

Summary
Mitochondria constitute an important topic of biomedical enquiry (one paper in every 154 indexed in PubMed since 1998 is retrieved by the keyword ‘mitochondria’) because of widespread recognition of their importance in cell physiology and pathology. Mitochondrial dysfunction is widely implicated in ageing and in the diseases of ageing, through dysfunction in adenosine triphosphate (ATP) synthesis, Ca2+ homeostasis, central metabolic pathways or radical production. Nonetheless, the mechanisms and regulation of superoxide and hydrogen peroxide formation by mitochondria remain poorly described. Measurement of the capacities of different sites of superoxide and hydrogen peroxide production in isolated skeletal muscle mitochondria show that the maximum capacities of sites in complexes I, II and III and in several associated redox enzymes greatly exceed the native rates observed in the absence of respiratory chain inhibitors. In vitro, the native rates and the relative importance of different sites both depend on the substrate being oxidized, with sites IQ, IIF, GPDH, IF and IIIQo each being important with particular substrates. The techniques involved in measuring rates from each site should become applicable to cell cultures and in vivo in the future.
The importance of mitochondria in cell physiology and medicine
There has been explosive growth in the biomedical literature over the past few decades. It has grown tenfold from 100 000 papers per year in the 1950s to the current one million per year (Fig. 1). Studies of mitochondria had a golden age in the 1970s, when the mechanism of energy conservation and adenosine triphosphate (ATP) synthesis by mitochondria was first being worked out and the chemiosmotic theory of oxidative phosphorylation was being established and tested, culminating in the award of the Nobel Prize for Chemistry to Peter Mitchell in 1978. In 1973, at the peak of this revolution, 3229 papers on ‘mitochondria’ were published, comprising 1·4% of the biomedical literature that year, according to Pub-Med. You might expect that this intense focus on mitochondrial bioenergetics would have been the high water mark of the study of mitochondria, but since 1998 there has been a remarkable resurgence of activity and publications on mitochondria, as their roles in cell physiology, apoptosis, radical production, ageing and disease have become hot topics of research. So much so that there were nearly twice as many papers on mitochondria published in 2011 (5921) as there were in 1973. Indeed, the explosive growth of biomedical research has been matched and partly driven by papers on mitochondria: over the last 15 years an average of one paper in every 154 published in biomedicine was associated with ‘mitochondria’, that’s one every 20 min of the working day, each working day, every year.
Fig 1.
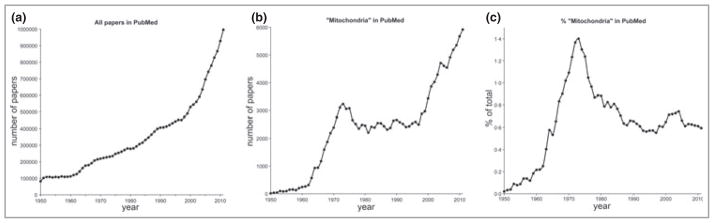
Papers in biomedicine and papers on ‘mitochondria’ 1950–2011. (a) All papers in PubMed each year. (b) Papers in PubMed each year retrieved using the keyword ‘mitochondria’. (c) Mitochondrial papers as a percentage of all papers in PubMed each year. Search date October 2012.
Of course, nobody has the time to read and digest all of these papers. Nevertheless, we can ask what topics the majority of them cover. A relatively simple way to do this is to see what other terms are linked to ‘mitochondria’ in PubMed. Table 1 shows that over the last 5 years, nearly all mitochondrial papers are also picked up by the keywords ‘cell’, ‘function’, ‘physiology’ and ‘metabolism’, indicating that their role in the general area of cell physiology is the main driver of the current huge interest in mitochondria. High on the list are terms showing that there is great interest in the relationship between mitochondria and ‘pathology’ and ‘medicine’, with ‘human’, ‘drug’, ‘treatment’, ‘disease’, ‘therapeutic’, ‘clinical’, ‘injury’, ‘target’ and ‘pediatric’ featuring very prominently. The role of mitochondria in programmed cell death is highlighted by the linkage to ‘death’, ‘apoptosis’, ‘caspase’ and ‘survival’, while the importance of shape and movement is picked out by the terms ‘morphology’ and ‘translocation’. Their