

Abstract
Methamphetamine (METH) is a central nervous system psychostimulant with a high potential for abuse. At high doses, METH causes a selective degeneration of dopaminergic terminals in the striatum, sparing other striatal terminals and cell bodies. We previously detected a deficit in parkin after binge METH in rat striatal synaptosomes. Parkin is an ubiquitin-protein E3 ligase capable of protecting dopamine neurons from diverse cellular insults. Whether the deficit in parkin mediates the toxicity of METH and whether parkin can protect from toxicity of the drug is unknown. The present study investigated whether overexpression of parkin attenuates degeneration of striatal dopaminergic terminals exposed to binge METH. Parkin overexpression in rat nigrostriatal dopamine system was achieved by microinjection of adeno-associated viral transfer vector 2/6 encoding rat parkin (AAV2/6-parkin) into the substantia nigra pars compacta. The microinjections of AAV2/6-parkin dose-dependently increased parkin levels in both the substantia nigra pars compacta and striatum. The levels of dopamine synthesizing enzyme, tyrosine hydroxylase, remained at the control levels; therefore, tyrosine hydroxylase immunoreactivity was used as an index of dopaminergic terminal integrity. In METH-exposed rats, the increase in parkin levels attenuated METH-induced decreases in striatal tyrosine hydroxylase immunoreactivity in a dose-dependent manner, indicating that parkin can protect striatal dopaminergic terminals against METH neurotoxicity.
Keywords: methamphetamine, toxicity, parkin, neuroprotection, dopamine, striatum
Introduction
Methamphetamine (METH) is a psychostimulant drug, which is widely abused in the United States and worldwide. When taken at high doses, METH has toxic effects on the dopaminergic (DAergic) system in the brain in experimental animals and humans. In rats, administration of binge high-dose METH causes persistent deficits in DAergic markers in striatal DAergic terminals but has little effect on other DAergic nerve endings, such as those terminating in the nucleus accumbens (Broening, et al., 1997, Cass, 1997, Haughey, et al., 1999, Morgan and Gibb, 1980), and on DA cell bodies in the substantia nigra pars compacta (SNc) from which striatal DAergic terminals originate (Harvey, et al., 2009, Hotchkiss and Gibb, 1980, Ricaurte, et al., 1982). Chronic METH users do not display classic Parkinsonian motor impairments; however, they are at higher risk for developing Parkinson’s disease (PD) than non-users (Callaghan, et al., 2010, Callaghan, et al., 2011). Abuse of high doses of METH can cause impairment of cognitive skills, agitation, violent behavior, anxiety, confusion, and paranoia. Despite years of active research in the area of METH abuse and its related neurotoxicity, to date, there are no specific medications that counteract the damaging effects of METH on the brain. Thus, there is a compelling need to discover new molecular drug targets in order to develop novel pharmaceuticals that can protect the CNS from the toxic effects of acute METH overdose.
Parkin is an ubiquitin-protein E3 ligase; its primary function is to add polyubiquitin chains to proteins destined for degradation by the 26S proteasome (Moore, 2006, Shimura, et al., 2000, Zhang, et al., 2000). A decrease in parkin function leads to toxic accumulation of unwanted proteins and has been implicated in the etiology of various neurodegenerative disorders, including PD (Buneeva and Medvedev, 2006, Lim, 2007, Olzmann, et al., 2008). Conversely, overexpression of parkin protects DA neurons against a variety of cellular insults in vitro and in vivo, most importantly against those involved in mediating METH neurotoxicity, such as DA-induced oxidative stress, inhibition of mitochondrial function, and impairment of the proteasome (Darios, et al., 2003, Jiang, et al., 2012, Jiang, et al., 2004, Kirik and Bjorklund, 2005, Lo Bianco, et al., 2004, Oluwatosin-Chigbu, et al., 2003, Petrucelli, et al., 2002, Yang, et al., 2005, Yang, et al., 2011). These findings suggest the importance of parkin in the functioning and maintenance of DA neurons.
Our previous in vivo study demonstrated that binge METH was followed by a rapid decrease in parkin levels in rat striatal synaptosomes, which persisted for a minimum of 24 h after METH administration (Moszczynska and Yamamoto, 2011). Whether the deficit in parkin mediates the toxicity of METH is not known and no studies, to date, have examined whether parkin could protect DAergic terminals against neurotoxicity of the drug. Thus, the major aim of the present study was to determine whether overexpression of parkin in the nigrostriatal DA system protects DAergic terminals in the striatum against binge high-dose METH. In order to gain further insight into the role of parkin in METH neurotoxicity and potential mechanisms of parkin neuroprotection, we also investigated the effects of parkin overexpression on the levels of tyrosine hydroxylase (TH) (a rate-limiting enzyme in DA synthesis), the activity of 20S proteasome, and METH-induced hyperthermia. We are the first to report that the overexpression of parkin in the nigrostriatal DA system attenuates METH toxicity to striatal DAergic terminals in a dose-dependent manner. These results, together with our previous findings, suggest that a parkin deficit mediates, in part, toxicity of METH to striatal DAergic terminals in vivo.
