

Abstract
The identity of radical species associated with particulate formed from the oxidative pyrolysis of 1-methylnaphthalene (1-MN) was investigated using low temperature matrix isolation electron paramagnetic resonance spectroscopy (LTMI-EPR), a specialized technique that provided a method of sampling and analysis of the gas-phase paramagnetic components. A superimposed EPR signal was identified to be a mixture of organic radicals (carbon and oxygen-centered) and soot. The carbon-centered radicals were identified as a mixture of the resonance-stabilized indenyl, cyclopentadienyl, and naphthalene 1-methylene radicals through the theoretical simulation of the radical’s hyperfine structure. Formation of these radical species was promoted by the addition of Fe(III)2O3 nanoparticles. Enhanced formation of resonance stabilized radicals from the addition of Fe(III)2O3 nanoparticles can account for the observed increased sooting tendency associated with Fe(III)2O3 nanoparticle addition.
Keywords: Environmentally Persistent Free Radicals (EPFRs), Particulate, Nanoparticles, Cryogenic trapping, EPR, Annealing studies
1. Introduction
Association of particulate matter with adverse human health effects and global climate change requires mechanistic studies of formation pathways in order to mitigate the adverse effects associated with anthropogenic emissions. Electron Paramagnetic Resonance (EPR) spectroscopy is the standard tool for the detection of paramagnetic species and has been widely used to elucidate the nature of combustion products produced from various sources including: propane [1], hexane [2–4], 1-methynapthalene [5], anthracene [6], diesel [4,7], jet fuel [4,8], coal [9–11], ash [12], ambient particulate [13–17], and cigarettes [18]. The EPR signal of particulate, often only a broad featureless singlet, has been proposed to be the superposition of multiple radical species based solely on the broadness of the signal and the detection of multiple radical decays [1,5,10,19]. However, the lack of hyperfine structure and broadness of the signal impedes spectral assignment and only reveals the persistent nature of radicals associated with particulate. Variation in reported line widths associated with combustion generated particulate matter can result from the superposition of multiple radical species leading to a broad featureless EPR signal [3,4,7,8,10,15–17]. These broad features make identification of radical species present in particulate matter difficult and often lead to the generalization of these species as “soot radicals.”
From an EPR perspective, soot is a well characterized fraction of particulate matter defined by its persistent nature, relatively high spin density (1018–1020 spins/g) [4,6,18], g-factors ranging from 2.0028 to 2.0030 [7,10,16,17,20], and a ΔHp–p ~ 2–8G [3,4,7,8,10,15,16,21]. However, since soot is composed of carbon, the spectral width (ΔHp–p) of soot resulting from electron spin-spin interactions should only be between 1 and 2 G [22]. The reported variation in spectral width (ΔHp–p) suggests the presence of multiple radical species.
These radical species generalized as “soot radicals” are active participants in the molecular growth of soot. Recent studies have reported increased radical formation and subsequent formation of soot from the introduction of Fe(III)2O3 nanoparticles to a high-temperature flow reactor containing 1-methylnaphthalene (1-MN) fuel under oxidative pyrolysis conditions [5]. Redox-active transition metal nanoparticles, such as Fe(III)2O3, have been identified as active sites in the mediation and formation of surface-stabilized radicals [23–25]. Hence, surface-stabilized radicals were hypothesized to undergo subsequent surface-mediated, radical–radical, and/or radical–molecule, molecular growth pathways to contribute to polycyclic aromatic hydrocarbon (PAH) coalescence, soot particle inception, and soot surface growth [5]. This correlation, increased radical intensity as a function of iron concentration, has also been reported for atmospheric particulate [13] and soot generated in the presence of iron pentacarbonyl [19].
Iron oxide nanoparticles, generated from iron pentacarbonyl and ferrocene, have been reported to exhibit dichotomous behavior in their ability to both enhance soot formation and promote soot oxidation. Introduction of iron precursors has been shown to enhance soot formation through increased soot number density, particle size, PAH concentrations, and decreased time of appearance of soot in the flame [26–28] while decreasing sooting tendency [29] and enhancing soot oxidation rates through more efficient burnout rather than inhibition [30,31]. Although increased soot formation was observed from the introduction of Fe(III)2O3 nanoparticles to a 1-MN fuel [5], the broadness of the EPR signal hindered spectral assignment of the radicals associated with soot and questions still remained about the speciation of iron oxide nanoparticles prior to and after soot formation. The current study was performed to addresses the chemical speciation of paramagnetic centers observed in soot as superimposed features, the speciation of iron oxide nanoparticles, and the effect of iron oxide addition on radical formation.
The study of iron oxides influence on the soot formation is not limited to laboratory studies but has larger implications for soot formation from internal combustion engines. Metal oxides are available as minor components of fuel, oil, and result from normal engine wear [32]. Used motor oil was found to contain ppm quantities of Fe, Cu, and Zn. [33,34] that begin to enter the combustion cylinders through leaking piston rings as an engine ages. Similarly, a strong correlation has been established between metals present in diesel fuel and their incorporation into diesel particulate matter [35].
In this study, low temperature matrix isolation in conjunction with electron paramagnetic resonance spectroscopy (LTMI-EPR) [36] was employed for the detection of paramagnetic species formed during the growth of soot from a high-sooting 1-MN fuel in the presence and absence of iron oxide nanoparticles. This technique provided a means of gas-phase sampling of paramagnetic species without impacting the overall reaction chemistry.
2. Experimental procedures
These experiments were designed to gain an understanding of the chemical speciation of paramagnetic centers observed as superimposed features in EPR spectroscopy, the speciation of iron oxide, and the effect of iron oxide addition on radical formation.
2.1. Generation of iron oxide nanoparticles and soot
A two-zone, fused silica, heterogeneous-flow reactor system was utilized to generate soot from the oxidative pyrolysis of 95% 1-MN (Sigma–Aldrich M56808-100G) in the presence of iron oxide nanoparticles (cf. Fig. 1). The Zone 1 furnace was constructed of a CRFC helically wound ceramic ribbon heater with vestibules measuring 30 cm in length (8.8 cm outer diameter (OD), 3.8 cm inner diameter (ID), and 2.5 cm vestibule opening). A 33 cm fused silica tube with a 2.5 cm OD, tapered to 0.6 cm on both ends, was placed within the furnace. The Zone 2 furnace was constructed of a CRWS helically wound ceramic ribbon heater measuring 30 cm in length (8 cm OD and 3.8 cm ID). Connected to the Zone 1 reactor, a 0.6 cm OD, fused silica tube (51 cm in length) was inserted within the furnace and extended 10 cm outside of the Zone 2 reactor in both directions. The reactor (outside the furnace prior to Zone 2) created a cool zone to condense the iron oxide nanoparticles prior to introduction to 1-MN.
