DNA binding domain-containing protein | n.p. | n.p. | 263 | 432 |
| gi|496684679 | Conjugal transfer coupling protein TraG | n.p. | n.p. | 22 | 8 |
| gi|190573650 | Transposase | n.p. | n.p. | 218 | 206 |
| gi|190572552 | Transposase IS3 | n.p. | n.p. | 189 | 156 |
| gi|497209012 | Transposase | n.p. | n.p. | 0 | 0 |
| gi|495918616 | Integrase | n.p. | n.p. | 187 | 126 |
| gi|37958842 | Putative transposase | n.p. | n.p. | 202 | 452 |
| gi|386063806 | Conjugal transfer protein TrbJ | n.p. | n.p. | 29 | 5 |
| gi|446923490 | Conjugal transfer protein TrbL | n.p. | n.p. | 66 | 40 |
| gi|410690825 | Transposase | n.p. | n.p. | 837 | 2156 |
| gi|319765135 | Transposase TniA | n.p. | n.p. | 874 | 1657 |
| gi|491446119 | TniQ | n.p. | n.p. | 448 | 546 |
| gi|490385992 | Transposase | n.p. | n.p. | 26 | 30 |
| gi|497303964 | Integrating conjugative element protein | n.p. | n.p. | 4 | 6 |
| gi|497081867 | Conjugative transfer region protein | n.p. | n.p. | 2 | 2 |
| gi|497303936 | Integrating conjugative element protein | n.p. | n.p. | 0 | 0 |
| gi|497081861 | Integrating conjugative element protein | n.p. | n.p. | 13 | 11 |
| gi|493535069 | Integrating conjugative element protein. PFL_4709 family | n.p. | n.p. | 19 | 19 |
| gi|497081686 | Integrase family protein | n.p. | n.p. | 223 | 259 |
| gi|133756449 | TniA | n.p. | n.p. | 498 | 866 |
| gi|289064112 | TniQ transposition protein | n.p. | n.p. | 57 | 45 |
| gi|410609201 | Transposase | n.p. | n.p. | 2789 | 2351 |
| gi|446985433 | Integrase | n.p. | n.p. | 0 | 1 |
| PA4797 | Probable transposase | n.p. | n.p. | 1294 | 879 |
| gi|496684684 | Conjugal transfer protein TrbE | n.p. | n.p. | 28 | 27 |
| gi|496684684 | Conjugal transfer protein TrbE | n.p. | n.p. | 13 | 15 |
| gi|496684684 | Conjugal transfer protein TrbE | n.p. | n.p. | 7 | 12 |
| gi|496684685 | Conjugal transfer protein TrbJ | n.p. | n.p. | 1 | 0 |
| gi|496684685 | Conjugal transfer protein TrbJ | n.p. | n.p. | 14 | 14 |
| gi|330503729 | Transposase IS4 | n.p. | n.p. | 91 | 80 |
| gi|496684687 | Conjugal transfer protein TrbL | n.p. | n.p. | 79 | 43 |
| gi|256367798 | Transposase | n.p. | n.p. | 15674 | 13666 |
| gi|496684681 | Conjugal transfer protein TrbB | n.p. | n.p. | 1 | 1 |
| gi|116050177 | Transposase Tn4652 | n.p. | n.p. | 596 | 487 |
| gi|116050169 | Recombinase | n.p. | n.p. | 553 | 1182 |
| gi|152984407 | TnpT protein | n.p. | n.p. | 180 | 266 |
| gi|12698413 | Transposase B | n.p. | n.p. | 50 | 71 |
| gi|498341535 | Integrase [Pseudomonas fragi] | n.p. | n.p. | 453 | 462 |
| gi|489250021 | Conjugal transfer protein TrbL | n.p. | n.p. | 1 | 0 |
| gi|496684681 | Conjugal transfer protein TrbB | n.p. | n.p. | 1 | 2 |
| gi|496684682 | Conjugal transfer protein TrbC | n.p. | n.p. | 0 | 2 |
| gi|497207593 | Integrase | n.p. | n.p. | 3508 | 2908 |
| gi|489182829 | Conjugative transfer protein TrbI | n.p. | n.p. | 87 | 59 |
| gi|386063802 | Conjugative transfer protein TrbG | n.p. | n.p. | 25 | 15 |
| gi|386063802 | Conjugative transfer protein TrbG | n.p. | n.p. | 29 | 14 |
| gi|386063803 | Conjugal transfer protein TrbF | n.p. | n.p. | 22 | 3 |
| gi|386063804 | Conjugal transfer protein TrbL | n.p. | n.p. | 4 | 0 |
| gi|505461140 | Shufflon-specific recombinase | n.p. | n.p. | 339 | 328 |
| gi|497074269 | Integrating conjugative element protein pill. pfgi-1 | n.p. | n.p. | 7 | 5 |
| gi|485834156 | Transposase | n.p. | n.p. | 632 | 428 |
| gi|446195994 | Transposase | n.p. | n.p. | 374 | 259 |
| gi|496684690 | Conjugal transfer protein TrbI | n.p. | n.p. | 14 | 9 |
| gi|496684690 | Conjugal transfer protein TrbI | n.p. | n.p. | 15 | 23 |
| gi|496684689 | Conjugal transfer protein TrbG | n.p. | n.p. | 8 | 14 |
| gi|495242526 | Conjugal transfer protein TrbF | n.p. | n.p. | 11 | 4 |
| gi|17547297 | Conjugal transfer protein TrbL | n.p. | n.p. | 1 | 0 |
| gi|3688518 | Putative transposase | n.p. | n.p. | 968 | 1026 |
| gi|491446843 | Integrase | n.p. | n.p. | 48 | 67 |
| gi|152987984 | Conjugal transfer protein TrbD | n.p. | n.p. | 2 | 5 |
| gi|493518183 | Conjugal transfer protein TrbC | n.p. | n.p. | 3 | 1 |
| gi|489201924 | Conjugal transfer protein TrbB | n.p. | n.p. | 3 | 1 |
| gi|330824177 | Conjugal transfer protein TrbB | n.p. | n.p. | 8 | 2 |
| gi|489194682 | Conjugal transfer protein TraG | n.p. | n.p. | 6 | 8 |
| gi|92112121 | TnpA transposase | n.p. | n.p. | 2857 | 1238 |
| gi|505462488 | Transposase mutator family protein | n.p. | n.p. | 483 | 234 |
| gi|472324076 | Site-specific recombinase XerC | n.p. | n.p. | 187 | 201 |
| gi|512557653 | Transposase | 55 | 51 | 57 | 30 |
| gi|489232756 | Transposase component | 7 | 8 | 13 | 5 |
| gi|495332841 | Transposase | 159 | 157 | n.p. | n.p. |
| gi|410693866 | Transposase of ISThsp18. IS1182 family | 2730 | 2154 | n.p. | n.p. |
