

Abstract
Parkinson's disease (PD) is characterized by selective degeneration of dopaminergic neurons. Although the etiology of PD remains incompletely understood, oxidative stress has been implicated as an important contributor in the development of PD. Oxidative stress can lead to oxidation and functional perturbation of proteins critical to neuronal survival. Glutaredoxin 1 (Grx1) is an evolutionally conserved antioxidant enzyme that repairs protein oxidation by reversing the oxidative modification of cysteine known as S-glutathionylation. We aimed to explore the regulatory role of Grx1 in PD. We first examined the levels of Grx1 in postmortem midbrain samples from PD patients, and observed that Grx1 content is decreased in PD, specifically within the dopaminergic neurons. We subsequently investigated the potential role of Grx1 deficiency in PD pathogenesis by examining the consequences of loss of the Caenorhabditis elegans Grx1 homolog in well-established worm models of familial PD caused by overexpression of pathogenic human LRRK2 mutants G2019S or R1441C. We found that loss of the Grx1 homolog led to significant exacerbation of the neurodegenerative phenotype in C. elegans overexpressing the human LRRK2 mutants. Re-expression in the dopaminergic neurons of the active, but not a catalytically inactive form of the Grx1 homolog rescued the exacerbated phenotype. Loss of the Grx1 homolog also exacerbated the neurodegenerative phenotype in other C. elegans models, including overexpression of human α-synuclein and overexpression of tyrosine hydroxylase (a model of sporadic PD). Therefore, our results reveal a novel neuroprotective role of glutaredoxin against dopaminergic neurodegeneration in models of familial and sporadic PD.
Introduction
Parkinson's disease (PD) is the second most common neurodegenerative disease in the world, characterized by selective loss of dopaminergic neurons in the substantia nigra of the midbrain. Although the etiology of PD is not completely understood, perturbation of redox homeostasis leading to oxidative stress appears to be an important contributor in the development of both sporadic and familial forms of the disease (1–5). Dopaminergic neurons are particularly vulnerable to oxidative stress due to the propensity of dopamine to auto-oxidize, generating reactive oxygen species (ROS). The sensitivity of dopaminergic neurons to ROS driven by excessive dopamine turnover has been used as a model of sporadic PD. Direct injection of dopamine into rat brains striatum resulted in neuronal cell death (6), and treatment of model dopaminergic neurons (SH-SY5Y cells in culture) with dopamine resulted in both apoptosis and autophagy (7,8). Studies on human populations have identified several gene alterations linked to familial PD. Among these alterations, missense mutations in leucine-rich repeat kinase 2 (LRRK2) and multiplication of the α-synuclein gene (SNCA) are autosomal dominant causes of the disease (9). Overexpression of mutant LRRK2 protein and wild–type (WT) α-synuclein in cell culture have been observed to induce oxidative stress (10–13).
Oxidative stress can lead to oxidation and functional perturbation of proteins critical to neuronal survival. S-Glutathionylation is a prevalent oxidative modification involving mixed disulfide bond formation between glutathione (GSH) and specific cysteine residues on proteins (protein-SSG) (14). Glutaredoxin 1 (Grx1) is an evolutionally conserved antioxidant enzyme that repairs protein oxidation by catalyzing the reversal of S-glutathionylation. Reversible protein S-glutathionylation serves two key functions, homeostatic protection of proteins from irreversible oxidation and redox signal transduction. Thus, conversion of protein-SOH to protein-SSG and reversal by Grx1 precludes further irreversible oxidation to protein-SO3 (15). Protein-SSG formation also represents an important intermediate in redox regulation of signaling pathways, resulting in changes in protein activity and/or subcellular localization (16). By regulating the dynamic status of protein S-glutathionylation, Grx1 has been shown to play a key role in cell survival in various contexts, including models of PD.
We previously reported that knockdown of Grx1 via siRNA resulted in increased apoptosis of model dopaminergic neurons (SHSY-5Y cells) (4). Others have shown that overexpression of Grx1 in the SK-N-SH dopaminergic cell line attenuates cell death caused by the oxidative stressors paraquat and 6-hydroxydopamine (6-OHDA) (17). In addition to direct evidence of Grx1-mediated neuronal protection in cell culture, other studies have suggested a neuroprotective role of Grx1 in vivo. Knockdown of Grx1 via antisense RNA in mice revealed that Grx1 provided protection against inactivation of mitochondrial complex I in the striatum and frontal cortex when mice were acutely treated with the toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (18). However, no data were presented regarding alterations in dopaminergic viability (18). It has also been reported that female mice express higher levels of Grx1 protein within the midbrain compared with male mice. Correlatively, female mice are more resistant to MPTP toxicity compared with their male counterparts (19). While these reports suggest a potential neuroprotective role for Grx1, no direct evidence has been presented definitively showing that Grx1 modulates dopaminergic survival in vivo.
In the present study, we first examined whether Grx1 levels were altered in brain tissue from PD patients. Analysis of homogenates of postmortem midbrain samples from PD and non-PD patients showed a diminished level of total Grx1 protein for the PD samples. Furthermore, analysis of fixed midbrain tissue revealed selective loss of Grx1 in dopaminergic neurons in PD samples compared with non-PD controls. These analyses provided initial evidence of Grx1 deficiency in human PD brain tissue. In order to elucidate the regulatory role of Grx1 in dopaminergic viability under basal and PD-relevant stress conditions in an in vivo setting, we took advantage of the model organism Caenorhabditis elegans. The nematode C. elegans has been used effectively to study neurodegenerative diseases, because of the conserved dopaminergic pathway, the ease of genetic manipulation and short lifespan (20–22). We previously demonstrated that dopaminergic expression of familial PD forms of LRRK2 (either G2019S-LRRK2 or R1441C-LRRK2) in C. elegans results in a PD-like phenotype characterized by age-dependent impairment of dopamine-dependent behavior and loss of dopaminergic neurons (23). Also, other models of PD have been characterized in C. elegans. For example, dopaminergic overexpression of either human α-synuclein or nematode tyrosine hydroxylase results in neurodegeneration (24,25). Overexpression of α-synuclein in C. elegans is a model of PD that has characteristics of both familial and sporadic PD. This dual feature is based on the role of α-synuclein in Lewy body formation, a characteristic of sporadic PD, whereas multiplication of the SNCA causes a form of familial PD (11,26,27). Addition of excess dopamine has been shown to drive oxidative stress and neuronal cell death (6–8), prominent features of sporadic PD. Thus tyrosine hydroxylase overexpression leading to increased dopamine production in the dopaminergic neurons of C. elegans represents an in vivo model of sporadic PD. To simulate a deficiency in Grx1 in vivo analogous to the observations with human PD brain samples, we used C. elegans lacking GLRX-10, the homolog of Grx1. We crossed the glrx-10 null line with several distinct C. elegans models of PD. We found that loss of GLRX-10 exacerbated neurodegenerative phenotypes in three different models of PD namely overexpression of pathogenic LRRK2, α-synuclein or tyrosine hydroxylase. Re-expression of WT GLRX-10, but not the catalytically dead form of the enzyme, in the dopaminergic neurons reversed the effect of the glrx-10 knockout. These data suggest that disruption of redox homeostasis in the dopaminergic neurons via loss of glutaredoxin predisposes to neurodegeneration, suggesting a neuroprotective role for glutaredoxin in both sporadic and familial PD.