Fig. 1.
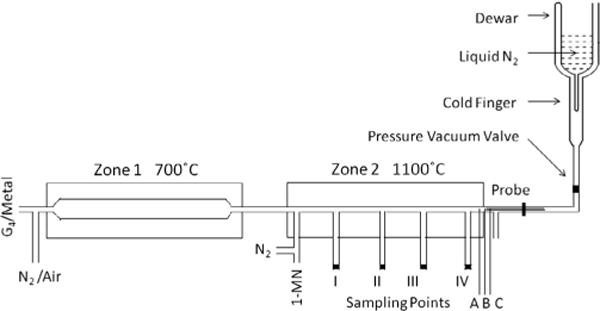
Dual-zone reactor capable of generating metal oxide nanoparticles (Zone 1) in the presence of a high sooting 1-MN fuel (Zone 2) coupled with a cold finger for condensation of paramagnetic species. Soot was collected through a moveable probe stationed at sampling ports denoted I–IV in the isothermal region as well as sampling points A (700 °C) and B (475 °C) in the quenching zone of reactor, and sampling point C (80 °C) in the exhaust stream.
Zone 1 generated metal oxide nanoparticles in situ from the oxidation of a polypropylenimine tetra-hexacontaamine dendrimer complexed with iron(III) nitrate nonahydrate under stoichiometric quantities of air diluted in nitrogen. A methanolic solution of the dendrimer-metal complex was delivered at 85 μL/h with a syringe pump into reactor 1 maintained at 700 °C and 1 atm. The gas-phase residence time in Zone 1 was maintained at 60 s. This resulted in the formation of iron oxide nanoparticles approximately 5 nm in diameter as determined by transmission electron microscopy [5]. These iron oxide nanoparticles were continually introduced into Zone 2 of the reactor to a high sooting 1-MN fuel at a fuel/air equivalence ratio (φ) of 2.5. 1-MN was introduced with a syringe pump into Zone 2 at 240 μL/h. These iron oxide nanoparticles (less than 1 ppm) could then influence the otherwise homogenous soot formation process. Zone 2 of the dual zone reactor was maintained at 1100 °C and 1 atm. with a gas-phase residence time of 1.0 s. This dual zone construction also allowed particulate to be generated in the absence of iron oxide nanoparticles.
2.2. LTMI-EPR technique
The LTMI-EPR technique has been previously and extensively described in the literature [36]. This technique makes possible the accumulation and detection of trace quantities of radicals from the gas phase and allows the kinetic behavior of these radicals produced during the reaction of many classes of organic precursors to be determined [37]. The dual-zone thermal reactor was interfaced to a liquid nitrogen-cooled Dewar to condense combustion products on the cold finger at 77 K for further EPR analysis (cf. Fig. 1). The Dewar was equipped with a special PTFE pressure–vacuum valve (PV-ANV, Wilmad) and threaded stopcock, which allowed the Dewar to be separated from the dual zone reactor and maintained at liquid nitrogen temperature at 10−4 torr for further EPR measurement. A rotary pump was used to maintain the pressure at <0.5 torr to transport the by-products to the cold finger from the thermo-electrically heated, oxidative-pyrolysis zone of the reactor (cf. Fig. 1, Zone 2). Using a moveable probe, sampling of the particulate was conducted through an orifice (i.d. from 100 to 300 μm) at distances of 8, 13, 18, and 23 cm from the entrance in the isothermal region (above sampling points I–IV) and the non-isothermal sampling points A (30 cm at 700 °C), B (31.5 cm at 475 °C) in the quenching zone of reactor, and C (37.5 cm at 80 °C) in the external exhaust stream.
The sampling orifice directed a portion of the effluent from the reaction zone onto a cold finger. The expansion of the gas-mixture in an orifice region resulted in a rapid decrease of temperature which suppressed further chemical and physical changes of the collected species. Carbon dioxide was introduced as a supporting matrix at 77 K to optimize the condensation of pyrolysis products and increase the resolution of EPR spectra. Depending on the orifice size, all samples were accumulated on the Dewar cold finger for 25 min or less.
2.3. EPR measurements
EPR spectra were recorded using a Bruker EMX-20/2.7 EPR spectrometer (X-band) with dual cavities, modulation and microwave frequencies of 100 kHz and 9.516 GHz, respectively. Typical parameters were: sweep width of 5000 G, EPR microwave power of 10 mW, modulation amplitude of 2 G, time constant of 40.96 ms, and sweep time of 167.77 s. Values of the g-tensor were calculated using Bruker’s WIN-EPR SimFonia 2.3 program, which allowed control of the Bruker EPR spectrometer, data-acquisition, automation routines, tuning, and calibration programs. The exact g-factors for key spectra were determined by comparison with a 2,2-diphenyl-1-picrylhydrazyl (DPPH) standard.
2.4. Annealing and annihilation studies
Gradual warming of the Dewar resulted in the annealing of the matrix and selective annihilation of the more reactive radicals, such that the spectra of the more persistent, individual radicals in the mixture could be discerned. By removing liquid nitrogen with a bubbling stream of N2 gas, the cold finger of the Dewar was slowly warmed. This resulted in some of the initially observed reactive radicals being annihilated by radical–radical recombination. The liquid N2 was then re-added to the Dewar to quickly refreeze the matrix at 77 K.
2.5. Hyperfine splitting constants calculations
Radicals formed during the oxidative pyrolysis of 1-MN were investigated using Density Functional Theory (DFT). The carbon-centered radical’s geometry was optimized with the B3LYP hybrid functional and EPR-II basis set. Hyperfine splitting constants were computed with the Gaussian 09 program packages with the B3LYP hybrid functional and aug-cc-pvdz basis set. These calculations were calibrated against hyperfine splitting constants of known radical standards (benzyl and phenyl) reported in the literature from experiments and computation. EPR signals with hyperfine structure were simulated from calculated hyperfine splitting constants with Bruker’s WIN-EPR SimFonia 2.3 program with Lorenzian lineshapes.