| gi|392420288 | Integrating conjugative element ParB | 28 | 75 | 4 | 11 |
| gi|330824345 | Integrating conjugative element protein | 83 | 168 | 11 | 25 |
| gi|330824346 | Integrase | 31 | 67 | 36 | 24 |
| gi|512557590 | Integrating conjugative element protein PilL. PFGI-1 class | 8 | 6 | n.p. | n.p. |
| gi|490375364 | Integrating conjugative element protein | 15 | 35 | n.p. | n.p. |
| gi|339493279 | Conjugal transfer protein TraG | 109 | 96 | n.p. | n.p. |
| gi|498491306 | Integrating conjugative element protein | 6 | 2 | 9 | 9 |
| gi|497303938 | Integrating conjugative element membrane protein | 3 | 8 | 7 | 2 |
| gi|392420336 | Conjugal transfer protein | 8 | 9 | n.p. | n.p. |
| gi|330824396 | Integrating conjugative element protein | 26 | 22 | n.p. | n.p. |
| gi|330824398 | Integrating conjugative element protein | 13 | 18 | n.p. | n.p. |
| gi|497303929 | Conjugative transfer ATPase | 73 | 44 | 32 | 19 |
| gi|498491292 | Integrating conjugative element protein | 39 | 22 | 4 | 16 |
| gi|410471275 | Phage-related integrase | 442 | 603 | n.p. | n.p. |
| PA3738 | Integrase/recombinase XerD | 177 | 231 | 122 | 275 |
| gi|489224124 | Integrase | 1148 | 1068 | n.p. | n.p. |
| gi|489224128 | Integrase | 1072 | 1106 | n.p. | n.p. |
| gi|498248343 | Transposase | 124 | 98 | n.p. | n.p. |
| gi|496762510 | Conjugal transfer protein TrbJ | 359 | 253 | n.p. | n.p. |
| gi|489229539 | Conjugal transfer protein TrbL | 417 | 610 | n.p. | n.p. |
| gi|489229536 | Conjugal transfer protein TrbJ | 479 | 538 | n.p. | n.p. |
| gi|489229532 | Integrase | 634 | 593 | n.p. | n.p. |
| gi|157420224 | TnpA | 0 | 0 | 1224 | 1485 |
| gi|489225709 | Integrase | 230 | 160 | 137 | 63 |
| gi|353334486 | IncI1 plasmid conjugative transfer ATPase PilQ | 241 | 176 | 84 | 59 |
| gi|148807320 | Site-specific recombinase | 348 | 392 | n.p. | n.p. |
| gi|152985999 | Conjugal transfer protein TraG | 565 | 416 | 207 | 167 |
| gi|490477548 | Conjugal transfer protein | 98 | 44 | 19 | 16 |
| gi|498491413 | Conjugative transfer ATPase | 510 | 328 | 218 | 122 |
| gi|386064726 | Tyrosine recombinase XerC | 431 | 444 | 237 | 196 |
| gi|386056644 | Integrase | 71 | 85 | 44 | 61 |
| gi|446985433 | Integrase | 109 | 80 | 172 | 178 |
| gi|163856484 | Transposase | 115 | 68 | n.p. | n.p. |
| gi|496684672 | Conjugal transfer protein TraF peptidase | 9 | 2 | n.p. | n.p. |
| gi|187940137 | Phage integrase family protein | 187 | 135 | n.p. | n.p. |
| gi|121595234 | Conjugal transfer coupling protein TraG | 36 | 26 | 97 | 110 |
| gi|386063809 | Conjugal transfer protein TrbB | 14 | 12 | 12 | 21 |
| gi|492686504 | Conjugal transfer protein Trbc | 5 | 4 | n.p. | n.p. |
| gi|387129929 | Conjugal transfer protein TrbD | 2 | 2 | 6 | 3 |
| gi|497202778 | Conjugal transfer protein TrbE | 55 | 58 | 47 | 31 |
| gi|489201921 | Conjugal transfer protein TrbJ | 47 | 28 | 11 | 7 |
| gi|491446728 | Conjugal transfer protein TrbL | 869 | 474 | 16 | 8 |
| gi|152984687 | Transposase | 709 | 388 | n.p. | n.p. |
| gi|489221152 | Integrase | 645 | 569 | n.p. | n.p. |
| gi|489184774 | Integrase | 352 | 296 | n.p. | n.p. |
| gi|512563762 | P-type conjugative transfer protein TrbL | 15 | 3 | n.p. | n.p. |
| gi|94310290 | conjugal transfer protein TrbF | 17 | 22 | 23 | 4 |
| gi|512587667 | P-type conjugative transfer protein TrbG | 31 | 31 | 20 | 14 |
| gi|489200991 | Conjugative transfer protein TrbI | 76 | 67 | 38 | 55 |
| gi|490275574 | IS5 Transposase. Partial | 796 | 610 | n.p. | n.p. |
| gi|218891103 | Integrase | 362 | 278 | 111 | 97 |
| gi|392983596 | Integrase | 3054 | 1923 | 476 | 858 |
| gi|489349534 | Transposase IS3 | 327 | 133 | n.p. | n.p. |
| gi|187939496 | Transposase | 453 | 382 | 560 | 641 |
| PA1534 | Recombination protein RecR | 390 | 500 | 231 | 278 |
| PA5280 | Site-specific recombinase Sss | 320 | 320 | 112 | 113 |
| gi|148807435 | Phage integrase | 115 | 87 | n.p. | n.p. |
| gi|148807434 | Phage integrase | 371 | 303 | n.p. | n.p. |
| gi|148807431 | Site-specific recombinase | 598 | 366 | 488 | 488 |
| gi|489214965 | Integrase | 999 | 937 | 226 | 488 |
| gi|148807465 | Phage integrase | 121 | 139 | n.p. | n.p. |
| gi|392420677 | Transposase IS5 | 420 | 266 | n.p. | n.p. |
| gi|94311234 | Integrase | 83 | 88 | 12 | 4 |
| gi|493264878 | Conjugal transfer protein TraG | 81 | 100 | 101 | 58 |
| gi|489180720 | Conjugal transfer protein | 6 | 1 | 2 | 2 |
| gi|386717922 | Conjugal transfer protein | 76 | 77 | 54 | 53 |
| gi|489215831 | Integrase | 347 | 368 | 1107 | 3025 |
| gi|498490909 | Integrating conjugative element relaxase. PFGI-1 class | 212 | 126 | 181 | 129 |
| gi|493264934 | Transposase IS204 | 332 | 292 | n.p. | n.p. |
| gi|493265821 | Transposase | 303 | 257 | n.p. | n.p. |
| gi|489180871 | Integrase | 521 | 582 | 181 | 240 |
n.p. – gene sequence is not present in this strain.