Materials and Methods
Animals
Adult male Sprague-Dawley rats (Harlan, Indianapolis, IN, USA) weighing 175-220 g at the time of the arrival were housed two per cage under a 12h light/dark cycle in a temperature-controlled room (21-23°C). Food and water were available ad libitum. Temperature of the rats was measured via a rectal probe digital thermometer (Thremalert TH-5; Physitemp Instruments, Clifton, NJ, USA). All animal procedures were conducted in strict accordance with the NIH Guide for Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee at Wayne State University (# A 06-03-10). All surgery was performed under anesthesia, and all efforts were made to minimize suffering of animals.
Adeno-associated viral transfer vectors
The AAV2/6 gene transfer vectors, non-coding AAV2/6 (with a DNA segment cloned upstream of the pgk promoter to adapt the size of vector genome to AAV packaging capacity) and rat parkin-encoding AAV2/6 (AAV2/6 and AAV2/6-parkin), were kindly gifted by Dr. Patrick Aebischer at Swiss Federal Institute of Technology Lausanne (EPFL), Switzerland. The cDNA encoding rat parkin was cloned into the pAAV-pgk-MCS transfer vector, and serotype 6 adeno-associared viral particles (AAV2/6) were produced and titered as described previously (Dusonchet, et al., 2009). The virus titers for AAV2/6-parkin and non-coding AAV2/6 were 4.7×1010 TUs/ml (transducing units/mL) and 4.4×1010 TUs/ml, respectively. Virus-containing suspensions of 1×107 or 2×107 TUs in 2 μL volume were microinjected into the brain.
Experimental Design
Following 1 week of acclimation, rats were stereotaxically injected with non-coding (1×107 or 2×107 TUs) or parkin-coding (1×107 or 2×107 TUs) transfer vector suspensions into the left SNc. After 3 weeks, the rats were sacrificed by decapitation without treatment or treated with binge METH or saline and sacrificed by perfusion a week later. Dissected or sliced brains were stored at −80°C until assayed. The untreated rats were used for validation and evaluation of parkin overexpression in the nigrostriatal system and for evaluation of its effects on activity of the 20S proteasome. Saline- and METH-treated rats were used to assess the effects of parkin-overexpression on the extent of METH toxicity and for evaluation of METH-induced hyperthermia. TH levels were assessed at 3 weeks after microinjections (western blotting) and at 4 weeks after microinjections (immunohistochemistry). The experimental design is illustrated in Fig.1.
Figure 1.
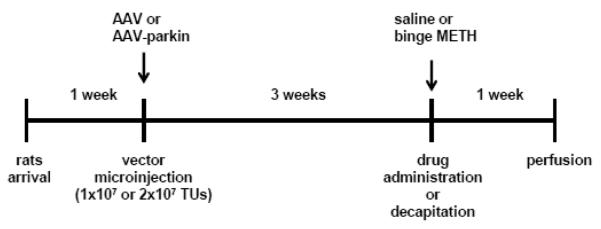
Experimental design. Following 1 week of acclimation, adult male Sprague-Dawley rats were stereotaxically injected with non-coding or parkin-coding adeno-associated transfer vectors (AAV2/6 and AAV2/6-parkin) into the left substantia nigra pars compacta (SNc). After 3 weeks, the rats were sacrificed without treatment or treated with binge saline (1mL/kg × 4., every 2 h, i.p.) or binge METH (7.5 mg/kg × 4, every 2 h). Untreated rats were used for evaluation and validation of parkin overexpression. To assess the effects of parkin-overexpression on the extent of METH toxicity, saline and METH-treated rats were sacrificed by perfusion 1 week after the day of METH and saline treatment. The arrows indicate stereotaxic microinjection (AAV2/6 vectors) and drug injection (METH or saline) times. Abbreviations: AAV, adeno-associated virus; TUs, transducing units; METH, methamphetamine.
Stereotaxic surgery
Rats were anesthetized with an intraperitoneally (i.p.) administered mixture of xylazine (20 mg/ml) and ketamine hydrochloride (100 mg/ml). When necessary, supplementary doses of ketamine were administered to maintain surgical levels of anesthesia. AAV2/6 vectors (AAV2/6 or AAV2/6-parkin) were unilaterally injected into the left SNc with the following coordinates: −5.6 mm (AP), −2 mm (ML), −7.6 mm (V) from the dura according to the Paxinos and Watson’s rat brain atlas. Viral suspensions were injected with a 5 μl Hamilton syringe at a rate of 0.15 μL/min using a syringe pump (Harvard Apparatus, Holliston, MA, USA). The syringe was left in place for 5 minutes at −7.6 mm than withdrawn to −6.6 mm and −5.5 mm, staying in each location for 5 minutes, then raised slowly out of the brain over 2 minutes.