3. Results
The aim of the current research study is to utilize the LTMI-EPR technique to provide chemical speciation of individual radical species present in particulate that appear as broad featureless spectrum in EPR analysis of particulate. This data will be applied to understand the promotion effect of iron oxide nanoparticle addition on the soot formation process. The paramagnetic centers generated from the oxidative pyrolysis of 1-MN in the absence of iron oxide nanoparticles will be presented first for clarity and then the influence of iron oxide nanoparticle addition will be presented.
3.1. Paramagnetic centers from the oxidative pyrolysis of 1-MN
At least four separate and distinct radical species have been suggested to be associated with combustion-generated particulate: (i) the “soot radical”, (ii) organic-carbon centered radicals, (iii) oxygen-centered radicals, (iv) and bound Environmentally Persistent Free Radicals (EPFRs) (cf. Fig. S1). Often referred to as “soot radicals,” this radical is observed as a sharp singlet with a g-factor = 2.0028–2.0030 [7,10,16,17,20] and a theoretical ΔHp–p = 2 G [22]. This radical signal is believed to originate from a delocalized electron in the carbon matrix that originate from the PAH species that compose the soot framework [1,22]. Organic carbon-centered radicals, also referred to as σ or π-type radicals, result from the cleavage of the parent polycyclic aromatic hydrocarbon (PAH). These organic-carbon centered radicals are defined by their hyper-fine splitting constants [38] but may also appear as a broad featureless spectrum depending on the environment [20]. Oxygen-centered radicals retain their aromatic structure while incorporating a substituent oxygen atom and exhibit g-factors in the range of 2.0029–2.0040 [39] and often appear as broad featureless spectrum. EPFRs are radical species formed on the surfaces of transition metal-containing particles by chemisorption of a molecular precursor and subsequent electron transfer from the organic to the metal [25,40]. This electron transfer results in a reduction of the metal and the formation of the EPFR. These species have long-lived (hour to days) EPR signals in the presence of air with g-factors in the range of 2.0024–2.0065 [25]. Since EPFRs can be generated from a multitude of precursors, these radicals can be both car-bon-and oxygen-centered species depending on the molecular precursor.
EPR analysis of particulate accumulated from the reactor exhaust at sampling point C reveals new paramagnetic species. The radical signal initially appeared to be consistent with the broadened signal (Fig. 2A) often associated with the soot radical with a relatively standard g-factor of 2.0030 [3,4,7,8,10,15,21]. However, the EPR spectrum expanded in a 100G window exhibited hyperfine structure, a spectral feature not typically observed in soot (cf. Fig. 2B). Since soot is composed of carbon, which has no nuclear moment (C12), there should be no hyperfine structure arising from the carbon atoms, or from other constituent atoms [22]. In addition, the spectral width (ΔHp–p) of carbonaceous material, which results from electron spin–spin interactions, will only be between 1 and 2 G. [22]. This hyperfine structure must result from paramagnetic intermediates embedded in or attached to the carbon soot matrix.
Fig. 2.
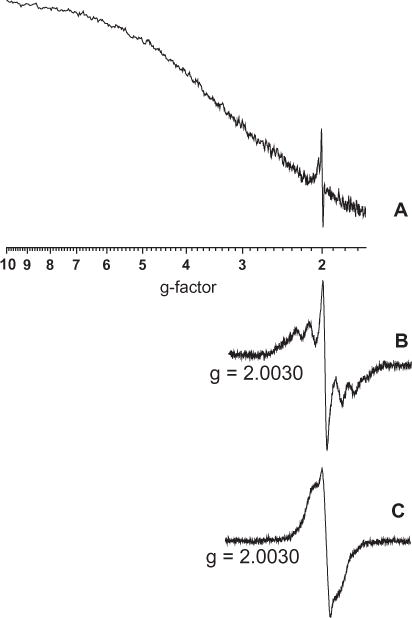
(A) EPR spectra of particulate accumulated at sampling port C using the cryogenic method. (B) Spectrum A on expanded scale to show detail. (C) EPR spectra of particulate collected on a cellulose-ester filter at the same sampling position.
The EPR signal obtained utilizing the LTMI-EPR technique was compared against soot collected on cellulose ester filters at the same sampling location to determine if the LTMI-EPR technique revealed new details previously not reported for soot. Although EPR spectrum of soot collected on cellulose-ester filters exhibited a g-factor consistent with the soot radical of 2.0030, the ΔHp–p varied between 2–8 G and exhibited small side features (cf. Fig. 2C). Most often the side features presented in Fig. 2C, of soot on cellulose ester filters, were merged and the EPR signal appeared as a broad featureless spectrum. This broadened featureless spectrum has been reported in the literature for soot from a variety of other anthropogenic sources [4,10,11,15,17]. Since soot collected on the cellulose ester filters exhibited similar unresolved features as the spectral features observed in soot collected with the LTMI-EPR technique, it is inferred that the hyperfine structure observed with the LTMI-EPR technique is the previously unresolved and speculated paramagnetic intermediates associated with soot. This demonstrates collection of soot on filter media results in the merging of spectral lines, broadening of the EPR signal, and subsequent loss of hyperfine structure. Thus the use of the LTMI-EPR technique provided new details about the previously unresolved EPR signal of soot.
Annealing experiments were performed to determine the stability of the paramagnetic species and if the EPR signal originated from more than one radical species [36]. Recombination of the more active intermediates should lead to a change in the EPR signal and confirm the existence of multiple radical species. Gradual warming of the matrix resulted in the loss of hyperfine structure and conversion of the EPR signal presented in spectrum 3A to spectrum 3B. Since the system was maintained in a an isolated environment, loss of the reactive paramagnetic species resulted from radical recombination at elevated temperatures and subsequent hyperfine structure loss, shift in g-factor to 2.0034, and decrease in the total width of spectrum from 48 G to 10 G, while maintaining the ΔHp–p = 2 G for the central soot signal. This g-factor in Fig. 3B is typical of an oxygen-centered radical [39]. Further annealing of the matrix resulted in the appearance of a symmetrical singlet line with a g-factor of 2.0030, ΔHp–p = 2 G, and ΔHtotal -≤ 10.0 G (cf. Fig. 3C), characteristic of the soot radical and its theoretical spectral width [22]. Any further annealing resulted in no significant change in the EPR spectrum, even at room temperature. Spectrum 3D is a residue spectrum, a subtraction of A–B, which represents a mixture of the annihilated organic radicals (see below).