Results
Grx1 protein levels are diminished in midbrain dopaminergic neurons of PD
We and others have shown that Grx1 plays an important role in maintaining cell viability in model dopaminergic neurons in culture (4,17). However, the relative levels of Grx1 in PD tissue have not been reported previously. Therefore, we compared Grx1 protein levels in postmortem brains of PD patients and non-PD subjects (see subject information in Supplementary Material, Table S1). Representative western blot analysis of midbrain homogenates from PD and control cases probed with antibodies against Grx1 and actin (loading control) are shown in Figure 1A (with corresponding full scans of western blots in Supplementary Material, Fig. S1A). Relative Grx1 levels (normalized to actin) for all samples analyzed (n = 9 for control, and n = 8 for PD) are depicted graphically in Figure 1B. We observed a significant decrease in Grx1 content in the midbrain samples of PD patients relative to non-PD controls (Fig. 1B). To analyze the Grx1 content specifically within the dopaminergic neurons, consecutive sections of paraffin-embedded midbrain tissue from PD and non-PD controls (see subject information in Supplementary Material, Table S2) were immunohistochemically labeled with antibodies against Grx1 and tyrosine hydroxylase (TH, a marker for dopaminergic neurons). Owing to the appearance of higher molecular weight bands observed on the western blot (Supplementary Material, Fig. S1A), the suitability of the Grx1 antibody for immunohistochemical analysis was examined. Thus, we performed parallel staining with the Grx1 antibody that was either untreated or pre-absorbed with recombinant Grx1. Pre-absorption resulted in substantial reduction in staining compared with the non-absorbed antibody (Supplemental Material, Fig. S1B). These data indicate a high degree of selectivity of the antibody for Grx1 in immunohistochemical applications. Representative immunostaining from one PD patient and one control subject are shown in Figure 1C. Cells identified with solid arrows represent neurons stained for both TH and Grx1, whereas cells identified with hollow arrows represent neurons that stained for TH, but not Grx1. We then quantified the number of TH-positive/Grx1-negative neurons relative to TH-positive/Grx1-positive neurons for all immunostained cases (n = 4 for control and n = 5 for PD; Fig. 1D). Our analysis showed that the PD samples had a significantly higher fraction of Grx1-deficient dopaminergic (TH-positive) neurons compared with controls (Fig. 1D). It is conceivable that a protein other than Grx1 that reacts with the anti-Grx1 antibody is selectivity diminished in TH-positive neurons of midbrain samples from PD patients. However, it is much more likely that the diminution in signal corresponds to a diminution in human Grx1, the antigen that was shown to be selectively reactive with the Grx1 antibody (Supplemental Material, Fig. S1B). Hence, we conclude that the data of Figure 1C and D reveal decreased Grx1 content in the surviving dopaminergic neurons in human PD samples. These results provide novel evidence of Grx1 deficiency in human PD.
Figure 1.
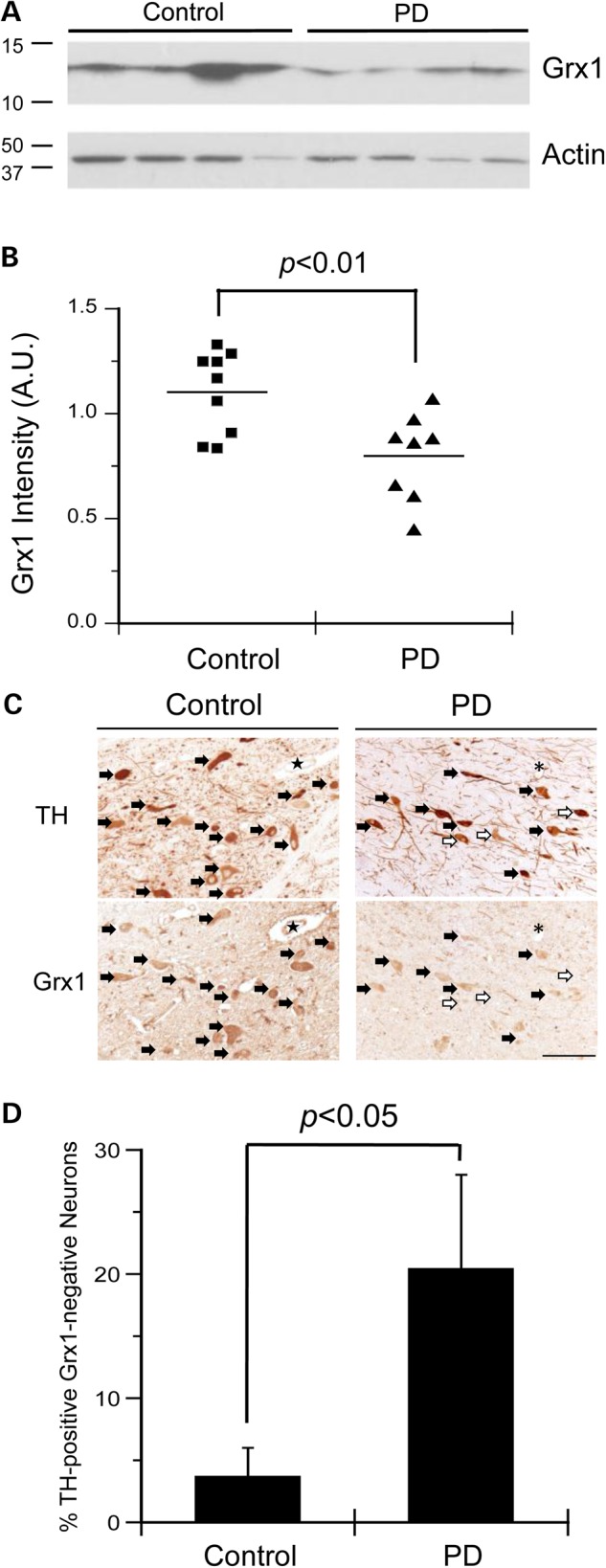
Grx1 protein is decreased in human PD postmortem brain tissue, and specifically in dopaminergic neurons. (A) Representative western blot analysis of midbrain homogenate from PD and control samples stained with anti-Grx1 or anti-actin antibodies, respectively. (B) Quantification of Grx1 protein expression (normalized to actin) in the midbrain of PD and control samples. n = 9 control cases and n = 8 PD cases. P < 0.01. Statistical analysis was completed using Student's t-test. (C) Representative immunohistochemical staining for TH and Grx1. Closed arrows indicate TH-positive/Grx1-positive neurons and open arrows indicate TH-positive/Grx1-negative neurons. ★ and * represent landmarks. Scale bar = 100 μm. (D) Bar graph showing the percentage of TH-positive/Grx1-negative neurons in the midbrain of PD and control patients. Error bars represent SEM. n = 4 control cases and n = 5 PD cases. P < 0.05 via Student's t-test.
Lack of Grx1 homolog exacerbates LRRK2-mediated dopaminergic neurodegeneration in C. elegans
To assess the potential role of Grx1 deficiency in the integrity of dopaminergic neurons in vivo under basal and PD-relevant conditions, C. elegans was chosen as a model organism. Caenorhabditis elegans was selected because of the simplicity in dopaminergic neuron morphology and availability of multiple genetic models of PD. In addition, C. elegans contains several evolutionally conserved Grxs homologous to human Grxs (Supplementary Material, Table S3). To simulate Grx1 deficiency, we utilized C. elegans lacking GLRX-10, the worm homolog of Grx1, using an isogenic strain with a null allele of glrx-10 due to deletion of exon 1 (glrx-10−/−; Fig. 2A). Genetic crosses were made between the glrx-10−/− strain and transgenic C. elegans lines that either express GFP alone (as WT control) or co-express GFP and familial forms of human LRRK2 (G2019S-LRRK2 or R1441C-LRRK2, hereafter referred to as G2019S and R1441C, respectively) in the dopaminergic neurons (23) ( Table 1). Confirmation of these crosses was obtained via PCR genotyping of glrx-10 or LRRK2 and western blot analysis of GLRX-10 in individual isogenic lines (Fig. 2B and C). In order to test if loss of GLRX-10 changed LRRK2 expression, semi-quantitate RT–PCR for LRRK2 mRNA was conducted. Loss of GLRX-10 had no effect on the relative levels of either R1441C or G2019S-LRRK2 mRNA (Supplementary Material, Fig. S2).
Figure 2.

Loss of C. elegans Grx1 homolog GLRX-10 exacerbates LRRK2-mediated impairment of dopamine-dependent behavior and dopaminergic neurodegeneration. (A) Diagram of the C. elegans glrx-10 gene including PCR primer positions and the region of deleted DNA in glrx-10−/− lines. (B) Representative PCR analysis of LRRK2 and either WT (∼500 bp, upper band) or mutant (∼300 bp, lower band) glrx-10 DNA in indicated C. elegans lines (see strain designation in Table 1). (C) Representative western blot analysis of GLRX-10 protein in indicated C. elegans lines. The blot was probed with the polyclonal anti-Grx1 antibody generated against human Grx1 (as described in Materials and Methods), exhibiting effective cross-reactivity with the C. elegans homolog GLRX-10. Actin was used as loading control. (D) Basal slowing data demonstrating that loss of GLRX-10 exacerbates LRRK2-mediated behavioral dysfunction. #P < 0.01, comparing control with either R1441C or G2019S; *P < 0.01, comparing R1441C; glrx-10−/− to R1441C; $P < 0.01, comparing G2019S; glrx-10−/− to G2019S. Statistical analyses were completed using a two-tailed t-test comparing corresponding days in adulthood (e.g. Day 0 and Day 0) between groups as indicated. Error bars represent SEM for n = 3–4 independent assays for all days, ∼10 worms per line per experiment. (E) Dopaminergic neuron analysis demonstrating that loss of GLRX-10 exacerbates LRRK2-mediated dopaminergic degeneration. &P < 0.05, comparing control; glrx-10−/− with control, and comparing control with R1441C; #P < 0.01, comparing control with either R1441C or G2019S; *P < 0.01, comparing R1441C; glrx-10−/− with R1441C; $ P < 0.01, comparing G2019S; glrx-10−/− with G2019S. Error bars represent SEM. n = 3–4 independent groups of worms for all days, ∼30 worms per genotype per experiment. Statistical analyses were completed using a two-tailed t-test comparing corresponding days in adulthood (e.g. Day 2 and Day 2) between groups as indicated.
