

Abstract
Facioscapulohumeral muscular dystrophy (FSHD) is one of the most common neuromuscular disorders. The major form of the disease (FSHD1) is linked to decrease in copy number of a 3.3-kb tandem repeated macrosatellite (D4Z4), located on chromosome 4q35. D4Z4 deletion alters chromatin structure of the locus leading to aberrant expression of nearby 4q35 genes. Given the high variability in disease onset and progression, multiple factors could contribute to the pathogenesis of FSHD. Among the FSHD candidate genes are double homeobox 4 (DUX4), encoded by the most telomeric D4Z4 unit, and FSHD region gene 1 (FRG1). DUX4 is a sequence-specific transcription factor. Here, we located putative DUX4 binding sites in the human FRG1 genomic area and we show specific DUX4 association to these regions. We found also that ectopically expressed DUX4 up-regulates the endogenous human FRG1 gene in healthy muscle cells, while DUX4 knockdown leads to a decrease in FRG1 expression in FSHD muscle cells. Moreover, DUX4 binds directly and specifically to its binding site located in the human FRG1 gene and transactivates constructs containing FRG1 genomic regions. Intriguingly, the mouse Frg1 genomic area lacks DUX4 binding sites and DUX4 is unable to activate the endogenous mouse Frg1 gene providing a possible explanation for the lack of muscle phenotype in DUX4 transgenic mice. Altogether, our results demonstrate that FRG1 is a direct DUX4 transcriptional target uncovering a novel regulatory circuit contributing to FSHD.
Introduction
Facioscapulohumeral muscular dystrophy (FSHD) is one of the most prevalent neuromuscular diseases (1,2). It is an autosomal dominant disease characterized by a unique pattern of affected musculature, typically arising with a reduction of facial and shoulder girdle muscle mass followed by weakness of the lower extremities muscles (3). The major FSHD locus (FSHD1, MIM #158900) was mapped to the subtelomeric portion of human chromosome 4q35 (4), where is located a 3.3-kb macrosatellite called D4Z4 (5). In healthy individuals, the number of D4Z4 repeats varies between 11 and more than 100, while FSHD patients carry from 1 to 10 repeats. In FSHD, the reduction in D4Z4 copy number is associated with a Polycomb/Trithorax switch (6) leading to aberrant expression of as many as 17 different protein-coding genes located at 4q35 (7–16). However, several studies failed to confirm these results (17–22). Nevertheless, some 4q35 gene appears to be affected mainly during skeletal muscle development justifying why it has not been identified by studies using adult FSHD samples (15,16). Moreover, several 4q35 genes display very close homologues on other chromosomes and, as reported elsewhere (23,24), microarray probes do not target specifically their 4q35 copy.
The leading FSHD candidate gene is double homeobox 4 (DUX4) (12,25). It is encoded from an open reading frame that is present within each D4Z4 repeat, but only the most telomeric unit generates a stable DUX4 transcript (12,13). While there is strong indication that DUX4 has a causative role in the disease (12,26), it appears insufficient to fully explain FSHD pathogenesis. Indeed, it has been shown that DUX4 can be similarly overexpressed in cells and muscle tissues derived from unaffected individuals and FSHD patients (16,27). Moreover, transgenic mice displaying a DUX4 expression pattern similar to FSHD patients do not show any muscle phenotype (28). Also, DUX4 ectopic expression in Zebrafish causes developmental muscle abnormalities (29), however muscular degeneration is not consistent with a dystrophic phenotype. Altogether, these results suggest that DUX4 expression per se is not sufficient for FSHD muscle pathology and rise the possibility that additional factors might contribute with DUX4 leading to disease progression.
Another FSHD candidate gene is FSHD region gene 1 (FRG1) (30). FRG1 has been shown to be selectively overexpressed in FSHD patients (7,14,16,31), although with inconsistent results (20–22,24). FRG1 is a dynamic nuclear and cytoplasmic shuttling protein that, in skeletal muscle, is also localized to the sarcomere (32). In the nucleus, FRG1 is localized in nucleoli, Cajal bodies and actively transcribed chromatin (33,34) where it regulates RNA splicing (35–38) and the activity of the histone methyltransferase SUV4-20H1 (14). FRG1 overexpression leads to muscle stem cell defects (39,40) and the development of FSHD-like phenotypes in mice (36,40), Xenopus laevis and Caenorhabditis elegans (41–43).
To date, the molecular pathogenesis of FSHD has not been completely elucidated. The peculiar nature of the mutation at the basis of FSHD and its complex effect on the chromatin surrounding the 4q35 genomic locus make it unlikely that the development of the disease could be attributed to a single gene. Thus, in FSHD multiple 4q35 genes could contribute to the final result. Here, we report that the transcription factor DUX4 binds in vitro and in vivo specifically to the human FRG1 genomic region. Accordingly, ectopic DUX4 overexpression mediates FRG1 up-regulation in human healthy myoblasts, while DUX4 knockdown leads to FRG1 down-regulation in FSHD muscle cells. Moreover, we demonstrate that a 31-bp sequence from the human FRG1 genomic region is sufficient to recapitulate transcriptional activation by DUX4. Notably, mouse Frg1 is not regulated by DUX4. Based on our results, we propose that a direct DUX4 and FRG1 interplay could contribute to the development of FSHD muscular dystrophy.
Results
DUX4 is a transcriptional activator of FRG1 in human muscle cells
To identify DUX4 targets, microarrays and chromatin immunoprecipitation coupled with ultra-high-throughput sequencing (ChIP-seq) studies following ectopic DUX4 overexpression in control human muscle cells were previously performed (44). In this work, a 2-fold cut-off was set for the microarrays results. Hence, the genes whose transactivation was lower than the threshold imposed were not reported. We accessed the entire microarrays dataset available on the Gene Expression Omnibus database (GSE33799) and its inspection revealed a 1.8-fold (adjusted P-value 1.24 × 10−9) FRG1 up-regulation in control muscle cells ectopically expressing DUX4. While the FRG1 up-regulation level was not very high, we decided to further investigate the matter due to its relevance for FSHD. To confirm that DUX4 regulates the endogenous human FRG1 gene, we electroporated control human muscle cells with the pCIneo-DUX4 expression vector or with the empty vector pCIneo (Supplementary Material, Fig. S1). Real-time quantitative Reverse transcription polymerase chain reaction (RT-PCR) (RT-qPCR) analysis indicated that ectopic DUX4 expression significantly increased the expression of the endogenous FRG1 gene by 2.8-fold, compared with the empty vector control (Fig. 1A). As positive controls, we used RFPL2 (ret finger protein-like 2) and TRIM48 (tripartite motif containing 48), two bona fide DUX4 targets previously identified upon ectopic DUX4 overexpression in human control myoblasts (44) (Fig. 1A).
Figure 1.
