

Abstract
Traditionally, scientific research has focused on studying individual events, such as single mutations, gene function or the effect of the manipulation of one protein on a biological phenotype. A range of technologies, combined with the ability to develop robust and predictive mathematical models, is beginning to provide information that will enable a holistic view of how the genomic and epigenetic aberrations in cancer cells can alter the homeostasis of signalling networks within these cells, between cancer cells and the local microenvironment, at the organ and organism level. This systems biology process needs to be integrated with an iterative approach wherein hypotheses and predictions that arise from modelling are refined and constrained by experimental evaluation. Systems biology approaches will be vital for developing and implementing effective strategies to deliver personalized cancer therapy. Specifically, these approaches will be important to select those patients most likely to benefit from targeted therapies as well as for the development and implementation of rational combinatorial therapies. Systems biology can help to increase therapy efficacy or bypass the emergence of resistance, thus converting the current (often short term) effects of targeted therapies into durable responses, ultimately to improve quality of life and provide a cure.
To ensure the survival of the organism, millions of signals are sent and received every second between cells, tissues, or organs and the external environment, and they are integrated into responses at multiple levels and scales in the body. Robust homeostatic mechanisms must be in place to fine-tune responses and, in particular, allow the organism to deal with potentially toxic environmental perturbations, such as those derived from pathogens, food, air (tobacco smoke, diesel fumes), or drugs. These mechanisms must also be resistant to the effects of spontaneous somatic mutations and the many germline mutations that ultimately alter the biological function of proteins.
In a cancer cell, genomic and epigenetic deregulation results in marked disruption of these homeostatic mechanisms.1,2 Developing an integrated view of the mechanisms by which genomic and epigenetic aberrations disrupt normal cellular function to induce malignant transformation and tumour progression remains a tremendous challenge.
Advances in high-throughput, cost-effective, and tissue-sparing technologies that can analyse tumours at multiple levels, combined with high-speed computing resources, has facilitated a transition in the research paradigm toward an integrated systems biology approach.3–7 These new technologies have greatly expanded our ability to develop robust datasets and to integrate the data into a holistic view that is much more than the sum of the parts. Although the traditional research approach is instrumental in the acquisition of basic detailed knowledge of the function of specific genes and proteins, the appreciation of the wider cellular signalling networks between cells, stroma, organs, and the entire organism through an integrated ‘system biology’ approach is crucial for the development and implementation of effective anti-cancer strategies. Systems biology is a ‘data-driven’ science requiring large amounts of high-quality data, crossing multiple scales and concepts in order to build robust predictive models. Indeed, one of the key precepts of systems biology is that the complexity of the cell and its environment can be “modeled” and that these models can be both robust and predictive. It allows an unbiased analysis of the data and has the important advantage that it is also hypothesis deriving. Several aspect of clinical research can benefit from the use of systems biology, including understanding of (emergent) drug resistance, prediction of effective combination therapies and identification of predictive biomarkers to increase the response rate to (targeted) treatments. In this review we will cover an overview of systems biology approaches and resources that have been developed, focusing on some of the clinical and basic science studies that have benefitted from using a systems approach to uncover novel concepts and properties. We will also discuss some of the challenges and how this relatively young filed can make a positive impact on cancer.
Cancer systems biology
Cancer systems biology, therefore, represents the application of systems biology approaches to the analysis of how the intracellular networks of normal cells are perturbed during carcinogenesis to develop effective predictive models that can assist scientists and clinicians in the validations of new therapies and drugs. These perturbations are caused by the massive and ongoing genomic and epigenetic instability in tumors altering the functions of many different molecules and networks in a single cell and further complicated by the alterations in the interactions with the local environment as well as the individual as a whole through the tumorigenic process itself. Cancer systems biology approaches are therefore based on the use of computational and mathematical methods to decipher the complexity in cancer heterogeneity
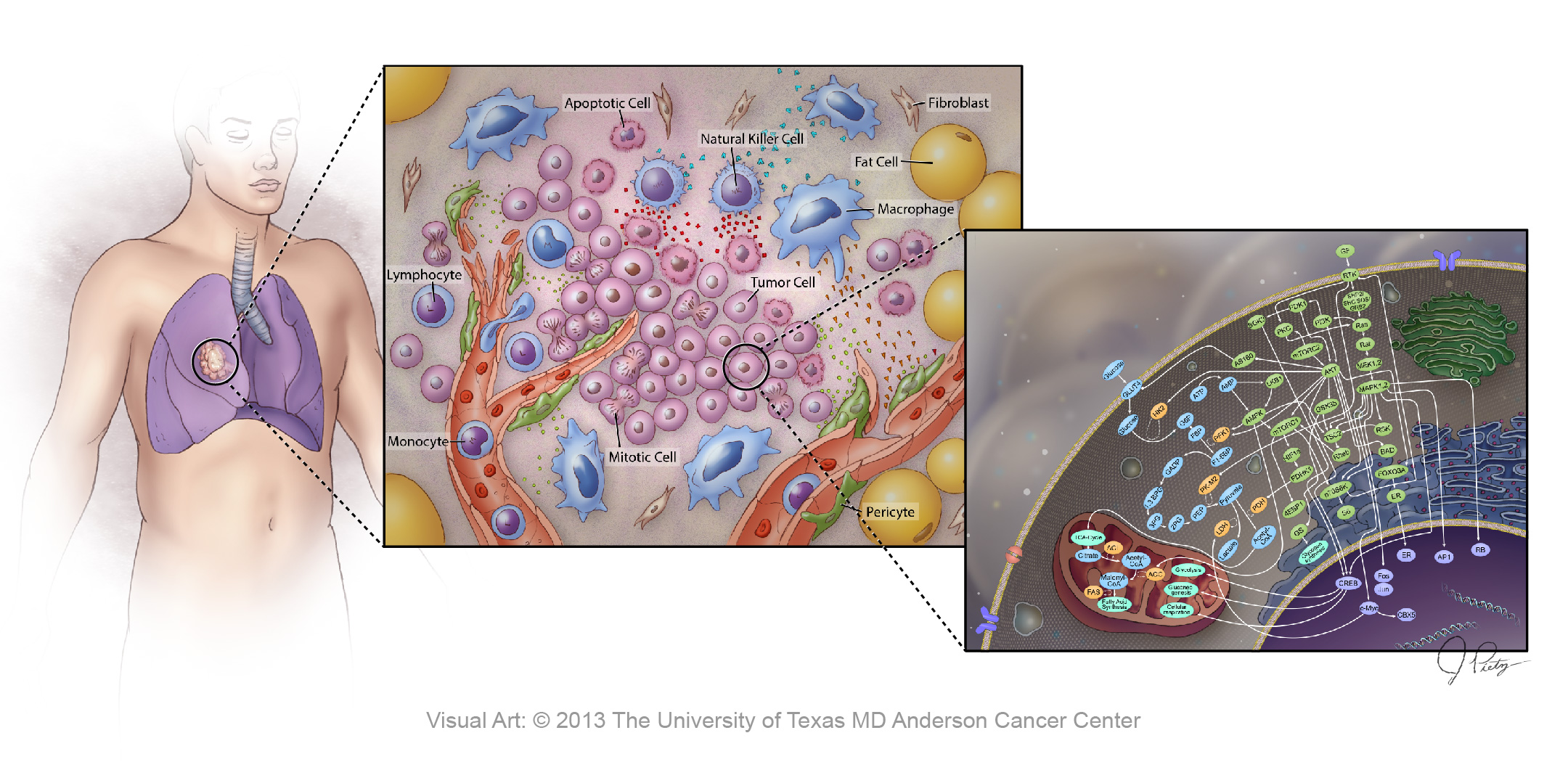
Projects such as The Cancer Genome Atlas (TCGA)8, and the International Cancer Genomics Consortium (ICGC)9, that build on the success of the Human Genome Project, are providing the ‘parts list’ of cancer; while (cancer) systems biology will provide the regulatory logic.10 Multiple data types are used and integrated, including clinical data. Just as the spectrum of input cancer biology data is wide, so are the computational approaches used in cancer systems biology, including new mathematical and computational algorithms that reflect the dynamic interplay between experimental biology and the quantitative sciences. A cancer systems biology approach can be applied at the level of an individual cell or to a tissue, to a patient with a primary tumour and possible metastases, or to any combination of these (Figure 1). This approach can integrate the molecular characteristics of tumours at different levels (DNA, RNA, protein, epigenetic, imaging) and different intervals (seconds vs. days) with multidisciplinary analysis. One of the major challenges to its success, besides the challenge posed by the heterogeneity of cancer per se, resides in acquiring high-quality data describing clinical characteristics, pathology, treatment, and outcomes and integrating the data into robust predictive models.