Table 1.
Caenorhabditis elegans strains used in this study.
| Designation | Genotype | Strain | Reference |
|---|---|---|---|
| N2 | WT | N2 | CGC |
| glrx-10−/− | glrx-10(tm4634)I | tm4634 | NBP |
| Control (GFP) | lin-15(n765ts)X; cwrIs730[Pdat-1::GFP, lin-15(+)] | SGC730 | (23) |
| R1441C | lin-15(n765ts)X; cwrIs851[Pdat-1::GFP, Pdat-1::LRRK2(R1441C), lin-15(+)] | SGC851 | (23) |
| G2019S | lin-15(n765ts)X; cwrIs856[Pdat-1::GFP, Pdat-1::LRRK2(G2019S), lin-15(+)] | SGC856 | (23) |
| Control; glrx-10−/− | glrx-10(tm4634)I; cwrIs730 | SGC301 | This study |
| R1441C; glrx-10−/− | glrx-10(tm4634)I; cwrIs851 | SGC302 | This study |
| G2019S; glrx-10−/− | glrx-10(tm4634)I; cwrIs856 | SGC303 | This study |
| R1441C; glrx-10−/−; Vector | glrx-10(tm4634)I; cwrIs851; Ex[Punc-54::GFP] | SGC310 | This study |
| R1441C; glrx-10−/−; GLRX-10 | glrx-10(tm4634)I; cwrIs851; Ex[Punc-54::GFP, Pdat-1 ::GLRX-10] | SGC320 | This study |
| R1441C; glrx-10−/−; C22S | glrx-10(tm4634)I; cwrIs851; Ex[Punc-54::GFP, Pdat-1 ::GLRX-10(C22S)] | SGC330 | This study |
| α-Syn | BaIn11[Pdat-1::GFP, Pdat-1::a-synuclein] | UA44 | (24) |
| α-Syn; glrx-10−/− | glrx-10(tm4634)I; BaIn11 | SGC401 | This study |
| CAT-2 | BaIs4[Pdat-1::GFP, Pdat-1::CAT-2] | UA57 | (23,24) |
| CAT-2; glrx-10−/− | glrx-10(tm4634)I; BaIs4 | SGC402 | This study |
CGC, Caenorhabditis Genetics Center (USA); NBP, National Bioresource Project (Japan).
We first examined functional integrity of dopaminergic neurons in these C. elegans lines. Worms require dopaminergic neurons to mechanically sense the availability of bacterial food via the detection of its texture, and subsequently decrease their locomotion as a foraging strategy (28). This dopamine-dependent behavioral response, termed basal slowing, is a well-established phenotypic readout of dopaminergic function in C. elegans, and it is assayed most sensitively in young adults (adult Days 0–2, post L4 larval stage) (28). In agreement with our previous findings (23), transgenic expression of pathogenic LRRK2 (either R1441C or G2019S) in dopaminergic neurons resulted in adult-onset and age-dependent diminution of the basal slowing response in comparison to the GFP control (Fig. 2D, first, third and fifth sets of bars). The presence of the glrx-10−/− allele in the GFP control line caused no change in basal slowing response (Fig. 2D, first and second set of bars), indicating that lack of GLRX-10 alone does not compromise overall dopaminergic function under basal conditions. In contrast, the presence of the glrx-10−/− allele in R1441C- and G2019S-expressing lines led to a significantly greater loss of basal slowing response (Fig. 2D, third and fourth, and fifth and sixth sets of bars, respectively). Therefore, lack of GLRX-10 exacerbated dopaminergic dysfunction caused by pathogenic LRRK2 mutants.
We next examined morphological integrity of dopaminergic neurons by analysis of neuronal loss in young and aged animals via visualization of the GFP-labeled dopaminergic neurons (29). As we reported previously (23), expression of pathogenic LRRK2 mutants G2019S or R1441C resulted in age-dependent and progressive loss of dopaminergic neurons when compared with the GFP control (Fig. 2E, first, third and fifth sets of bars). Consistent with the basal slowing assay, the presence of the glrx-10−/− allele in the GFP control line resulted in essentially no change in the number of surviving dopaminergic neurons in corresponding age groups (Fig. 2E, first and second set of bars). In contrast, the presence of the glrx-10−/− allele in LRRK2 mutant-expressing lines led to increased loss of dopaminergic neurons, starting on adult Day 0 for the R1441C-expressing and on adult Day 6 for the G2019S-expressing lines (Fig. 2E, third and fourth, and fifth and sixth sets of bars, respectively). Therefore, loss of GLRX-10 apparently does not affect the survival of dopaminergic neurons under non-pathological conditions. The loss of GLRX-10, however, increased dopaminergic vulnerability to neurodegeneration mediated by G2019S and R1441C. Taken together, these data provide evidence that loss of GLRX-10 exacerbates ongoing dopaminergic neurodegeneration in vivo, thus implicating GLRX-10 as a neuroprotective protein in C. elegans.
Re-expression of GLRX-10 selectively in dopaminergic neurons attenuates mutant LRRK2-mediated phenotype
In order to test directly whether GLRX-10 confers a protective role in limiting LRRK2 mutant-mediated dopaminergic neurotoxicity, we engineered the R1441C-expressing and glrx-10 null worms (R1441C; glrx-10−/−) to re-express GLRX-10 exclusively in the dopaminergic neurons. We directed dopaminergic re-expression of GLRX-10 in the R1441C; glrx-10−/− worms by placing the glrx-10 cDNA under the control of the promoter of the dopamine transporter dat-1 for germline transformation via DNA microinjection, generating the GLRX-10 re-expression line (Fig. 3A). We also generated a control re-expression line from mock transformation of the R1441C; glrx-10−/− worms in the same manner, except that exogenous glrx-10 cDNA was omitted. To validate the GLRX-10 re-expression strategy, we used PCR to confirm the deletion of endogenous glrx-10 DNA and the presence of the exogenous glrx-10 cDNA (Fig. 3B). Using RT–PCR, we documented glrx-10 mRNA expression in the GLRX-10 re-expression worms but not in the corresponding control line (Fig. 3C). We analyzed basal slowing response and dopaminergic neurodegeneration in three independent sets of C. elegans lines with GLRX-10 re-expression and the corresponding control.
Figure 3.

Re-expression of GLRX-10 selectively in dopaminergic neurons rescues exacerbation of the LRRK2-mediated PD phenotypes in the glrx-10−/− background. (A) Diagram of glrx-10 allele and re-expression construct indicating primer positions used for PCR amplification. (B) Representative PCR analysis of WT glrx-10 (∼1100 bp), re-expressed glrx-10 (∼300 bp) and LRRK2 DNA. (C) Representative RT–PCR analysis of glrx-10 and gapdh (loading control) RNA. (D) Basal slowing data demonstrating that re-expression of WT GLRX-10 in dopaminergic neurons rescues the exacerbated LRRK2-mediated behavioral dysfunction in the glrx-10−/− background. *P < 0.01, comparing R1441C; glrx-10−/− with R1441C; glrx-10−/−; GLRX-10 re-expression. n = 3–4 independent experiments per bar, ∼10 worms per genotype per experiment, except for the data for the GLRX-10 re-expression lines where the mean represents pooled data from three independently generated lines. Error bars represent SEM. Statistical analyses were completed using an ANOVA followed by the Tukey's post hoc test for significance. (E) Dopaminergic neuron analysis demonstrating that re-expression of WT GLRX-10 rescues the exacerbated LRRK2-mediated dopaminergic degeneration in the glrx-10−/− background. *P < 0.01, comparing R1441C; glrx-10−/− with R1441C; glrx-10−/−; GLRX-10 re-expression lines. n = 3–4 independent experiments per bar, ∼30 worms per genotype per experiment, except for the GLRX-10 re-expression lines where the mean represents pooled data from three independently generated lines. Error bars represent SEM. Statistical analyses were completed using an ANOVA followed by the Tukey's post hoc test for significance.