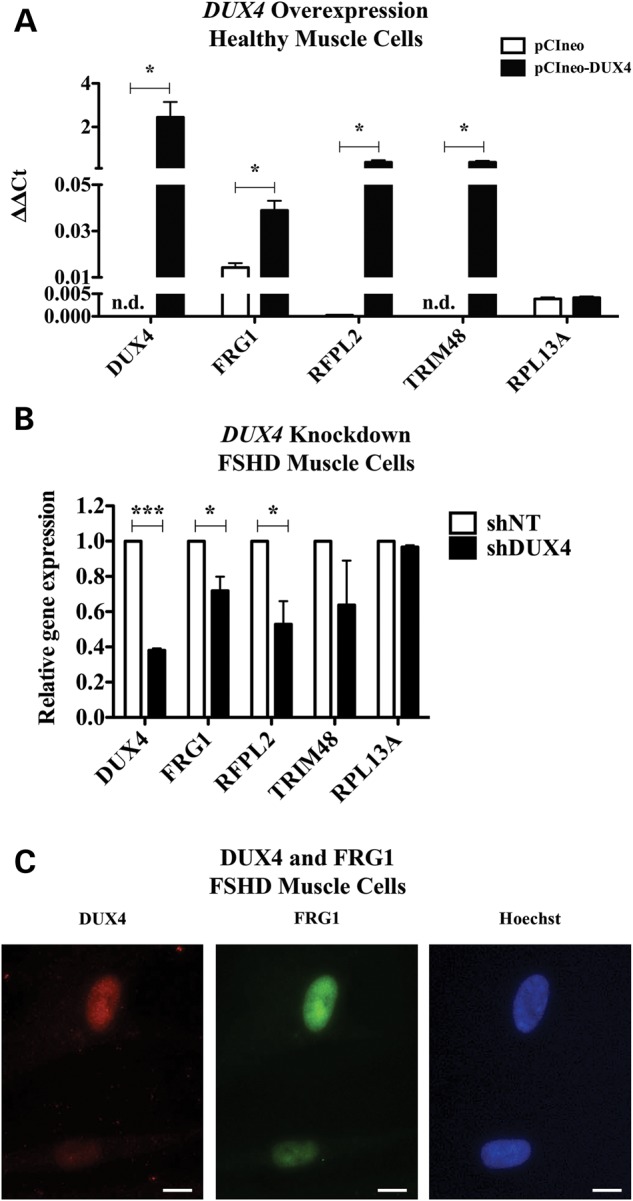
DUX4 regulates the endogenous FRG1 gene in human muscle cells. (A) Control human muscle cells were electroporated with an expression vector either encoding DUX4 (pCIneo-DUX4) or insertless (pCIneo). Expression levels of FRG1 are enhanced upon DUX4 overexpression. RFPL2 and TRIM48, bona fide DUX4 targets, were used as positive controls and RPL13A as negative control (paired t-test, *P < 0.05, n = 3, mean ± SEM). N.d. is not detected. (B) FSHD human muscle cells were infected with retroviruses containing a non-targeting sequence (shNT) or a DUX4-targeting sequence (shDUX4). Upon DUX4 knockdown, FRG1 expression levels were reduced. RFPL2 and TRIM48 were used as positive control, while RPL13A was used as negative control (paired t-test, *P < 0.05, ***P < 0.001, n = 3, mean ± SEM). (C) Immunofluorescence for DUX4 and FRG1 in FSHD muscle cells. Scale bar is 10 μm.
It has to be noted that RFPL2 and TRIM48 genes are silent or expressed at very low levels in myoblasts, because they are normally expressed only in primate neocortex during development and in pre-implantation embryos, respectively (45,46). Because of their extremely low basal expression levels, RFPL2 and TRIM48 were highly up-regulated by DUX4 overexpression (44). In contrast FRG1 is expressed at substantial levels ubiquitously (30,31), justifying its relatively mild activation by DUX4 overexpression.
Next, we tested the effect of DUX4 knockdown on the expression level of the endogenous human FRG1. We infected FSHD human muscle cells with retroviruses expressing either non-silencing or DUX4-targeting shRNAs. Upon DUX4 silencing, FRG1 expression was significantly reduced, as well as the positive control RFPL2 (Fig. 1B). TRIM48 expression was also impaired, while expression of the negative control gene RPL13A was not altered (Fig. 1B). To further support DUX4 role in positive regulation of FRG1, we performed immunofluorescence for DUX4 and FRG1 on FSHD muscle cells. As previously reported (13,27,47–49), we observed variable DUX4 expression at single cell level. Interestingly, we found that DUX4 and FRG1 expression are positively correlated. Figure 1C illustrates that DUX4-positive cells have higher FRG1 levels compared with DUX4-negative cells.
Collectively, these findings demonstrate that DUX4 positively regulates the expression of the endogenous FRG1 gene in human muscle cells.
DUX4 associates with FRG1 genomic regions in human muscle cells
As mentioned before, ChIP-seq experiments upon ectopic DUX4 overexpression in control human muscle cells were previously performed (44). We loaded the ChIP-seq results (GSE33838) on the UCSC genome browser and we found that two DUX4 ChIP-seq peaks were present in the human FRG1 genomic area. The first peak (afterwards referred as FRG1 Peak1) was located inside the second intron of the FRG1 gene, while the second peak (FRG1 Peak2) was located at the 3′ end of the gene (Fig. 2A). Intriguingly, inspection of the ChIP-seq tracks belonging to the ENCODE Enhancer- and Promoter-associated histone marks (50,51) revealed that FRG1 Peak1 (but not FRG1 Peak2) displayed a relative enrichment for Enhancer-associated histone marks (H3K4me1 and H3K27Ac) (Supplementary Material, Fig. S2), suggesting that it might belong to an enhancer regulating FRG1 expression. To verify the DUX4 association to the above regions, we electroporated control human muscle cells with a Myc-tagged form of DUX4 (pCMV-Myc-N-DUX4) or the control pCMV-Myc-N empty vector. Next, we performed chromatin immunoprecipitation followed by quantitative PCR (ChIP-qPCR) using anti-DUX4 or anti-Myc tag antibodies (to support the specificity of the ChIP-qPCR signal derived from DUX4), and with control IgG. As shown in Figure 2B and C, with both anti-Myc and anti-DUX4 antibodies we detected a clear DUX4 enrichment over FRG1 Peak1 and Peak2, with respect to control cells. Intriguingly, while DUX4 enrichment in FRG1 Peak1 was comparable to that of the positive controls RFPL2 and TRIM48 (Fig. 2B and D and E, respectively), FRG1 Peak2 signal was the lowest (Fig. 2C).
Figure 2.

DUX4 associates to the FRG1 genomic area. (A) Schematic draw of the FRG1 genomic area. White boxes represent the exons of the FRG1 gene. ChIP-seq peak regions identified by Geng et al. (44) are shown as boxes with stripes. The first peak (FRG1 Peak1) is located inside the second intron, while the second peak (FRG1 Peak2) is at the 3′ end of the gene. Black arrows represent the position of the primers employed for the ChIP-qPCR analysis. (B) Control human muscle cells were electroporated with an expression vector either encoding a Myc-tagged form of DUX4 (pCMV-Myc-N-DUX4) or the corresponding empty vector (pCMV-Myc-N). DUX4 was immunoprecipitated with either α-Myc or α-DUX4, and the signal was detect showing enrichment at FRG1 Peak1 region. (C) DUX4 was present also in FRG1 Peak2 region as well as in the genomic region of the two positive controls (D) RFPL2 and (E) TRIM48. One representative experiment out of three independent experiments is shown. Error bars indicate standard error of the mean.
Thus, our results strongly suggest that DUX4 associates with FRG1 genomic regions to activate its expression in human muscle cells.
DUX4 mediates transactivation of FRG1 peak1 region
To investigate whether DUX4 might function as a FRG1 transcriptional activator through the identified genomic regions, we cloned FRG1 Peak1 or Peak2 regions upstream of the Firefly Luciferase reporter gene (Fig. 3A). We co-transfected the constructs together with pCIneo-DUX4 expression vector or with the corresponding pCIneo empty vector, and with a Renilla Luciferase expression vector to normalize the results. Surprisingly, we found that DUX4 was able to transactivate selectively FRG1 Peak1 region, but not FRG1 Peak2 region (Fig. 3B). Interestingly, inspection of the nucleotide sequence underlying FRG1 Peak regions revealed that the previously described DUX4 consensus binding site (TAAYBBAATCA, IUPAC nomenclature) (44) was present in FRG1 Peak1 (TAATTCAATCA), while FRG1 Peak2 displayed only a partial sequence (TAATGTA), providing a plausible explanation for the differential activity of the two regions. To investigate the functional relevance of our findings, we mutated the DUX4 core motif present in FRG1 Peak1 region. Mutations of FRG1 Peak1 region (Fig. 3A) ablated the transactivation mediated by DUX4 (Fig. 3B) supporting the sequence-specificity of DUX4-mediated transcriptional activation of FRG1 gene expression.
Figure 3.