Identifiers state PAO1 gene IDs or RefSeq accession where applicable. Transcriptional activity in tap water (T) and waste water (W) was normalized for the two strains independently and is shown as normalized pseudocounts.
Transcriptome analyses
In order to investigate the impact of waste water and tap water on the transcriptional activities, we performed RNA sequencing on both the sensitive and multi-resistant P. aeruginosa strain. The de novo assembled genomes were used as references for the mapping of reads obtained from RNA sequencing. In total, 95% of the reads mapped to the genome reference, which is comparable to results for RNA sequencing of known genomes and indicates a high quality and completeness of the two assembled genomes (Table S3). Between 1.5 and 3.6 million reads mapped uniquely to coding regions, yielding a median read count per gene of 54 to 130 and was sufficient for an in depth analysis.
Both strains, PA30 and PA49, were exposed to tap water and waste water, and differential gene expression was analysed between the different water matrices as well as between the two strains. Upon exposure to waste water, 222 genes were at least fourfold differentially expressed in strain PA30 (94 upregulated, 128 downregulated) as compared with tap water exposure (Table S4). Most of the differentially expressed genes encode for hypothetical proteins. Investigating whether any functional groups of genes were significantly over-represented among the differentially expressed genes, we performed an enrichment analysis of gene ontology (GO) terms. Genes that were associated with ‘copper ion binding’ (GO:0005507) and ‘potassium-transporting ATPase activity’ (GO:0008556) were significantly over-represented with six (out of 17) and three (out of three) genes being differentially expressed respectively. In strain PA49, 144 (51 upregulated, 93 downregulated, Table S5) gene showed differential expression upon exposure to the different water matrices, but no significant enrichment of GO terms was observed. A comparison of the expression of orthologous genes between PA30 and PA49 revealed a differential in the expression of 32 genes in tap water (e.g. some phenazine biosynthesis genes and a potassium-transporting ATPase, kdpABC, were upregulated in PA30), while only five gene coding for hypothetical proteins were found to be differentially expressed in waste water. This low number of differentially expressed genes between the two strains indicates a high similarity in their response to these specific environments.
The four horizontally acquired antibiotic resistance genes found in strain PA49 (Table 1) were transcriptionally active independent from the water matrix and therefore most likely are a main cause of the observed resistance towards a wide spectrum of aminoglycoside and beta-lactam antibiotics (Table 2). Genes associated with antibiotic resistance (not including multi-drug efflux pumps) showed a higher average expression as compared with the rest of the genome in PA49 (Fig. 4), which is obviously a result of the generally high expression of horizontally acquired resistance genes (Table 2). This tendency was independent from the water matrix and not found in the transcriptome of PA30 (Fig. 4), which lacks such additional resistance genes (Fig. 2). A common cause of antibiotic resistance in P. aeruginosa is the overexpression of multi-drug efflux pumps (usually termed ‘Mex’ pumps). Indeed, the genes encoding the MexCD-OprJ efflux pump were overexpressed in PA49 in response to waste water (Table 5 and Table S5). This pump system can confer resistance towards a broad spectrum of antibiotics (Poole et al., 1996) and thus may further contribute to the multi-resistance phenotype of PA49. The induced expression of this efflux pump specifically in waste water is indicating a specific stimulation presumably by one or multiple of antibiotics found in the used waste water or via so far unknown waste water components. However, since the expression of specific resistance genes and presence of resistance-related target mutations already sufficiently explains the broad resistance phenotype in PA49 (Table 1), the exact contribution of a MexCD-OprJ overexpression remains unclear. It should be again pointed out that the expression of resistance genes (with the exception of MexCD-OprJ) in PA49 was independent from the water matrix. Similarly, the antibiotic sensitive strain PA30 does not show any difference in expression of the intrinsic resistance determinants. Thus, the exposure of both strains to polluted waste water and oligotrophic tap water resulted in similar expression profiles of resistance genes. It seems to be obvious that changes in environmental milieus result in rather unspecific transcriptional responses than selected and stimuli-specific gene regulation.
Figure 4.
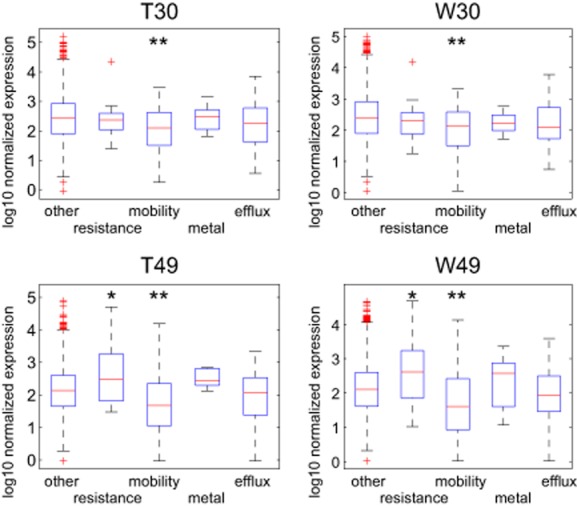
Expression of specific genes in strain PA30 and PA49 in different water matrices. Absolute gene expression values are depicted as box plots for the different samples of strain PA30 and PA49 cultivated in tap water (T) and waste water (W). Genes were manually selected by their functional classification as resistance (associated with modification and deactivation of antibiotics), mobility (associated with horizontal gene transfer and recombination), metal (associated with heavy metal tolerance), efflux (associated with multi-drug efflux pumps) or other (not included in any other class). Asterisks indicate a significant difference in the medians of the particular gene class and the other genes determined with the Mann–Whitney–Wilcoxon test (*P < 0.05; **P < 0.001).
Table 5.