Like its cognate R1441C; glrx-10−/− strain, the mock-transformed control line showed a severely impaired basal slowing response when compared with the R1441C-expressing worms with the WT glrx-10 background (Fig. 3D, second versus fourth sets of bars). Notably, GLRX-10 re-expression in the R1441C; glrx-10−/− worms markedly improved their basal slowing response in all age groups (adult Days 0–2; Fig. 3D, third versus fifth sets of bars). Consistent with the behavioral phenotype, GLRX-10 re-expression in the R1441C; glrx-10−/− worms significantly enhanced their survival of dopaminergic neurons on adult Days 4, 6 and 8 (Fig. 3E, third versus fifth sets of bars). These data indicate that selective re-expression of GLRX-10 in the dopaminergic neurons attenuates the exacerbated neurotoxicity induced by mutant LRRK2 expression in worms lacking GLRX-10.
Catalytic function of GLRX-10 is necessary for neuroprotection
The CPYC sequence motif (amino acids 22–25) represents the catalytic site of human Grx1. Alignment of amino acid sequences of Grx1 homologs from humans to worms shows complete conservation of the four-residue motif (Fig. 4A). In particular, cysteine 22 is the catalytic nucleophile required for Grx1 activity (30). To determine whether its catalytic activity is necessary for GLRX-10-mediated neuroprotection, we generated a C22S mutant representing an enzymatically dead version of GLRX-10 for re-expression in the dopaminergic neurons of the R1441C; glrx-10−/− worms, using the same strategy as described above. Using PCR, we confirmed the presence of exogenous cDNA for glrx-10 and LRRK2 in the C. elegans lines in which C22S GLRX-10 was expressed (Fig. 4B). Moreover, using RT–PCR we confirmed mRNA expression for C22S GLRX-10 (Fig. 4C). We then assessed dopaminergic function in the C. elegans lines in which C22S GLRX-10 was expressed and the corresponding mock-transformed controls, using the basal slowing assay. Unlike the rescuing effects of WT GLRX-10 (Fig. 3D), re-expression instead of the C22S mutant did not attenuate dopaminergic dysfunction of the R1441C; glrx-10−/− worms (Fig. 4D, second versus fourth sets of bars). In addition, re-expression of the C22S mutant failed to rescue the enhanced dopaminergic degeneration in the R1441C; glrx10−/− lines (Fig. 4E, second versus fourth set of bars). These data indicate that GLRX-10 catalytic activity is necessary for neuroprotection in vivo.
Figure 4.

Catalytic activity is necessary for GLRX-10-mediated protection from LRRK2-induced neurotoxicity. (A) Sequence alignment of Grx1 and its homologs ranging from human to C. elegans. Bolded region represents the conserved Grx1 catalytic motif, asterisk indicates nucleophilic cysteine 22. (B) Representative PCR analysis of WT glrx-10 allele (∼1100 bp), C22S glrx-10 cDNA (∼300 bp), and LRRK2 cDNA. Primer scheme is equivalent to that shown in Figure 3A. (C) Representative RT–PCR analysis of glrx-10 and gapdh RNA, respectively. (D) Basal slowing data demonstrating that expression of C22S GLRX-10 (C22S) in the dopaminergic neurons fails to rescue the exacerbated LRRK2-mediated behavioral phenotype in the glrx-10−/− background. n.s., no statistical difference, comparing R1441C; glrx-10−/−; C22S re-expression with R1441C; glrx-10−/−. n = 3–4 independent experiments, ∼10 worms per line per experiment except for C22S re-expression containing pooled data from three independently generated lines. Error bars represent SEM. Statistical analyses were completed using an ANOVA. (E) Dopaminergic neuron analysis demonstrating that re-expression of C22S GLRX-10 does not rescue the exacerbated neurodegeneration driven by loss of GLRX-10 in the R1441C background. n.s, no statistical difference compared with R1441C; glrx10−/− via ANOVA. Error bars represent SEM. n = 3 independent groups of worms for all days, ∼30 worms per genotype per experiment.
Lack of GLRX-10 exacerbates dopaminergic dysfunction in multiple models of PD in C. elegans
Considering the important role of Grx1 in regulating redox homeostasis, we sought to investigate whether deficiency of the Grx1 homolog would exacerbate phenotypes in other models of PD previously linked to redox imbalance. Accordingly, we crossed the glrx-10−/− null worms with two additional C. elegans models of PD, characterized by overexpression of TH homolog CAT-2 and human α-synuclein, respectively (Table 1). Like LRRK2, both TH and α-synuclein have been shown to be involved in increased generation of ROS, which is understood to play a key role in PD. Within dopaminergic neurons TH catalyzes the rate-limiting step in dopamine synthesis by converting tyrosine to l-DOPA, an immediate precursor of dopamine. Redox cycling of l-DOPA and dopamine, and metabolism of dopamine by monoamine oxidase B, all result in generation of ROS (31). Likewise, overexpression of α-synuclein in cell culture results in increased ROS generation, although the underlying mechanism is unclear (32). All three PD models in C. elegans, namely overexpression of pathogenic LRRK2, nematode CAT-2/TH or human α-synuclein, have been shown to result in age-dependent dopaminergic neurodegeneration, respectively (23–25).
Caenorhabditis elegans lines overexpressing either CAT-2/TH or human α-synuclein specifically in the dopaminergic neurons in the glrx-10−/− background were confirmed via PCR analysis of glrx-10 genotype and western blot analysis of GLRX-10 protein (Fig. 5A and B). We assessed dopaminergic function in the glrx-10−/− crossed lines and their corresponding controls using the basal slowing assay. The CAT-2 and α-synuclein expressing lines manifested age-dependent loss of basal slowing response for all age groups assessed (Fig. 5C, first, second and forth sets of bars). Notably, overexpression of α-synuclein resulted in a more severe phenotype compared with overexpression of CAT-2. Lack of GLRX-10 significantly exacerbated loss of basal slowing response caused by CAT-2 overexpression for all ages assessed (Fig. 5C, second versus third sets of bars). Absence of GLRX-10 in the α-synuclein overexpressing worms led to an analogous pattern of increased loss of basal slowing response relative to the WT glrx-10 background (Fig. 5C, fourth versus fifth sets of bars), although the difference was statistically significant only on adult Day 0. Loss of GLRX-10 had no effect on the relative levels of CAT-2 mRNA but increased the relative levels of α-synuclein mRNA (Supplementary Material, Fig. S2). Taken together, these data indicate that lack of GLRX-10 exacerbates dopaminergic neurodegeneration in multiple C. elegans models of PD, suggesting that glutaredoxin may act as a global neuroprotective factor against inducers of both familial and sporadic PD.
Figure 5.

Loss of GLRX-10 exacerbates dopaminergic dysfunction mediated by α-synuclein and CAT-2 overexpression. (A) Representative PCR analysis of either WT (∼500 bp) or mutant (∼300 bp) glrx-10 DNA in indicated C. elegans lines. (B) Representative western blot analysis of GLRX-10 protein in indicated C. elegans lines; actin was used as loading control. (C) Basal slowing data indicating that loss of GLRX-10 exacerbates dopaminergic behavioral dysfunction caused by overexpression of CAT-2 or α-synuclein. *P < 0.05, comparing CAT-2; glrx-10−/− with CAT-2. #P < 0.05, comparing α-synuclein; glrx-10−/− to α-synuclein. Statistical analyses were completed using a two-tailed t-test comparing corresponding days in adulthood (e.g. Day 0 and Day 0) between two groups as indicated. Error bars represent SEM for n = 3–4 independent experiments for all days; ∼10 worms per genotype per experiment.
Discussion
The molecular mechanisms that link sporadic and familial PD are incompletely understood, representing a significant gap in the PD field. Sporadic PD has long been associated with perturbed redox homeostasis, including elevated levels of ROS and reduced levels of antioxidants within the brain (33,34). Many of the proteins associated with familial and/or sporadic PD, including α-synuclein and LRRK2, have been implicated in disruption of redox homeostasis in vitro (12,13). Our findings indicate a decreased level of the redox regulatory enzyme Grx1 in the dopaminergic neurons of PD patients, and PD-like phenotypes in several C. elegans models of familial and/or sporadic PD were exacerbated by the absence of the Grx1 homolog. These data suggest that dysregulation of cellular redox homeostasis through loss of glutaredoxin represents a common mechanism linking sporadic and familial PD.