roles in ‘signaling’ and in ‘growth’ and ‘development’ are also high on the list. Continued interest in the basic mechanisms of mitochondrial biochemistry is suggested by ‘enzyme’, ‘membrane’, ‘bioenergetics’, ‘electron’, ‘oxidoreductase’, ‘membrane potential’, ‘cytochrome’, ‘energy’, ‘ATP’, ‘structure’, ‘calcium’ and so on, although many of these links may also indicate how these mechanisms play out in cell physiology and disease. The relationship of mitochondria to reactive oxygen species (ROS) production and damage is very strong and marked by terms including ‘oxidative stress’, ‘reactive’, ‘antioxidant’, ‘redox’, ‘ROS’, ‘superoxide’, ‘peroxide’, ‘glutathione’, ‘disorder’, ‘dysfunction’, ‘DNA’, ‘mutation’, ‘damage’ and ‘toxicity’. Significantly, many specific diseases are linked to mitochondria, including ‘cancer’, ‘diabetes’, ‘ischemia’, ‘reperfusion’ (injury), ‘Parkinson’s’, ‘neurodegeneration’, ‘infection’, ‘obesity’, ‘dementia’, ‘trauma’, ‘Alzheimer’, ‘immunity’, ‘sepsis’, ‘epileptic’ and ‘Huntington’. The links to ageing are marked by ‘ageing’, ‘elderly’ and ‘aged’; links to life-style are marked by ‘alcohol’, ‘exercise’ and ‘training’. The species of interest include ‘human’, ‘mouse’, ‘rat’, ‘bovine’, ‘fish’, ‘drosophila’ and ‘parasite’, while the tissues and organs of interest include ‘muscle’, ‘cerebral’, ‘brain’, ‘heart’, ‘liver’, ‘in vivo’, ‘neuron’, ‘cardiovascular’, ‘fibroblast’, ‘nerve’, ‘lung’, ‘kidney’, ‘stem cell’, ‘cortex’, ‘hippocampus’, ‘embryo’, ‘striatum’ and ‘skin’. Overall, Table 1 gives a compelling snapshot of active current research that is considering roles of mitochondria in a wide range of physiological and pathological situations, as might be expected given their great functional importance in energy metabolism and other processes.
Table 1.
Numbers of papers in the last 5 years associated with both ‘mitochondria’ and the terms listed (chosen using background knowledge; queried in PubMed in October 2012)
| Mitochondria | 28 275 |
| Cell | 26 032 |
| Function | 25 407 |
| Physiology | 25 568 |
| Metabolism | 23 064 |
| Human | 14 581 |
| Enzyme | 13 703 |
| Morphology | 11 428 |
| Genetics | 10 635 |
| Drug | 10 635 |
| Membrane | 9735 |
| Treatment | 8827 |
| Bioenergetics | 8153 |
| Apoptosis | 7875 |
| Oxygen | 7195 |
| Oxidative | 7050 |
| Pathology | 6880 |
| Stress | 6320 |
| Mouse | 5991 |
| Medicine | 5835 |
| DNA | 5707 |
| Death | 5682 |
| Reactive | 5578 |
| Disorder | 5556 |
| Dysfunction | 5514 |
| Electron | 5417 |
| Disease | 5303 |
| Signaling | 5238 |
| Oxidative stress | 4889 |
| Cancer | 4856 |
| Therapeutic | 4851 |
| Oxidoreductase | 4845 |
| Membrane potential | 4597 |
| Cytochrome | 4580 |
| Translocation | 4505 |
| Growth | 4340 |
| Lipid | 4301 |
| Antioxidant | 4228 |
| Caspase | 3982 |
| Development | 3895 |
| Energy | 3855 |
| Muscle | 3800 |
| Damage | 3708 |
| Inhibition | 3635 |
| Survival | 3606 |
| ATP | 3567 |
| Redox | 3453 |
| RNA | 3446 |
| Phosphorylation | 3285 |
| Cerebral | 3198 |
| ROS | 3180 |
| Rat | 3116 |
| Brain | 3102 |
| Heart | 2853 |
| Liver | 2851 |
| Toxicity | 2833 |
| Oxidation | 2768 |
| Mutation | 2658 |
| Structure | 2630 |
| In vivo | 2621 |
| Calcium | 2545 |
| Neuron | 2491 |
| Cardiovascular | 2463 |
| Respiration | 2436 |
| Injury | 2423 |
| Clinical | 2307 |
| Target | 2085 |
| Ageing | 2069 |
| Superoxide | 2019 |
| Resistance | 1884 |
| Homeostasis | 1849 |
| Permeability | 1841 |
| Peroxide | 1678 |
| Deficiency | 1627 |
| Protective | 1543 |
| Phenotype | 1438 |
| Elderly | 1406 |
| Glutathione | 1397 |
| Diabetes | 1365 |
| Aged | 1357 |
| NADH | 1333 |
| Chronic | 1330 |
| Ischemia | 1319 |
| Evolution | 1286 |
| Biogenesis | 1269 |
| Acute | 1240 |
| Fibroblast | 1202 |
| Autophagy | 1115 |
| Reperfusion | 1047 |
| Coenzyme | 1141 |
| ATPase | 1127 |
| Pore | 1117 |
| Hypoxia | 1076 |
| Uncoupling | 984 |
| Radical | 958 |
| Parkinson’s | 919 |
| Transgenic | 916 |
| Alcohol | 880 |
| Neurodegeneration | 868 |
| Nerve | 863 |
| Proton | 833 |
| Steroid | 804 |
| Lung | 792 |
| Movement | 784 |
| Infection | 779 |
| Kidney | 771 |
| Neuroprotective | 717 |
| Stem cell | 700 |
| Fusion | 679 |
| Cortex | 654 |
| Embryo | 654 |
| Cytoskeleton | 629 |