Fig. 3.
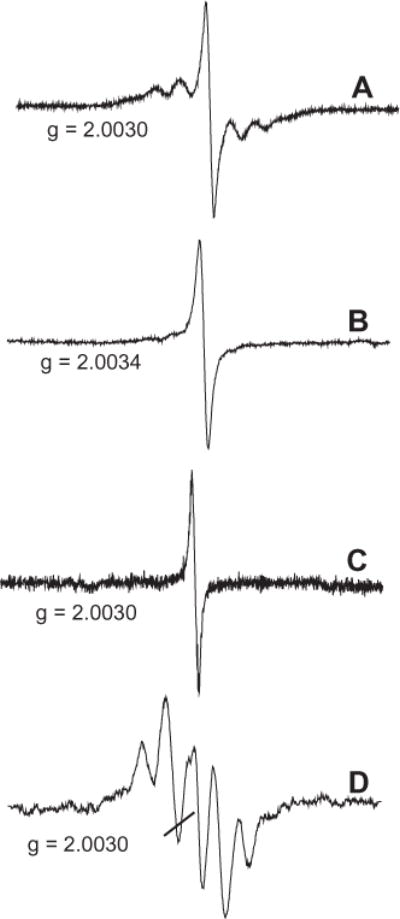
(A) EPR spectra of particulate collected on Dewar cold finger. (B) EPR spectra after gradual annealing of the matrix. (C) EPR spectra after further annealing. (D) The subtraction spectrum, A and B, provides a residue spectrum of the mixture of annihilated organic radicals.
Power dependence studies of EPR signals reflect the spin–lattice relaxation time, a measure of the rate of energy transfer between the electron spin and its surroundings. As the microwave power is increased, the spin–lattice relaxation slows and the population difference needed to maintain the electron distribution is lost. This results in no net absorption of the electron and is referred to as saturation [41]. Since not all components of an EPR spectrum have the same microwave power dependence, this can be used to distinguish the presence of multiple components in one signal. The EPR spectrum of particulate (Fig. 2B) as a function of the microwave power is presented in Fig. 4. By varying the microwave power, the signal can be clearly shown to be composed of two features. At low microwave power (0.1 mW), the organic radical is clearly resolved (cf. Fig. 4). As the microwave power is increased the organic radicals signal begins to saturate and the soot radical emerges. Removing of the soot radical by subtraction of the spectrum obtained at 161 mW from the spectrum obtained at 0.1 mW results in a spectrum similar to the spectrum in Fig. 3D. These microwave power dependence studies provided an alternative method to distinguish the presence of multiple components in the complex EPR signal obtained from the oxidative pyrolysis of 1-MN.
Fig. 4.
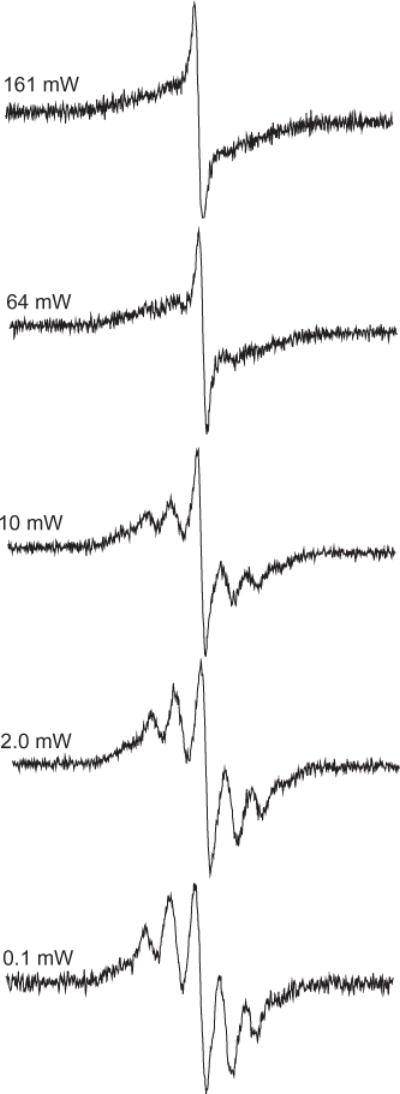
Power dependence of spectra of particulate collected on Dewar cold finger. At low microwave power (0.1 mW), the organic radical is resolved.
Since the hyperfine structure was observed in particulate collected at sampling location C in the dual zone reactor, where the temperature was low, the formation of reactive intermediates was investigated at various positions in Zone 2, and thus different reaction times during the oxidative pyrolysis of 1-MN. Moving the probe closer to the exit of Zone 2 (sampling points I–IV) resulted in a singlet spectrum with g = 2.0030 and ΔHp–p = 2 G and a gradual increase in the signal intensity due to increased soot yield. Position IV exhibited a signal an order of magnitude higher than position I. However, the EPR spectrum at position IV differed slightly from the singlet EPR signal at position I by the presence of hyperfine splitting consistent with what was observed at sampling port C in the exhaust stream. The hyperfine splitting began to appear at sampling port IV (800 °C) and continues to grow as the temperate decreased (cf. Fig. 5). Since the hyperfine structure was not observed in the EPR spectrum of particulate collected in the isothermal region through sampling ports I–III maintained at 1100 °C, it appears the concentration of these radicals was much lower at elevated temperatures because of acceleration of their decomposition pathways. Consequently, at lower temperatures in the quenching zone these organic radicals may be the dominant species as soot yields increase significantly.
Fig. 5.