Perturbations in key regulators of redox systems have been shown to increase susceptibility to chemical agents that are used to simulate PD in animal and cellular models, including MPP+ (the active metabolite of MPTP) and 6-OHDA (3,35). These inducers of dopaminergic neuron degeneration are used in part to increase levels of ROS, which is associated with PD in humans. Increased ROS has been shown to drive protein glutathionylation and concomitant changes in protein function/subcellular location, leading to cell toxicity (34). Grx1 is a critical regulator of cell viability, regulating redox homeostasis via deglutathionylation of proteins, thereby restoring them to their steady-state functions. As alluded to above, we have previously shown that knock down of Grx1 results in cell death of human model dopaminergic neurons (4), and overexpression of Grx1 has been reported to protect model dopaminergic neurons against paraquat and 6-OHDA-induced toxicity (17). Grx1 has also been reported to regulate expression of specific familial PD proteins. For example, shRNA-mediated knockdown of Grx1 in mouse model dopaminergic neurons led to decreased protein content of DJ-1 (36). These observations prompted us to investigate the status of Grx1 in midbrain samples from human PD cases relative to non-PD controls. We have documented a deficiency of Grx1 within the dopaminergic neurons in postmortem PD brains compared with controls (Fig. 1). Despite the small sample size, this is the first report suggesting that Grx1 may be decreased in dopaminergic neurons in PD.
To test the in vivo effects of Grx1 deficiency on dopaminergic neuron viability, we used C. elegans as a model system. Within the context of the non-stressed organism, Grx1 appears to be dispensable for dopaminergic neuron survival since loss of the Grx1 homolog GLRX-10 alone did not result in neurodegenerative phenotypes (Fig. 2). Several well-known C. elegans models of PD were crossed with the glrx-10-null worms to investigate the role of GLRX-10 in mediating neuronal survival. These previously characterized models included overexpression of pathogenic LRRK2 mutants in the dopaminergic neurons, representing familial PD; overexpression of the worm homolog of TH in the dopaminergic neurons, representing sporadic PD, and overexpression of α-synuclein within the dopaminergic neurons, representing both sporadic and familial PD since α-synuclein has been shown to play a role in both sporadic and familial PD (26,27). In all of these cases concomitant lack of GLRX-10 exacerbated the PD-like phenotypes (Figs 2 and 5). Notably, impairment of dopamine-dependent movement appeared prior to/and more robustly than the loss of dopaminergic neurons. This is explained by the fact that dopamine-dependent movement is a functional assay based on the physiological role of dopamine, whereas dopaminergic degeneration is a morphological assay based on the disappearance of dopaminergic neurons. Indeed, we have previously reported that in C. elegans models of PD, the timeline for loss of dopamine-dependent movement is a more sensitive measure and precedes obvious loss of neuron structure (23). Re-expression of WT, but not catalytically dead GLRX-10, selectively in the dopaminergic neurons of R1441C-LRRK2; glrx-10-null worms reversed the exacerbated behavioral and neurodegenerative phenotypes (Figs 3 and 4), suggesting that GLRX-10, via its catalytic activity, plays a key role in meditating neuronal protection against the adverse effects of mutant LRRK2 in vivo. In addition, overexpression of GLRX-10 in R1441C-expressing worms in the WT background did not prevent LRRK2-mediated toxicity (data not shown), indicating that endogenous levels of GLRX-10 are sufficient for dopaminergic protection. These data further suggest that mutant LRRK2-mediated dopaminergic toxicity is likely driven through several mechanisms, at least one of which is disruption of redox homeostasis regulated by glutaredoxin. Taken together, our present study implicates a critical role for glutaredoxin in mediating dopaminergic protection during PD pathogenesis. Building on this foundation, future studies are necessary to validate the protective role of Grx1 in mammalian animal models.
Our in vivo data indicate that depletion of Grx1 in the face of increased ROS results in increased dopaminergic cell death. Grx1 is one of several key enzymes that regulate redox homeostasis within cells. Manipulation of other key redox regulating proteins has demonstrated the importance of redox homeostasis in cell viability within the context of PD. For example, knockdown of peroxiredoxin-3 has been shown to exacerbate mutant LRRK2-mediated toxicity in model dopaminergic cells, and expression of human peroxiredoxin-3 blunts mutant LRRK2-mediated dopaminergic neuron death in Drosophila (37). Overexpression of thioredoxin-1 in mice was found to diminish the toxic effect on TH-positive dopaminergic neurons due to acute treatment with MPTP (38). Collectively, these results highlight the importance of redox homeostasis in dopaminergic cell viability in models of sporadic or familial PD. Our current data which identifies a deficiency of Grx1 in dopaminergic neurons of PD patients along with showing exacerbation of PD-like phenotypes in vivo due to loss of Grx1 homolog advance the concept that dysregulation of redox homeostasis likely represents a common mechanism connecting sporadic and familial PD. Thus, an important aspect of redox homeostasis that may be altered in familial and sporadic PD is oxidative protein modification, resulting in functional changes in key regulatory proteins in survival/death pathways in DA neurons. To our knowledge, this is the first study to link dysregulation of a redox factor (Grx1) to exacerbation of both sporadic and familial PD using in vivo models. In future studies, these novel C. elegans lines can be used to characterize critical oxidative protein modifications.
Pharmacological treatment of PD has not changed substantially since the introduction of levodopa (l-DOPA) into clinical practice. While l-Dopa alleviates PD symptoms temporarily, progressive decline of the remaining dopaminergic neurons continues (39). Currently, there are no PD treatments that slow the progressive dopaminergic cell death. In recent years, efforts have focused on bolstering antioxidant defenses and increasing activity of protective redox systems (40,41). Treatment aimed at increasing antioxidant defense has been sufficiently promising so that a Phase 2 clinical trial was conducted using N-acetylcysteine as a GSH precursor, even though the results of this study have not yet been published. Therapeutic intervention restoring redox homeostasis would potentially allow for treatment of both sporadic and familial PD. It is important to proceed with caution, however, as such a treatment would affect the redox status of non-neuronal cells also, potentially leading to some unintended side effects. Our study supports the concept that enhancement of proteins regulating redox homeostasis, including Grx1, represents a promising approach for therapeutic intervention for PD.
Technological advances with the C. elegans model have recently made high-throughput drug screening possible. Using C. elegans, 364 000 compounds were screened for potential inhibitors of the transcription factor SKN-1 (42). Ultimately, 128 compounds were found to inhibit the GFP-reporter expression in vivo at concentrations <10 μm (42). We have previously reported that small molecule inhibitors of LRRK2 can be used in C. elegans to attenuate mutant LRRK2-induced dopaminergic toxicity (43), demonstrating the utility of C. elegans for discovering potential PD therapeutics. The new models of dysregulated redox homeostasis characterized in the present study simulate pathological conditions occurring in PD. These new C. elegans lines represent useful tools for in vivo screenings for agents that would restore redox homeostasis in dopaminergic neurons. Discovering such agents may facilitate development of PD treatments to mitigate the progressive loss of dopaminergic neurons.
In summary, perturbations in redox homeostasis have been demonstrated to drive toxicity in models of PD, and are thought to play a role in PD pathogenesis. Our data indicate that the redox regulator Grx1 is decreased in the dopaminergic neurons in human PD and that loss of the Grx1 homolog in vivo exacerbates toxicity in three distinct models of PD, reversible by re-expression of catalytically active form of glutaredoxin. Together, these findings indicate that dysregulation of redox homeostasis through loss of glutaredoxin exacerbates both sporadic and familial models of PD in vivo. These new models are expected to serve as an effective in vivo platform for drug discovery, leading to novel therapeutic strategies for PD, a debilitating disease that affects millions of people worldwide.
Materials and Methods
Western blot analysis of human tissue
Postmortem brain tissue samples from histopathologically diagnosed PD and non-PD cases (see Supplementary Material, Table S1) were used for this study according to approved IRB protocols. Specifically, frozen tissues of brainstem or pons were obtained from the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland (Baltimore, MD, USA), and from the Case Western Reserve University/University Hospitals of Cleveland Brain Bank (Cleveland, OH, USA). Areas of the frozen brainstem samples containing the substantia nigra were carefully dissected and were homogenized with 1× cell lysis buffer (Cell Signaling) plus 1 mM phenylmethanesulfonylfluoride (Sigma) and Protease Inhibitor Cocktail (Sigma). Equal amounts of total protein extract (10 μg) were resolved by SDS–PAGE and transferred to Immobilon-P (Millipore). Following blocking with 10% non-fat dry milk, primary and secondary antibodies were applied as previously described (44), and the blots were developed with Immobilon Western Chemiluminescent HRP Substrate (Millipore). Primary antibodies used included anti-actin (Sigma) and anti-Grx1. Rabbit polyclonal anti-Grx1 was generated by Genscript against recombinant human Grx1 produced in Escherichia coli (45). Band densities were quantified using QuantityOne software (Bio-Rad).