| Exercise | 616 |
| Obesity | 602 |
| Import | 594 |
| Efficiency | 590 |
| Catalase | 569 |
| Bovine | 541 |
| Dementia | 549 |
| Training | 501 |
| Fish | 498 |
| Ubiquinone | 492 |
| Hippocampus | 488 |
| Defective | 487 |
| Trauma | 467 |
| Pediatric | 448 |
| Drosophila | 418 |
| Parasite | 377 |
| Alzheimer | 372 |
| Estrogen | 346 |
| Preconditioning | 342 |
| Immunity | 337 |
| Striatum | 230 |
| Sepsis | 164 |
| Epileptic | 150 |
| Huntington | 147 |
| Skin | 144 |
Functions of mitochondria
The functions of mitochondria obviously include oxidative phosphorylation to produce cellular ATP, but they also have important roles in ion homeostasis, in several metabolic pathways, in apoptosis and programmed cell death, and in ROS production and consumption. All of these functions may be significant in ageing and/or disease. Damage may cause mitochondria to accumulate dysfunctional components. This damage may be caused directly by radicals produced by the mitochondria themselves. It may be caused by sequence or regulatory errors following mutation of nuclear or mitochondrial DNA1,2 as a result of a wide range of internal or environmental insults, such as exposure of the skin to ultraviolet radiation. These effects can be exacerbated by degradation of the quality control machinery that normally limits the build-up of dysfunctional mitochondria by targeting poorly performing constituents of the mitochondrial network for destruction.3,4
The classic role of mitochondria is oxidative phosphorylation, which generates ATP by utilizing the energy released during the oxidation of the food we eat. ATP is used in turn as the primary energy source for most biochemical and physiological processes, such as growth, movement and homeostasis. We turn over approximately our own body weight in ATP each day, and almost all of this is generated by mitochondria, primarily within muscle, brain, liver, heart and gastrointestinal tract.5 The pre-eminent role of eating is to provide the fuel for mitochondria, and the pre-eminent role of breathing is to provide the oxygen and to remove the carbon dioxide produced during oxidative phosphorylation by mitochondria. Similarly, a major role of the cardiovascular system is to deliver the substrates (glucose, fatty acids, oxygen) and remove the products (carbon dioxide) of mitochondrial activity.
As a result of intensive study, particularly since the 1950s, the mechanism of oxidative phosphorylation is very well understood, both in general principle and detailed biochemistry. The general principle is chemiosmotic coupling (Fig. 2),6 in which the oxidation of respiratory substrates by oxygen, catalysed by the mitochondrial electron transport chain, causes proton extrusion across the mitochondrial inner membrane. The proton-motive force set up by this proton pumping drives protons back into the mitochondrial matrix through the ATP synthase to generate ATP. The proton-motive force also drives the uptake of ADP and phosphate and the efflux of ATP to deliver the synthesized ATP to the cytosol where it is consumed. It is also crucial for uptake and efflux of Ca2+, and hence for ionic homeostasis in the cytosol and matrix and for Ca2+-related signalling pathways. The crystal structures of most of the electron transport chain complexes have been solved (Fig. 2) and the detailed mechanisms of the coupling of electron transport to proton pumping in complexes III and IV are well understood.7,8 The mechanisms of proton pumping in complex I9 and the ATP synthase10 are known in outline but have yet to be worked out in molecular detail. In addition to ATP synthesis, the proton-motive force is coupled directly to uptake of substrates such as pyruvate, glutamate and ornithine and to export of products such as citrulline across the mitochondrial inner membrane,11 to proton leak pathways through the adenine nucleotide translocase and specific uncoupling proteins that provide thermogenesis and regulation of radical production,6 to calcium transporters that regulate matrix and cytosolic calcium concentrations,12 and to the nicotinamide nucleotide transhydrogenase that maintains the reduction state of the matrix glutathione pool.13
Fig 2.