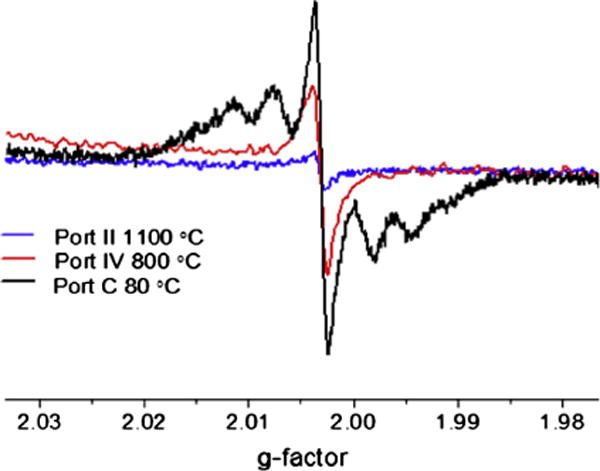
Normalized EPR spectra of soot particles detected in Zone 2, in the quenching zone of the reactor (ports A and B) and in the exhaust stream, port C.
Therefore, accumulation of particulate from the exhaust stream of the dual zone reactor revealed a mixture of paramagnetic centers including organic radicals and soot. The hyperfine structure associated with the organic radical provided a method to identify the species through simulation of theoretical EPR spectrum. DFT calculations were performed to determine the hyperfine splitting constants (isotropic Fermi contact couplings) for possible radicals associated with the oxidative pyrolysis of 1-MN based on previous mechanistic studies [42–46]. The indenyl and cyclopentadienyl radicals were identified as the dominant stable radicals from the oxidation of 1-MN. These calculations were calibrated against the benzyl radical to ensure the level of computational theory resulted in both computationally and experimentally determined hyperfine splitting constants for the benzyl radical. Once calibrated, the hyperfine splitting constants of other similar radicals were calculated and the EPR spectrum was simulated. The calculated hyperfine splitting constants for all radicals are presented in Table S1. These optimized geometries and simulated EPR spectrum radicals associated with the oxidative pyrolysis of 1-MN are presented in Fig. 6. Comparison of the experimentally obtained hyperfine structure with each individual simulated EPR spectra revealed no singular match to the unknown hyperfine splitting indicating a mixture of radical species.
Fig. 6.
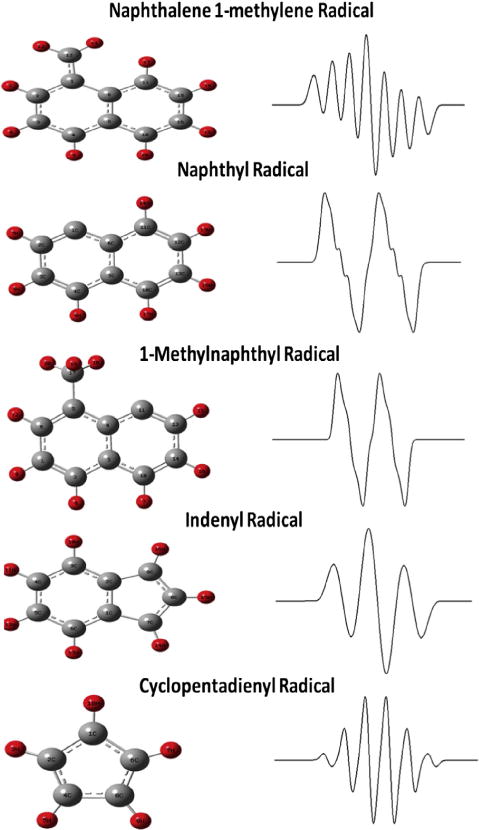
Optimized geometries of radicals mentioned in the text; C atoms are gray, H atoms are red. On the right side; simulated spectra of radicals formed from the oxidative pyrolysis of 1-MN. The Win-EPR Simfonia simulation software was used with parameters: ΔHp–p for each individual line − 2.5 G, Lorenzian lineshape, g = 2.0030. The hyperfine splitting constants for cyclopentadienyl radical (with five equivalent protons) was 6.2 G [36]. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Bruker’s WIN-EPR SimFonia 2.3 program was utilized to superimpose multiple radical signals through addition of the simulated EPR spectra. A simulated spectrum composed of the indenyl, cyclopentadienyl, and naphthalene 1-methylene radical EPR spectrum in proportions of 57.5%, 30.0%, and 12.5%, respectively resulted in an EPR spectrum which best fit with the experimental EPR spectrum (cf. Fig. 7). This mixture of resonance stabilized radicals exhibited the exact spectral width (ΔHp–p) as experimentally obtained radicals. Based on this comparison, the radicals associated with the oxidative pyrolysis of 1-MN are predominantly the resonance stabilized indenyl, cyclopentadienyl, and naphthalene 1-methylene radicals.
Fig. 7.
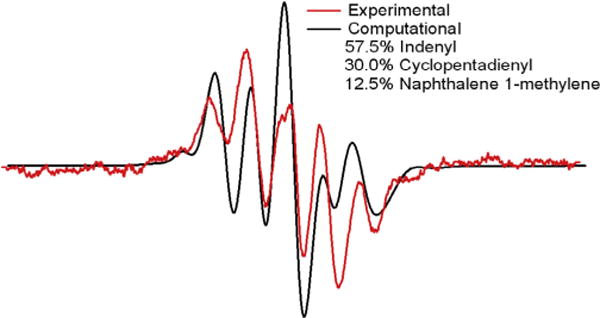
Comparison of experimental EPR spectra with simulated spectrum composed of indeyl, cyclopentadienyl, and naphthalene 1-methylene radical. The Win-EPR Simfonia simulation software was used with parameters: ΔHp–p for each individual line − 2.5 G, Lorenzian lineshape, g = 2.0030.
3.2. Influence of Fe(III)2O3 Addition on Paramagnetic Centers from 1-MN
The influence of Fe(III)2O3 nanoparticle addition on the formation of radical species was also investigated. Fe(III)2O3 nanoparticles were introduced to 1-MN prior to soot inception. The same multi-line spectrum was detected in both the presence and absence of Fe(III)2O3 nanoparticles (cf. Fig. 8). However, Fe(III)2O3 nanoparticle introduction resulted in increased radical signal intensity, when introduced to 1-MN prior to soot inception. Control experiments were performed to confirm the dendrimer template in methanol did not give rise to the increased signal intensity. This indicates Fe(III)2O3 nanoparticles enhanced formation of carbon-centered radical species without affecting their structure. Introduction of Fe(III)2O3 nanoparticles prior to soot inception results in enhanced formation of the resonance stabilized indenyl, cyclopentadienyl, and naphthalene 1-methylene radicals.
Fig. 8.