Expression of genes encoding (putative) multi-drug efflux pumps (MEX pumps)
| PA30 | PA49 | ||||
|---|---|---|---|---|---|
| Gene ID/accession | Product | T | W | T | W |
| gi|15596434 | Multi-drug resistance efflux pump | n.p.a | n.p. | 8 | 6 |
| gi|491814602 | Macrolide efflux protein | n.p. | n.p. | 69 | 85 |
| gi|386336514 | RND transporter | n.p. | n.p. | 104 | 91 |
| gi|307130252 | RND efflux membrane fusion protein | n.p. | n.p. | 29 | 42 |
| PA1237 | Probable multi-drug resistance efflux pump | 9 | 6 | 10 | 0 |
| PA3719 | Anti-repressor for MexR, ArmR | 20 | 15 | 10 | 33 |
| PA4990 | SMR multi-drug efflux transporter | 103 | 79 | 21 | 15 |
| PA4374 | Probable Resistance-Nodulation-Cell Division (RND) efflux membrane fusion protein precursor | 499 | 444 | 233 | 251 |
| PA4375 | Probable Resistance-Nodulation-Cell Division (RND) efflux transporter | 1024 | 643 | 439 | 293 |
| PA3522 | Probable Resistance-Nodulation-Cell Division (RND) efflux transporter | 1090 | 102 | 381 | 82 |
| PA3523 | Probable Resistance-Nodulation-Cell Division (RND) efflux membrane fusion protein precursor | 355 | 56 | 140 | 27 |
| gi|489214414 | Outer membrane component of multi-drug efflux pump. partial | 0 | 0 | 1 | 1 |
| gi|15596434 | Multi-drug resistance efflux pump | 0 | 0 | 10 | 0 |
| PA0427 | Major intrinsic multiple antibiotic resistance efflux outer membrane protein OprM precursor | 2722 | 2351 | 991 | 1137 |
| PA0426 | Resistance-Nodulation-Cell Division (RND) multi-drug efflux transporter MexB | 7087 | 5994 | 2277 | 3917 |
| PA0425 | Resistance-Nodulation-Cell Division (RND) multi-drug efflux membrane fusion protein MexA precursor | 3129 | 2318 | 1098 | 1904 |
| PA0424 | Multi-drug resistance operon repressor MexR | 1305 | 520 | 331 | 321 |
| PA1238 | Probable outer membrane component of multi-drug efflux pump | 4 | 7 | 0 | 3 |
| PA1435 | Probable Resistance-Nodulation-Cell Division (RND) efflux membrane fusion protein precursor | 44 | 40 | 16 | 23 |
| PA1436 | Probable Resistance-Nodulation-Cell Division (RND) efflux transporter | 158 | 181 | 70 | 61 |
| PA0158 | Resistance-Nodulation-Cell Division (RND) triclosan efflux transporter, TriC | 2826 | 2341 | 1241 | 539 |
| PA0157 | Resistance-Nodulation-Cell Division (RND) triclosan efflux membrane fusion protein, TriB | 398 | 433 | 200 | 118 |
| PA0156 | Resistance-Nodulation-Cell Division (RND) triclosan efflux membrane fusion protein, TriA | 530 | 529 | 367 | 177 |
| PA2019 | Resistance-Nodulation-Cell Division (RND) multi-drug efflux membrane fusion protein precursor | 939 | 1323 | 79 | 70 |
| PA2018 | Resistance-Nodulation-Cell Division (RND) multi-drug efflux transporter | 3969 | 4876 | 321 | 240 |
| PA2528 | Probable Resistance-Nodulation-Cell Division (RND) efflux membrane fusion protein precursor | 508 | 452 | 349 | 690 |
| PA2527 | Probable Resistance-Nodulation-Cell Division (RND) efflux transporter | 603 | 529 | 345 | 572 |
| gi|489252367 | Multi-drug transporter | 23 | 35 | 16 | 8 |
| PA2526 | Probable Resistance-Nodulation-Cell Division (RND) efflux transporter | 504 | 542 | 594 | 449 |
| gi|489215922 | Resistance-Nodulation-Cell Division (RND) efflux transporter. partial | 31 | 28 | n.p. | n.p. |
| PA2522 | Outer membrane protein precursor CzcC | 80 | 17 | 116 | 15 |
| PA2521 | Resistance-Nodulation-Cell Division (RND) divalent metal cation efflux membrane fusion protein CzcB precursor | 184 | 47 | 147 | 20 |
| PA2520 | Resistance-Nodulation-Cell Division (RND) divalent metal cation efflux transporter CzcA | 369 | 77 | 430 | 51 |
| PA2495 | Multi-drug efflux outer membrane protein OprN precursor | 91 | 111 | 13 | 6 |
| PA2494 | Resistance-Nodulation-Cell Division (RND) multi-drug efflux transporter MexF | 1021 | 767 | 126 | 82 |
| PA2493 | Resistance-Nodulation-Cell Division (RND) multi-drug efflux membrane fusion protein MexE precursor | 110 | 106 | 35 | 61 |
| PA1541 | Probable drug efflux transporter | 38 | 104 | 148 | 1088 |
| PA5160 | Drug efflux transporter | 225 | 208 | 191 | 128 |
| PA4206 | Probable Resistance-Nodulation-Cell Division (RND) efflux membrane fusion protein precursor | 49 | 54 | 11 | 80 |
| PA4207 | Probable Resistance-Nodulation-Cell Division (RND) efflux transporter | 175 | 146 | 58 | 99 |
| PA4597 | Multi-drug efflux outer membrane protein OprJ precursor | 42 | 142 | 28 | 197 |
| PA4598 | Resistance-Nodulation-Cell Division (RND) multi-drug efflux transporter MexD | 153 | 297 | 83 | 964 |
| PA4599 | Resistance-Nodulation-Cell Division (RND) multi-drug efflux membrane fusion protein MexC precursor | 16 | 66 | 31 | 583 |
| PA3676 | Probable Resistance-Nodulation-Cell Division (RND) efflux transporter | 385 | 503 | 141 | 233 |
| PA3677 | Probable Resistance-Nodulation-Cell Division (RND) efflux membrane fusion protein precursor | 24 | 72 | 23 | 70 |
n.p. – gene sequence is not present in this strain.
Identifiers state PAO1 gene IDs or RefSeq accession where applicable. Transcriptional activity in tap water (T) and waste water (W) was normalized for the two strains independently and is shown as normalized pseudocounts.
A small set of genes associated with heavy metal tolerance was also found in the genomes of PA30 and PA49 (Fig. 3B; Table 3). However, no differential expression in the two water matrices was detected. Comparing the average expression of these genes with genes not related to metal tolerance also showed no general difference, independent of strain background and water matrix (Fig. 4).