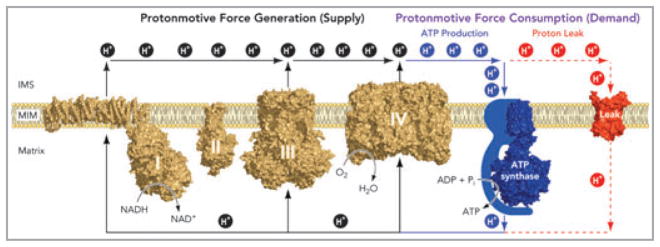
Chemiosmotic coupling of oxidative phosphorylation in mitochondria. Electrons harvested from oxidizable substrates are passed through the respiratory chain in an exergonic process that drives proton pumping by respiratory complexes I, III and IV. The resulting electrochemical proton gradient across the mitochondrial inner membrane can be dissipated in two ways: (i) through the FOF1–ATP synthase, where relieving the proton-motive force drives ADP phosphorylation, and (ii) via proton leak pathways that do not generate ATP, but regulate physiological processes including nonshivering thermogenesis and perhaps glucose-stimulated insulin secretion and protection from oxidative damage. Proton leak pathways are structurally represented by ANT, which can mediate both basal and inducible proton conductance. The structures depicted are: complex I from Thermus thermophilus (PDB ID: 3M9S); complex II from porcine heart (PDB ID: 1ZOY); dimeric complex III from bovine heart (PDB ID: 1BGY); dimeric complex IV from bovine heart (PDB ID: 2OCC); F1c10 ATP synthase complex from Saccharomyces cerevisiae (PDB ID: 2XOK) and carboxyatractyloside-inhibited ANT from bovine heart (PDB ID: 1OKC). Reproduced from Divakaruni and Brand.6 ADP, adenosine diphosphate; ANT, adenine nucleotide translocase; ATP, adenosine triphosphate.
Mitochondria have several critical roles in metabolism,14 even in organisms that live anaerobically and do not use their mitochondria for ATP synthesis.15 They are the central player in carbon metabolism. As well as their well-known catabolic role in oxidation of sugars (pyruvate), fats (palmitoylcarnitine) and proteins (glutamine, glutamate, alanine, and so on), they have a critical anabolic role, providing the carbon skeletons for the biosynthesis of most biomolecules, particularly glucose, fatty acids and amino acids. They are a major player in 1-carbon metabolism. They are central in nitrogen metabolism, metabolizing the glutamate used in transamination reactions and the glutamine used to shuttle nitrogen around the body, as well as the site of half of the reactions of the urea cycle. They are also essential in the synthesis of haem and iron-sulphur clusters.
As reviewed extensively elsewhere,16–20 mitochondria are central players in programmed cell death. They activate caspases in the cytosol through the release of cytochrome c and other factors from the intermembrane space when pro-apoptotic stimuli trigger Bcl-2 family members and the permeability transition pore.
Dysfunction in any of these pathways may contribute to the pathologies that develop with age and stress. In the following sections the mitochondrial sources of ROS that may contribute to such dysfunction will be examined.
The mitochondrial free radical theory of ageing and disease
Mitochondria generate ROS during oxidative metabolism. In the mitochondrial free radical theory of ageing,21 these ROS are the primary cause of damage to proteins, lipids and nucleic acids. Some damage is not repaired (perhaps because it is not repairable), causing failure of cellular machinery and leading to ageing- related diseases and to ageing itself. In the strictest version of the theory, the damage is self-reinforcing: damaged mitochondrial DNA codes for dysfunctional electron transport complexes that generate even more radicals than usual, leading to a vicious cycle of exponentially increasing damage and dysfunction. There is a substantial amount of evidence both supporting and against the theory.21–31 The current consensus is best summarized by the view that radicals generated by mitochondria can be an important contributor to ageing, and are particularly important in age-related diseases, including Alzheimer disease, 32,33 cardiomyopathy34–36 and cancer.37,38 However, mitochondrial radical production is not the sole cause of ageing, and may be most prominent only in particular model organisms and conditions of husbandry. Mitochondrial radical production may contribute to many of the symptoms of ageing, such as frailty and loss of elasticity in skin.
To understand mitochondrial ROS generation and fully establish its true role in ageing and age-related diseases, it is necessary to identify, quantify and ultimately manipulate the specific mitochondrial electron transport chain sites that generate ROS within cells.
The production of reactive oxygen species by mitochondria in vitro: maximum capacities of different sites
The generation of hydrogen peroxide by isolated mitochondria was first reported and characterized in the early 1970s.39,40 Most of this hydrogen peroxide is produced initially as superoxide and is then converted to hydrogen peroxide by a very active superoxide dismutase in the mitochondrial matrix.41 Subsequent work by many research groups42–46 has identified a number of sites of superoxide and hydrogen peroxide production in the citric acid cycle and the electron transport chain of mammalian mitochondria (Table 2).
Table 2.
Sites of superoxide and hydrogen peroxide production associated with oxidative phosphorylation in mitochondria
| Site | References |
|---|---|
| Citric acid cycle | |
| Pyruvate dehydrogenase | 55 |
| 2-oxoglutarate dehydrogenase | 55–57 |
| Dihydrolipoamide dehydrogenase | 58 |
| Ubiquinone reduction pathways | |
| Complex II flavin (site IIF) | 59 |
| Electron transferring flavoprotein/ETF:Q oxidoreductase | 50 |
| Glycerol 3-phosphate dehydrogenase | 47,60 |
| Dihydroorotate dehydrogenase | 47 |
| Electron transport chain | |
| Complex I flavin (site IF) | 61 |
| Complex I ubiquinone (site IQ) | 61–63 |
| Complex III outer ubiquinone binding site (site IIIQo) | 40,64 |
The maximum capacities of these sites under standard conditions in mitochondria isolated from skeletal muscle are illustrated in Figure 3. Site IIIQo has the greatest capacity for superoxide/hydrogen peroxide production, followed by sites IQ and IIF. Other sites have lower maximum capacities. Of course, these absolute maximum capacities and their relative importance will differ between tissues and species as the concentrations of the relevant redox centres differ. For example, the hydrogen peroxide-generating capacity of glycerol 3-phosphate dehydrogenase is much less than that of complex II in skeletal muscle mitochondria (Fig. 3), or in heart or brain mitochondria, but significantly exceeds the capacity of complex II in mitochondria from brown adipose tissue, where glycerol 3-phosphate dehydrogenase is much more highly expressed.47
Fig 3.