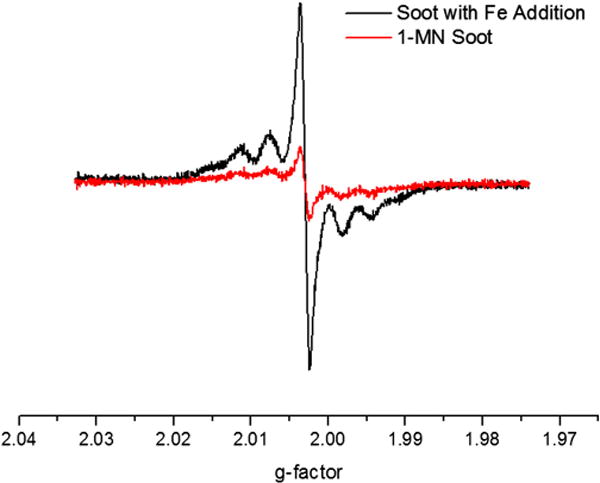
Observed enhancement in radical formation as a result of Fe(III)2O3 nanoparticle introduction prior to soot inception.
3.3. Fe(III)2O3 nanoparticles in Zone 2
Although Fe(III)2O3 nanoparticles were expected to be formed in the Zone 1 oxidative regime, the exact species of iron oxide nanoparticles generated in the flow reactor proved difficult to study due to the small particle size and low concentration. Fe(III), with an electronic configuration [Ar]3d5 with s = 5/2 and s = 1/2, is well suited for EPR analysis because the ground state of the paramagnetic ions split into a number of components resulting in a fine structure EPR spectrum [13]. By comparing this EPR fine structure to the fine structure for other electronic configurations, the oxidation state of iron in the nanoparticles can be identified (cf. Fig. 9).
Fig. 9.
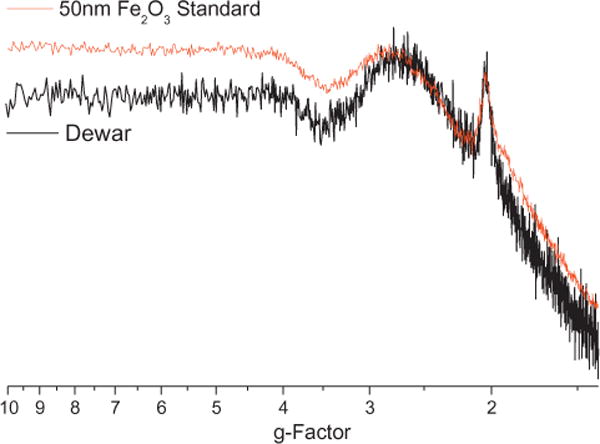
Accumulated Fe(II)2O3 nanoparticles on the Dewar cold-finger exhibited the same spectral features as a ng quantity of an Fe(II)2O3 standard.
As experiments continued, the Dewar background signal was continually monitored for a background signal and to ensure no particulate remained on the Dewar cold-finger. The EPR Dewar background signal began to change as Fe(III)2O3 nanoparticles began accumulating on the Dewar cold-finger during the course of the studies of the influence of Fe(III)2O3 nanoparticle addition on radical formation. These accumulated iron oxide nanoparticles were then compared against a standard of Fe(III)2O3 nanoparticles and were found to exhibit the same spectral features (cf. Fig. 9). These experiments inadvertently confirmed the generation of Fe(III)2O3 nanoparticles in the oxidative regime as previously proposed [5].
3.4. Partial reduction of Fe(III)2O3 nanoparticles
The addition of Fe(III)2O3 nanoparticles to a high sooting 1-MN fuel resulted in the enhanced formation of carbon-centered radical species and loss of spectral details associated with the Fe(III)2O3 nanoparticles. The spectral features previously observed at g = 2.00 were presumably masked by the formation of carbon-centered radicals but the spectral features at g = 4.30 were no longer present. However in some experiments, the introduction of iron-oxide nanoparticles resulted in the presence of a broad peak at g ~ 9.2, along with soot spectral features (cf. Fig. S2). This peak is associated with the presence of Fe3O4, a mixture of Fe(II)O species and Fe(III)2O3 [11]. Although Fe(II)O is EPR silent with an electronic configuration [Ar]3d4 with s = 2, Fe(II)O in the presence of Fe(III)2O3 results in a EPR active species. The EPR spectrum for Fe(II) as Fe3O4 is characterized by a broad signal at a low magnetic field (~1000 G, g ~ 9.0) [14]. This broad peak has been observed in the literature in magnetite, Fe3O4 [47]. The presence of a broad peak at g ~ 9.2, soot spectral features, and the loss of the spectral features associated with Fe(III)2O3 nanoparticles indicate a possible reduction of the Fe(III)2O3 upon the formation of soot.
4. Discussion
The LTMI-EPR technique provided new details pertaining to the chemical speciation of radical species associated with “soot radicals” often observed as broad EPR signals. The previously reported broad EPR signal associated with soot was found to be the superposition of multiple radicals including: organic carbon-centered radicals; oxygen-centered radicals; and soot [5]. The indenyl, cyclopentadienyl, and naphthalene 1-methylene radicals were identified from the oxidative pyrolysis of 1-MN with the LTMI-EPR through simulation of the hyperfine structure. These species were found as major intermediate products from the oxidation of 1-MN [48,49]. The enhanced stability provided by the resonance-stabilization had led to the general acceptance of the species as key contributors in the formation of the first aromatic ring, molecular growth of PAHs, and soot formation [48–51]. For instance, the cyclopentadienyl radical and the propargyl radical have been identified in the formation of the first benzene ring [52]. Molecular growth proceeding phenanthrene and anthracene formation has been shown to result from reactions of naphthalene with both cyclopentadienyl and idenyl radicals [13,48,49]. These were two of the major products found from the oxidative pyrolysis of 1-MN in the dual zone flow reactor [5].