Figure 1. Application of cancer systems biology to decipher complex interactions in multiple dimensions.

This figure shows the complexity of interactions between different molecular networks within the cell that regulate the multitude of cellular functions. The intracellular machinery must coordinate with surrounding cells and the extracellular microenvironment to achieve homeostasis. Working within the body as a whole, each of these physiologically functional units is tightly coupled and regulated. Accumulated aberrations at the molecular level can decouple any one of these complex interactions and lead to a neoplastic phenotype. The promise of cancer systems biology is the ability to integrate these multi-scale characteristics into cohesive predictive models that can uncover emergent properties to be used in making cancer history.
Modelling approaches tackling various aspects of cancer have been successful in predicting the behaviour of cancer cells and tissues in the context of drug response and angiogenesis, understanding stem cell heterogeneity, the integration of large omics data as well as modelling cellular behaviours at the tissue level.11–15 The urgency for the implementation of a successful cancer systems biology programme has been highlighted by the emergence of targeted therapies as cancer treatment. Although targeted therapies have many advantages owing to their ability to specifically target one process, the response rates have not as yet fulfilled their early promise and hype.16–19 The use of cancer systems biology and integrated network analyses has the potential to support the understanding of the relationship between molecular markers and response to (targeted) therapies, facilitating the identification of the subgroup of patients most likely to benefit from a particular therapy, or the identification of those combinations of therapies that will result in an increased efficacy or will be able to bypass drug resistance. Resistance to therapy remains the most frequent cause of treatment failure. A systems analysis of the molecular characteristics of non-responsive or resistant tumours is an exciting opportunity to elucidate mechanisms underlying resistance and to identify ways to overcome it (Figure 2).
Figure 2. Integrating tumour molecular characteristics with pharmacogenomics for precision.

Cancer systems biology can bridge the vast amount of molecular characteristics of the tumour with pharmacogenomics to deliver on the promise of personalized therapy. The use of systems models to integrate patient specific data sets with drug response profiles can enable the prediction of effective patient-specific therapeutic options.
Because of the extreme heterogeneity and genomic instability of cancer cells, one of the emerging challenges is to distinguish ‘driver’ aberrations, which directly alter the behaviour of the tumour, and thus represent potential targets or biomarkers, from ‘passenger’ aberrations that do not affect the cancer cell (and thus are not effective targets).20,21 It is important to emphasize that passenger aberrations can occur within cancer genes, those genes that clearly play a role in cancer pathophysiology, and that different aberrations within cancer genes can have distinct effects on cellular functions and response to targeted therapeutics. Systems-based computational methods have been useful in identifying low frequency mutations in impure and heterogenous samples, which is related directly to the correct identification of subclonal drivers and, therefore, our understanding of tumour progression and treatment resistance.22 Thus, a systems biology approach has the potential to provide the infrastructure to allow a more-efficient discrimination between drivers and passengers mutations.
Cancer systems biology resources
High-throughput technologies have been developed to enable comprehensive genomic analyses of mutations, rearrangements, copy number variations, and methylation at the cellular and tissue levels, as well as robust analysis of RNA and microRNA expression data, protein levels and metabolite levels.3,20,21,23–27 In parallel, predictive data-driven mathematical and computational methods have been developed to analyse the large amounts of data arising from the new analysis platforms.28–33 Unfortunately, none of the platforms alone is sufficient to capture the complexity of changes in the genome of a cancer cell or to discriminate optimally between cancer driver and passenger mutations.34,35 In addition, all methods generate noise and have their own intrinsic weaknesses, thereby hampering interpretation. Beyond the previously mentioned problem of obtaining high-quality clinical and phenotypic data to be integrated with the ‘omics’ platforms, a major challenge will be the integration of data across the different platforms.
So far, the ability to integrate and interpret data across platforms has substantially lagged behind the ability to generate the data;36 however, integrative genomic studies can provide a new paradigm for the discovery of cancer aberrations and interactions, novel treatments, and resistance mechanisms. Large-scale projects have generated terabytes of ‘omics’ data that have been curated and made available to the international scientific community (Table 1). To integrate the data and present them in a way that biological translations can be generated,37 large programs have developed novel computational tools and databases of interactions that are accessible to the larger community (table 1). Furthermore, independent of these large-scale projects, many online resources contain annotated high-throughput data, computing tools and visualization, databases of interaction and quantitative biological information (Table 1).
Table 1.