FRG1 Peak1 region is sufficient for specific DUX4 transactivation. (A) Schematic representation of the constructs employed in luciferase assays. Genomic fragments of the FRG1 gene containing DUX4 putative binding sites were cloned upstream of the SV40 promoter, which controlled the expression of the Luciferase reporter gene. DUX4 core binding motifs within FRG1 genomic regions are reported. The three nucleotides of FRG1 Peak1 region that were mutated to generate FRG1 Peak1-mutated region are underlined. (B) FRG1 Peak1 region is able to transactivate the Luciferase reporter gene in the presence of DUX4, while the mutation in key DUX4 consensus sequence nucleotides abolishes the transactivation. DUX4 is not able to transactivate the Luciferase gene through FRG1 Peak2 region (paired t-test, *P < 0.05, n = 3, mean ± SEM).
DUX4 directly binds to FRG1 peak1 region
Our results indicated that DUX4 could bind specifically to FRG1 Peak1 to transactivate FRG1 expression. To evaluate this hypothesis, oligonucleotide probes containing the regions of interest were tested in an electrophoretic mobility shift assay (EMSA). When a labelled FRG1 Peak1 probe was incubated with an in vitro translated DUX4 protein, a specific, slower migrating protein–DNA complex was formed (Fig. 4A). Competition with an excess of unlabelled FRG1 Peak1 oligo abolished binding, whereas competition with unlabelled FRG1 Peak1 oligo mutated in the DUX4 consensus sequence was ineffective (Fig. 4A). A similar band shift was obtained using the positive control TRIM48 (Fig. 4B), previously shown to be directly bound by DUX4 (44). Intriguingly, no band shift was obtained using FRG1 Peak2 (Fig. 4B), further explaining the lack of activity of this region.
Figure 4.

DUX4 directly binds to FRG1 Peak1. (A) In vitro translated DUX4 protein binds to the labelled FRG1 Peak1 probe containing the TAATTCAATCA core sequence. Competition with unlabelled FRG1 Peak1 probe abolishes DUX4 binding, while competition with an excess of unlabelled FRG1 Peak1-mutated region probe (mutated core sequence TACTTCTATGA) does not. (B) DUX4 incubation with TRIM48 Peak region probe produces a shifted band that indicates direct DNA–protein binding, while FRG1 Peak2 region probe, containing a partial DUX4 core motif (TAATGTA), is not bound by DUX4. One representative experiment out of three independent experiments is shown.
Altogether, our results show that DUX4 binds directly and specifically to human FRG1 Peak1 to activate the expression of this relevant FSHD candidate gene.
DUX4 does not regulate the mouse Frg1 gene
Because DUX4 does not activate the same gene network in mouse muscle cells, it has been suggested that the failure of DUX4 transgenic mice to develop a muscle phenotype could be due to lack of activation of key target genes (28,52). Intriguingly, overexpression of DUX4 does not cause any significant change in the expression of the endogenous Frg1 gene in mouse muscle cells (28,52) or in DUX4 transgenic mice (Supplementary Material, Fig. S3), while the positive control WAP four-disulfide core domain 3 (Wfdc3) gene was up-regulated (28,52) and (Supplementary Material, Fig. S3). Accordingly, the mouse Frg1 genomic locus does not contain any putative DUX4 binding site (data not shown). On the basis of these results, it is tempting to speculate that the failure to activate mouse Frg1 could be one of the reasons underlying lack of muscle phenotype in DUX4 transgenic mice.
Discussion
Despite its extensive study, FSHD pathogenesis remains still unclear and controversial. All current models predict that reduction in D4Z4 copy number results in altered expression of the gene(s) located in the 4q35 genomic locus, leading to the disease (1,53). While the most accepted FSHD candidate gene is DUX4, the molecular mechanism following its up-regulation and leading to the disease remains elusive. Moreover, the level of DUX4 is not consistently altered in FSHD (13,16,18,27). It has been shown that only 50% of FSHD samples display DUX4 overexpression (13,16,18) and that DUX4 transcript and protein can be similarly expressed in cells and muscle tissues derived from FSHD affected subjects and unaffected controls (16,27). Notably, DUX4 expression in FSHD-derived cells is extraordinarily infrequent. Indeed, only 1 in 1000 cells is expressing a relatively abundant amount of DUX4 at any given time (13). Recently, transgenic mice overexpressing DUX4 through a D4Z4 array from an FSHD patient have been reported (28). They recapitulate DUX4 epigenetic regulation and expression pattern typical of FSHD muscles, DUX4 sporadic expression in muscle nuclei and its high expression levels in the germline (28). However, DUX4 mice do not develop any obvious muscle phenotype (28).
Another gene that has been reported overexpressed in FSHD patients is FRG1 (7,14,16,31), but also in this case there is inconsistency (19–22,24).
Intriguingly, transgenic mice overexpressing FRG1 selectively in the skeletal muscle develop a disease with physiological, histological, ultrastructural and molecular features of FSHD. FRG1 overexpressing mice display a progressive muscle dystrophy with differential involvement of muscle types that is strikingly similar to that observed in FSHD, but not in other muscular dystrophies (36,38–40,54). In addition, studies conducted in X. laevis and C. elegans reveal that frg1 is required for normal muscle development and its overexpression causes muscle defects and vascular abnormalities correlated with the clinical findings from FSHD patients (41–43). Further support for an involvement of FRG1 in FSHD is provided by studies of individuals harbouring large genomic deletions in the 4q35 region. For example, some FSHD patients carry deletions that include not only the D4Z4 repeat array, but also eliminates FRG2 and DUX4C (two other FSHD candidate genes) from the disease allele (55,56). This indicates that DUX4C and FRG2 play a minor role in FSHD pathogenesis. In contrast, individuals carrying larger deletion including FRG1 have no phenotypic consequences (57), consistent with a requirement of FRG1 for FSHD.
FSHD is a very heterogeneous disease characterized by an extreme variability in disease onset, progression and severity. An interesting possibility is that the complexity of the disease could reflect heterogeneity in gene expression at the basis of it. Indeed, it is unlikely that the root cause of a so much heterogeneous disease could be attributed to a single gene. Here, we propose that both DUX4 and FRG1 could be both required to fully develop the disease.
While the genetic defect at the basis of FSHD is present in every tissue, an unknown mechanism dictates the disease pathology is predominantly observed in skeletal muscle. Hence, it is relevant to investigate the molecular interaction between DUX4 and FRG1 in this context. We have inspected microarrays and ChIP-seq datasets obtained by ectopically overexpressing DUX4 in control human muscle cells (44). In this setting, DUX4 overexpression leads to a modest, but highly significant, increase in FRG1 expression. Accordingly, our results show that DUX4 overexpression increases the endogenous FRG1 expression in control muscle cells, while DUX4 knockdown causes a reduction in FRG1 expression levels in FSHD muscle cells. So far, all the studies describing DUX4 target genes have been entirely based on ectopic DUX4 manipulation (10,26,28,44,58). Instead, our data indicate that the expression of DUX4 and FRG1 is positively correlated in FSHD muscle cells under native conditions.
The inspection of the human FRG1 genomic locus identified a single consensus DUX4 binding site located inside the second intron of the gene (FRG1 Peak1), a region likely corresponding to an enhancer for the FRG1 gene as determined by the ENCODE Consortium (50,51,59). Accordingly, EMSA and reporter assays show that DUX4 specifically binds and transactivates FRG1 Peak1. Collectively, our results promote FRG1 as direct DUX4 transcriptional target placing the two major FSHD candidate genes in the same circuitry.
In human muscle cells, DUX4 directly activates several genes including germline and early stem cell program genes (44). Notably, it has been shown that DUX4 does not activate the same gene network in mouse muscle cells (28,52). On the basis of these results, it has been suggested that the failure of DUX4 transgenic mice to develop a muscle phenotype could be due to failure to activate key target genes (28,52). Intriguingly, the mouse Frg1 genomic locus is devoid of putative DUX4 binding sites. Accordingly, we and others (28,52) have found that DUX4 is unable to activate mouse Frg1. Consequently, it is tempting to hypothesize that one of the reasons underlying lack of muscle phenotype in DUX4 transgenic mice is the failure to activate mouse Frg1.