Hydrogen peroxide generation at different sites in isolated muscle mitochondria. Maximum capacities of sites defined using different combinations of substrates and inhibitors are in blue, actual overall rates during oxidation of glutamate plus malate in the absence of respiratory inhibitors are in red. All rates were either measured in the presence of 1-chloro-2,4-dinitrobenzene to greatly attenuate losses of hydrogen peroxide in the matrix by endogenous glutathione-linked peroxidases,65 or corrected to such measurements using the equations in Quinlan et al.48 and Treberg et al.65 St 4, state 4 (no ATP synthesis); st 3, state 3 (maximum ATP synthesis); OGDH, 2-oxoglutarate dehydrogenase; ETF, electron-transferring flavoprotein and ETF:Q oxidoreductase; DHODH, dihydroorotate dehydrogenase; GPDH, glycerol 3-phosphate dehydrogenase; glut + mal, glutamate plus malate; IF, IQ, IIF, IIIQo, see Table 2. Data are from unpublished observations and references 47, 48 and 59. Values are means ± SEM (n ≥ 3).
Figure 3 also shows the native rates of hydrogen peroxide generated by skeletal muscle mitochondria during oxidation of the conventional respiratory substrates glutamate plus malate. Under resting conditions (state 4, with no ATP synthesis), the native rate is much lower than the maximum capacity of any of the individual major sites. Under active conditions (state 3, with ADP added to allow ATP synthesis), the native rate is even lower. Clearly, knowledge of the maximum capacities of different sites in the presence of respiratory inhibitors to allow full reduction of the site is important, but it is not sufficient to allow prediction of which sites are actually operating under native conditions in the absence of respiratory inhibitors, as they will normally be in cells or in vivo.
The production of reactive oxygen species by mitochondria in vitro: rates from different sites under native conditions
The sites responsible for hydrogen peroxide production by mitochondria in cells or in vivo remain unknown44,45 because inhibiting or genetically modifying a candidate site disrupts normal electron flow and alters the redox states of remaining sites, which can dramatically alter their rates of superoxide or hydrogen peroxide production. To help solve this problem, we have devised methods to evaluate the rates of superoxide and hydrogen peroxide production from different sites in isolated mitochondria. To do this, we use internally calibrated endogenous reporters of the redox states of the different sites [the redox state of endogenous NAD(P)H to report site IF and the redox state of cytochrome b566 to report site IIIQo] together with selective inhibition of specific subsidiary sites such as IIF.48
Figure 4 shows the native rates of superoxide and hydrogen peroxide production from different sites during oxidation of glutamate plus malate under native conditions in state 4 in the absence of respiratory inhibitors, i.e. it dissects the small native rate shown in red in Figure 3 into its component sites.48 Figure 4 also shows the same analysis during oxidation of other commonly used substrates: succinate, glycerol 3-phosphate and palmitoylcarnitine plus carnitine. There are two striking results. Firstly, the native rates differ greatly between substrates, suggesting that the rates of ROS production by mitochondria in cells and in vivo are likely to depend very strongly on what substrate is being oxidized. Secondly, the contributions of different sites are very different with different substrates. During succinate oxidation, the major site of superoxide production is site IQ, with small contributions from IF and IIIQo. However, with glutamate plus malate as substrate, site IQ makes little contribution. With palmitoylcarnitine as substrate, site IIF becomes a significant contributor, and with glycerol 3-phosphate as substrate, five different sites all contribute, including glycerol 3-phosphate dehydrogenase. Thus, which sites contribute to ROS production in cells and in vivo is likely to depend very strongly on the substrates being oxidized.
Fig 4.

The contributions of different sites to native rates of hydrogen peroxide production in mitochondria isolated from muscle during oxidation of different substrates in state 4. All rates were either measured in the presence of 1-chloro-2,4-dinitrobenzene to greatly attenuate losses of hydrogen peroxide in the matrix by endogenous glutathione-linked peroxidases,65 or corrected to such measurements using the equations in Quinlan et al.48 and Treberg et al.65 Open bars, observed total rates; coloured stacks of bars, calculated contributions of different sites to the observed rates, as indicated. Values are means ± SEM (n ≥ 3). Data are from unpublished observations and references 47 and 48.