| Project | Website |
|---|---|
| TCGA (The Cancer Genome Project) | http://cancergenome.nih.gov/ |
| TCGA performs comprehensive genomic characterisation and analysis of multiple cancer types to drive the understanding of the molecular basis of cancer | |
|
| |
| ICGC (International Cancer Genome Consortium) | http://icgc.org/ |
| ICGC is focused on a comprehensive understanding of the genomic abnormalities in cancer | |
|
| |
| SU2C (Stand Up to Cancer) | http://www.standup2cancer.org/ |
| SU2C aims to drive innovative cancer research through interdisciplinary science | |
|
| |
| LINCS (Library of Integrated Network-based Cellular Signatures) | http://www.lincsproject.org/ |
| LINCS aims to enhance a network-based understanding of biology, cataloging changes in gene expression and other cellular processes in response to perturbations | |
|
| |
| Sanger Institute databases | http://www.sanger.ac.uk/resources/databases |
| The institute has developed a suite of databases to manage and interpret large-scale data | |
|
| |
| cBioPortal for Cancer Genomics | http://www.cbioportal.org/public-portal/ |
| CBIO is a platform to visualise, analyse and download large scale genomic data sets | |
|
| |
| The Human Protein Atlas | http://www.proteinatlas.org/ |
| The atlas aims to explore the human proteome systematically through antibody-based proteomics | |
|
| |
| The Cancer proteome atlas | http://app1.bioinformatics.mdanderson.org/tcpa/_design/basic/index.html |
| The atlas aims to provide a comprehensive resource for accessing, visualising, and analysing cancer functional proteomics | |
|
| |
| TCGA data portal | https://tcga-data.nci.nih.gov/tcga/ |
| A platform for researchers to search, download, and analyse data sets generated by TCGA | |
|
| |
| Cytoscape | http://www.cytoscape.org/ |
| Cytoscope is an open source platform for visualising complex networks and integrating the networks with other data types | |
|
| |
| Vcell | http://www.nrcam.uchc.edu/ |
| The Virtual cell is a computational platform to model and simulate cell biology | |
|
| |
| COPASI | http://www.copasi.org/tiki-view_articles.php |
| COPASI is a platform to simulate and analyse networks and their dynamics | |
|
| |
| Netwalker | https://netwalkersuite.org/ |
| Netwalker is a platform to assist in functional analyses of large-scale genomics datasets focused on molecular networks | |
|
| |
| SageBio | http://sagebase.org/ |
| SageBio creates platforms to enable collaboration on data and data sharing. | |
| They also run challenges on complex biomedical problems | |
|
| |
| STRING | http://string-db.org/ |
| String is a database of known and predicted physical and functional protein interactions | |
|
| |
| GeneCards | http://www.genecards.org/ |
| GeneCards provides a database of human genes with comprehensive information on all known and predicted human genes. | |
|
| |
| Pathway Commons | http://www.pathwaycommons.org/about/ |
| Pathway Commons is a portal to access biological pathway information collected from public pathway databases | |
| DREAM (Dialogue for Reverse Engineering Assessments and Methods) | http://www.the-dream-project.org/ |
| DREAM aims to be a catalyser for the interaction between experiment and theory focused on cellular network inference and quantitative model building | |
|
| |
| ICBP (Integrated Cancer Biology Program) | http://icbp.nci.nih.gov/ |
| ICBP develops and implements computational models of cancer processes | |
The existing systems biology resources contain different types of data that are synergistic and complementary and form the spokes of the cancer systems biology wheel (Fig. 3). On one side, TCGA, ICGC, and Stand Up to Cancer projects have focused on the collection of tumour samples and on the characterization of their molecular profiles together with data on patient outcomes. On the other side, CCLE, LINCS, and ICBP projects have focused on obtaining a detailed molecular characterization of the responses of several cancer cell lines to multiple perturbations. Online databases and computing resources contain detailed information on genes, proteins, and molecular interactions as well as online computing software and tools to visualize and analyze large datasets (Table 1).
The data collection and computing efforts through many programmes have provided a rich resource for the implementation of cancer systems biology and have supported the progression of the field. Moreover, adapting new efforts, such as ‘crowd sourcing’ to large data analysis, have already provided an exciting and novel way to analyse large system-level biological data.38,39 Projects such as the Dialogue for Reverse Engineering Assessments and Methods (DREAM challenge), have made ‘big data’, collected through efforts such as the ICBP, available to the international community of computational biologists and mathematicians (Table 1).
Cancer systems biology approaches
In spite of extensive research investigating and uncovering many important aspects of cancer biology, we are still far from an integrated understanding of how the genomic and epigenetic abnormalities that occur in cancer cells mediate their functional consequences. Many groups have assessed large clinical cohorts by measuring one molecular data type (such as gene expression, mutation, single-nucleotide polymorphism) in relation to a measurable outcome to derive correlations between a particular molecular event and disease subtypes. These observational studies are important, but they are insufficient to demonstrate causality. Furthermore, the interactions between genes, proteins, and metabolites are coordinated in intracellular and intercellular networks to enable effective interaction with the microenvironment and with all organ systems in our body, necessitating both the acquisition and integration of data across multiple levels. The cellular networks that are perturbed in cancer are, in general, the same cellular pathways used by normal cells to perceive and respond to the environment, although not always with the same interactions or spatio-temporal dynamics, owing to the rewiring of the networks as a result of molecular aberrations. Furthermore, in some cases, gene mutations or fusions can result in new functions that are not mediated by the parental molecule. These neomorphic functions engendered by genomic aberrations in cancer cells are particularly difficult to deconvolute and integrate into cellular pathways and networks. Because aberrations can occur at various levels and lead to changes in multiple parts of a signalling network, integrating multiple data types from the same tumour becomes necessary in order to derive a more global or ‘systemic’ understanding of the molecular drivers of the cancer.
The TCGA Research Network has developed perhaps the most comprehensive data set encompassing exon and whole-genome sequencing data combined with analysis of DNA copy number, mRNA, microRNA, promoter methylation and protein expression.8 In each tumour lineage examined, new observations have been reported. For example, the genomic analysis of high-grade serous ovarian cancer in the North American and European populations has revealed the existence of TP53 mutations across virtually all tumours and the dominant effect of DNA copy number aberrations, Furthermore, this analysis identified specific microRNA, mRNA, or methylation subtypes and the activation of the NOTCH and FOXM1 pathways as key features of ovarian carcinoma.25 Similar analyses in breast cancer increased our understanding of previously identified subsets, highlighted the recurrence of mutations in TP53, PIK3CA, and GATA3 genes as well as specific mutations within subtypes, such as PIK3CA in luminal tumours, and led to the identification of new subtypes of breast cancer that were not obvious from previous analyses.3 Interestingly, integrated analyses identified dominant signalling pathways driven by HER2 or EGFR activity.3 based on the downstream phosphorylation of the EGFR HER2 signaling network stressing the differential treatment of patients within this subgroup moving forward. In endometrial, colon, and rectal cancer, hypermutated tumours seemed to be a result of microsatellite instability, with a new type of instability driven by pole gene mutations resulting in ultramutated tumors.24 Integrated analyses also revealed MYC-directed activation in aggressive colorectal carcinoma.24 The use of this type of analysis in clear-cell renal cell carcinoma, identified cellular oxygen sensing, chromatin remodeling/histone methylation, and metabolic shifts in the tricarboxylic acid (TCA) cycle as key processes in this pathology.20 Of note, integrated analyses across different cancer types highlighted some striking similarities in terms of molecular characteristics for basal-like breast cancer, high-grade serous ovarian cancer, and serous endometrial cancer.21 Systems approaches integrating data across multiple diseases in a ‘pan cancer’ effort aimed at improving our knowledge of the molecular pathogenesis of cancer whereby data is analyzed across tumor types to identify molecular characteristics common to a range of cancer types versus disease sub-types. The greatest opportunities associated with such analysis are only beginning to emerge. The multiplatform data sets on highly characterized patient samples provide the information necessary for the development of robust and predictive models. However, the importance of an iterative approach wherein hypotheses and predictions arising from the modelling are refined and constrained by experimental evaluation must be emphasized. In addition to such large consortial efforts, several groups have employed systems approaches using novel computational methods to integrate different data types to elicit novel principles. Carro et al.,11 using gene expression and network approaches, identified key transcriptional factors, such as CEBP and Stat3, as master regulators of the mesenchymal transformation in glioblastoma. Importantly, they demonstrated that the expression of these transcription factors could predict poor clinical outcome. In another approach the integration of protein and phosphoproteomic interactions with transcription factor-DNA binding data led to the discovery of novel features of cellular responses to stress and growth factors.12,29 Similarly, systems-level analyses of proteomic and metabolomic data revealed novel regulatory functions between these two important cellular networks that would not have been achieved otherwise.40 Furthermore, the integration, through network analysis, of mRNA expression data with proteomic, phosphoproteomic and clinical outcomes data, allowed the identification of key regulators of EGFR signalling and correlated these events with patient survival.41 Whole-genome sequencing and proteomics were also used to identify new driver genes in endometrial cancer and to identify a novel functional association between ARID1A and PI3K pathway activity.42
Systems approaches for tissue complexity
In any tissue in the body, there is extensive interaction between cells within the organ and the microenvironment, giving rise to an additional layer of complexity whereby multiple networks must work together cohesively. These same interactions occur between the tumour and its microenvironment, regulating the process of tumorigenesis. Many of these interactions are contextual, with markedly different effects at different stages of tumour development. For example, it is clear that TGFβ in the tumour microenvironment can mediate both tumour growth and inhibition. However, an understanding of the mechanisms underlying these disparate processes has remained elusive. This complex functionality may in fact only be solved via a systems biology approach. The molecular aberrations in cancer can significantly alter the normal dynamics of (neo)vascularisation and hypoxia response.. Furthermore, the role of the microenvironment in tumour dormancy and evading therapeutic interventions that allows oestrogen receptor-positive breast cancer to recur years later remains elusive.