Methamphetamine administration
Three weeks after AAV2/6 or AAV2/6-parkin microinjection, rats were randomly divided into two groups and injected (i.p.) with either (+)methamphetamine hydrochloride (Sigma-Aldrich, St. Louis, MO, USA) (7.5 mg/kg × 4) or vehicle saline (1mL/kg × 4) at 2 hours intervals. This METH regimen produces a marked neurotoxic effect to the DAergic terminals in rats (Tata, et al., 2007). Each treatment group was composed of 5-7 animals. Rectal temperature was taken at 1 hour after each saline and METH injection.
Immunohistochemistry
One week after binge high-dose METH or saline treatment, rats were anesthetized with a mixture of xylazine and ketamine hydrochloride and transcardially perfused with 0.01M phosphate buffer solution (PBS) at pH 7.4 and 4% paraformaldehyde in 0.01M PBS. Brains were removed and postfixed in 4% paraformaldehyde for 2 hours and then placed in 10% glycerol and 20% glycerol at 4°C for overnight. Brains were frozen in isopentane and kept at −80°C for further immunohistochemistry procedures.
Coronal brain sections (30 μm-thick) from the entire SNc and a large part of the striatum were sliced on a cryostat (Thermo Fisher Scientific Inc. Waltham, MA) and collected in 6-well plate, every 6th section per well. Sections containing the SNc and striatum were selected according to Paxinos & Watson’s rat brain atlas, with the rostro-caudal extent of the SNc being between −4.8 mm and −6.12 mm and the rostro-caudal extent of the striatum being between +2.16 mm and −0.24 mm with respect to Bregma. Free-floating staining procedures were conducted to reveal TH and/or parkin using an immunoperoxidase or immunofluorescence techniques. Briefly, sections were washed in PBS-Triton X-100 (0.2%) for 3×10 min. Brain sections were then rinsed in 0.3% hydrogen peroxide to quench the endogenous peroxide. Citrate buffer (1x) was used to retrieve parkin antigen during parkin staining process. After blocking the unspecific binding with 5% normal goat serum (NGS) in PBS-Triton X-100, sections were incubated in rabbit anti-TH (Ab152, 1:2000, Millipore Co., Billerica, MA) or rabbit anti-parkin (AB5112, 1:1000, Millipore Co.) antibodies at 4°C overnight. Sections were then incubated in biotinylated goat anti-rabbit antibody (BA1000, Vector Laboratories Inc., Burlingame, CA) diluted 1:200 in 5% NGS-PBS-Triton-X100. The biotinylated antibody was detected using ABC kit (PK-6100, Vector Laboratories) and revealed by peroxidase reaction with diaminobenzidine (DAB) as the chromagen. Sections were mounted onto the slides, dehydrated through a series of dilutions of ethanol (70%, 95% and 100%), cleared with xylene and coversliped. For the fluorescence multiple labeling, sections were incubated in mouse anti-TH (MAB318, 1:2000, Millipore Co., Billerica, MA) and rabbit anti-parkin (AB5112, 1:1000, Millipore Co.) antibodies at 4°C overnight, and then incubated in Alexa Fluor 488 goat anti-mouse and Alexa Fluor 596 goat anti-rabbit secondary antibodies (Invitrogen Co., Carlsbad, CA) at a dilution of 1:200 in PBS-Triton-X-100 for 2 hours at room temperature. After counterstaining in DAPI (300 nM), brain sections were mounted and coversliped. In the control incubation, the primary or biotinylated secondary antibodies were individually omitted; it demonstrated that the immunoreaction could not be a result of non-specific reactivity. To distinguish between injected and non-injected hemisphere on floating sections, small hole was made with a needle through the left cortex in each brain before its cryosectioning.