Waste waters are already known to stimulate genetic transfer due to the sublethal noxa of pharmaceutical residues (e.g. antibiotics, heavy metal ions) or other xenobiotics. But, the expression of mobile genetic element was also found to be induced after exposure to tap water. Here, physiological shifts to oligotrophic habitats and/or starvation might be responsible for the genetic activities and might contribute to horizontal gene transfer, as discussed in Davies and colleagues (2006). The genomic analysis identified a large number of mobile genomic elements in the genomes of both PA30 and PA49 (Fig. 3C; Table 4). The genes that can be directly associated with horizontal gene transfer and recombination (recombinases, integrases, transposases and genes related to conjugative transfer) were mostly found to be actively expressed in both strains and independent from the water matrix. On average, these ‘mobility genes’ were expressed on a lower level than the ‘other’ genes of the genome (Fig. 4). However, their expression is insensitive to the strain background and to the water matrix.
In conclusion, the multi-drug resistance of strain PA49 can be attributed to the presence and expression of genes encoding a set of antibiotic-modifying enzymes located both in the core genome and on mobile genetic elements that were presumably acquired by horizontal gene transfer. Thus, the multi-drug resistant phenotype of PA49 seems directly linked with this set of resistance determinants. The impact of one overexpressed efflux pump being induced in waste water on the resistance characteristics of PA49 is so far an open question. Both, the antibiotic resistant and the sensitive strain, showed similar transcriptomic responses to the different water matrices but no strain-specific stress responses to both matrices (with exception to one efflux pump).
Experimental procedures
Isolation and cultivation of P. aeruginosa strains PA30 and PA49
Bacterial strains were enriched and isolated from a German waste water treatment plant compartment as described in a previous study (Schwartz et al., 2006). For routine culturing, bacteria were grown on agar plates containing BM2 minimal medium (Yeung et al., 2011) supplemented with 15 g l−1 agar (Merck, Darmstadt, Germany). For overnight cultures, a colony from the agar plate was inoculated in BM2 minimal medium as well as BHI (Merck, Darmstadt, Germany) broth (1:4 diluted) and incubated at 37°C. The growth behavior of the strains was observed by diluting overnight cultures to an optical density (OD) of 0.1 in BM2 and BHI medium, incubation at 37°C with gentle agitation for a time span of 24 h and monitoring the OD over time for each strain (Infinite 200 PRO, Tecan, Männedorf, Switzerland). No difference in growth behavior between the two isolates was observed in BM2 and BHI broth respectively (data not shown).
Antibiotic susceptibility testing PA30 and PA49
Resistance characterization for GM (10 μg disc−1), CIP (5 μg disc−1), IPM (10 μg disc−1), CAZ (30 μg disc−1), AN (30 μg disc−1), AZ (75 μg disc−1) and PT (100/10 μg disc−1) was evaluated using agar diffusion test according to Clinical Laboratory Standards Institute (CLSI) guidelines, wherein the zone of growth inhibition on Miller Hinton agar (Merck) was measured after 18 h incubation at 37°C. The P. aeruginosa strain PA30 was found to be sensitive for GM, CIP, IPM, CAZ, AN, AZ and PT. In contrast, PA49 was found to be resistant against all mentioned antibiotics (Schwartz et al., 2006).
Incubation in tap water and waste water
Distinct colonies of each strain were inoculated in 25 ml BHI medium (Merck, Darmstadt, Germany) diluted 1:4 with distilled water in a 50 ml sterile tube (Falcon, Nürtingen, Germany) and incubated on a shaker at 37°C at 100 rpm overnight. A volume of 2.5 ml of this overnight culture was used to inoculate 25 ml of 1:4 diluted BHI medium and incubated on a shaker at 37°C at 100 rpm. At an optical density (OD600nm) of 1.0 (early stationary growth phase), bacterial suspension were pelleted at 5000 g at 20°C for 15 min. Pellets were re-suspended in 20 ml sterile tap water (T) or sterile filtered waste water (W) collected from the influent of a municipal WWTP. The OD of these suspensions with PA30 and PA49 were adjusted at 0.5. The samples were incubated on a shaker (80 rpm) at 22°C for 3 h.
The tap water conditioned from groundwater at the municipal waterworks met the requirements of the German drinking water guideline. The average total organic carbon value was measured as 0.9 mg l−1. The chemical and physical characteristics of the final conditioned drinking water are listed in Jungfer et al. (2013; see reference waterworks).
The used waste water originated from the effluent of a municipal waste water treatment plant of a city with 445 000 inhabitants and is equipped with a conventional three treatment process (nitrification, denitrification, phosphor elimination). Chemical analyses demonstrated the presences of different classes of antibiotics (e.g. clarithromycin, roxithromycin, erythromycin, sulfamethoxazol, and trimehoprim) in a range of 0.5–1.5 μg l−1 (unpublished data).
DNA extraction and purification
Previous to the DNA extraction 25 ml BHI was inoculated with a single colony of PA30 and PA49, respectively, and cultivated at 37°C and 150 rpm on a rotary shaker until ODs reached 1.0 value. An aliquot of 5 ml of each culture was pelleted at 3000 g for 10 min. Subsequent DNA extraction was performed according to the protocol of QIAGEN Genomic-tip 100/G kit system (Qiagen, Germany). The concentration and purity of the obtained DNA was determined using the NanoDrop 1000 Spectrophotometer (Thermo Scientific, Germany). The quality of the genomic DNA was also controlled by agarose gel electrophoresis.
RNA extraction and purification
Ribonucleic isolation of the samples was performed in quadruplicates that were pooled before sequencing. One millilitre of each of the four independent bacterial suspensions (T or W) was mixed with 1 ml of RNA protect (Qiagen, Hilden, Germany) and incubated for 5 min at room temperature. The bacteria were pelleted at 12.000 g for 10 min, and the supernatant was discarded. Prior to RNA extraction from bacteria, four replicate cultures from parallel experiments (tap water and waste water) from each type (PA30 and PA49) were combined. Ribonucleic acid isolation was performed using the RNeasy extraction kit (Qiagen, Hildern, Germany) according to the manufacturer's protocol, and the RNA was eluted in 50 μl RNase-free water. To eliminate residual DNA contamination, the RNA was treated with TURBO Desoxyribonuclease (DNase, Ambion Inc., Kaufungen, Germany). Five microlitres of 10× TURBO DNase buffer and 1 μl of TURBO DNase were added to 50 μl RNA solution and incubated at 37°C for 30 min. Desoxyribonuclease inactivation reagent (5 μl) was added to the RNA solution and incubated under occasional mixing for 5 min. The sample was centrifuged at 10 000 rpm for 1.5 min, and the RNA was transferred to a new tube, and RNA concentration was measured in triplicate using the Nanodrop ND1000 spectrophotometer (PeqLab Biotechnology GmbH, Erlangen, Germany). The integrities of all RNA samples were tested using the Agilent 2100 Bioanalyzer (Agilent Technologies Sales & Services GmbH & Co.KG, Waldbronn, Germany).