Owing to the complexity of the tumour microenvironment interactions, many other aspects of cancer biology have not been explained by traditional scientific approaches and can benefit by the integrated analysis supported by system biology.2,43,44 Imaging of cells and their interactions with the extracellular environment can provide the high-quality data needed for the development of predictive models. To distinguish a stromal signature from a tumour signature and to understand the influence of different (metastatic) microenvironments, or how stromal expression changes can influence tumour progression, several strategies have been used. An extensive analysis of gene-expression patterns in primary and metastatic tumours, applying novel statistical analytical methods, led to the discovery of genes that were reprogrammed in brain metastases, but not in other metastatic sites. This reprogramming event was a function of the local microenvironment, likely regulated by promoter methylation.45 Moreover, Finak et al.46 purified tumour stromal cells by laser capture microdissection and compared gene-expression data of the tumour stroma to healthy stroma, and developed a stromal signature with clear prognostic impact independent of tumour subtypes. Thus, using systems-based methods, several groups have shown that the stroma has an important interplay with the tumour and has clear effects on disease progression and possibly also on effective treatment options. Developing computational models to fit the complex dynamics of the cellular microenvironment can provide potential hypotheses to be tested experimentally and aid in the interpretation of the results. This iterative approach provides the basis for a truly integrative modelling and experimental platform. Several experimental approaches are well adapted to feed data for this high-complexity integrative platform.
Three-dimensional culture systems provide a potential approach to more realistically mimic the in vivo situation. Multiple articles have shown that in contrast to 2D culture, cells grown in 3D respond differently to chemotherapy and radiotherapy,47,48 and also have markedly different gene-expression patterns related to the extracellular matrix and cell adhesion.49 Moreover, microfluidic systems, where tissues and even multiple different cell lineages can be studied in relatively high-throughput (micro)models, provide another experimental approach to support the modelling of the interactions between the tumour and microenvironment.50 These approaches have demonstrated critical environmental and spatial aspects of cellular responses within the tumour microenviroment.51,52,53,54 These in vitro approaches, therefore, have the potential to provide tractable experimental systems to develop hypotheses to test in vivo and also to experimentally test mathematical models developed from in vivo studies. Integrating in vitro and in vivo modeling approaches can play a major role in understanding how spatial orientation and interactions with the microenvironment can affect diverse tumour behaviour.
Response to (targeted) treatment and tumor progression are intrinsically linked to immunological pathways. At a single platform level, the complex interplay from the multitude of players cannot be completely captured. Using a pathway recognition algorithm and multilevel data from TCGA, Kristensen et al.55 showed that tumors can be stratified by their immune signatures. This stratification is shown to be relevant for patient survival and targeted treatment response. Further underlining the importance of the relationship between the tumor and the microenvironment, multiscale analysis showed the tumor can recreate its immune environment throughout cancer progression56.
Several investigators have developed modelling approaches to study the relationship between intracellular pathways and the behaviour of cells at the tissue level.57 Recently, some researchers modelled myc and p53 pathways during the proliferation of lymphoma cells in the context of surrounding tissue, growth factors, and angiogenesis.58 In addition, models considering the effect of cell division rates as well as interactions between cancer cells and the microenvironment predicted that the order in which targeted and chemotherapy approaches are delivered, is important to optimizing benefit whereby the proliferation rates and migration capabilities of different cell types could alter the tissue level response to therapy.59 Critically, these models will help to predict how effects at small intervals within the cell relate to long-term effects observed at the tissue or whole body level. Mathematical models of the spatio-temporal heterogeneity of the extracellular matrix have provided unexpected insights into how, for example, the urokinase plasminogen activation system impacts on cell invasion.60 The aforementioned studies primarily used ‘omics’ data derived from analyses of tumour lysates without the consideration of spatial orientation, one of the key aspects of the tumour microenvironment is spatial constraints. To incorporate spatial information, imaging of tumour cells and their environment is of paramount importance, particularly in the context of angiogenesis. The differential imaging of blood vessels and cell types using fluorescent probes as well as intravital imaging of tumours in animal models and cell cultures are techniques that provide the data necessary to develop ‘tissue-level’ computational models that can incorporate all these facets. For example, two predictive models for angiogenesis and cell-cell interactions have been developed, by incorporating imaging data, in order to study in vivo aspects of the tumour biology and drug response, respectively.70,71 Furthermore, Haeno et al.61 used a dissemination dynamics model (including well-annotated patient data) to predict the type and sequential order of treatments most likely to benefit patients with pancreatic cancer. This model can have important clinical implications for this disease that has a notoriously poor prognosis. Importantly, Kim and colleagues highlighted the applicability and individual benefits of different types of modelling in combination with in vitro and patient data to study spatio temporal drug distribution within the tumor to optimize drug selection and delivery.62
Systems approaches for cell heterogeneity
Cancer stem-like cells, also known as tumour-initiating cells, have been intensely studied in recent years. These studies have been driven in part by the discovery that the presence of cancer stem-like cells likely contributes to drug resistance and poor outcome.63,64 Interestingly, characteristics previously attributed to epithelial-mesenchymal transition (EMT), including EMT markers and mesenchymal morphology, can contribute to or be associated with a cancer stem cell-like state and drug resistance.65 The ability to transition in and out of an EMT-like state is critical for a cell to initiate and complete the metastatic process.66,67 Systems-based analysis of prostate cancer identified a molecular signature associated with patient outcome.68 Moreover, the results from this study highlighted an extensive plasticity of basal cells, supporting a model in which cells of origin can generate distinct molecular subtypes of prostate cancer.68
Different models of the spatiotemporal dynamics of stem cells in both healthy cells and tumour cells have been published to evaluate normal olfactory epithelium,69 bowel crypts,70 and solid tumours71 in an attempt to understand the regulatory strategies of cell renewal. These models give rise to the hypothesis that stem cells are not cell types as such, but rather a behavioural state imposed on the cell by feedback mechanisms. Such mechanisms are still in place in tumours, although they are susceptible to perturbations, including those induced by treatment. Changes in tumour size can alter cell proliferation and death by disturbing the feedback mechanisms and shift the balance towards cell renewal. These models could help in predicting which approaches might successfully shrink the tumour and benefit patients.71
Intratumoural and intertumoural heterogeneity contribute to patient prognosis and predicts response to therapy.67,72 Intratumoural heterogeneity may represent the greatest challenge to deliver effective personalized cancer therapy. Tyson et al.73 developed novel automated tools to capture and quantitate multiplexed imaging features to develop predictive models of the heterogeneity of response of individual cells to perturbations and were able to show heterogeneous cell fates upon exposure to anti-proliferative drugs. In another study, Yankeelov et al.,74 used quantitative tumour imaging methods and developed a model of interactions between cancer cells, stroma and immune cells, vascularisation and the extracellular matrix, that was able to predict treatment response and tumour progression. These modeling approaches have been very useful in enabling us to understand cellular heterogeneity and response to perturbations and lay the foundation of how we can use such information to develop novel methods to treat tumors which are inherently heterogeneous.