In healthy subjects high D4Z4 copy number leads to chromatin compaction at 4q35, while in FSHD patients low D4Z4 copy number is associated with more accessible chromatin. Nevertheless, none of the proteins that have been found associated with the FSHD locus is capable to directly regulate transcription. Hence, one or more transcriptional activators must be involved in directly increasing the transcriptional output of 4q35 genes in FSHD. Our results strongly suggest that DUX4 acts downstream of 4q35 chromatin relaxation to activate FRG1 expression in FSHD, pointing towards a unifying molecular pathway to FSHD pathogenesis.
Materials and Methods
Microarray and ChIP-seq data analysis
Microarray datasets (44) were available on the Gene Expression Omnibus database under accession number GSE33799. To analyze microarrays data, GEO2R web tool (www.ncbi.nlm.nih.gov/geo/geo2r) was used. ChIP-seq dataset (44) was available on the Gene Expression Omnibus database under accession number GSE33838. Custom tracks were analyzed using UCSC Genome Browser (www.genome.ucsc.edu).
Cell culture
All procedures involving human samples were approved by Fondazione San Raffaele del Monte Tabor Ethical Committee.
Immortalized human myoblasts were obtained from the University of Massachusetts Medical School Senator Paul D. Wellstone Muscular Dystrophy Cooperative Research Center for FSHD (http://www.umassmed.edu/wellstone) in Worcester, MA, USA. The immortalized control myoblasts (WS161, 01Ubic CT#5) were derived from the biceps of a healthy 46-year-old male (60). The immortalized FSHD-patient myoblasts (WS227, 15Abic CT#1) were derived from the biceps of a 67-year-old male with mild muscle weakness (60). Immortalized human myoblasts were grown in lox-hygro-hTERT Cdk4-neo (LHCN) medium [4:1 Dulbecco's Modified Eagle's Medium (DMEM):Medium 199 (DMEM-HIGH DMEM, high glucose with sodium pyruvate and l-glutamine; Euroclone) (Gibco) supplemented with 15% fetal bovine serum (FBS) (Gibco), 0.02 m Hydroxyethyl-Piperazine Ethanesulafonic Acid (HEPES), pH 7.2, 0.03 μg/ml ZnSO4, 1.4 μg/ml vitamin B12 (Sigma-Aldrich), 0.055 μg/ml dexamethasone (Sigma-Aldrich), 1% penicillin/streptomycin (100 U/ml final concentration), 2.5 ng/ml hepatocyte growth factor (PeproTech) and 10 ng/ml basic fibroblast growth factor (PeproTech)]. When 90% confluent, cells were switched to differentiation medium [4:1 DMEM:Medium 199 supplemented with 2% horse serum (Euroclone) and 1% penicillin/streptomycin] for 4 days. Medium was replaced with fresh differentiation medium every day. Muscle cells were routinely cultured in a humidified atmosphere at 37°C with 5% O2 and 5% CO2. The culture dishes were coated with 0.1% gelatin (Sigma-Aldrich) (60).
Chinese hamster ovary (CHO) cells were obtained from American Type Culture Collection (ATCC). Cells were maintained in DMEM-HIGH supplemented with 10% FBS and 1% penicillin/streptomycin. HEK293T cells were cultured in Iscove's Modified Dulbecco's Medium (IMDM; Sigma-Aldrich) supplemented with 10% FBS and 1% penicillin/streptomycin. CHO and HEK293T cells were maintained at 37°C in a 5% CO2 incubator.
Cell electroporation
To mediate ectopic DUX4 overexpression, immortalized human control myoblasts were electroporated using Neon Transfection System (Life Technologies) following the manufacturer's protocol. For expression analysis, 500 000 myoblasts were resuspended in 100 μl of buffer R (Life Technologies) and transferred to a tube containing 10 μg of pCIneo or pCIneo-DUX4 expression vectors. For chromatin immunoprecipitation, one million and a half of myoblasts were resuspended in 100 μl of buffer R and transferred to a tube containing 15 μg of pCMV-Myc-N or pCMV-Myc-N-DUX4 expression vectors. Two pulses at 1150 V for 30 ms were performed. After electroporation, 500 000 myoblasts were seeded in 10 cm dish in LHCN medium without antibiotics. Cells were collected after 24 h.
Retrovirus production and transduction
HEK293T cells were seeded in 15 cm dishes 24 h before transfection. The medium was changed 2 h before transfection. The plasmid DNA mix for a dish was prepared by adding 7 μg of VSV-G envelope-encoding plasmid, 16.25 μg of pCMV-gagpol (MLV) plasmid and 35 μg of transfer vector pMLP retroviral shRNA-mir non-targeting control or targeting DUX4 (RLGH-GU19952) (transOMIC). The plasmid solutions were made up to a final volume of 1125 μl with tris-EDTA (TE) and 125 μl of 2.5 m CaCl2 was added. The mixes were incubated for 5 min at RT on rotation. The 2× HEPES-buffered saline, pH 7.12 solution (280 mm NaCl, 100 mm HEPES, 1.5 mm Na2HPO4) was added dropwise to the DNA–TE–CaCl2 mixtures while vortexing at full speed and immediately added to the cells. The medium was replaced after 14 h. Viral supernatants were collected after 30 h, filtered and concentrated by one round of ultracentrifugation for 2 h at 20 000 rpm at 4°C. The viruses were resuspended in phosphate buffer solution (PBS).
For viral transduction, 600 000 immortalized human FSHD myoblasts were seeded in 10 cm dish. Cells were incubated overnight with the virus supplemented with polybrene (final concentration 8 μg/μl; Sigma-Aldrich). The day after, cells were splitted and 48 h post-infection Puromycin selection (final concentration 0.3 μg/ml) was started and it was continued until all non-infected control cells died. Cells were differentiated into myotubes and collected for RNA extraction.
Immunofluorescence
Human primary myoblasts were cultured as previously reported (27). Cells were propagated on gelatin-coated glasses until 90% confluence and then switched to differentiation for 4 days. Immunofluorescence was performed as described in (27). Briefly, cells were rinsed two times with PBS, fixed with 2% paraformaldehyde for 7 min at RT, washed three times with PBS, permeabilized with 1% Triton-X100 in PBS for 15 min and incubated in blocking solution [2% horse serum (Gibco), 2% goat serum (Sigma), 2% bovine serum albumin (BSA) (Sigma) in PBS + 0.1% Triton-X100] for 45 min at RT. Cells were incubated overnight at 4°C with a 1:50 dilution in blocking solution of the E5-5 antibody (61) for DUX4 and a 1:500 dilution of the FRG1 antibody (Abnova, cat. H00002483-B01). The day after, a 1:500 dilution of Alexa Fluor (Life Technologies) secondary antibodies was employed.
RNA extraction, RT-PCR and RT-qPCR
All animal procedures were approved by the Institutional Animal Care and Use Committee of Fondazione San Raffaele del Monte Tabor and were communicated to the Ministry of Health and local authorities according to Italian law.
Total RNA was isolated from tongue muscle using Trizol reagent (Life Technologies). Tissue was disrupted and homogenized using TissueLyser (Qiagen) for 5 min at 50 Hz. Then, RNA extraction was performed by purification with RNA spin columns (PureLink RNA Mini kit, Ambion).
RNA extraction from cells was performed by purification with RNA spin columns (PureLink RNA Mini kit, Ambion), including the digestion with DNAseI (Life Technologies), following the manufacturer's instructions.
Retrotranscription was performed employing SuperScript III first-strand synthesis system for RT-PCR (Life Technologies). Two micrograms of total RNA were mixed with oligo(dT) and the RT-PCR reaction was performed following the manufacturer's protocol. For gene expression analysis, real-time PCR with Sybr GreenER qPCR kit (Life Technologies) was used. Aliquots of 1 or 2 μl of cDNA were used for each reaction. Each sample was run in triplicate and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as housekeeping gene for sample normalization. Real-time PCRs were conducted as follows: initial denaturation 10 min at 95°C, denaturation 30 s at 95°C, annealing 30 s at 58°C, extension 30 s at 72°C and repeated for 40 cycles. The specificity of the amplified products was monitored by performing melting curves at the end of each amplification reaction. Sequence of the primers used are listed in Supplementary Material, Table S1.