Removal of the ribosomal RNA
Removal of ribosomal RNA (rRNA) was performed with each sample. Fourteen microlitres of total RNA was mixed with 1 μl of RNase inhibitor SUPERase IN (Ambion). Ribosomal RNA was removed with the MICROBExpress KIT (Ambion) according to the manufacturer's protocol. Purified RNA was re-suspended in 25 μl TE buffer (1 mM EDTA, 10 mM Tris, pH: 8.0). The resulting purified mRNA yields were quantified with Nanodrop ND1000.
Library preparation and Illumina sequencing
Deoxyribonucleic acid sequencing libraries were produced from 1 μg of genomic DNA and RNA libraries from 50 ng of rRNA depleted RNA, following the recommendations of the TruSeq DNA and TruSeq RNA protocols (Illumina) respectively. Briefly, the quality and quantity of ribosomal depleted RNA were assessed with the Bioanalyzer 2100 (Agilent), and the RNA-seq libraries were fragmented chemically, purified with AMPure XP beads (Beckman Coulter) and ligated to adapters with specific DNA barcode for each sample following the Illumina protocol. For DNA-seq libraries, genomic DNA was sheared to 200 bp fragments by sonication with a Covaris S2 instrument using the following settings: peak incidence power 175 W, duty factor 10%, cycle per burst 200, time 430 s. Sizes and concentrations of both RNA and DNA sequencing libraries were determined on a Bioanalyzer 2100 (DNA1000 chips, Agilent). Paired-end sequencing (2 × 50 bp) was performed on two lanes on a Hiseq1000 (Illumina) platform using TruSeq PE Cluster KIT v3 – cBot – HS and TruSeq SBS KIT v3 – HS. Cluster detection and base calling were performed using rtav1.13 and quality of reads assessed with casava v1.8.1 (Illumina). The sequencing resulted in at least 40 million pairs of 50 nt long reads for each sample, with a mean Phred quality score > 35 (Tables S1 and S3). These sequence data have been submitted to the GenBank Sequence Read Archive and are available under the accession numbers SRP041029 (PA30 genome), SRP041030 (PA49 genome), SRP041150 (PA30 transcriptomes) and SRP041151 (PA49 transcriptomes).
Genome assembly and annotation
Raw sequence reads were trimmed and filtered using the fastq-mcf tool of the ea-utils software package (Aronesty, 2011) with a Phred quality cut-off of 20 and the appropriate Illumina TruSeq adapter sequences to detect and remove adapters. The genomes of strains PA30 and PA49 were assembled independently using the idba_hybrid assembler (Peng et al., 2012) with a range of k-mer sizes from 20 to 50 bp (step size 10 bp). The genome reference used to guide the hybrid assembly included the full genomic sequence of P. aeruginosa PAO1 and the sequences of 13 genomic islands and one plasmid (Table S2). To confirm the resulting contigs, the filtered and trimmed reads were mapped against the contigs using Bowtie 2 (Langmead and Salzberg, 2012), and the read coverage of each contig was calculated by dividing the number of reads mapping to that contig by its length. Contigs with a length of less than 250 bp or with a read coverage of less than one read per base pair were discarded for being too short and/or potentially artefacts. Thereby, 207 (out of 775) and 269 (out of 887) contigs remained for the genomes of PA30 and PA49 respectively (Table S1). The overlap of the contigs with the reference sequences was determined by multi-sequence alignment using the PROmer function of the MUMmer 3.0 software package (Kurtz et al., 2004).
The filtered contigs were scanned for protein coding genes using MetaGeneMark (Trimble et al., 2012) with default parameters yielding 6572 and 6781 genes for PA30 and PA49 respectively (Table S1). The genes were annotated in two steps by using a blastp search (Camacho et al., 2009) first against the P. aeruginosa PAO1 genome with protein sequences taken from the Pseudomonas genome database (Winsor et al., 2011) and then for the remaining unidentified protein sequences against the database of nr protein sequences available for download from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/blast/db). Thereby, 65 and 97 of the predicted genes of PA30 and PA49 remained unidentified. Additionally, the predicted protein sequences were compared by blastp to the CARD (McArthur et al., 2013) to identify putative resistance genes.
To enable direct comparisons between the genomes of PA30 and PA49, the orthologous genes were determined by reciprocal alignment of the protein sequences using blast. Genes in both genomes that reciprocally yielded the highest blast score for each other in the alignment with at least 80% sequence identity were considered to be orthologs.
Analysis of gene expression
Raw reads generated from complementary DNA were trimmed and filtered using the fastq-mcf tool of the ea-utils software package with a Phred quality cut-off of 20 and the appropriate Illumina TruSeq adapter sequences to detect and remove adapters. Gene expression was determined by mapping the reads to the newly assembled and contigs using Bowtie 2 and counting the reads that uniquely overlapped with the annotated genes. Differential gene expression was analysed using the r-package DESeq (Anders and Huber, 2010). Gene were considered to be differentially expressed, when the absolute log2 fold change was greater than 2 (equivalent to fourfold upregulation or downregulation) with a P-value (adjusted for multiple hypothesis testing) below 0.05. A GO enrichment analysis was performed using the Blast2GO software (Conesa et al., 2005; Götz et al., 2008), testing for the enrichment of GO terms in the set of differentially expressed genes with a false discovery rate (FDR) of less than 0.05.
Multilocus Sequence Typing
The sequence types of the strains PA30 and PA49 were determined following the multilocus sequence typing (MLST) scheme described by Curran and colleagues (2004), by comparing the sequences of seven variable genes commonly found in P. aeruginosa (acsA, aroE, guaA, mutL, nuoD, ppsA, trpE) with the public online database PubMLST, http://www.pubmlst.org (Jolley and Maiden, 2010). The sequences of the seven MLST regions were concatenated for both strains and aligned with the concatenated MLST sequences of eight previously published genomes (P. aeruginosa strains PAO1, PA14, PA7, PASC2, LESB58, NCGM2, DK2 and M18, all taken from the Pseudomonas genome database http://www.pseudomonas.com (Winsor et al., 2011) ) using ClustalW (Larkin et al., 2007). A phylogenetic tree was constructed from the alignment using ClustalW and TreeView (Page, 1996).
Conflict of Interest
None declared.