In these experiments, introduction of Fe(III)2O3 nanoparticles to a high sooting 1-MN fuel resulted in enhanced formation of the resonance-stabilized indenyl, cyclopentadienyl, naphthalene 1-methylene radicals and promotion of soot. Increased formation of resonance-stabilized radicals suggests Fe(III)2O3 nanoparticles can promote the formation of intermediate radicals and potentially stabilize these radicals at the nanoparticle surface. These Fe(III)2O3 nanoparticles provide a surface for condensation reactions to occur. Similarly, a threefold increase in soot and PAH formation was reported when 200 ppm of iron was doped into the fuel of a laminar, sooting, premixed ethlylene/oxygen/nitrogen flame [53]. Equilibrium calculations predicted the metal to exist as volatile free metal in the flame but condense as metallic FeOx in the post-flame region. It was hypothesized that metallic Fe condensed on growing soot particles and catalyzed formation of PAH via acetylene addition reactions [53]. Although both studies report an increase in PAH and soot formation, the detection of the resonance-stabilized indenyl, cyclopentadienyl, and naphthalene 1-methylene radicals suggests an alternative pathway associated with the catalyzed formation of PAHs instead of previously hypothesized molecular growth by acetylene addition, a much slower pathway [53]. In other studies, iron oxide was found to nucleate prior to soot formation in a diffusion flame doped with ferrocene vapor [54]. Ferrocene was shown to produce condensation nuclei for condensation reactions to occur [54] and was hypothesized to induce nucleation of soot though surface reactions [55].
A partial reduction in the Fe(III)2O3 nanoparticles was observed by the formation of Fe3O4, a mixture of Fe(II)O and Fe(III)2O3, following soot formation. A partial reduction of the metal oxide, upon the addition of a hydrocarbon soot precursor, further suggests the EPFR-based model may be applicable to the molecular growth of soot in the presence of redox-active transition metal nanoparticles. In a similar study, a partial reduction was observed at the surface of Fe(III)2O3 nanoparticles at high soot concentrations in an ethylene–air diffusion flame seeded with iron pentacarbonyl and acetylene [56]. Other similar studies prepared carbon encapsulated iron nanoparticles and observed different states of iron within the carbon matrix [57,58].
5. Conclusion
The LTMI-EPR technique was utilized to experimentally determine the superimposed signal of particulate from the oxidative pyrolysis of 1-MN. The EPR spectrum of particulate was found to be a mixture of organic carbon-centered radicals, oxygen-centered radicals, and soot. The carbon-centered radicals were identified through simulation of the hyperfine structure to be the resonance-stabilized indenyl, cyclopentadienyl, and naphthalene 1-methylene radicals. Introduction of Fe(III)2O3 nanoparticles to a high sooting 1-MN fuel resulted in enhanced formation of the resonance-stabilized indenyl, cyclopentadienyl, naphthalene 1-methylene radicals and enhanced soot formation. The detection of resonance-stabilized radicals in the cool zone of the reactor is consistent with the EPFR based model in which organic radicals are stabilized by transition metal oxides [23–25]. In the EPFR based model, the metal oxide is reduced upon the chemisorption of an organic radical. This pathway is further supported by the partial reduction in the Fe(III)2O3 nanoparticles to form Fe3O4. These current studies suggest Fe(III)2O3 nanoparticles can mediate the formation and stabilization of resonance stabilized radicals and subsequent reactions can result in the molecular growth of PAHs into soot.
Supplementary Material
Acknowledgments
This work was partially supported by the NIEHS Superfund Research Program under Grant 5 P42 ES013648-03 and the Patrick F. Taylor Chair.
Appendix A. Supplementary material
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.combustflame.2013.06.025.
References
- 1.Mansurov Z, Ongarbaev E, Tutkabaeva TT. Combust Flame. 1999;118(4):741–743. [Google Scholar]
- 2.Akhter M, Chughtai A, Smith D. Appl Spectrosc. 1985;39(1):143–153. [Google Scholar]
- 3.Chughtai A, Atteya M, Kim J, Konowalchuk B, Smith D. Carbon. 1998;36(11):1573–1589. [Google Scholar]
- 4.Jones CC, Chughtai AR, Murugaverl B, Smith DM. Carbon. 2004;42(12–13):2471–2484. [Google Scholar]
- 5.Paul Herring M, Potter PM, Wu H, Lomnicki S, Dellinger BP. Combust Inst. 2012;34(1):1749–1757. doi: 10.1016/j.proci.2012.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Solum MS, Veranth JM, Yi-Jin Jiang AM, Sarofim AF, Pugmire RJ. Energy Fuels. 2003;17(3):738–743. [Google Scholar]
- 7.Abi-Aad E. Top Catal. 2001;16/17(1–4):263–268. [Google Scholar]
- 8.Yan S, Jiang YJ, Marsh ND, Eddings EG, Sarofim AF, Pugmire RJ. Energy Fuels. 2005;19(5):1804–1811. [Google Scholar]
- 9.Dalal N. Fuel and Energy Abstracts, Elsevier. 1997;51(9):1429–1431. [Google Scholar]
- 10.Tian LW, Koshland CP, Yano JK, Yachandra VK, Yu ITS, Lee SC, Lucas D. Energy Fuels. 2009;23:2523–2526. doi: 10.1021/ef8010096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wawros A. Pol J Environ Std. 2003;12(5):619–627. [Google Scholar]
- 12.Valavanidis A, Iliopoulos N, Gotsis G, Fiotakis K. J Hazard Mater. 2008;156(1–3):277–284. doi: 10.1016/j.jhazmat.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 13.Ledoux F. Atmos Environ. 2002;(36):939–947. [Google Scholar]