Endpoint PCR
For DUX4 full-length isoforms amplification, 0.5 μl of cDNA was employed as template. PCR amplifications were performed with Expand Long Range, dNTPack (Roche) following manufacturer's instructions. Dimethyl sulfoxide was employed in the reaction mix at a final concentration of 8%. PCRs were conducted as follows: initial denaturation 5 min at 94°C, denaturation 30 s at 94°C, annealing 30 s at 62°C, extension 2.5 min at 68°C and repeated for 30 cycles. A single final extension for 7 min at 68°C was included. Previously documented (13) primers 15A and 175 were employed. The amplified products were loaded on a 1% agarose gel.
Chromatin immunoprecipitation
Immortalized human control myoblasts were electroporated with pCMV-Myc-N (Clontech) and pCMV-Myc-N-DUX4 expression vectors. In order to collect chromatin, cells were briefly washed once with PBS and immediately fixed for 10 min at RT in 1% formaldehyde in PBS (from a 37.5% formaldehyde/10% methanol stock; Sigma-Aldrich). After formaldehyde quenching with glycine (final concentration 125 mm) (Sigma-Aldrich) for 2 min, cells were washed three times for 5 min in PBS with gentle swirl. Cells were harvested by using a silicon scraper and cold PBS. Cells were collected and centrifuged at 1350 g for 5 min at 4°C. Two million cells were lysed in 600 μl of LB1 solution [50 mm HEPES–KOH, pH 7.5, 140 mm NaCl, 1 mm ethylenediaminetetraacetic acid (EDTA), 10% glycerol, 0.5% NP-40, 0.25% Triton X-100] and incubated for 10 min on ice. The samples were centrifuged at 1350 g for 5 min at 4°C. The resulting pellet was washed in 600 μl of LB2 solution [10 mm Tris–HCl, pH 8; 200 mm NaCl, 1 mm EDTA, 0.5 mm ethyleneglycolbistetra acetate (EGTA)] with gentle swirl 10 min at RT. Next, samples were centrifuged at 1350 g for 5 min at 4°C and the resulting pellet was lysed in 600 μl of LB3 solution (10 mm Tris–HCl, pH 8; 100 mm NaCl, 1 mm EDTA, 0.5 mm EGTA, 0.1% Na-deoxycholate, 0.5% N-lauroylsarcosine). LB1, LB2 and LB3 solutions were supplemented with protease inhibitor (complete EDTA-free protease inhibitor cocktail tablets; Roche). Lysates were sonicated with Bioruptor (Diagenode). Briefly, 300 μl aliquots of LB3 lysates in eppendorf tubes were sonicated for 10 min (high intensity, 30 s on 30 s off).
An aliquot (55 μl) of the sonicated material was collected to determine the quality of the chromatin by adding 0.1 m NaHCO3, 1% sodium dodecyl sulfate (SDS) (100 μl), 5 μl of Proteinase K (20 mg/ml; Promega) and incubated for 1 h at 55°C for cross-link reversal. Next, samples were precipitated with 5 m LiCl (3.2 μl) and 1 ml 100% EtOH by centrifuging for 30 min at 4°C at 16 360 g. The pellet was washed in 70% EtOH and centrifuged for 10 min at 4°C at 16 360 g. After air dry, the pellet was resuspended in H2O and loaded on 1% agarose gel for electrophoresis. We considered good a chromatin enriched in fragments of 300–500 bp. The samples were quantified with Nanodrop spectrophotometer to determine the concentration of chromatin.
Before starting the ChIP, Triton X-100 was added to chromatin samples at a final concentration of 1% and a centrifugation step of 10 min at 4°C at 16 360 g followed. Fifty micrograms of chromatin were used for each ChIP. For each ChIP, 50 μl of Dynabeads protein G (Life Technologies) were washed three times with 0.5% BSA in PBS and incubated with 10 μg of antibody [anti-c-Myc clone 9E10 (Covance), anti-DUX4 E5-5 (ab124699, Abcam) (61)] in 250 μl of 0.5% BSA in PBS for 2–3 h on rotation at 4°C. Next, the beads–antibody complex was washed three times with 0.5% BSA in PBS and resuspended in 50 μl of 0.5% BSA in PBS. Chromatin and beads–antibody complexes were incubated on rotation overnight at 4°C. The day after, before starting the washes, 5% of the total ChIP volume was taken from the control IgG supernatant as input fraction. Next, six washes of 5 min at 4°C in RIPA buffer (50 mm HEPES–KOH, pH 7.6, 500 mm LiCl, 1 mm EDTA, 1% NP-40, 0.7% Na-deoxycholate) were performed. An additional wash of 5 min on rotation at 4°C in TE buffer (10 mm Tris–HCl, pH 8; 1 mm EDTA) with 50 mm NaCl was performed. Next, samples were centrifuged for 3 min at 1000 g at 4°C. The supernatant was discarded and to the beads–antibody–chromatin complex were added 240 μl of elution buffer (TE buffer with 2% SDS). Samples were incubated in a thermo mixer for 15 min at 65°C with shaking and then centrifuged for 1 min at RT at 16 360 g. The eluted supernatant was transferred to a new tube and samples, together with the input fractions to which three volumes of elution buffer were added, were crosslink-reverted overnight at 65°C. For sample purification, the QIAquick PCR purification kit was used (Qiagen), following the manufacturer's recommendations. DNA was eluted in 50 μl of TE buffer and 1 μl was used in real-time PCR with the qPCR Mix (Promega). Real-time PCR conditions were the same reported in RT-qPCR section. Sequence and annealing temperature of the employed primers are listed in Supplementary Material, Table S2.
Constructs and cloning procedures
Primers used for cloning are listed in Supplementary Material, Table S3. PCR amplifications were performed with Expand Long Range, dNTPack (Roche) or Expand High Fidelity Plus PCR System, dNTPack (Roche). PCR products were digested with the restriction enzymes (New England BioLabs) listed in Supplementary Material, Table S3. For pCMV-Myc-N-DUX4, pCS2-mkgDUX4 expression vector (Addgene) (25) was employed as template. pCMV-Myc (Clontech) was digested with EcoRI and XhoI and DUX4 insert was ligated with T4 DNA Ligase (Promega). For luciferase assays, the pGL3-promoter vector (Promega), which contains the SV40 promoter, was digested with BglII and HindIII (New England BioLabs) and ligated in a previously digested and dephosphorylated pGL4.14 [luc2/Hygro] vector (Promega). FRG1 Peak1 and Peak2 regions were cloned employing human genomic DNA as template. PCR products were digested with the restriction enzymes listed in Supplementary Material, Table S3 and ligated in the pGL4.14-SV40 promoter vector previously digested and dephosphorylated. To clone FRG1 Peak1-mutated region, a mutagenesis by PCR-driven overlap extension approach was used. Briefly, two flanking primers that marked the 5′ ends of both strands and two internal overlapping primers that contained the desired mutations were designed. An initial PCR generated overlapping gene segments containing the desired mutations that were then used as template DNA for another PCR to create a full-length product. All the inserts were fully sequenced.
Luciferase reporter assay
CHO cells were transfected with Lipofectamine LTX (Life Technologies) following manufacturer's protocol. Specifically, cells were co-transfected with combinations of three different plasmids: pGL4.14-SV40 promoter Firefly Luciferase Reporter vector (empty or carrying FRG1 Peak1, FRG1 Peak1-mutated or FRG1 Peak2 regions upstream of SV40 promoter), pCIneo or pCIneo-DUX4 expression vectors (10) and pGL4.73 [hRluc/SV40] Renilla Luciferase Reporter vector (Promega) in a ratio of 3:1:1, respectively. Twenty-four hours after transfection, cells were lysed in Passive Lysis Buffer (Promega) and both the Firefly and Renilla Luciferase activities were quantified using Dual-Luciferase Reporter Assay System (Promega). Biological triplicates were performed. Luciferase data for each region were normalized to the corresponding samples transfected with pCIneo vector.
EMSA
For band-shift assays, 3′ end biotin-labelled oligonucleotides listed in Supplementary Material, Table S4 were employed. Complementary oligonucleotides were annealed by mixing together at a 1:1 molar ratio and incubated in boiling water for 5 min. Then, they were slowly cooled to RT.