Every 6th section containing the SNc was immunostained with an immunoperoxidase or immunofluorescence method and examined for parkin immunoreactivity. Three or five representative sections per rat were used in quantifications. Nigral sections stained using the immunoperoxidase procedure were examined with EVOS xl microscope (Advanced Microscopy Group, Bothell, WA). Parkin-immunoreactive neurons in the SNc from 3 representative sections per rat (n=5-6 rats) were counted in 3 equal-size non-overlapping 10× microscopic fields per section. The numbers of parkin-immunoreactive cells from nine locations per rat (3 fields per section × 3 sections per rat hemisphere) were averaged and expressed as positive cells per field (10x). The numbers of parkin-immunoreactive neurons in the microinjected SNc were expressed as the percentages of parkin-immunoreactive neurons in the contralateral non-injected side. The staining densities of parkin-immunoreactive cells from 30 nigral cells expressing parkin (light brown, non-injected hemisphere) and 30 nigral cells overexpressing parkin (medium-to dark-brown, injected hemisphere) (10 cells per section × 3 sections per rat hemisphere) were quantified using NIH Image J 1.4 imaging program, averaged and expressed as a fold change in the relation to the contralateral non-injected side. The number of dark brown cells (high parkin overexpression) within the injected SNc (1×107 and 2×107 TUs of AAV2/6-parkin) was estimated as following: total number and number of dark brown parkin-positive neurons were counted in 5 equal non-overlapping areas (40x) within SNc boundary in 5 slices per animal (n=6 rats/group). Numbers were expressed as sum of cells per slice; dark brown cells were expressed as percentage of total parkin-positive cells per slice and averaged for each animal. To calculate the number of DA neurons in the SNc in injected and non-injected hemispheres, the number of DAPI-positive nuclei of TH-positive nigral cells was counted (4 slices per animal, 3 areas per slice, 4 equal squares per area, n=6 rats/group), expressed as number of nuclei per unit of area per animal.
Several areas of the striatum were analyzed at several different coronal planes along its rostro-caudal extent. Three methods were used to quantify TH-immunoreactive terminals in the striatum (4-5 sections per brain), performed independently by two experimenters. In the first method, the optical densities of DAB-visualized TH immunoreactivities were measured in 2 equal-size striatal areas per slice (2 areas encompassing almost entirely left and right striatum) and quantified using NIH ImageJ 1.4 program. Background staining was measured in the neighboring corpus callosum. After background subtraction, the optical densities were averaged in each animal. In the second method, 3 images per hemisphere in each striatal section stained for TH using immunoperoxidase method were taken. The optical densities of 3 equal-size non-overlapping 10× microscopic fields per image were quantified using NIH ImageJ 1.4 program. TH-immunoreactive density was evaluated based upon the optical density in the cortex of each section. The optical densities of TH-immunoreactive area in the striatum from 9-12 locations per animal hemisphere (3 fields per section × 3-4 sections per rat) were averaged to give one value per hemisphere of the animal. In the third method, 3 different striatal sections were immunolabelled for TH using immunofluorescence method. As in the second method, 3 images per hemisphere from in each striatal section were taken. TH immunofluorescence was quantified using NIH ImageJ 1.4 program and averaged to give one value per hemisphere per animal. In all methods, 5-6 animals were included in each group. Sections stained using the immunoperoxidase procedures were examined with EVOS xl microscope, whereas sections stained with the immunofluorescence procedure were examined with EVOS fl microscope (Advanced Microscopy Group). Each of these methods has advantages and disadvantages. The first method quantifies TH immunoreactivity in almost entire striatal area on a slice whereas other two methods quantify striatal sections that do not cover the entire striatal area. The advantage of immunofluorescence is lack of the background staining, which can be a confounding factor in the immunoperoxidase immunostaining if the background is uneven. In addition, DAB method is not linear due to saturation in the color development.
Preparation of synaptosomes
Crude synaptosomal fractions were prepared from striatal tissues via differential centrifugation as described previously (Riddle, et al., 2002). Briefly, striata were dissected, homogenized with a hand glass homogenizer in 0.5 mL of ice-cold 0.32 M sucrose and centrifuged (800 × g for 24 min; 4°C) to remove nuclei and large debris (P1). The supernatant (S1) was centrifuged at high speed (22,000 × g for 17 min; 4°C), and the pellet (P2) retained was the crude total synaptosomal fraction. This fraction was re-suspended in ice-cold distilled deionized water (ddH2O). Synaptosomal preparations were analyzed for protein concentration by the method of Bradford (Bradford, 1976) using bovine serum albumin as the standard.
Western Blot Analysis
Striatal synaptosomes were prepared from rats killed by decapitation and subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), blocking for 1 hr at room temperature with 5% nonfat dried milk dissolved in TBST (10 mM Tris, 150 mM NaCl, and 0.5% Tween-20), and western blotting with either a primary monoclonal antibody against parkin (1:1,000; 1 hr at room temperature) (#4211; Cell Signaling Technology, Danfers, MA) or polyclonal rabbit antibody against TH (1:1,000; 1 hr at room temperature) (AB152, EMD Millipore Corp., Billeric, MA) followed by incubation with appropriate HRP-conjugated secondary antibodies. Blots were developed using ECL detection and LAS4000 bioimager (GE Healthcare, Piscataway, NJ). To standardize across blots, each blot contained all experimental groups. Actin was used as a loading control. The western blot data was expressed as relative optical density units on each gel normalized to saline controls. This approach normalized differences in the development of the blot and across blots.