Immunohistochemistry
Formalin fixed brainstem samples (4 control cases and 5 neuropathologically confirmed PD cases; Supplementary Material, Table S2) were obtained from Case Western Reserve University/University Hospitals of Cleveland Brain Bank, according to approved IRB protocols. Formalin fixed brain samples were embedded in paraffin and 6 μm sections were cut using a microtome and placed on coated slides. Immunohistochemistry was performed using the peroxidase anti-peroxidase (PAP) method as previously described (44). Briefly, sections were deparaffinized in two changes of xylene, dehydrated through descending series of ethanol immersions, and finally transferred into Tris-buffered saline (TBS: 50 mm Tris, 150 mm NaCl, pH = 7.6). Endogenous peroxidase was quenched with 30 min incubation in 3% H2O2. Antigen retrieval was performed using citrate buffer and pressure cooking (Biocare). After blocking for 30 min in 10% normal goat serum (NGS), primary antibodies were applied and incubated overnight at 4°C. After rinsing with 1% NGS and blocking for 10 min in 10% NGS, secondary goat anti-rabbit antibodies were applied for 30 min. After rinsing again, rabbit PAP complexes were applied for 1 h. After washing with three changes of TBS buffer, the staining was developed with DAB (Dako) and slides were rinsed in TBS, dehydrated and affixed with Permount and coverslips. Polyclonal antibodies to tyrosine hydroxylase (TH, Abcam) and Grx1 (as described above) were used to stain adjacent serial sections. Following tissue landmarks on the adjacent serial sections, images were obtained of identical fields of the substantia nigra stained for either TH or Grx1 using a Zeiss Axiocam. All TH-positive neurons were noted and the staining intensity measuring the level of Grx1 was determined in each of those cells using Axiovision image analysis software. Neurons from three fields were measured (n = 14–22 neurons per case).
Caenorhabditis elegans strains and maintenance
A list of C. elegans strains with their designation, corresponding genotype and strain nomenclature is shown in Table 1. C. elegans strains were maintained on nematode growth medium (NGM) plates with E. coli OP50 as a bacterial food, as previously described (23).
Single worm PCR
Individual worms were placed in PCR tubes containing 1× PFU Turbo Master Mix buffer (Agilent), 0.5 mg/ml proteinase K (Sigma) and were frozen on dry ice for 15 min. Worms were then lysed by incubation at 65°C for 90 min followed by 95°C for 30 min and frozen at −80°C until PCR reactions were performed. PCR amplifications were performed using OneTaq polymerase following manufacturer's instructions (New England BioLabs). Identification of WT and mutant glrx-10 DNA was accomplished using the primers F1: 5′-CTCGCCACTCTCTCATTTCCC-3′ and R1: 5′-CAGAACCCCGTGACAGATTT-3′.
Caenorhabditis elegans western blot analysis
Protein samples used for western blotting experiments were obtained from two 10 cm petri dishes of worms. Bacteria were removed by washing the worms three times in M9 buffer. Worms were then placed in 1.5 ml ice-cold lysis buffer containing 1% Triton X-100, 137 mm NaCl, 2.7 mm KCl and protease inhibitors (Roche). Worms were homogenized using mini-Beadbeater and 0.7 mm Zirconia beads (BioSpec Products) at a setting of 4800 oscillations/min for 60 s per cycle, for a total of three cycles with cooling on ice between the cycles. Lysates were spun down at 10 000g for 30 min at 4°C and supernatants were collected. Protein concentration was determined using a micro BCA kit following manufacturer's instructions (Pierce). Samples containing 12.5 μg of protein were run on a 15% SDS–PAGE gel, transferred at 15 V for 45 min onto PVDF membrane (Millipore) using a semi-dry transfer system (Bio-Rad). Blots were blocked in 5% non-fat milk made in TBS containing 0.1% Tween 20 (TBST) for 1 h prior to antibody addition. To reduce non-specific binding and for detection of C. elegans GLRX-10, the Grx1 antibody was pre-absorbed with the lysate from the glrx-10 null strain at a ratio of 1 : 4 (v/v) at 4°C for 2 h, followed by dilution to 1 : 1000 with 5% non-fat milk in TBST. Blots were incubated with pre-absorbed anti-Grx1 or anti-actin (Sigma, 1 : 5000 dilution) primary antibodies overnight. Blots were then washed three times in TBST for 15 min each and incubated in 5% non-fat milk in TBST with HRP conjugated secondary antibodies for 1 h. Following the washes with TBST, protein bands were visualized using ECL (Millipore).
Basal slowing assay
The basal slowing assay was performed as previously described (23). Briefly, NGM assay plates were seeded with a ring of E. coli OP50 bacteria the night before the assay. Well-fed, age-synchronized worms were transferred to a non-seeded NGM plate and washed twice in S basal buffer (100 mm NaCl, 10 μg/ml cholesterol, 50 mm potassium phosphate, pH 6.0) to remove bacteria. Worms were then transferred to either seeded or non-seeded assay plates, allowed to recover for 5 min, and their body bends were recorded in 20-s intervals.
Dopaminergic neuron counting
Approximately 30 age-synchronized adult worms were placed into 15 μl of 10 mm tetramizole (Sigma) on agar-seeded microscope slides. GFP neurons were visualized with a Nikon microscope at ×20 zoom. Dopaminergic CEP and ADE head neurons were assessed for degeneration in a blinded fashion, according to published protocols (23,29). Neurons with detectable GFP signal were counted as positive, neurons lacking cell body GFP signal were counted as negative.
Generation of Pdat-1::GLRX-10 plasmid
glrx-10 cDNA (clone Y34D9A.K) was obtained from Open Biosystems. glrx-10 with additional BamH1 (5′) and EcoR1 (3′) sites was amplified via PCR using the following primers F2: 5′-CGATCAGAATTCATGTCAAAAGCCTTTGTCGACG -3′ and R2: 5′-TCGTCAGGATCCCTATAGAGCTCCGGTTTCCTTG-3′. glrx-10 cDNA and the Pdat-1::GFP vector (23) were digested for 1 h at 37°C using BamH1 and EcoR1 (Invitrogen). Digested DNA was run on a 1% agarose gel and eluted using the QIAquick gel extraction kit following manufacturer's instructions (Qiagen). Constructs were ligated overnight at 16°C with T4 ligase following manufacturer's instructions (Invitrogen). Cloning was confirmed via DNA sequencing (Biotic Solutions).
Site-directed mutagenesis of GLRX-10
Guanine 65 was mutated to cytosine 65 (resulting in a cysteine to serine (C22S) mutation in GLRX-10) in the Pdat-1::GLRX-10 using the QuikChange II XL site-directed mutagenesis kit following manufactures instructions (Agilent). Primers used in the mutagenesis were F3: 5-TCGTATTCAGCAAATCGTACTCCCCGTACTGCC-3′ and R3: 5′-GGCAGTACGGGGAGTACGATTTGCTGAATACGA-3′. C22S mutation was confirmed via DNA sequencing (Biotic Solutions).
Generation of GLRX-10 re-expressing transgenic lines
Transgenic C. elegans were generated by directly injecting R1441C-LRRK2; glrx-10−/− worms with a mixture of Pdat-1::GLRX-10 (either WT or C22S), Punc-54::GFP, or pBluescript empty vector as previously described (23). Transgenic animals expressing muscle GFP were isolated and allowed to self-fertilize. glrx-10 cDNA (Figs 3B and 4B) was confirmed via PCR as described above using primers F4: 5′-CGATCAGAATTCATGTCAAAAGCCTTTGTCGACG-3′ and R4: 5′-TCGTCAGGATCCCTATAGAGCTCCGGTTTCCTTG. Independent lines of WT GLRX-10 re-expression (3 lines), C22S GLRX-10 re-expression (4 lines) and control (1 line) were generated.