The cell-free expression system TNT SP6 High-Yield Wheat Germ Protein Expression System (Promega) was used to produce the recombinant DUX4 protein. The plasmids employed as templates were the empty pCS2+ and the pCS2-mkgDUX4 expression vectors (Addgene) (25). For protein synthesis, 5 μg of the appropriate DNA template were used according to manufacturer's instructions. In vitro produced DUX4 protein was verified by sodium dodecyl sulfate polyacrylamide gel electrophoresis, followed by immunoblotting. Twenty femtomoles of probes were incubated with 5 μl of in vitro translated in the presence of 1 μg of poly(dI:dC) and 5 μg of herring sperm in a buffer containing 10 mm Tris, pH 7.5, 0.1 mm EDTA, 1 mm DTT, 100 mm KCl, 3 mm MgCl2, 12% glycerol (62). For oligonucleotide competition assays, two hundred times wild-type or mutated double-stranded unlabelled probes were added to the sample before complex formation.
After 30 min incubation at RT, DNA–protein complexes were separated by electrophoresis in 10% (w/v) acrylamide gels formed in 0.5X TBE (Sigma-Aldrich) and ran in the same buffer at 4 mA at 4°C. Then, the gels were transferred on a Biodyne nylon membrane (Thermo Scientific) at 380 mA for 45 min at 4°C. The membrane was crosslinked at 120 mJ/cm2 with an UV-Stratalinker and the biotin-labelled DNA was detected by chemiluminescence using the LightShift Chemiluminescent EMSA Kit (Thermo Scientific) following the manufacturer's protocol. Autoradiography films were used to detect biotin signal.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 5.0a (GraphPad Software, San Diego, USA). Statistical significance was calculated by Student's t-test on at least three independent experiments; P-value: *P < 0.05; **P < 0.01; ***P < 0.001. Details of each dataset are provided in the corresponding figure legends.
Authors' Contributions
G.F. designed and performed experiments, analyzed results and wrote the manuscript. C.H.H. grew control and FSHD muscle cells. R.C. handled mice. D.G. designed experiments, analyzed results and wrote the manuscript.
Supplementary Material
Funding
This work was supported by Italian Telethon Foundation, Association Française contre les Myopathies, ERA-Net for Research on Rare Diseases (E-Rare-2), European Research Council, Italian Epigenomics Flagship Project, Italian Ministry of Health, FSHD Global Research Foundation and Stichting FSHD. Funding to pay the Open Access publication charges for this article was provided by Italian Telethon Foundation.
Supplementary Material
Acknowledgements
This work is a partial fulfilment of G.F. and C.H.H. International PhD Course in Molecular Medicine, Curriculum in Cellular and Molecular Biology, Vita-Salute San Raffaele University, Milan, Italy. We thank D. Cesana and P. Gallina for technical assistance during retroviral vectors production and M. Riba for the assistance during microarrays analysis. We are grateful to M.V. Neguembor and San Raffaele Alembic Bio Imaging Center for assistance during immunofluorescence images acquisition. D.G. is a Dulbecco Telethon Institute Senior Scientist.
Conflict of Interest statement. None declared.
References
- 1.Cabianca D.S., Gabellini D. The cell biology of disease: FSHD: copy number variations on the theme of muscular dystrophy. J. Cell. Biol. 2010;191:1049–1060. doi: 10.1083/jcb.201007028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deenen J.C., Arnts H., van der Maarel S.M., Padberg G.W., Verschuuren J.J., Bakker E., Weinreich S.S., Verbeek A.L., van Engelen B.G. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology. 2014;83:1056–1059. doi: 10.1212/WNL.0000000000000797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pandya S., King W.M., Tawil R. Facioscapulohumeral dystrophy. Phys. Ther. 2008;88:105–113. doi: 10.2522/ptj.20070104. [DOI] [PubMed] [Google Scholar]
- 4.Wijmenga C., Frants R.R., Brouwer O.F., Moerer P., Weber J.L., Padberg G.W. Location of facioscapulohumeral muscular dystrophy gene on chromosome 4. Lancet. 1990;336:651–653. doi: 10.1016/0140-6736(90)92148-b. [DOI] [PubMed] [Google Scholar]
- 5.van Deutekom J.C., Wijmenga C., van Tienhoven E.A., Gruter A.M., Hewitt J.E., Padberg G.W., van Ommen G.J., Hofker M.H., Frants R.R. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum. Mol. Genet. 1993;2:2037–2042. doi: 10.1093/hmg/2.12.2037. [DOI] [PubMed] [Google Scholar]
- 6.Cabianca D.S., Casa V., Bodega B., Xynos A., Ginelli E., Tanaka Y., Gabellini D. A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in FSHD muscular dystrophy. Cell. 2012;149:819–831. doi: 10.1016/j.cell.2012.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gabellini D., Green M.R., Tupler R. Inappropriate gene activation in FSHD: a repressor complex binds a chromosomal repeat deleted in dystrophic muscle. Cell. 2002;110:339–348. doi: 10.1016/s0092-8674(02)00826-7. [DOI] [PubMed] [Google Scholar]
- 8.Rijkers T., Deidda G., van Koningsbruggen S., van Geel M., Lemmers R.J., van Deutekom J.C., Figlewicz D., Hewitt J.E., Padberg G.W., Frants R.R., et al. FRG2, an FSHD candidate gene, is transcriptionally upregulated in differentiating primary myoblast cultures of FSHD patients. J. Med. Genet. 2004;41:826–836. doi: 10.1136/jmg.2004.019364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laoudj-Chenivesse D., Carnac G., Bisbal C., Hugon G., Bouillot S., Desnuelle C., Vassetzky Y., Fernandez A. Increased levels of adenine nucleotide translocator 1 protein and response to oxidative stress are early events in facioscapulohumeral muscular dystrophy muscle. J. Mol. Med. 2005;83:216–224. doi: 10.1007/s00109-004-0583-7. [DOI] [PubMed] [Google Scholar]
- 10.Dixit M., Ansseau E., Tassin A., Winokur S., Shi R., Qian H., Sauvage S., Matteotti C., van Acker A.M., Leo O., et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc. Natl. Acad. Sci. USA. 2007;104:18157–18162. doi: 10.1073/pnas.0708659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ansseau E., Laoudj-Chenivesse D., Marcowycz A., Tassin A., Vanderplanck C., Sauvage S., Barro M., Mahieu I., Leroy A., Leclercq I., et al. DUX4c is up-regulated in FSHD. It induces the MYF5 protein and human myoblast proliferation. PLoS One. 2009;4:e7482. doi: 10.1371/journal.pone.0007482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lemmers R.J., van der Vliet P.J., Klooster R., Sacconi S., Camano P., Dauwerse J.G., Snider L., Straasheijm K.R., van Ommen G.J., Padberg G.W., et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010;329:1650–1653. doi: 10.1126/science.1189044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Snider L., Geng L.N., Lemmers R.J., Kyba M., Ware C.B., Nelson A.M., Tawil R., Filippova G.N., van der Maarel S.M., Tapscott S.J., et al. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet. 2010;6:e1001181. doi: 10.1371/journal.pgen.1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neguembor M.V., Xynos A., Onorati M.C., Caccia R., Bortolanza S., Godio C., Pistoni M., Corona D.F., Schotta G., Gabellini D. FSHD muscular dystrophy region gene 1 binds Suv4-20h1 histone methyltransferase and impairs myogenesis. J. Mol. Cell Biol. 2013;5:294–307. doi: 10.1093/jmcb/mjt018. [DOI] [PubMed] [Google Scholar]