Chymotrypsin-like activity of 20S proteasome assay
Striatal synaptosomes were prepared from parkin overexpressing rats (dose: 2 × 107 TUs of AAV2/6-parkin) and assayed for maximal velocity (Vmax) of chymotrypsin-like activity of the 20S proteasome. The activity was determined in the absence of ATP in a 96-well fluorimetric plate reader (Synergy H4 Hybrid, BioTek, Winooski, VT) (Ex 350 nm, Em 440 nm). The activity was monitored at 37°C for 10 min after the addition of 200 μM of the enzyme substrate, a fluorogenic peptide Suc-Leu-Leu-Val-Tyr-7-amido-4-methylcoumarin (LLVY-AMC) (P-802; Sigma-Aldrich, St. Louis, MO) (Bulteau, et al., 2001). Contribution of non-proteasomal proteases in each preparation was assessed by measuring the activity in the presence of epoxomicin (Sigma-Aldrich, St. Louis, MO), a proteasome inhibitor, and subsequently subtracted from the total activity. Assays were performed in a total volume of 50 μL. The assay buffer was composed of 25 mM Tris-HCl at pH 7.5, 2 μM epoxomicin in DMSO or DMSO, and the peptide substrate. The maximal velocities were calculated using GraphPad Prism program (GraphPad Software Inc., La Jolla, CA). The standard curve for relative 20S activity quantification was generated using 0-10 μM AMC (Sigma-Aldrich). The data were expressed as nmols of AMC released/min/mg protein.
Statistical Analysis
Data with two treatment groups were analyzed using Student’s t-test. To determine significant differences between more than two groups, one-way ANOVA with Student-Newman-Keuls post-hoc test was employed. Two-way ANOVA (factors: drug treatment and parkin dose) followed by Bonferroni post-hoc test was used to analyze striatal TH immunoreactivity data. Two-way repeated measures ANOVA analysis with Student-Newman-Keuls post-hoc test was performed on temperature data. All data are expressed as mean ± SEM. The statistically significance was set at <0.05.
Results
Evaluation and validation of parkin overexpression in the nigrostriatal system
The substantia nigra pars compacta
The placement of microinjection needles was evaluated by microinjecting 0.1 μL of Evans blue into the left SNc of 4 animals at the following co-ordinates: −5.6 mm (AP) from Bregma, −2 mm (ML) and −7.6 mm (V) from the dura (Fig.2A,B). Fig.2C demonstrates that the injection sites using these co-ordinates were within the SNc. Fig.2D shows 4 representative coronal sections from far boundaries of the SNc (−4.48 to −6.12 mm with respect to Bregma) immunolabelled for parkin, demonstrating the level of parkin overexpression along the rostro-caudal axis. As expected, parkin overexpression was the highest close to the injection site and lowest at the ends of the SNc.
Figure 2.

Evaluation of the microinjection sites in the substantia nigra pars compacta (SNc). (A, B) Rat brains (n = 7) were stereotaxically injected on the left side with 0.1 μL of Evans blue to validate microinjection co-ordinates: −5.2 mm (AP) from Bregma, −2 mm (ML) from Bregma, −7.6 (V) mm from the dura. (C) A representative anatomical location of the microinjection sites in the SNc and its magnification. (D) Representative coronal sections from far ends of the SNc along the rostro-caudal axis (−4.48 to −6.12 mm with respect to Bregma) immunolabelled for parkin, demonstrating the highest parkin overexpression close to the injection site and the lowest at the ends of the SNc. Bars: 1000 μm. Abbreviations: AP, anterior-posterior; ML, medio-lateral; V, ventral.
To evaluate transduction efficiency of AAV2/6 transfer vector, two doses of AAV2/6-parkin (1×107 TUs and 2×107 TUs) were stereotaxically microinjected into the left SNc generating two levels of parkin overexpression. Control animals received a microinjection of AAV2/6 (non-coding vector, 1×107 TUs or 2×107 TUs). The immunoreactivity of parkin in the SNc was examined 3 weeks after the procedure (n=5-6 rats/group). Microinjection of the non-coding AAV2/6 vector did not affect endogenous parkin levels (Fig.3Aa-d, 3Da, 3Ea). Microinjection of AAV2/6-parkin resulted in dose-dependent increase in parkin levels in the left SNc as compared to the right SNc (Fig.3B,C, Db,c, Eb,c). Thus, the average number of parkin-positive cells in the injected SNc was 112% and 154% in relation to the non-injected SNc for 1×107 and 2×107 TUs of AAV2/6-parkin, respectively (Fig.3Db,c). The average optical density of parkin overexpressing cells (medium to dark brown) was 2.8 fold and 3.3 fold higher than optical density of parkin-positive cells in the non-injected SNc (light brown), for 1×107 and 2×107 TUs of AAV2/6-parkin, respectively (Fig.3Eb,c). There was 2.1% more of dark brown parkin-positive cells in nigras injected with higher dose than in those injected with lower dose of parkin-coding transfer vector (2×107 TUs: 17.3 ± 2.6% of total parkin-positive cells, 1×107 TUs: 15.2 ± 1.6% of total parkin-positive cells, n=6 rats/group).