It is important to note that the sites differ markedly in the proportion of the superoxide or hydrogen peroxide they produce to the matrix or to the intermembrane space,45,47,49,50 with essentially all of the ROS from sites IF, IQ and IIF being directed to the matrix, but about half that from sites IIIQo and GPDH being directed to the intermembrane space. Thus the amount of damage in the matrix compartment and the strength of cytosolic ROS signalling will be different with different substrates, even at identical overall rates of mitochondrial ROS production.
The production of reactive oxygen species by mitochondria in cells and in vivo
Extension of the principles used above to measure the native rates of ROS production by different sites in isolated mitochondria may also prove useful in intact cells, whole tissues and in vivo. The reduction states of mitochondrial NAD and ETF can be measured in cells.51–53 Measurement of the redox state of cytochrome b566 in cells or in vivo is more difficult, but has been reported.54 The ability to make such measurements suggests that assessment of the rates of ROS production from specific mitochondrial sites in cells and in vivo using endogenous reporters may ultimately be feasible. This would provide a method to quantify ROS production in situ from different mitochondrial sites in health, ageing and disease, and allow the efficacy of treatments designed to suppress oxidative stress originating at the mitochondria to be assessed.
What’s already known about this topic?
Mitochondrial dysfunction is widely implicated in ageing and its diseases.
Mitochondria are well understood; one paper in every 154 indexed in PubMed since 1998 has studied mitochondrial function.
Nonetheless, the mechanisms and regulation of mitochondrial reactive oxygen species (ROS) formation remain poorly described.
What does this study add?
The capacities and in vitro rates of different sites of mitochondrial ROS production are being established.
The techniques involved are applicable to isolated cells.
Acknowledgments
Funding sources
Experimental work described in this paper was supported by the National Institutes of Health (USA) (P01 AG025901, PL1 AG032118, R01 AG033542 and TL1 AG032116) and The Ellison Medical Foundation (AG-SS-2288-09). Funding for the publication of this supplement was provided by Proctor & Gamble.
We thank David Nicholls for suggesting the ideas explored in Figure 1 and Table 1.
Footnotes
Conflicts of interest
None declared.
References
- 1.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–95. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Larsson NG. Somatic mitochondrial DNA mutations in mammalian aging. Annu Rev Biochem. 2010;79:683–706. doi: 10.1146/annurev-biochem-060408-093701. [DOI] [PubMed] [Google Scholar]
- 3.Tatsuta T, Langer T. Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J. 2008;27:306–14. doi: 10.1038/sj.emboj.7601972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker BM, Haynes CM. Mitochondrial protein quality control during biogenesis and aging. Trends Biochem Sci. 2011;36:254–61. doi: 10.1016/j.tibs.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Rolfe DFS, Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev. 1997;77:731–58. doi: 10.1152/physrev.1997.77.3.731. [DOI] [PubMed] [Google Scholar]
- 6.Divakaruni AS, Brand MD. The regulation and physiology of mitochondrial proton leak. Physiology (Bethesda) 2011;26:192–205. doi: 10.1152/physiol.00046.2010. [DOI] [PubMed] [Google Scholar]
- 7.Hong S, Victoria D, Crofts AR. Inter-monomer electron transfer is too slow to compete with monomeric turnover in bc1 complex. Biochim Biophys Acta. 2012;1817:1053–62. doi: 10.1016/j.bbabio.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamashita T, Voth GA. Insights into the mechanism of proton transport in cytochrome c oxidase. J Am Chem Soc. 2012;134:1147–52. doi: 10.1021/ja209176e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Efremov RG, Sazanov LA. The coupling mechanism of respiratory complex I – a structural and evolutionary perspective. Biochim Biophys Acta. 2012;1817:1785–95. doi: 10.1016/j.bbabio.2012.02.015. [DOI] [PubMed] [Google Scholar]
- 10.Baker LA, Watt IN, Runswick MJ, et al. Arrangement of subunits in intact mammalian mitochondrial ATP synthase determined by cryo-EM. Proc Natl Acad Sci USA. 2012;109:11675–80. doi: 10.1073/pnas.1204935109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palmieri F, Pierri CL. Mitochondrial metabolite transport. Essays Biochem. 2010;47:37–52. doi: 10.1042/bse0470037. [DOI] [PubMed] [Google Scholar]
- 12.Rizzuto R, De Stefani D, Raffaello A, et al. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012;13:566–78. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- 13.Yin F, Sancheti H, Cadenas E. Silencing of nicotinamide nucleotide transhydrogenase impairs cellular redox homeostasis and energy metabolism in PC12 cells. Biochim Biophys Acta. 2012;1817:401–9. doi: 10.1016/j.bbabio.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nelson DL, Cox MM. Lehninger Principles of Biochemistry. 5. New York: W. H. Freeman; 2008. [Google Scholar]