- 15.Caruso N., Herberth B., Bartoli M., Puppo F., Dumonceaux J., Zimmermann A., Denadai S., Lebosse M., Roche S., Geng L., et al. Deregulation of the protocadherin gene FAT1 alters muscle shapes: implications for the pathogenesis of facioscapulohumeral dystrophy. PLoS Genet. 2013;9:e1003550. doi: 10.1371/journal.pgen.1003550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Broucqsault N., Morere J., Gaillard M.C., Dumonceaux J., Torrents J., Salort-Campana E., Maues De Paula A., Bartoli M., Fernandez C., Chesnais A.L., et al. Dysregulation of 4q35- and muscle-specific genes in fetuses with a short D4Z4 array linked to facio-scapulo-humeral dystrophy. Hum. Mol. Genet. 2013;22:4206–4214. doi: 10.1093/hmg/ddt272. [DOI] [PubMed] [Google Scholar]
- 17.Winokur S.T., Barrett K., Martin J.H., Forrester J.R., Simon M., Tawil R., Chung S.A., Masny P.S., Figlewicz D.A. Facioscapulohumeral muscular dystrophy (FSHD) myoblasts demonstrate increased susceptibility to oxidative stress. Neuromuscul. Disord. 2003;13:322–333. doi: 10.1016/s0960-8966(02)00284-5. [DOI] [PubMed] [Google Scholar]
- 18.Tsumagari K., Chang S.C., Lacey M., Baribault C., Chittur S.V., Sowden J., Tawil R., Crawford G.E., Ehrlich M. Gene expression during normal and FSHD myogenesis. BMC Med Genomics. 2011;4:67. doi: 10.1186/1755-8794-4-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Winokur S.T., Chen Y.W., Masny P.S., Martin J.H., Ehmsen J.T., Tapscott S.J., van der Maarel S.M., Hayashi Y., Flanigan K.M. Expression profiling of FSHD muscle supports a defect in specific stages of myogenic differentiation. Hum. Mol. Genet. 2003;12:2895–2907. doi: 10.1093/hmg/ddg327. [DOI] [PubMed] [Google Scholar]
- 20.Jiang G., Yang F., van Overveld P.G., Vedanarayanan V., van der Maarel S., Ehrlich M. Testing the position-effect variegation hypothesis for facioscapulohumeral muscular dystrophy by analysis of histone modification and gene expression in subtelomeric 4q. Hum. Mol. Genet. 2003;12:2909–2921. doi: 10.1093/hmg/ddg323. [DOI] [PubMed] [Google Scholar]
- 21.Klooster R., Straasheijm K., Shah B., Sowden J., Frants R., Thornton C., Tawil R., van der Maarel S. Comprehensive expression analysis of FSHD candidate genes at the mRNA and protein level. Eur. J. Hum. Genet. 2009;17:1615–1624. doi: 10.1038/ejhg.2009.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Masny P.S., Chan O.Y., de Greef J.C., Bengtsson U., Ehrlich M., Tawil R., Lock L.F., Hewitt J.E., Stocksdale J., Martin J.H., et al. Analysis of allele-specific RNA transcription in FSHD by RNA-DNA FISH in single myonuclei. Eur. J. Hum. Genet. 2010;18:448–456. doi: 10.1038/ejhg.2009.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arashiro P., Eisenberg I., Kho A.T., Cerqueira A.M., Canovas M., Silva H.C., Pavanello R.C., Verjovski-Almeida S., Kunkel L.M., Zatz M. Transcriptional regulation differs in affected facioscapulohumeral muscular dystrophy patients compared to asymptomatic related carriers. Proc. Natl. Acad. Sci. USA. 2009;106:6220–6225. doi: 10.1073/pnas.0901573106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Osborne R.J., Welle S., Venance S.L., Thornton C.A., Tawil R. Expression profile of FSHD supports a link between retinal vasculopathy and muscular dystrophy. Neurology. 2007;68:569–577. doi: 10.1212/01.wnl.0000251269.31442.d9. [DOI] [PubMed] [Google Scholar]
- 25.Snider L., Asawachaicharn A., Tyler A.E., Geng L.N., Petek L.M., Maves L., Miller D.G., Lemmers R.J., Winokur S.T., Tawil R., et al. RNA transcripts, miRNA-sized fragments and proteins produced from D4Z4 units: new candidates for the pathophysiology of facioscapulohumeral dystrophy. Hum. Mol. Genet. 2009;18:2414–2430. doi: 10.1093/hmg/ddp180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yao Z., Snider L., Balog J., Lemmers R.J., Van Der Maarel S.M., Tawil R., Tapscott S.J. DUX4-induced gene expression is the major molecular signature in FSHD skeletal muscle. Hum. Mol. Genet. 2014;20:5342–5352. doi: 10.1093/hmg/ddu251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones T.I., Chen J.C., Rahimov F., Homma S., Arashiro P., Beermann M.L., King O.D., Miller J.B., Kunkel L.M., Emerson C.P., Jr., et al. Facioscapulohumeral muscular dystrophy family studies of DUX4 expression: evidence for disease modifiers and a quantitative model of pathogenesis. Hum. Mol. Genet. 2012;21:4419–4430. doi: 10.1093/hmg/dds284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krom Y.D., Thijssen P.E., Young J.M., den Hamer B., Balog J., Yao Z., Maves L., Snider L., Knopp P., Zammit P.S., et al. Intrinsic epigenetic regulation of the D4Z4 macrosatellite repeat in a transgenic mouse model for FSHD. PLoS Genet. 2013;9:e1003415. doi: 10.1371/journal.pgen.1003415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitsuhashi H., Mitsuhashi S., Lynn-Jones T., Kawahara G., Kunkel L.M. Expression of DUX4 in zebrafish development recapitulates facioscapulohumeral muscular dystrophy. Hum. Mol. Genet. 2012;3:568–577. doi: 10.1093/hmg/dds467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Deutekom J.C., Lemmers R.J., Grewal P.K., van Geel M., Romberg S., Dauwerse H.G., Wright T.J., Padberg G.W., Hofker M.H., Hewitt J.E., et al. Identification of the first gene (FRG1) from the FSHD region on human chromosome 4q35. Hum. Mol. Genet. 1996;5:581–590. doi: 10.1093/hmg/5.5.581. [DOI] [PubMed] [Google Scholar]
- 31.Bodega B., Ramirez G.D., Grasser F., Cheli S., Brunelli S., Mora M., Meneveri R., Marozzi A., Mueller S., Battaglioli E., et al. Remodeling of the chromatin structure of the facioscapulohumeral muscular dystrophy (FSHD) locus and upregulation of FSHD-related gene 1 (FRG1) expression during human myogenic differentiation. BMC Biol. 2009;7:41. doi: 10.1186/1741-7007-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanel M.L., Sun C.Y., Jones T.I., Long S.W., Zanotti S., Milner D., Jones P.L. Facioscapulohumeral muscular dystrophy (FSHD) region gene 1 (FRG1) is a dynamic nuclear and sarcomeric protein. Differentiation. 2011;81:107–118. doi: 10.1016/j.diff.2010.09.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Koningsbruggen S., Dirks R.W., Mommaas A.M., Onderwater J.J., Deidda G., Padberg G.W., Frants R.R., van der Maarel S.M. FRG1P is localised in the nucleolus, Cajal bodies, and speckles. J. Med. Genet. 2004;41:e46. doi: 10.1136/jmg2003.012781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun C.Y., van Koningsbruggen S., Long S.W., Straasheijm K., Klooster R., Jones T.I., Bellini M., Levesque L., Brieher W.M., van der Maarel S.M., et al. Facioscapulohumeral muscular dystrophy region gene 1 is a dynamic RNA-associated and actin-bundling protein. J. Mol. Biol. 2011;411:397–416. doi: 10.1016/j.jmb.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Koningsbruggen S., Straasheijm K.R., Sterrenburg E., de Graaf N., Dauwerse H.G., Frants R.R., van der Maarel S.M. FRG1P-mediated aggregation of proteins involved in pre-mRNA processing. Chromosoma. 2007;116:53–64. doi: 10.1007/s00412-006-0083-3. [DOI] [PubMed] [Google Scholar]
- 36.Gabellini D., D'Antona G., Moggio M., Prelle A., Zecca C., Adami R., Angeletti B., Ciscato P., Pellegrino M.A., Bottinelli R., et al. Facioscapulohumeral muscular dystrophy in mice overexpressing FRG1. Nature. 2006;439:973–977. doi: 10.1038/nature04422. [DOI] [PubMed] [Google Scholar]