Figure 3.

Validation of parkin overexpression in the substantia nigra pars compacta (SNc) by color immunohistochemistry. Non-coding or parkin-coding adeno-associated virus 2/6 transfer vectors (AAV2/6) were stereotaxically injected in the left SNc of adult male Sprague-Dawley rats with the following coordinates: −5.2 (AP) and −2 (ML) from Bregma, −7.6 (V) mm from the dura. After 3 weeks, the rats were sacrificed by perfusion. (A-C) Representative images of parkin immunolabeling (DAB) in the SNc of rats microinjected with (A) the non-coding AAV2/6 (a-d), (B) 1×107 TUs of AAV2/6-parkin (e-h), and (C) 2×107 TUs of AAV2/6-parkin (i-l), respectively (left: injected and right: non-injected; magnification: a, d, e, h, i, l - 40x; b, c, f, g, j, k - 10x). Injected hemispheres show higher parkin immunostaining than non-injected hemispheres (e, f vs. g, h and i, j vs. k, l), indicating successful dose-dependent overexpression of parkin. Microinjections of the non-coding vector suspensions (1×107 or 2×107 TUs) did not affect endogenous parkin levels (a-d). (D) Quantification of parkin overexpression in the SNc by counting of parkin-positive cells. There was no difference in number of parkin-positive cells between the non-coding AAV2/6-injected and non-injected SNc (p>0.05, unpaired two-tailed Student’s t-test, t=0.1190, df=10, n=6). SNc injected with parkin-coding AAV2/6 (1×107 or 2×107 TUs of AAV2/6-parkin) showed dose-dependent increase in number of parkin-positive cells (+12% and +54%, respectively, as compared to contralateral SNc); however, the 12% increase was not statistically significant (unpaired one-tailed Student’s t-test; 1×107: t=0.6077, df=8, p>0.05; 2×107: t=2.444, df=8, p<0.05; n=5). (E) Quantification of parkin overexpression in the SNc by measurement of color density in parkin-positive cells. Injection of the non-coding AAV2/6 did not affect color density in parkin-positive cells (p>0.05, unpaired two-tailed Student’s t-test, t=0.1965, df=10, n=6). SNc injected with parkin-coding AAV2/6 (1×107 or 2×107 TUs of AAV2/6-parkin) showed dose-dependent increase in color density in parkin-overexpressing cells (medium to light brown) (2.5 fold and 3.3 fold, respectively) as compared to parkin-positive cells contralateral SNc (endogenous parkin: light brown) (unpaired one-tailed Student’s t-test; 1×107: t=2.340, df=8, p<0.05; 2×107: t=4.850, df=8, p<0.01; n=5). *p<0.05, **p<0.01 injected vs. corresponding non-injected SNc. Data are presented as mean ± SEM. Bar in a,d,e,h,i,l: 100 μm; bar in b,c,f,g,j,k: 400 μm. Abbreviations: TUs, transducing units.
Double immunostaining for parkin and TH, a DAergic marker, of brain slices containing SN confirmed the results from DAB-mediated parkin immunostaining. Thus, parkin levels were higher in the left SNc (injected) than in the right SNc (non-injected) (Fig.4, f,n vs. b,j). Some TH-positive cells overexpressed parkin, some did not. This was observed on the same coronal plane (Fig.4h,p) as well as at different coronal planes (Fig.4A,B and C) with less parkin overexpressing DA neurons located away from the injection site (Fig.4 u-z) than those located closer (Fig.4 r-t) along the rostro-caudal axis. Parkin overexpression was also observed in some TH-negative nigral cells. The endogenous parkin was hardly detectable by this technique (Fig.4b,j). Controls in which primary or secondary antibodies were omitted showed no staining (not shown).
Figure 4.