- 15.Muller M, Mentel M, Van Hellemond JJ, et al. Biochemistry and evolution of anaerobic energy metabolism in eukaryotes. Microbiol Mol Biol Rev. 2012;76:444–95. doi: 10.1128/MMBR.05024-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 17.Martinou JC, Youle RJ. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell. 2011;21:92–101. doi: 10.1016/j.devcel.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Estaquier J, Vallette F, Vayssiere JL, et al. The mitochondrial pathways of apoptosis. Adv Exp Med Biol. 2012;942:157–83. doi: 10.1007/978-94-007-2869-1_7. [DOI] [PubMed] [Google Scholar]
- 19.Huttemann M, Helling S, Sanderson TH, et al. Regulation of mitochondrial respiration and apoptosis through cell signaling: cytochrome c oxidase and cytochrome c in ischemia/reperfusion injury and inflammation. Biochim Biophys Acta. 2012;1817:598–609. doi: 10.1016/j.bbabio.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith MA, Schnellmann RG. Calpains, mitochondria, and apoptosis. Cardiovasc Res. 2012;96:32–7. doi: 10.1093/cvr/cvs163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20:145–7. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 22.Beckman KB, Ames BN. The free radical theory of aging matures. Physiol Rev. 1998;78:547–81. doi: 10.1152/physrev.1998.78.2.547. [DOI] [PubMed] [Google Scholar]
- 23.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–47. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 24.Golden TR, Hinerfeld DA, Melov S. Oxidative stress and aging: beyond correlation. Aging Cell. 2002;1:117–23. doi: 10.1046/j.1474-9728.2002.00015.x. [DOI] [PubMed] [Google Scholar]
- 25.Schriner SE, Linford NJ, Martin GM, et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–11. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 26.Loeb LA, Wallace DC, Martin GM. The mitochondrial theory of aging and its relationship to reactive oxygen species damage and somatic mtDNA mutations. Proc Natl Acad Sci USA. 2005;102:18769–70. doi: 10.1073/pnas.0509776102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muller FL, Lustgarten MS, Jang Y, et al. Trends in oxidative aging theories. Free Radic Biol Med. 2007;43:477–503. doi: 10.1016/j.freeradbiomed.2007.03.034. [DOI] [PubMed] [Google Scholar]
- 28.Van Remmen H, Jones DP. Current thoughts on the role of mitochondria and free radicals in the biology of aging. J Gerontol A Biol Sci Med Sci. 2009;64:171–4. doi: 10.1093/gerona/gln058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee HY, Choi CS, Birkenfeld AL, et al. Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metab. 2010;12:668–74. doi: 10.1016/j.cmet.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Speakman JR, Selman C. The free-radical damage theory: accumulating evidence against a simple link of oxidative stress to ageing and lifespan. BioEssays. 2011;33:255–9. doi: 10.1002/bies.201000132. [DOI] [PubMed] [Google Scholar]
- 31.Dai DF, Chen T, Johnson SC, et al. Cardiac aging: from molecular mechanisms to significance in human health and disease. Antioxid Redox Signal. 2012;16:1492–526. doi: 10.1089/ars.2011.4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Massaad CA, Pautler RG, Klann E. Mitochondrial superoxide: a key player in Alzheimer’s disease. Aging (Albany NY) 2009;1:758–61. doi: 10.18632/aging.100088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Massaad CA, Washington TM, Pautler RG, et al. Overexpression of SOD-2 reduces hippocampal superoxide and prevents memory deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2009;106:13576–81. doi: 10.1073/pnas.0902714106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song Y, Du Y, Prabhu SD, et al. Diabetic cardiomyopathy in OVE26 mice shows mitochondrial ROS production and divergence between in vivo and in vitro contractility. Rev Diabet Stud. 2007;4:159–68. doi: 10.1900/RDS.2007.4.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581– 609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aon MA, Cortassa S, O’Rourke B. Redox-optimized ROS balance: a unifying hypothesis. Biochim Biophys Acta. 2010;1797:865–77. doi: 10.1016/j.bbabio.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weinberg F, Hamanaka R, Wheaton WW, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA. 2010;107:8788–93. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ladiges W, Wanagat J, Preston B, et al. A mitochondrial view of aging, reactive oxygen species and metastatic cancer. Aging Cell. 2010;9:462–5. doi: 10.1111/j.1474-9726.2010.00579.x. [DOI] [PubMed] [Google Scholar]
- 39.Loschen G, Flohe L, Chance B. Respiratory chain linked H2O2 production in pigeon heart mitochondria. FEBS Lett. 1971;18:261–4. doi: 10.1016/0014-5793(71)80459-3. [DOI] [PubMed] [Google Scholar]
- 40.Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J. 1973;134:707–16. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCord JM, Fridovich I. The biology and pathology of oxygen radicals. Ann Intern Med. 1978;89:122–7. doi: 10.7326/0003-4819-89-1-122. [DOI] [PubMed] [Google Scholar]