- 37.Pistoni M., Shiue L., Cline M.S., Bortolanza S., Neguembor M.V., Xynos A., Ares M., Jr., Gabellini D. Rbfox1 downregulation and altered calpain 3 splicing by FRG1 in a mouse model of facioscapulohumeral muscular dystrophy (FSHD) PLoS Genet. 2013;9:e1003186. doi: 10.1371/journal.pgen.1003186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sancisi V., Germinario E., Esposito A., Morini E., Peron S., Moggio M., Tomelleri G., Danieli-Betto D., Tupler R. Altered Tnnt3 characterizes selective weakness of fast fibers in mice overexpressing FSHD region gene 1 (FRG1) Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014;306:R124–R137. doi: 10.1152/ajpregu.00379.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen S.C., Frett E., Marx J., Bosnakovski D., Reed X., Kyba M., Kennedy B.K. Decreased proliferation kinetics of mouse myoblasts overexpressing FRG1. PLoS One. 2011;6:e19780. doi: 10.1371/journal.pone.0019780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xynos A., Neguembor M.V., Caccia R., Licastro D., Nonis A., Di Serio C., Stupka E., Gabellini D. Overexpression of facioscapulohumeral muscular dystrophy region gene 1 causes primary defects in myogenic stem cells. J. Cell. Sci. 2013;126:2236–2245. doi: 10.1242/jcs.121533. [DOI] [PubMed] [Google Scholar]
- 41.Hanel M.L., Wuebbles R.D., Jones P.L. Muscular dystrophy candidate gene FRG1 is critical for muscle development. Dev. Dyn. 2009;238:1502–1512. doi: 10.1002/dvdy.21830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wuebbles R.D., Hanel M.L., Jones P.L. FSHD region gene 1 (FRG1) is crucial for angiogenesis linking FRG1 to facioscapulohumeral muscular dystrophy-associated vasculopathy. Dis. Model. Mech. 2009;2:267–274. doi: 10.1242/dmm.002261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Q., Jones T.I., Tang V.W., Brieher W.M., Jones P.L. Facioscapulohumeral muscular dystrophy region gene-1 (FRG-1) is an actin-bundling protein associated with muscle-attachment sites. J. Cell. Sci. 2010;123:1116–1123. doi: 10.1242/jcs.058958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Geng L.N., Yao Z., Snider L., Fong A.P., Cech J.N., Young J.M., van der Maarel S.M., Ruzzo W.L., Gentleman R.C., Tawil R., et al. DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev. Cell. 2012;22:38–51. doi: 10.1016/j.devcel.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bonnefont J., Nikolaev S.I., Perrier A.L., Guo S., Cartier L., Sorce S., Laforge T., Aubry L., Khaitovich P., Peschanski M., et al. Evolutionary forces shape the human RFPL1,2,3 genes toward a role in neocortex development. Am. J. Hum. Genet. 2008;83:208–218. doi: 10.1016/j.ajhg.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stanghellini I., Falco G., Lee S.L., Monti M., Ko M.S. Trim43a, Trim43b, and Trim43c: novel mouse genes expressed specifically in mouse preimplantation embryos. Gene Expr. Patterns. 2009;9:595–602. doi: 10.1016/j.gep.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ferreboeuf M., Mariot V., Bessieres B., Vasiljevic A., Attie-Bitach T., Collardeau S., Morere J., Roche S., Magdinier F., Robin-Ducellier J., et al. DUX4 and DUX4 downstream target genes are expressed in fetal FSHD muscles. Hum. Mol. Genet. 2014;23:171–181. doi: 10.1093/hmg/ddt409. [DOI] [PubMed] [Google Scholar]
- 48.Tassin A., Laoudj-Chenivesse D., Vanderplanck C., Barro M., Charron S., Ansseau E., Chen Y.W., Mercier J., Coppee F., Belayew A. DUX4 expression in FSHD muscle cells: how could such a rare protein cause a myopathy? J. Cell. Mol. Med. 2013;17:76–89. doi: 10.1111/j.1582-4934.2012.01647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krom Y.D., Dumonceaux J., Mamchaoui K., den Hamer B., Mariot V., Negroni E., Geng L.N., Martin N., Tawil R., Tapscott S.J., et al. Generation of isogenic D4Z4 contracted and noncontracted immortal muscle cell clones from a mosaic patient: a cellular model for FSHD. Am. J. Pathol. 2012;181:1387–1401. doi: 10.1016/j.ajpath.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heintzman N.D., Hon G.C., Hawkins R.D., Kheradpour P., Stark A., Harp L.F., Ye Z., Lee L.K., Stuart R.K., Ching C.W., et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heintzman N.D., Stuart R.K., Hon G., Fu Y., Ching C.W., Hawkins R.D., Barrera L.O., Van Calcar S., Qu C., Ching K.A., et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 52.Sharma V., Harafuji N., Belayew A., Chen Y.W. DUX4 differentially regulates transcriptomes of human rhabdomyosarcoma and mouse C2C12 cells. PLoS One. 2013;8:e64691. doi: 10.1371/journal.pone.0064691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van der Maarel S.M., Tawil R., Tapscott S.J. Facioscapulohumeral muscular dystrophy and DUX4: breaking the silence. Trends Mol. Med. 2011;17:252–258. doi: 10.1016/j.molmed.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.D'Antona G., Brocca L., Pansarasa O., Rinaldi C., Tupler R., Bottinelli R. Structural and functional alterations of muscle fibres in the novel mouse model of facioscapulohumeral muscular dystrophy. J. Physiol. 2007;584:997–1009. doi: 10.1113/jphysiol.2007.141481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deak K.L., Lemmers R.J., Stajich J.M., Klooster R., Tawil R., Frants R.R., Speer M.C., van der Maarel S.M., Gilbert J.R. Genotype-phenotype study in an FSHD family with a proximal deletion encompassing p13E-11 and D4Z4. Neurology. 2007;68:578–582. doi: 10.1212/01.wnl.0000254991.21818.f3. [DOI] [PubMed] [Google Scholar]
- 56.Lemmers R.J., Osborn M., Haaf T., Rogers M., Frants R.R., Padberg G.W., Cooper D.N., van der Maarel S.M., Upadhyaya M. D4F104S1 deletion in facioscapulohumeral muscular dystrophy: phenotype, size, and detection. Neurology. 2003;61:178–183. doi: 10.1212/01.wnl.0000078889.51444.81. [DOI] [PubMed] [Google Scholar]
- 57.Tupler R., Berardinelli A., Barbierato L., Frants R., Hewitt J.E., Lanzi G., Maraschio P., Tiepolo L. Monosomy of distal 4q does not cause facioscapulohumeral muscular dystrophy. J. Med. Genet. 1996;33:366–370. doi: 10.1136/jmg.33.5.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Young J.M., Whiddon J.L., Yao Z., Kasinathan B., Snider L., Geng L.N., Balog J., Tawil R., van der Maarel S.M., Tapscott S.J. DUX4 binding to retroelements creates promoters that are active in FSHD muscle and testis. PLoS Genet. 2013;9:e1003947. doi: 10.1371/journal.pgen.1003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- 60.Homma S., Chen J.C., Rahimov F., Beermann M.L., Hanger K., Bibat G.M., Wagner K.R., Kunkel L.M., Emerson C.P., Jr., Miller J.B. A unique library of myogenic cells from facioscapulohumeral muscular dystrophy subjects and unaffected relatives: family, disease and cell function. Eur. J. Hum. Genet. 2012;20:404–410. doi: 10.1038/ejhg.2011.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Geng L.N., Tyler A.E., Tapscott S.J. Immunodetection of human double homeobox 4. Hybridoma (Larchmt) 2011;30:125–130. doi: 10.1089/hyb.2010.0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Knoepfler P.S., Bergstrom D.A., Uetsuki T., Dac-Korytko I., Sun Y.H., Wright W.E., Tapscott S.J., Kamps M.P. A conserved motif N-terminal to the DNA-binding domains of myogenic bHLH transcription factors mediates cooperative DNA binding with pbx-Meis1/Prep1. Nucleic Acids Res. 1999;27:3752–3761. doi: 10.1093/nar/27.18.3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.