Validation of parkin overexpression in the substantia nigra pars compacta (SNc) by immunofluorescence. Representative images of parkin and tyrosine hydroxylase (TH) immunofluorescent labeling in the SNc: (A) area adjacent to the VTA, (B) middle area of the SNc. In non-injected SNc, parkin immunostaining was not detectable (Ab and Bj). In microinjected SNc, immunolabelling for parkin (green) colocalized with immunolabeling for DAergic TH (red), indicating parkin overexpression in most of the DA neuronal cell bodies (Ah and Bp, white arrows). Parkin-positive neurons were also observed in the ventral tegmental area (Ah). (C) Two coronal planes of the SNc along the rostro-caudal axis. More parkin was overexpressed in DA neurons located closer to the injection site (upper panel) than in DA neurons located away from the site (lower panel). Bars: 200 μm.
The striatum
As expected (D’Agata, et al., 2002, Vercammen, et al., 2006), the endogenous levels of parkin were undetectable in the right striatum (non-injected side) (Fig.5A right). Similarly to the results from the SNc, single staining for parkin in the striatum showed higher parkin levels on the side ipsilateral to the microinjected SNc than on the contralateral side. Parkin co-localized with TH in the left striatum, indicating its overexpression in striatal DAergic terminals (Fig.5A left, arrows). Some striatal cell bodies also appeared to have higher than control parkin levels. The co-localization of TH with parkin was not clearly visible due to bright TH fluorescence. To confirm parkin overexpression in these terminals, striatal synaptosomal preparations from rats microinjected with the non-coding AAV2/6 or AAV2/6-parkin (n=5-6 rats/group) were subjected to SDS-PAGE and western blotting with antibody against parkin. Following intranigral microinjection of 1×107 and 2×107 TUs of AAV2/6-parkin, the levels of parkin increased 3 and 8 fold, respectively, in synaptosomes from the left striatum (ipsilateral to AAV2/6-parkin-microinjected SNc) as compared to synaptosomes from the right striatum (Fig.5B). Microinjections of non-coding AAV2/6 (1×107 or 2×107 TUs) did not result in statistically significant changes in striatal parkin levels (Fig.5B).
Figure 5.

Validation of parkin overexpression in the striatum. Rats were microinjected with the non-coding AAV2/6 (1×107 or 2×107 TUs) or AAV2/6-parkin (1×107 or 2×107 TUs) into the left SNc, and sacrificed 3 weeks later. (A) Representative image of double immunostaining for parkin and DAergic marker tyrosine hydroxylase (TH) and (B) quantification of parkin levels in striatal synaptosomes by western blotting. (A) Parkin immunostaining is not clearly visible in merged image because of the bright TH staining. Nevertheless, the co-localization of parkin and TH is apparent (A, white arrows). Bars: 100 μm. (B) Representative blots of parkin (52 kDa) and corresponding actin loading control from striatal synaptosomal preparations. Following intranigral microinjection of 1×107 and 2×107 TUs of AAV2/6-parkin, the levels of parkin increased 3 and 8.5 fold, respectively, in ipsilateral striatum as compared to the contralateral striatum (p<0.001, unpaired, one-tailed Student’s t-test; 1×107: t=11.92, df=8; 2×107: t=6.209, df=8; n=5). Microinjections of either dose of the non-coding AAV2/6 resulted in no statistically significant changes in striatal parkin levels (western blot: first 4 bands, 1×107 and 2×107 TUs, respectively) (p>0.05, unpaired two-tailed Student’s t-test, t=0.6062, df=12, n=7). The data are expressed as mean ± SEM. ***p<0.001 injected vs. corresponding non-injected striatum. Abbreviations: AAV vector, adeno-associated viral vector, I, injected; N, non-injected; TUs, transducing units.
Evaluation of parkin neuroprotection against METH neurotoxicity
Toxicity of high-dose METH to DAergic terminals is commonly demonstrated by decreased levels of DAergic markers such as TH, DA, and/or DA metabolites a week after METH treatment (Yamamoto, et al., 2010). We first evaluated whether TH could serve as an index for assessing METH-induced neurotoxicity to striatal DAergic terminals in parkin overexpressing rats. Evaluation of striatal TH levels by SDS-PAGE and western blotting in drug naïve rats injected with the non-coding AAV2/6 or AAV2/6-parkin revealed no statistically significant differences between left and right striatum (Fig.6). In contrast, evaluation of striatal TH levels by immunohistochemistry in saline-treated AAV2/6-parkin-injected rats detected differences between the left and right striatum. The first immunoperoxidase method (see Materials and Methods section) detected statistically significant mild decreases in striatal TH immunoreactivity in both saline- and METH-treated rats injected with the higher dose (2×107 TUs) of non-coding AAV2/6 (−13% and −21%, respectively) (Fig.7A). Microinjections of 1×107 TUs of non-coding AAV2/6 did not affect TH levels (Fig.7A). The second immunoperoxidase method, detected similar but non-significant decreases in TH levels with 2×107 TUs dose (−10% and 15% in saline and METH rats, respectively). Examination of TH immunofluorescence in the SNc from these rats revealed slightly lower levels of the TH in the left SNc as compared to the contralateral one (Fig.7B). To determine whether the decrease in TH immunoreactivity is due to DA neuronal degeneration, cells positive for both DAPI and TH were counted in the SNc; no difference was found between hemispheres (non-injected SNc: 100 ± 4%, injected SNc: 102 ± 3%, n=6 rats/group) (Fig.7C).