Supporting Information
Table S1. Genome sequencing and assembly statistics.
Table S2. Coverage of references sequences. Percentages indicate the fraction of the references sequences that overlapped with the newly assembled contigs. See also Fig. 1.
Table S3. Ribonucleic acid sequencing statistics.
Table S4. MS Excel table file (.xslx) of genes that were differentially expressed comparing the transcriptomes of isolate PA30 in waste water and tap water. The file contains two table sheets listing the genes that were upregulated or downregulated upon exposure to waste water as compared with exposure to tap water.
Table S5. MS Excel table file (.xslx) of genes that were differentially expressed comparing the transcriptomes of isolate PA49 in waste water and tap water. The file contains two table sheets listing the genes that were upregulated or downregulated upon exposure to waste water as compared with exposure to tap water.
References
- Alvarez-Ortega C, Wiegand I, Olivares J, Hancock RE. Martinez JL. Genetic determinants involved in the susceptibility of Pseudomonas aeruginosa to beta-lactam antibiotics. Antimicrob Agents Chemother. 2010;54:4159–4167. doi: 10.1128/AAC.00257-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S. Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronesty E. 2011. ea-utils: command-line tools for processing biological sequencing data. [WWW document]. URL http://code.google.com/p/ea-utils.
- Battle SE, Rello J. Hauser AR. Genomic islands of Pseudomonas aeruginosa. FEMS Microbiol Lett. 2009;290:70–78. doi: 10.1111/j.1574-6968.2008.01406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier SP, Lebeaux D, DeFrancesco AS, Valomon A, Soubigou G, Coppee JY, et al. Starvation, together with the SOS response, mediates high biofilm-specific tolerance to the fluoroquinolone ofloxacin. PLoS Genet. 2013;9:e1003144. doi: 10.1371/journal.pgen.1003144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breidenstein EB, de la Fuente-Nunez C. Hancock RE. Pseudomonas aeruginosa: all roads lead to resistance. Trends Microbiol. 2011;19:419–426. doi: 10.1016/j.tim.2011.04.005. [DOI] [PubMed] [Google Scholar]
- Bruchmann S, Dötsch A, Nouri B, Chaberny IF. Häussler S. Quantitative contributions of target alteration and decreased drug accumulation to Pseudomonas aeruginosa fluoroquinolone resistance. Antimicrob Agents Chemother. 2013;57:1361–1368. doi: 10.1128/AAC.01581-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K. Madden TL. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, Götz S, Garcia-Gomez JM, Terol J, Talon M. Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- Curran B, Jonas D, Grundmann H, Pitt T. Dowson CG. Development of a multilocus sequence typing scheme for the opportunistic pathogen Pseudomonas aeruginosa. J Clin Microbiol. 2004;42:5644–5649. doi: 10.1128/JCM.42.12.5644-5649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J. Davies D. Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev. 2010;74:417–433. doi: 10.1128/MMBR.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J, Spiegelman GB. Yim G. The world of subinhibitory antibiotic concentrations. Curr Opin Microbiol. 2006;9:445–453. doi: 10.1016/j.mib.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Donlan RM. Costerton JW. Biofilms: survival mechanisms of clinically relevant microorganisms. Clin Microbiol Rev. 2002;15:167–193. doi: 10.1128/CMR.15.2.167-193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dötsch A, Becker T, Pommerenke C, Magnowska Z, Jänsch L. Häussler S. Genome-wide identification of genetic determinants of antimicrobial drug resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2009;53:2522–2531. doi: 10.1128/AAC.00035-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dötsch A, Klawonn F, Jarek M, Scharfe M, Blöcker H. Häussler S. Evolutionary conservation of essential and highly expressed genes in Pseudomonas aeruginosa. BMC Genomics. 2010;11:234. doi: 10.1186/1471-2164-11-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajardo A, Martinez-Martin N, Mercadillo M, Galan JC, Ghysels B, Matthijs S, et al. The neglected intrinsic resistome of bacterial pathogens. PLoS ONE. 2008;3:e1619. doi: 10.1371/journal.pone.0001619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flemming HC. Wingender J. Relevance of microbial extracellular polymeric substances (EPSs) – part I: structural and ecological aspects. Water Sci Technol. 2001;43:1–8. [PubMed] [Google Scholar]
- Goh EB, Yim G, Tsui W, McClure J, Surette MG. Davies J. Transcriptional modulation of bacterial gene expression by subinhibitory concentrations of antibiotics. Proc Natl Acad Sci USA. 2002;99:17025–17030. doi: 10.1073/pnas.252607699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36:3420–3435. doi: 10.1093/nar/gkn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolley KA. Maiden MC. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics. 2010;11:595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungfer C, Friedrich F, Varela-Villarreal J, Brändle K, Gross HJ, Obst U. Schwartz T. Drinking water biofilms on copper and stainless steel exhibit specific molecular responses towards different disinfection regimes at waterworks. Biofouling. 2013;29:891–907. doi: 10.1080/08927014.2013.813936. [DOI] [PubMed] [Google Scholar]
- Klockgether J, Cramer N, Wiehlmann L, Davenport CF. Tümmler B. Pseudomonas aeruginosa genomic structure and diversity. Front Microbiol. 2011;2:150–167. doi: 10.3389/fmicb.2011.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung VL, Ozer EA. Hauser AR. The accessory genome of Pseudomonas aeruginosa. Microbiol Mol Biol Rev. 2010;74:621–641. doi: 10.1128/MMBR.00027-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C. Salzberg SL. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kümmerer K. Antibiotics in the aquatic environment – a review – part II. Chemosphere. 2009;75:435–441. doi: 10.1016/j.chemosphere.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Kwon DH. Lu CD. Polyamines induce resistance to cationic peptide, aminoglycoside, and quinolone antibiotics in Pseudomonas aeruginosa PAO1. Antimicrob Agents Chemother. 2006;50:1615–1622. doi: 10.1128/AAC.50.5.1615-1622.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B. Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- McArthur AG, Waglechner N, Nizam F, Yan A, Azad MA, Baylay AJ, et al. The comprehensive antibiotic resistance database. Antimicrob Agents Chemother. 2013;57:3348–3357. doi: 10.1128/AAC.00419-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez JL, Fajardo A, Garmendia L, Hernandez A, Linares JF, Martinez-Solano L. Sanchez MB. A global view of antibiotic resistance. FEMS Microbiol Rev. 2009;33:44–65. doi: 10.1111/j.1574-6976.2008.00142.x. [DOI] [PubMed] [Google Scholar]