Systems approaches for targeted therapy
One of the continual challenges in the use of targeted therapy is the low response rates observed in the clinical setting.16–19 In many cancer systems biology efforts, novel computational and mathematical methods are used to integrate and analyse patients’ molecular data to identify optimal markers and targets. Recent efforts focusing on colorectal and liver cancers have integrated whole-genome expression data with patient’s outcomes to classify patients and their therapy responses according to gene-expression patterns.75 Using unbiased analysis of these datasets, the investigators identified specific expression patterns and pathways that were associated to specific mutations in the tumour, and which altered drug response in the cancer cells.75 Furthermore, extensive computational analysis of a large metabolic network revealed key regulatory nodes, such as oxidative phosphorylation, glycolysis and citric acid cycle that could serve as optimal targets for therapy.76,77 Several groups have also integrated phosphoproteomic time-course data of cell line responses to growth factors and perturbations in epithelial tumours, developing predictive data-driven models to identify rational targets and combinations of drugs for effective therapy.13,33,78,79 In diseases with limited therapeutic options (such as pancreatic cancer) an extensive systems biology approach integrating gene-expression data from primary pancreatic ductal adenocarcinomas and from human or murine pancreatic adenocarcinoma cell lines, revealed novel classifiers, predicted potential subtype-specific therapies, and identified non-responders.80
Beyond pancreatic cancer, an area of intense research in terms of system biology has been breast cancer. Much work has in fact focused on molecular phenotypes and drug responses in breast cancer. These efforts used several systems-level approaches, including large ‘omics’ data acquisition, analysis of patients and responses to clinically approved drugs as well as large panels of cell line-based assays.26,40,81–85 Computational modeling and analysis of perturbation screens using non-coding RNA have also proven useful in defining network topologies and predicting combinations of targets based on mechanistic functions of subnetworks.7,86–88
Approaches for drug resistance
As mentioned before, drug resistance is one of the major challenges to effective cancer-treatments. Integration of thousands of ‘omics’ datapoints acquired from sensitive and resistant tumours into a systems-based therapeutic strategy has been shown to be a powerful tool for tackling resistance.75,89 For example., models of dynamic changes in signalling networks of receptor tyrosine kinases (RTK) families have facilitated the classification of RTKs and their network activation and led to the identification of points of intervention to delay or overcome drug resistance.78,90 Furthermore, Komurov et al.,91 used a novel network-based analysis of gene expression and proteomics coupled to ErbB2-positive patient survival data, and showed that in breast tumours with acquired resistance to lapatinib, the drug was still able to block EGFR/ErbB2 signalling, but that upregulation of glucose metabolism, unfolded protein response, and endoplasmic reticulum (ER) stress pathways mitigated the ability of lapatinib to induce cell death, suggesting that coordinated targeting of metabolic networks and signalling networks has the potential to improve patient outcomes.91,92 Beckman et al.93 illustrated that the potential effects on drug response and resistance acquisition induced by single cell heterogeneity and cellular dynamics can be mathematically modeled and, therefore, should be included in personalized treatment strategies. They demonstrated that a strategy targeting all cancer subpopulations including the precursors of resistant clones, rather than treatment targeting the clone predominantly present which therefore initially leads to the largest reduction in tumour size, may be more effective. As such counterintuitive strategies may be of advantage for patient outcome. Lastly, integrating patient data with murine models has proven a powerful systems approach to identify mechanisms to overcome resistance to androgen therapy in prostate cancers.94 All these different studies highlight how using systems biology approaches can improve targeted therapy and help to overcome drug resistance.
With the aim of improving effective combinatorial drug prediction in cancer, mathematical modelling combined with high-density time-dependent measurements showed that modulation of oncogenic pathways through sequential application of drug combinations is possible and can render cancer cells much more susceptible to the drugs, thus significantly increasing the efficacy of treatment.95 Context-specific metabolic modelling algorithms demonstrated feasibility in predicting drug targets and phenotypic tumour response, including off-target effects, making this an effective systems approach for the development of new (combinatorial) drugs therapy.96 For example, STK38 (Serine-threonine kinase 38,) was identified as an upstream regulator of MYC activity through a model of network dynamics(modulator inference by network dynamics (MINDy) Algorithm) aimed at predicting transcription factor modulators, and experimental manipulation of STK38 demonstrated inhibition of tumour growth in MYC-dependent tumors in vivo; and thus a potential novel means to target these tumours.97
Cancer systems biology has made an impact on drug discovery and development from the identification of targets up to the clinical trial stage. Computational analysis of the ErbB family network has revealed ErbB3 as a key node of ligand-induced ErbB2 activation.98 These results facilitated the engineering of a bispecific ErbB3 antibody, MM-111, that inhibits ligand-induced receptor activation in an ErbB2-overexpressed environment and as such bypasses the tumour ErbB3 escape route that is often activated in drug resistance.99 Computational modelling together with pharmacodynamic and tumour growth algorithms aimed at identifying optimal drug combinations (with MM-111) to prevent drug resistance.100 Of note, the MM-111 antibody, a drug that would not likely have been developed without a systems biology approach, is currently in clinical development.
The examples described in this Review highlight the potential of combining multiscale high-throughput data analysis with mathematical modelling to identify novel and sometimes unexpected principles. Importantly, a number of these models are being used to develop selection approaches for implementation into clinical trials.