Analysis of mRNA expression using RT–PCR (reverse transcription polymerase chain reaction)
Caenorhabditis elegans RNA was extracted using Trizol following manufacturer's instructions (Life Technologies). Briefly, bacteria were removed by washing the worms three times in M9 buffer. One milliliter of Trizol was added to ∼100 μl of settled worms and incubated at room temperature for 5 min. Then 200 μl of chloroform (Fisher) was added to the mixture, vigorously shaken for 15 s and incubated at room temperature for 5 min. Samples were spun at 12 000g for 15 min at 4°C. The upper aqueous phase was transferred to a tube containing 0.5 ml isopropanol (Fisher), shaken by hand for 30 s and incubated at room temperature for 10 min. Following centrifugation at 12 000g for 15 min at 4°C, the supernatant was removed. The pellet was washed in 75% RNase-free ethanol (Fisher) and centrifuged at 7500g for 5 min at 4°C. The ethanol was removed from the tube and the pellet was air dried for 10 min on ice. The pellet containing extracted RNA was then resuspended in 100 μl DEPC-treated water. cDNA synthesis was completed using SuperScript II reverse transcriptase with 1 μg of RNA as template following manufacturer's instructions (Life Technologies). PCR amplifications of glrx-10 cDNA and gapdh cDNA were accomplished using an aliquot of synthesized cDNA as template and exon-spanning primers F5: 5′-GTCGACGGACTTCTCCAATC-3′, R5: 5′-CTCCGGTTAGAGAGCCAAGA-3′ and F6: 5′-GAAACTGCTTCAACGCATCA-3′, R6: 5′CCTTGGCGACAAGAAGGTAG-3′ for glrx-10 and gapdh, respectively.
Statistical analysis
Statistics for the human data was performed using a two-tailed Student's t-test comparing Grx1 levels in brain homogenate and a one-tailed Student's t-test comparing the number of TH-positive and Grx1-negative neurons between PD and non-PD groups. Statistics for C. elegans data was performed using either ANOVA followed by Tukey's post hoc test or a Student's t-test as noted in the figure legends. All statistical analyses were run using the Microsoft Excel program or GraphPad Prism.
Supplementary Material
Funding
This work was supported in part by the National Institutes of Health (grant NS073170 to A.L.W. and S.G.C.; grant NS085503 to A.L.W., J.J.M. and S.G.C.; grant NS083498 to X.Z.; and predoctoral fellowship T32 GM008803 to W.M.J.), Parkinson's Disease Foundation (summer student fellowship PDF-SFW-1348 to W.M.J.), the Department of Veterans Affairs (merit review grant BX000290 to J.J.M.), the National Science Foundation Advance Institutional Transformation Program (ACES research opportunity grant to S.G.C.), and the Chinese Overseas, Hong Kong and Macao Scholars Collaborated Researching Fund (grant 81228007 to X.Z.).
Supplementary Material
Acknowledgements
We thank Dr Janet Duerr (Ohio University) for assistance with the generation of the GLRX-10 re-expressing worm lines. Part of the human tissue was obtained from the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland. The C. elegans glrx-10−/− strain (tm4634) was obtained from National Bioresource Project (Japan). The C. elegans WT reference strain N2 Bristol and E. coli strain OP50 were obtained from the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health Office of Research Infrastructure Programs (P40 OD010440).
Conflict of Interest statement. None declared.
References
- 1.Bai J., Nakamura H., Hattori I., Tanito M., Yodoi J. Thioredoxin suppresses 1-methyl-4-phenylpyridinium-induced neurotoxicity in rat PC12 cells. Neurosci. Lett. 2002;321:81–84. doi: 10.1016/s0304-3940(02)00058-7. [DOI] [PubMed] [Google Scholar]
- 2.Hu X., Weng Z., Chu C.T., Zhang L., Cao G., Gao Y., Signore A., Zhu J., Hastings T., Greenamyre J.T., et al. Peroxiredoxin-2 protects against 6-hydroxydopamine-induced dopaminergic neurodegeneration via attenuation of the apoptosis signal-regulating kinase (ASK1) signaling cascade. J. Neurosci. 2011;31:247–261. doi: 10.1523/JNEUROSCI.4589-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Simoni S., Goemaere J., Knoops B. Silencing of peroxiredoxin 3 and peroxiredoxin 5 reveals the role of mitochondrial peroxiredoxins in the protection of human neuroblastoma SH-SY5Y cells toward MPP+ Neurosci. Lett. 2008;433:219–224. doi: 10.1016/j.neulet.2007.12.068. [DOI] [PubMed] [Google Scholar]
- 4.Sabens E.A., Distler A.M., Mieyal J.J. Levodopa deactivates enzymes that regulate thiol-disulfide homeostasis and promotes neuronal cell death: implications for therapy of Parkinson's disease. Biochemistry. 2010;49:2715–2724. doi: 10.1021/bi9018658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson W.M., Wilson-Delfosse A.L., Mieyal J.J. Dysregulation of glutathione homeostasis in neurodegenerative diseases. Nutrients. 2012;4:1399–1440. doi: 10.3390/nu4101399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Filloux F., Townsend J.J. Pre- and postsynaptic neurotoxic effects of dopamine demonstrated by intrastriatal injection. Exp. Neurol. 1993;119:79–88. doi: 10.1006/exnr.1993.1008. [DOI] [PubMed] [Google Scholar]
- 7.Gomez-Santos C., Ferrer I., Santidrian A.F., Barrachina M., Gil J., Ambrosio S. Dopamine induces autophagic cell death and alpha-synuclein increase in human neuroblastoma SH-SY5Y cells. J. Neurosci. Res. 2003;73:341–350. doi: 10.1002/jnr.10663. [DOI] [PubMed] [Google Scholar]
- 8.Ham A., Kim D.W., Kim K.H., Lee S.J., Oh K.B., Shin J., Mar W. Reynosin protects against neuronal toxicity in dopamine-induced SH-SY5Y cells and 6-hydroxydopamine-lesioned rats as models of Parkinson's disease: reciprocal up-regulation of E6-AP and down-regulation of alpha-synuclein. Brain Res. 2013;1524:54–61. doi: 10.1016/j.brainres.2013.05.036. [DOI] [PubMed] [Google Scholar]
- 9.Singleton A.B., Farrer M.J., Bonifati V. The genetics of Parkinson's disease: progress and therapeutic implications. Mov. Disord. 2013;28:14–23. doi: 10.1002/mds.25249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gandhi P.N., Chen S.G., Wilson-Delfosse A.L. Leucine-rich repeat kinase 2 (LRRK2): a key player in the pathogenesis of Parkinson's disease. J. Neurosci. Res. 2009;87:1283–1295. doi: 10.1002/jnr.21949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singleton A.B., Farrer M., Johnson J., Singleton A., Hague S., Kachergus J., Hulihan M., Peuralinna T., Dutra A., Nussbaum R., et al. Alpha-synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 12.Wang X., Yan M.H., Fujioka H., Liu J., Wilson-Delfosse A., Chen S.G., Perry G., Casadesus G., Zhu X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 2012;21:1931–1944. doi: 10.1093/hmg/dds003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dryanovski D.I., Guzman J.N., Xie Z., Galteri D.J., Volpicelli-Daley L.A., Lee V.M., Miller R.J., Schumacker P.T., Surmeier D.J. Calcium entry and alpha-synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J. Neurosci. 2013;33:10154–10164. doi: 10.1523/JNEUROSCI.5311-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gallogly M.M., Starke D.W., Leonberg A.K., Ospina S.M., Mieyal J.J. Kinetic and mechanistic characterization and versatile catalytic properties of mammalian glutaredoxin 2: implications for intracellular roles. Biochemistry. 2008;47:11144–11157. doi: 10.1021/bi800966v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mieyal J.J., Gallogly M.M., Qanungo S., Sabens E.A., Shelton M.D. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid. Redox Signal. 2008;10:1941–1988. doi: 10.1089/ars.2008.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shelton M.D., Chock P.B., Mieyal J.J. Glutaredoxin: role in reversible protein S-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid. Redox Signal. 2005;7:348–366. doi: 10.1089/ars.2005.7.348. [DOI] [PubMed] [Google Scholar]