- 42.Brand MD, Affourtit C, Esteves TC, et al. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med. 2004;37:755–67. doi: 10.1016/j.freeradbiomed.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 43.Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 2005;70:200–14. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 44.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brand MD. The sites and topology of mitochondrial superoxide production. Exp Gerontol. 2010;45:466–72. doi: 10.1016/j.exger.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grivennikova VG, Kareyeva AV, Vinogradov AD. What are the sources of hydrogen peroxide production by heart mitochondria? Biochim Biophys Acta. 2010;1797:939–44. doi: 10.1016/j.bbabio.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Orr AL, Quinlan CL, Perevoshchikova IV, Brand MD. A refined analysis of superoxide production by mitochondrial sn-glycerol 3-phosphate dehydrogenase. J Biol Chem. 2012;287:42921–35. doi: 10.1074/jbc.M112.397828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Quinlan CL, Treberg JR, Perevoshchikova IV, et al. Native rates of superoxide production from multiple sites in isolated mitochondria measured using endogenous reporters. Free Radic Biol Med. 2012;53:1807–17. doi: 10.1016/j.freeradbiomed.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miwa S, Brand MD. The topology of superoxide production by complex III and glycerol 3-phosphate dehydrogenase in Drosophila mitochondria. Biochim Biophys Acta. 2005;1709:214–9. doi: 10.1016/j.bbabio.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 50.St-Pierre J, Buckingham JA, Roebuck SJ, et al. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. 2002;277:44784–90. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 51.Kuznetsov AV, Mayboroda O, Kunz D, et al. Functional imaging of mitochondria in saponin-permeabilized mice muscle fibers. J Cell Biol. 1998;140:1091–9. doi: 10.1083/jcb.140.5.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang S, Heikal AA, Webb WW. Two-photon fluorescence spectroscopy and microscopy of NAD(P)H and flavoprotein. Biophys J. 2002;82:2811–25. doi: 10.1016/S0006-3495(02)75621-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chorvat D, Jr, Kirchnerova J, Cagalinec M, et al. Spectral unmixing of flavin autofluorescence components in cardiac myocytes. Biophys J. 2005;89:L55–7. doi: 10.1529/biophysj.105.073866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sommer N, Pak O, Schorner S, et al. Mitochondrial cytochrome redox states and respiration in acute pulmonary oxygen sensing. Eur Respir J. 2010;36:1056–66. doi: 10.1183/09031936.00013809. [DOI] [PubMed] [Google Scholar]
- 55.Bunik VI, Sievers C. Inactivation of the 2-oxo acid dehydrogenase complexes upon generation of intrinsic radical species. Eur J Biochem. 2002;269:5004–15. doi: 10.1046/j.1432-1033.2002.03204.x. [DOI] [PubMed] [Google Scholar]
- 56.Tretter L, Adam-Vizi V. Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. J Neurosci. 2004;24:7771–8. doi: 10.1523/JNEUROSCI.1842-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Starkov AA, Fiskum G, Chinopoulos C, et al. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci. 2004;24:7779–88. doi: 10.1523/JNEUROSCI.1899-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kareyeva AV, Grivennikova VG, Cecchini G, et al. Molecular identification of the enzyme responsible for the mitochondrial NADH-supported ammonium-dependent hydrogen peroxide production. FEBS Lett. 2010;585:385–9. doi: 10.1016/j.febslet.2010.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Quinlan CL, Orr AL, Perevoshchikova IV, et al. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J Biol Chem. 2012;287:27255–64. doi: 10.1074/jbc.M112.374629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Drahota Z, Chowdhury SK, Floryk D, et al. Glycerophosphate-dependent hydrogen peroxide production by brown adipose tissue mitochondria and its activation by ferricyanide. J Bioenerg Biomembr. 2002;34:105–13. doi: 10.1023/a:1015123908918. [DOI] [PubMed] [Google Scholar]
- 61.Hansford RG, Hogue BA, Mildaziene V. Dependence of H2O2 formation by rat heart mitochondria on substrate availability and donor age. J Bioenerg Biomembr. 1997;29:89–95. doi: 10.1023/a:1022420007908. [DOI] [PubMed] [Google Scholar]
- 62.Lambert AJ, Brand MD. Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH:- ubiquinone oxidoreductase (complex I) J Biol Chem. 2004;279:39414–20. doi: 10.1074/jbc.M406576200. [DOI] [PubMed] [Google Scholar]
- 63.Lambert AJ, Brand MD. Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem J. 2004;382:511–7. doi: 10.1042/BJ20040485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Quinlan CL, Gerencser AA, Treberg JR, et al. The mechanism of superoxide production by the antimycin-inhibited mitochondrial Q-cycle. J Biol Chem. 2011;286:31361–72. doi: 10.1074/jbc.M111.267898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Treberg JR, Quinlan CL, Brand MD. Hydrogen peroxide efflux from muscle mitochondria underestimates matrix superoxide production – a correction using glutathione depletion. FEBS J. 2010;277:2766–78. doi: 10.1111/j.1742-4658.2010.07693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]