- 14.Ledoux F, Zhilinskaya EA, Courcot D, Aboukais A, Puskaric E. Atmos Environ. 2004;38(8):1201–1210. [Google Scholar]
- 15.Saathoff H, Moehler O, Schurath U, Kamm S, Dippel B, Mihelcic D. J Aerosol Sci. 2003;34:1277–1296. [Google Scholar]
- 16.Yardanov ND. Spectrochim Acta A. 2004;(60):1367–1370. doi: 10.1016/j.saa.2003.10.044. [DOI] [PubMed] [Google Scholar]
- 17.Yordanov N, Lubenova S, Sokolova S. Atmos Environ. 2001;35(5):827–831. [Google Scholar]
- 18.Lyons MJ, Spence JB. Br J Cancer. 1960;14(4):703–708. doi: 10.1038/bjc.1960.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ivanova N, Onischuk A, Vosel S, Purtov P, Kulik L, Rapatskiy L, Vasenin N, Anufrienko V. Appl Magn Reson. 2009;35(4):625–637. [Google Scholar]
- 20.Yamanaka C, Matsuda T, Ikeya M. Appl Radiat Isot. 2005;62:307–311. doi: 10.1016/j.apradiso.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 21.Mansurov ZA. J Therm Sci. 2001;10(3):269–280. [Google Scholar]
- 22.Ingram DJE. Free radicals as studied by electron spin resonance. London: Butterworths Scientific Publications; 1958. p. 210. [Google Scholar]
- 23.Dellinger B, Lomnicki S, Khachatryan L, Maskos Z, Hall RW, Adounkpe J, McFerrin C, Truong H. Proc Combust Inst. 2007;31(1):521–528. doi: 10.1016/j.proci.2006.07.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Truong H, Lomnicki S, Dellinger B. Environ Sci & Tech. 2010;44(6):1933–1939. doi: 10.1021/es902648t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dellinger B, Vejerano E, Lomnicki S. Environ Sci Technol. 2011;45(2):589–594. doi: 10.1021/es102841s. [DOI] [PubMed] [Google Scholar]
- 26.Hahn DW, Kim KB, Masiello KA. Combust Flame. 2008;154(1–2):164–180. [Google Scholar]
- 27.Linteris GT, Babushok VI. Proc Combust Inst. 2009;32:2535–2542. doi: 10.1016/j.proci.2018.06.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller A, Ahlstrand G, Kittelson D, Zachariah M. Combust Flame. 2007;149(1–2):129–143. [Google Scholar]
- 29.Marsh* ND, Preciado I, Eddings EG, Sarofim AF, Palotas AB, Robertson JD. Combust Sci Technol. 2007;179(5):987–1001. [Google Scholar]
- 30.Ritrievi KE, Longwell JP, Sarofim AF. Combust Flame. 1987;70(1):17–31. [Google Scholar]
- 31.Zhang J, Megaridis CM. Combust Flame. 1996;105(4):528–540. [Google Scholar]
- 32.Calixto de Campos R, Reis dos Santos H, Grinberg P. Spectrochim Acta Part B. 2002;57(1):15–28. [Google Scholar]
- 33.Yang Z, Hou X, Jones BT. Talanta. 2003;59(4):673–680. doi: 10.1016/S0039-9140(02)00580-5. [DOI] [PubMed] [Google Scholar]
- 34.Yaroshchyk P, Morrison RJ, Body D, Chadwick BL. Spectrochim Acta, Part B. 2005;60(7):986–992. [Google Scholar]
- 35.Sharma M, Agarwal AK, Bharathi K. Atmos Environ. 2005;39(17):3023–3028. [Google Scholar]
- 36.Khachatryan L, Adounkpe J, Maskos Z, Dellinger B. Environ Sci Technol. 2006;40(16):5071–5076. doi: 10.1021/es051878z. [DOI] [PubMed] [Google Scholar]
- 37.Mantashyan AA, Khachatryan LA, Niazyan OM, Arsentyev SD. Combust Flame. 1981;43:221–227. [Google Scholar]
- 38.Singer LS, Lewis IC. Appl Spectrosc. 1982;36(1):52–57. [Google Scholar]
- 39.Lewis IC, Singer LS. J Phys Chem – US. 1981;85(4):354–360. [Google Scholar]
- 40.Lomnicki S, Truong H, Vejerano E, Dellinger B. Environ Sci Technol. 2008;42(13):4982–4988. doi: 10.1021/es071708h. [DOI] [PubMed] [Google Scholar]
- 41.Gerson F, Huber W. Electron spin resonance spectroscopy of organic radicals. Wiley. com; 2006. [Google Scholar]
- 42.Goos E, Douce F, Djebaili-Chaumeix N, Braun-Unkhoff M, Slavinskaya N, Frank P, Paillard C. Proc Eur Combust Meet. 2003:223–232. [Google Scholar]
- 43.Leininger JP, Lorant F, Minot C, Behar F. Energy Fuels. 2006;20(6):2518–2530. [Google Scholar]
- 44.Mati K, Ristori A, Pengloan G, Dagaut P. Combust Sci Technol. 2007;179(7):1261–1285. [Google Scholar]
- 45.Somers ML, McClaine JW, Wornat MJ. Proc Combust Inst. 2007;31:501–509. [Google Scholar]
- 46.Yang J, Lu M. Environ Sci Technol. 2005;39(9):3077–3082. doi: 10.1021/es048537q. [DOI] [PubMed] [Google Scholar]
- 47.Guskos N, Papadopoulos G, Likodimos V, Patapis S, Yarmis D, Przepiera A, Przepiera K, Majszczyk J, Typek J, Wabia M. Mater Res Bull. 2002;37(6):1051–1061. [Google Scholar]
- 48.Lu M, Mulholland JA. Chemosphere. 2001;42(5–7):625–633. doi: 10.1016/s0045-6535(00)00236-8. [DOI] [PubMed] [Google Scholar]
- 49.Lu M, Mulholland JA. Chemosphere. 2004;55(4):605–610. doi: 10.1016/j.chemosphere.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 50.D’Anna A, Violi A, D’Alessio A. Combust Flame. 2000;121(3):418–429. [Google Scholar]
- 51.Mulholland JA, Lu M, Kim D-H. Proc Combust Inst. 2000;28(2):2593–2599. [Google Scholar]
- 52.Richter H, Howard JB. Prog Energy Combust Sci. 2000;26(4–6):565–608. [Google Scholar]
- 53.Feitelberg A, Longwell J, Sarofim A. Combust Flame. 1993;92(3):241–253. [Google Scholar]
- 54.Kasper M, Sattler K, Siegmann K, Matter U, Siegmann H. J Aerosol Sci. 1999;30(2):217–225. [Google Scholar]
- 55.Hirasawa T, Sung CJ, Yang ZW, Joshi A, Wang H. Combust Flame. 2004;139(4):288–299. [Google Scholar]
- 56.Jasinski J, Pinkerton KE, Kennedy IM, Leppert VJ. Sens Actuators, B. 2005;109(1):19–23. [Google Scholar]
- 57.Maldonado-Hodar F, Moreno-Castilla C, Rivera-Utrilla J, Hanzawa Y, Yamada Y. Langmuir. 2000;16(9):4367–4373. [Google Scholar]
- 58.Ozaki J, Watanabe T, Nishiyama Y. J Phys Chem. 1993;97(7):1400–1405. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.