Data sharing
Although large-scale collections of data is inherently associated with risks of errors, making these data sets available to the cancer research community and the public enables a community or crowdsourcing approach to data analysis as well as data forensics, facilitating the recognition of possible errors.101–103 Bilal et al.101 have proposed competition-based modelling using large datasets with omics and clinical information to improve survival predictions among patients with breast cancer. The fact that the data sets are available to the public, allows the models to evolve through interaction with the community and prevents the so-called ‘self-assessment trap’, where model building and testing are carried out within the same group with the risk of reporting (too) favourable results. A different crowdsourcing research study focused on developing prognostic models for breast cancer using genome-scale data (gene expression, copy number analysis, and clinical variables), and showed the approach to be capable of generating prognostic models of at least equal quality to previously reported studies,110 and consistent across multiple independent evaluations.38,39 Much as there have been different experimental approaches to understand cancer there have also been many different modeling approaches that have been developed. The modeling approaches utilise different algorithms and these are largely dependent on the type of data that has been available and the questions that are being answered
Conclusions and future directions
The era in which cancer was treated solely according to the organ of origin is coming to an end, leaving space for a greater understanding of the complex interactions between the genomic aberrations present in tumour cells, the intrinsic gene-expression patterns of the tissue of origin, and the tumour microenvironment (Figure 2). Cancer systems biology has the potential to usher in an era in which the effects of molecular aberrations and interactions within networks are integrated with molecular knowledge and pharmacogenomics. Improvements in our ability to image and measure quantitatively spatial localization of network activities and incorporate spatio-temporal information acquired in patients with ‘omics’ data and computational analysis are providing the right platform for systems biology to impact on patient outcomes. Cancer systems biology approaches can improve our understanding of how tumour heterogeneity, neovascularization, immune response, and changes in the tumour and its microenvironment over time and in response to therapeutic interventions contribute to treatment response. Cancer systems biology is a young field and clinical studies have not yet been reported based on systems approaches, nevertheless systems biology has already shown a great potential and it is clear that will have a major role in the further development of personalized therapy. In our opinion, the greatest opportunity for cancer systems biology in the near future is to elicit emergent mechanisms of resistance and identify rational combination approaches to prevent or bypass drug resistance. It will also be important to incorporate side effects and toxicities in the models. In this way, systems approaches will contribute to the development of the next generation of clinical trials and cancer therapy.
Supplementary Material
{kind=link}
{kind=link}
Review criteria.
A search for original articles was performed in MEDLINE and PubMed, with the search terms ‘integrative/systematic analysis’, ‘computational/mathematical modeling’, ‘multidimensional networks’, ‘genetic dynamics’, ‘cellular heterogeneity’, alone and in combination; with a strong emphasis on literature published in the last 3 years. All articles identified were English-language, full-text papers. We also searched the reference lists of identified articles for further relevant papers.
Key points.
Systems biology and its application to cancer research and therapy
Integration of multiple omics data types to determine systems properties that can exploited for cancer therapy
Integration of computational and mathematical models with patient and lab-based data to identify and target novel emergent properties
Use of systems biology approaches to make clinical impact in the future of cancer personalized therapy
Biographies
Dr HMJ Werner gained her MD in 2000 from Maastricht University, Maastricht, the Netherlands and currently works as a consultant gynaecologist at Haukeland University Hospital in Bergen, Norway. She is a PhD student in translational research in endometrial carcinoma at the University in Bergen during which she joined the department of Systems Biology at MD Anderson Cancer Center, Houston, USA for one year.
Dr. Prahlad T. Ram received his PhD from Tulane University, New Orleans, LA in 1997 and is currently Associate Professor in the Department of Systems Biology at The University of Texas MD Anderson Cancer Center and Co-Director of the Biostatistics, Bioinformatics and Systems Biology Program in the Graduate School of Biological Science, Houston, TX. Dr. Ram’s research lab is half experimental and half computational and utilizes an integrated systems approach to understand and target networks in cancer.
Dr. Gordon B. Mills received his MD in 1977 and PhD in 1984 from The University of Alberta, Canada and is currently Professor and Chairman of the Department of Systems Biology, the Co-Director of the Institute for Personalized Cancer Therapy, the Head of the Kleberg Center for Molecular Markers and co-Director of the Women’s Cancer Moonshot Program at the University of Texas MD Anderson Cancer Center, Houston, TX. Dr. Mills has published over 600 papers and has been on the forefront of cancer systems biology, molecular markers, targeted and personalized therapy in cancer.
Footnotes
Competing interests
The authors declare no competing interests
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gentles AJ, Gallahan D. Systems biology: confronting the complexity of cancer. Cancer Res. 2011;71:5961–4. doi: 10.1158/0008-5472.CAN-11-1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basu A, et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell. 2013;154:1151–61. doi: 10.1016/j.cell.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berns K, Bernards R. Understanding resistance to targeted cancer drugs through loss of function genetic screens. Drug Resist Updat. 2012;15:268–75. doi: 10.1016/j.drup.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 7.Whitehurst AW, et al. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature. 2007;446:815–9. doi: 10.1038/nature05697. [DOI] [PubMed] [Google Scholar]
- 8.Institute, T.N.C. The Cancer Genome Atlas. http://cancergenome.nih.gov/
- 9.ICGC. International Cancer Genome Consortium. http://icgc.org/
- 10.Lefebvre C, Rieckhof G, Califano A. Reverse-engineering human regulatory networks. Wiley Interdiscip Rev Syst Biol Med. 2012;4:311–25. doi: 10.1002/wsbm.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carro MS, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318–25. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang SS, et al. Linking proteomic and transcriptional data through the interactome and epigenome reveals a map of oncogene-induced signaling. PLoS Comput Biol. 2013;9:e1002887. doi: 10.1371/journal.pcbi.1002887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iadevaia S, Lu Y, Morales FC, Mills GB, Ram PT. Identification of optimal drug combinations targeting cellular networks: integrating phospho-proteomics and computational network analysis. Cancer Res. 2010;70:6704–14. doi: 10.1158/0008-5472.CAN-10-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pascal J, et al. Mechanistic patient-specific predictive correlation of tumor drug response with microenvironment and perfusion measurements. Proc Natl Acad Sci U S A. 2013;110:14266–71. doi: 10.1073/pnas.1300619110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swanson KR, et al. Quantifying the role of angiogenesis in malignant progression of gliomas: in silico modeling integrates imaging and histology. Cancer Res. 2011;71:7366–75. doi: 10.1158/0008-5472.CAN-11-1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dedes KJ, Wetterskog D, Ashworth A, Kaye SB, Reis-Filho JS. Emerging therapeutic targets in endometrial cancer. Nat Rev Clin Oncol. 2011;8:261–71. doi: 10.1038/nrclinonc.2010.216. [DOI] [PubMed] [Google Scholar]