- Mathee K, Narasimhan G, Valdes C, Qiu X, Matewish JM, Koehrsen M, et al. Dynamics of Pseudomonas aeruginosa genome evolution. Proc Natl Acad Sci USA. 2008;105:3100–3105. doi: 10.1073/pnas.0711982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlin C, Bonot S, Courtois S. Block JC. Persistence and dissemination of the multiple-antibiotic-resistance plasmid pB10 in the microbial communities of wastewater sludge microcosms. Water Res. 2011;45:2897–2905. doi: 10.1016/j.watres.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Morrison AJ. Wenzel RP. Epidemiology of infections due to Pseudomonas aeruginosa. Rev Infect Dis. 1984;6:627–642. doi: 10.1093/clinids/6.supplement_3.s627. [DOI] [PubMed] [Google Scholar]
- Nikaido H. Preventing drug access to targets: cell surface permeability barriers and active efflux in bacteria. Semin Cell Dev Biol. 2001;12:215–223. doi: 10.1006/scdb.2000.0247. [DOI] [PubMed] [Google Scholar]
- Nyström T. Stationary-phase physiology. Annu Rev Microbiol. 2004;58:161–181. doi: 10.1146/annurev.micro.58.030603.123818. [DOI] [PubMed] [Google Scholar]
- Page RD. TreeView: an application to display phylogenetic trees on personal computers. Comput Appl Biosci. 12:357–358. doi: 10.1093/bioinformatics/12.4.357. [DOI] [PubMed] [Google Scholar]
- Pedró L, Baños RC, Aznar S, Madrid C, Balsalobre C. Juárez A. Antibiotics shaping bacterial genome: deletion of an IS91 flanked virulence determinant upon exposure to subinhibitory antibiotic concentrations. PLoS ONE. 2011;6:e27606. doi: 10.1371/journal.pone.0027606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Leung HC, Yiu SM. Chin FY. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28:1420–1428. doi: 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- Pirnay JP, De Vos D, Mossialos D, Vanderkelen A, Cornelis P. Zizi M. Analysis of the Pseudomonas aeruginosa oprD gene from clinical and environmental isolates. Environ Microbiol. 2002;4:872–882. doi: 10.1046/j.1462-2920.2002.00281.x. [DOI] [PubMed] [Google Scholar]
- Poole K, Gotoh N, Tsujimoto H, Zhao Q, Wada A, Yamasaki T, et al. Overexpression of the mexC-mexD-oprJ efflux operon in nfxB-type multidrug-resistant strains of Pseudomonas aeruginosa. Mol Microbiol. 1996;21:713–724. doi: 10.1046/j.1365-2958.1996.281397.x. [DOI] [PubMed] [Google Scholar]
- Rizzo L, Manaia C, Merlin C, Schwartz T, Dagot C, Ploy MC, et al. Urban wastewater treatment plants as hotspots for antibiotic resistant bacteria and genes spread into the environment: a review. Sci Total Environ. 2013;447:345–360. doi: 10.1016/j.scitotenv.2013.01.032. [DOI] [PubMed] [Google Scholar]
- Ruiz J. Mechanisms of resistance to quinolones: target alterations, decreased accumulation and DNA gyrase protection. J Antimicrob Chemother. 2003;51:1109–1117. doi: 10.1093/jac/dkg222. [DOI] [PubMed] [Google Scholar]
- Schreiber K, Krieger R, Benkert B, Eschbach M, Arai H, Schobert M. Jahn D. The anaerobic regulatory network required for Pseudomonas aeruginosa nitrate respiration. J Bacteriol. 2007;189:4310–4314. doi: 10.1128/JB.00240-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz T, Volkmann H, Kirchen S, Kohnen W, Schon-Holz K, Jansen B. Obst U. Real-time PCR detection of Pseudomonas aeruginosa in clinical and municipal wastewater and genotyping of the ciprofloxacin-resistant isolates. FEMS Microbiol Ecol. 2006;57:158–167. doi: 10.1111/j.1574-6941.2006.00100.x. [DOI] [PubMed] [Google Scholar]
- Schweizer HP. Efflux as a mechanism of resistance to antimicrobials in Pseudomonas aeruginosa and related bacteria: unanswered questions. Genet Mol Res. 2003;2:48–62. [PubMed] [Google Scholar]
- Seiler C. Berendonk TU. Heavy metal driven co-selection of antibiotic resistance in soil and water bodies impacted by agriculture and aquaculture. Front Microbiol. 3:399–408. doi: 10.3389/fmicb.2012.00399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature. 2000;406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- Trautmann M, Lepper PM. Haller M. Ecology of Pseudomonas aeruginosa in the intensive care unit and the evolving role of water outlets as a reservoir of the organism. Am J Infect Control. 2005;33:41–49. doi: 10.1016/j.ajic.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Trimble WL, Keegan KP, D'Souza M, Wilke A, Wilkening J, Gilbert J. Meyer F. Short-read reading-frame predictors are not created equal: sequence error causes loss of signal. BMC Bioinformatics. 13:183. doi: 10.1186/1471-2105-13-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Masters S, Hong Y, Stallings J, Falkinham JO, Edwards MA. Pruden A. Effect of disinfectant, water age, and pipe material on occurrence and persistence of Legionella, mycobacteria, Pseudomonas aeruginosa, and two amoebas. Environ Sci Technol. 2012;46:11566–11574. doi: 10.1021/es303212a. [DOI] [PubMed] [Google Scholar]
- Winsor GL, Lam DK, Fleming L, Lo R, Whiteside MD, Yu NY, et al. Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res. 39:D596–D600. doi: 10.1093/nar/gkq869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung O, Law SP. Yau M. Treatment generalization and executive control processes: preliminary data from Chinese anomic individuals. Int J Lang Commun Disord. 2009;44:784–794. doi: 10.1080/13682820902929081. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genome sequencing and assembly statistics.
Table S2. Coverage of references sequences. Percentages indicate the fraction of the references sequences that overlapped with the newly assembled contigs. See also Fig. 1.
Table S3. Ribonucleic acid sequencing statistics.
Table S4. MS Excel table file (.xslx) of genes that were differentially expressed comparing the transcriptomes of isolate PA30 in waste water and tap water. The file contains two table sheets listing the genes that were upregulated or downregulated upon exposure to waste water as compared with exposure to tap water.
Table S5. MS Excel table file (.xslx) of genes that were differentially expressed comparing the transcriptomes of isolate PA49 in waste water and tap water. The file contains two table sheets listing the genes that were upregulated or downregulated upon exposure to waste water as compared with exposure to tap water.