- 17.Rodriguez-Rocha H., Garcia Garcia A., Zavala-Flores L., Li S., Madayiputhiya N., Franco R. Glutaredoxin 1 protects dopaminergic cells by increased protein glutathionylation in experimental Parkinson's disease. Antioxid. Redox Signal. 2012;17:1676–1693. doi: 10.1089/ars.2011.4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kenchappa R.S., Ravindranath V. Glutaredoxin is essential for maintenance of brain mitochondrial complex I: studies with MPTP. FASEB J. 2003;17:717–719. doi: 10.1096/fj.02-0771fje. [DOI] [PubMed] [Google Scholar]
- 19.Kenchappa R.S., Diwakar L., Annepu J., Ravindranath V. Estrogen and neuroprotection: higher constitutive expression of glutaredoxin in female mice offers protection against MPTP-mediated neurodegeneration. FASEB J. 2004;18:1102–1104. doi: 10.1096/fj.03-1075fje. [DOI] [PubMed] [Google Scholar]
- 20.Daigle I., Li C. apl-1, a Caenorhabditis elegans gene encoding a protein related to the human beta-amyloid protein precursor. Proc. Natl. Acad. Sci. USA. 1993;90:12045–12049. doi: 10.1073/pnas.90.24.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J., Le W. Modeling neurodegenerative diseases in Caenorhabditis elegans. Exp. Neurol. 2013;250:94–103. doi: 10.1016/j.expneurol.2013.09.024. [DOI] [PubMed] [Google Scholar]
- 22.Chakraborty S., Bornhorst J., Nguyen T.T., Aschner M. Oxidative stress mechanisms underlying Parkinson's disease-associated neurodegeneration in C. elegans. Int. J. Mol. Sci. 2013;14:23103–23128. doi: 10.3390/ijms141123103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yao C., El Khoury R., Wang W., Byrd T.A., Pehek E.A., Thacker C., Zhu X., Smith M.A., Wilson-Delfosse A.L., Chen S.G. LRRK2-mediated neurodegeneration and dysfunction of dopaminergic neurons in a Caenorhabditis elegans model of Parkinson's disease. Neurobiol. Dis. 2010;40:73–81. doi: 10.1016/j.nbd.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao S., Gelwix C.C., Caldwell K.A., Caldwell G.A. Torsin-mediated protection from cellular stress in the dopaminergic neurons of Caenorhabditis elegans. J. Neurosci. 2005;25:3801–3812. doi: 10.1523/JNEUROSCI.5157-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lakso M., Vartiainen S., Moilanen A.M., Sirvio J., Thomas J.H., Nass R., Blakely R.D., Wong G. Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alpha-synuclein. J. Neurochem. 2003;86:165–172. doi: 10.1046/j.1471-4159.2003.01809.x. [DOI] [PubMed] [Google Scholar]
- 26.Spillantini M.G., Crowther R.A., Jakes R., Hasegawa M., Goedert M. Alpha-synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taymans J.M., Cookson M.R. Mechanisms in dominant parkinsonism: the toxic triangle of LRRK2, alpha-synuclein, and tau. Bioessays. 2010;32:227–235. doi: 10.1002/bies.200900163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sawin E.R., Ranganathan R., Horvitz H.R. C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron. 2000;26:619–631. doi: 10.1016/s0896-6273(00)81199-x. [DOI] [PubMed] [Google Scholar]
- 29.Berkowitz L.A., Hamamichi S., Knight A.L., Harrington A.J., Caldwell G.A., Caldwell K.A. Application of a C. elegans dopamine neuron degeneration assay for the validation of potential Parkinson's disease genes. J. Vis. Exp. 2008;835:ii. doi: 10.3791/835. doi:10.3791/835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gallogly M.M., Starke D.W., Mieyal J.J. Mechanistic and kinetic details of catalysis of thiol-disulfide exchange by glutaredoxins and potential mechanisms of regulation. Antioxid. Redox Signal. 2009;11:1059–1081. doi: 10.1089/ars.2008.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bisaglia M., Greggio E., Beltramini M., Bubacco L. Dysfunction of dopamine homeostasis: clues in the hunt for novel Parkinson's disease therapies. FASEB J. 2013;27:2101–2110. doi: 10.1096/fj.12-226852. [DOI] [PubMed] [Google Scholar]
- 32.Liu Z., Yu Y., Li X., Ross C.A., Smith W.W. Curcumin protects against A53T alpha-synuclein-induced toxicity in a PC12 inducible cell model for Parkinsonism. Pharmacol. Res. 2011;63:439–444. doi: 10.1016/j.phrs.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 33.Nakabeppu Y., Tsuchimoto D., Yamaguchi H., Sakumi K. Oxidative damage in nucleic acids and Parkinson's disease. J. Neurosci. Res. 2007;85:919–934. doi: 10.1002/jnr.21191. [DOI] [PubMed] [Google Scholar]
- 34.Perry T.L., Godin D.V., Hansen S. Parkinson's disease: a disorder due to nigral glutathione deficiency? Neurosci. Lett. 1982;33:305–310. doi: 10.1016/0304-3940(82)90390-1. [DOI] [PubMed] [Google Scholar]
- 35.Lopert P., Day B.J., Patel M. Thioredoxin reductase deficiency potentiates oxidative stress, mitochondrial dysfunction and cell death in dopaminergic cells. PLoS ONE. 2012;7:e50683. doi: 10.1371/journal.pone.0050683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saeed U., Ray A., Valli R.K., Kumar A.M., Ravindranath V. DJ-1 loss by glutaredoxin but not glutathione depletion triggers Daxx translocation and cell death. Antioxid. Redox Signal. 2010;13:127–144. doi: 10.1089/ars.2009.2832. [DOI] [PubMed] [Google Scholar]
- 37.Angeles D.C., Ho P., Chua L.L., Wang C., Yap Y.W., Ng C., Zhou Z.D., Lim K.L., Wszolek Z.K., Wang H.Y., et al. Thiol peroxidases ameliorate LRRK2 mutant-induced mitochondrial and dopaminergic neuronal degeneration in Drosophila. Hum. Mol. Genet. 2014;23:3157–3165. doi: 10.1093/hmg/ddu026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bai J., Nakamura H., Kwon Y.W., Tanito M., Ueda S., Tanaka T., Hattori I., Ban S., Momoi T., Kitao Y., et al. Does thioredoxin-1 prevent mitochondria- and endoplasmic reticulum-mediated neurotoxicity of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine? Antioxid. Redox Signal. 2007;9:603–608. doi: 10.1089/ars.2006.1513. [DOI] [PubMed] [Google Scholar]
- 39.Olanow C.W., Jankovic J. Neuroprotective therapy in Parkinson's disease and motor complications: a search for a pathogenesis-targeted, disease-modifying strategy. Mov. Disord. 2005;20(Suppl. 11):S3–10. doi: 10.1002/mds.20457. [DOI] [PubMed] [Google Scholar]
- 40.Lou H., Jing X., Wei X., Shi H., Ren D., Zhang X. Naringenin protects against 6-OHDA-induced neurotoxicity via activation of the Nrf2/ARE signaling pathway. Neuropharmacology. 2014;79:380–388. doi: 10.1016/j.neuropharm.2013.11.026. [DOI] [PubMed] [Google Scholar]
- 41.Navarro-Yepes J., Zavala-Flores L., Anandhan A., Wang F., Skotak M., Chandra N., Li M., Pappa A., Martinez-Fong D., Del Razo L.M., et al. Antioxidant gene therapy against neuronal cell death. Pharmacol. Ther. 2014;142:206–230. doi: 10.1016/j.pharmthera.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leung C.K., Wang Y., Malany S., Deonarine A., Nguyen K., Vasile S., Choe K.P. An ultra high-throughput, whole-animal screen for small molecule modulators of a specific genetic pathway in Caenorhabditis elegans. PLoS ONE. 2013;8:e62166. doi: 10.1371/journal.pone.0062166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yao C., Johnson W.M., Gao Y., Wang W., Zhang J., Deak M., Alessi D.R., Zhu X., Mieyal J.J., Roder H., et al. Kinase inhibitors arrest neurodegeneration in cell and C. elegans models of LRRK2 toxicity. Hum. Mol. Genet. 2013;22:328–344. doi: 10.1093/hmg/dds431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu X., Babar A., Siedlak S.L., Yang Q., Ito G., Iwatsubo T., Smith M.A., Perry G., Chen S.G. LRRK2 in Parkinson's disease and dementia with Lewy bodies. Mol. Neurodegener. 2006;1:17. doi: 10.1186/1750-1326-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chrestensen C.A., Eckman C.B., Starke D.W., Mieyal J.J. Cloning, expression and characterization of human thioltransferase (glutaredoxin) in E. coli. FEBS Lett. 1995;374:25–28. doi: 10.1016/0014-5793(95)01066-n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.