- 17.Gore ME, Larkin JM. Challenges and opportunities for converting renal cell carcinoma into a chronic disease with targeted therapies. Br J Cancer. 2011;104:399–406. doi: 10.1038/sj.bjc.6606084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diaz LA, Jr, et al. Nature. 2012;486:537–40. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Janku F, et al. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol. 2012;30:777–82. doi: 10.1200/JCO.2011.36.1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cancer Genome Atlas Research N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–9. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cancer Genome Atlas Research N. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cibulskis K, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–9. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hennessy BT, et al. A Technical Assessment of the Utility of Reverse Phase Protein Arrays for the Study of the Functional Proteome in Non-microdissected Human Breast Cancers. Clin Proteomics. 2010;6:129–51. doi: 10.1007/s12014-010-9055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chin K, et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell. 2006;10:529–41. doi: 10.1016/j.ccr.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 27.Neve RM, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–27. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bodenmiller B, et al. Multiplexed mass cytometry profiling of cellular states perturbed by small-molecule regulators. Nat Biotechnol. 2012;30:858–67. doi: 10.1038/nbt.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hill SM, et al. Bayesian inference of signaling network topology in a cancer cell line. Bioinformatics. 2012;28:2804–10. doi: 10.1093/bioinformatics/bts514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pritchard JR, et al. Defining principles of combination drug mechanisms of action. Proc Natl Acad Sci U S A. 2013;110:E170–9. doi: 10.1073/pnas.1210419110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qiu P, et al. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat Biotechnol. 2011;29:886–91. doi: 10.1038/nbt.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sumazin P, et al. An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Cell. 2011;147:370–81. doi: 10.1016/j.cell.2011.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tentner AR, et al. Combined experimental and computational analysis of DNA damage signaling reveals context-dependent roles for Erk in apoptosis and G1/S arrest after genotoxic stress. Mol Syst Biol. 2012;8:568. doi: 10.1038/msb.2012.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bozic I, et al. Accumulation of driver and passenger mutations during tumor progression. Proc Natl Acad Sci U S A. 2010;107:18545–50. doi: 10.1073/pnas.1010978107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greenman C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–8. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mills GB. An emerging toolkit for targeted cancer therapies. Genome Res. 2012;22:177–82. doi: 10.1101/gr.136044.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chin L, Hahn WC, Getz G, Meyerson M. Making sense of cancer genomic data. Genes Dev. 2011;25:534–55. doi: 10.1101/gad.2017311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Derry JM, et al. Developing predictive molecular maps of human disease through community-based modeling. Nat Genet. 2012;44:127–30. doi: 10.1038/ng.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Norman TC, Bountra C, Edwards AM, Yamamoto KR, Friend SH. Leveraging crowdsourcing to facilitate the discovery of new medicines. Sci Transl Med. 2011;3:88mr1. doi: 10.1126/scitranslmed.3002678. [DOI] [PubMed] [Google Scholar]
- 40.Bordbar A, et al. Model-driven multi-omic data analysis elucidates metabolic immunomodulators of macrophage activation. Mol Syst Biol. 2012;8:558. doi: 10.1038/msb.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amit I, et al. A module of negative feedback regulators defines growth factor signaling. Nat Genet. 2007;39:503–12. doi: 10.1038/ng1987. [DOI] [PubMed] [Google Scholar]
- 42.Liang H, et al. Whole-exome sequencing combined with functional genomics reveals novel candidate driver cancer genes in endometrial cancer. Genome Res. 2012;22:2120–9. doi: 10.1101/gr.137596.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Swartz MA, et al. Tumor microenvironment complexity: emerging roles in cancer therapy. Cancer Res. 2012;72:2473–80. doi: 10.1158/0008-5472.CAN-12-0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lander AD, et al. What does the concept of the stem cell niche really mean today? BMC Biol. 2012;10:19. doi: 10.1186/1741-7007-10-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Park ES, et al. Cross-species hybridization of microarrays for studying tumor transcriptome of brain metastasis. Proc Natl Acad Sci U S A. 2011;108:17456–61. doi: 10.1073/pnas.1114210108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Finak G, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14:518–27. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- 47.Puigvert JC, et al. Cross-talk between integrins and oncogenes modulates chemosensitivity. Mol Pharmacol. 2009;75:947–55. doi: 10.1124/mol.108.051649. [DOI] [PubMed] [Google Scholar]
- 48.Storch K, et al. Three-dimensional cell growth confers radioresistance by chromatin density modification. Cancer Res. 2010;70:3925–34. doi: 10.1158/0008-5472.CAN-09-3848. [DOI] [PubMed] [Google Scholar]
- 49.Zschenker O, Streichert T, Hehlgans S, Cordes N. Genome-wide gene expression analysis in cancer cells reveals 3D growth to affect ECM and processes associated with cell adhesion but not DNA repair. PLoS One. 2012;7:e34279. doi: 10.1371/journal.pone.0034279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Whitesides GM. The origins and the future of microfluidics. Nature. 2006;442:368–73. doi: 10.1038/nature05058. [DOI] [PubMed] [Google Scholar]
- 51.Albeck JG, Mills GB, Brugge JS. Frequency-modulated pulses of ERK activity transmit quantitative proliferation signals. Mol Cell. 2013;49:249–61. doi: 10.1016/j.molcel.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kenny HA, Krausz T, Yamada SD, Lengyel E. Use of a novel 3D culture model to elucidate the role of mesothelial cells, fibroblasts and extra-cellular matrices on adhesion and invasion of ovarian cancer cells to the omentum. Int J Cancer. 2007;121:1463–72. doi: 10.1002/ijc.22874. [DOI] [PubMed] [Google Scholar]
- 53.Kenny PA, et al. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol Oncol. 2007;1:84–96. doi: 10.1016/j.molonc.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rejniak KA, et al. Linking changes in epithelial morphogenesis to cancer mutations using computational modeling. PLoS Comput Biol. 2010;6 doi: 10.1371/journal.pcbi.1000900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kristensen VN, et al. Integrated molecular profiles of invasive breast tumors and ductal carcinoma in situ (DCIS) reveal differential vascular and interleukin signaling. Proc Natl Acad Sci U S A. 2012;109:2802–7. doi: 10.1073/pnas.1108781108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Remark R, et al. Characteristics and Clinical Impacts of the Immune Environments in Colorectal and Renal Cell Carcinoma Lung Metastases: Influence of Tumor Origin. Clin Cancer Res. 2013;19:4079–4091. doi: 10.1158/1078-0432.CCR-12-3847. [DOI] [PubMed] [Google Scholar]
- 57.Enderling H, Hlatky L, Hahnfeldt P. Cancer Stem Cells: A Minor Cancer Subpopulation that Redefines Global Cancer Features. Front Oncol. 2013;3:76. doi: 10.3389/fonc.2013.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Frieboes HB, et al. An integrated computational/experimental model of lymphoma growth. PLoS Comput Biol. 2013;9:e1003008. doi: 10.1371/journal.pcbi.1003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Enderling H, et al. Paradoxical dependencies of tumor dormancy and progression on basic cell kinetics. Cancer Res. 2009;69:8814–21. doi: 10.1158/0008-5472.CAN-09-2115. [DOI] [PubMed] [Google Scholar]
- 60.Andasari V, Gerisch A, Lolas G, South AP, Chaplain MA. Mathematical modeling of cancer cell invasion of tissue: biological insight from mathematical analysis and computational simulation. J Math Biol. 2011;63:141–71. doi: 10.1007/s00285-010-0369-1. [DOI] [PubMed] [Google Scholar]
- 61.Haeno H, et al. Computational modeling of pancreatic cancer reveals kinetics of metastasis suggesting optimum treatment strategies. Cell. 2012;148:362–75. doi: 10.1016/j.cell.2011.11.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim M, Gillies RJ, Rejniak KA. Current Advances in Mathematical Modeling of Anti-Cancer Drug Penetration into Tumor Tissues. Front Oncol. 2013;3:278. doi: 10.3389/fonc.2013.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eyler CE, Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol. 2008;26:2839–45. doi: 10.1200/JCO.2007.15.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–51. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Biddle A, et al. Cancer stem cells in squamous cell carcinoma switch between two distinct phenotypes that are preferentially migratory or proliferative. Cancer Res. 2011;71:5317–26. doi: 10.1158/0008-5472.CAN-11-1059. [DOI] [PubMed] [Google Scholar]
- 66.Chaffer CL, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A. 2011;108:7950–5. doi: 10.1073/pnas.1102454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gupta PB, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146:633–44. doi: 10.1016/j.cell.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 68.Wang ZA, et al. Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nat Cell Biol. 2013;15:274–83. doi: 10.1038/ncb2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lander AD, Gokoffski KK, Wan FY, Nie Q, Calof AL. Cell lineages and the logic of proliferative control. PLoS Biol. 2009;7:e15. doi: 10.1371/journal.pbio.1000015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang L, Lander AD, Nie Q. A reaction-diffusion mechanism influences cell lineage progression as a basis for formation, regeneration, and stability of intestinal crypts. BMC Syst Biol. 2012;6:93. doi: 10.1186/1752-0509-6-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Youssefpour H, Li X, Lander AD, Lowengrub JS. Multispecies model of cell lineages and feedback control in solid tumors. J Theor Biol. 2012;304:39–59. doi: 10.1016/j.jtbi.2012.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lawrence MS, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–8. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tyson DR, Garbett SP, Frick PL, Quaranta V. Fractional proliferation: a method to deconvolve cell population dynamics from single-cell data. Nat Methods. 2012;9:923–8. doi: 10.1038/nmeth.2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yankeelov TE, et al. Clinically relevant modeling of tumor growth and treatment response. Sci Transl Med. 2013;5:187ps9. doi: 10.1126/scitranslmed.3005686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sadanandam A, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013;19:619–25. doi: 10.1038/nm.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hu J, et al. Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat Biotechnol. 2013;31:522–9. doi: 10.1038/nbt.2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nelander S, et al. Models from experiments: combinatorial drug perturbations of cancer cells. Mol Syst Biol. 2008;4:216. doi: 10.1038/msb.2008.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Saez-Rodriguez J, et al. Comparing signaling networks between normal and transformed hepatocytes using discrete logical models. Cancer Res. 2011;71:5400–11. doi: 10.1158/0008-5472.CAN-10-4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Alexopoulos LG, Saez-Rodriguez J, Cosgrove BD, Lauffenburger DA, Sorger PK. Networks inferred from biochemical data reveal profound differences in toll-like receptor and inflammatory signaling between normal and transformed hepatocytes. Mol Cell Proteomics. 2010;9:1849–65. doi: 10.1074/mcp.M110.000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Collisson EA, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17:500–3. doi: 10.1038/nm.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bussey KJ, et al. Integrating data on DNA copy number with gene expression levels and drug sensitivities in the NCI-60 cell line panel. Mol Cancer Ther. 2006;5:853–67. doi: 10.1158/1535-7163.MCT-05-0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Heiser LM, et al. Integrated analysis of breast cancer cell lines reveals unique signaling pathways. Genome Biol. 2009;10:R31. doi: 10.1186/gb-2009-10-3-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Heiser LM, et al. Subtype and pathway specific responses to anticancer compounds in breast cancer. Proc Natl Acad Sci U S A. 2012;109:2724–9. doi: 10.1073/pnas.1018854108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim N, et al. Systematic analysis of genotype-specific drug responses in cancer. Int J Cancer. 2012;131:2456–64. doi: 10.1002/ijc.27529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tabchy A, Eltonsy N, Housman DE, Mills GB. Systematic identification of combinatorial drivers and targets in cancer cell lines. PLoS One. 2013;8:e60339. doi: 10.1371/journal.pone.0060339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Banko MR, et al. Chemical genetic screen for AMPKalpha2 substrates uncovers a network of proteins involved in mitosis. Mol Cell. 2011;44:878–92. doi: 10.1016/j.molcel.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jiang H, Pritchard JR, Williams RT, Lauffenburger DA, Hemann MT. A mammalian functional-genetic approach to characterizing cancer therapeutics. Nat Chem Biol. 2011;7:92–100. doi: 10.1038/nchembio.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lu Y, et al. Kinome siRNA-phosphoproteomic screen identifies networks regulating AKT signaling. Oncogene. 2011;30:4567–77. doi: 10.1038/onc.2011.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nieman KM, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;17:1498–503. doi: 10.1038/nm.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wagner JP, et al. Receptor tyrosine kinases fall into distinct classes based on their inferred signaling networks. Sci Signal. 2013;6:ra58. doi: 10.1126/scisignal.2003994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Komurov K, et al. The glucose-deprivation network counteracts lapatinib-induced toxicity in resistant ErbB2-positive breast cancer cells. Mol Syst Biol. 2012;8:596. doi: 10.1038/msb.2012.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Csibi A, et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell. 2013;153:840–54. doi: 10.1016/j.cell.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Beckman RA, Schemmann GS, Yeang CH. Impact of genetic dynamics and single-cell heterogeneity on development of nonstandard personalized medicine strategies for cancer. Proc Natl Acad Sci U S A. 2012;109:14586–91. doi: 10.1073/pnas.1203559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lunardi A, et al. A co-clinical approach identifies mechanisms and potential therapies for androgen deprivation resistance in prostate cancer. Nat Genet. 2013;45:747–55. doi: 10.1038/ng.2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lee MJ, et al. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell. 2012;149:780–94. doi: 10.1016/j.cell.2012.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chang RL, Xie L, Xie L, Bourne PE, Palsson BO. Drug off-target effects predicted using structural analysis in the context of a metabolic network model. PLoS Comput Biol. 2010;6:e1000938. doi: 10.1371/journal.pcbi.1000938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bisikirska BC, et al. STK38 is a critical upstream regulator of MYC’s oncogenic activity in human B-cell lymphoma. Oncogene. 2012 doi: 10.1038/onc.2012.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schoeberl B, et al. Therapeutically targeting ErbB3: a key node in ligand-induced activation of the ErbB receptor-PI3K axis. Sci Signal. 2009;2:ra31. doi: 10.1126/scisignal.2000352. [DOI] [PubMed] [Google Scholar]
- 99.McDonagh CF, et al. Antitumor activity of a novel bispecific antibody that targets the ErbB2/ErbB3 oncogenic unit and inhibits heregulin-induced activation of ErbB3. Mol Cancer Ther. 2012;11:582–93. doi: 10.1158/1535-7163.MCT-11-0820. [DOI] [PubMed] [Google Scholar]
- 100.Kirouac DC, et al. Computational Modeling of ERBB2-Amplified Breast Cancer Identifies Combined ErbB2/3 Blockade as Superior to the Combination of MEK and AKT Inhibitors. Sci Signal. 2013;6:ra68. doi: 10.1126/scisignal.2004008. [DOI] [PubMed] [Google Scholar]
- 101.Bilal E, et al. Improving breast cancer survival analysis through competition-based multidimensional modeling. PLoS Comput Biol. 2013;9:e1003047. doi: 10.1371/journal.pcbi.1003047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Challenge, t.D. the Dream Challenge.
- 103.Shi L, et al. The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models. Nat Biotechnol. 2010;28:827–38. doi: 10.1038/nbt.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.