Figure 6.

Evaluation of striatal tyrosine hydroxylase (TH) levels by SDS-PAGE and western blotting in drug naïve rats injected with the non-coding AAV2/6 or AAV2/6-parkin. Representative blots of TH (62 kDa) and corresponding actin loading control from striatal synaptosomal preparations. Intranigral microinjection of non-coding AAV2/6 (1×107 or 2×107 TUs) or AAV2/6-parkin (1×107 or 2×107 TUs) revealed no statistically significant differences between left and right striatum (p>0.05; unpaired two-tailed Student’s t-test; non-coding AAV2/6: t=2.993, df=12; 1×107: t=0.2508, df=8; 1×107: t=0.8294, df=8; n=5-7). The data are expressed as mean ± SEM). Abbreviations: AAV vector, adeno-associated viral vector, I, injected; N, non-injected; TUs, transducing units.
Figure 7.

The effect of the non-coding AAV2/6 injection on the levels of tyrosine hydroxylase (TH) in the striatum and substantia nigra pars compacta (SNc) by immunohistochemistry. (A) Quantification of TH immunoreactivity in DAB-stained striatal slices. Microinjections of the non-coding AAV2/6 vector at the dose of 2×107 TUs induced mild decreases in striatal TH immunoreactivity in both saline- and METH-treated rats (−13% and −21%, respectively). Data was analyzed by one-way ANOVA followed by the Student-Newman-Keuls post-hoc test (F(5,22) =10.739, p<0.001). ***p<0.001, **p<0.01, *p<0.05, significant difference between saline and METH; #p<0.05, tp=0.051, significant difference or a trend between injected and non-injected; mean ± SEM, n=4-6. (B) Representative images of TH and DAPI immunolabeling in the SNc injected with 2×107 TUs of the non-coding AAV2/6. AAV2/6-injected SNc had slightly lower TH levels than contralateral non-injected SNc. Bar: 1000 μm. (C) Evaluation of nigral DA cell death. Quantification of DAPI-positive (blue) nuclei in TH-positive (red) nigral cells resulted in the same number of nuclei/area in AAV2/6-injected as in non-injected SNc (p>0.05, unpaired two-tailed, Student’s t-test, t=0.3926, df=10, n=6). Abbreviations: AAV vector, adeno-associated viral vector; METH, methamphetamine.
Striatal TH levels did not differ between hemispheres in parkin-overexpressing rats; therefore, TH immunoreactivity was used as index of DAergic terminal integrity in the striatum. To determine whether parkin overexpression protects DAergic terminals against METH neurotoxicity, separate groups of rats were microinjected with 1×107 and 2×107 TUs of AAV2/6-parkin, treated with binge METH or saline 3 weeks later, and sacrificed a week after the drug administration. TH immunoreactivity measured in DAB-stained striatal slices did not differ between left and right striatum in saline controls (Fig.8A, a and c, Fig.8B). METH induced a 33% and 36% decrease in TH immunoreactivity in the right striatum in rats microinjected on the left side with 1×107 and 2×107 TUs of AAV2/6-parkin (n=5-6 rats/group), respectively. Parkin overexpression in the left striata dose-dependently and significantly attenuated METH-induced decreases in TH immunoreactivity in these rats, from −37% to −23% and from −42% to −19%, respectively, thus protecting 14% and 23% of total DAergic terminals (Fig.8B). The 1×107 and 2×107 AAV2/6-parkin-injected striata showed 20% and 47% higher TH levels than non-injected striata in METH-treated rats. When compared to non-coding 2×107 AAV2/6-injected METH-treated rats, 2×107 AAV2/6-parkin-injected METH-treated rats expressed 44% higher TH levels in the left striatum. Assessing TH immunoreactivity by other two immunochemistry methods produced similar results as those described above. Thus, by the second immunoperoxidase method, higher parkin dose protected 18%, whereas by the immunofluorescence method 22% of DAergic terminals. Regardless of the method of detection used; the extent of parkin protection differed from slice to slice, with the protection being more pronounced closer to the injection site.
Figure 8.

Overexpression of parkin in the substantia nigra pars compacta (SNc) protects dopaminergic (DAergic) nerve terminals in the striatum against METH toxicity. (A) Immunostaining for tyrosine hydroxylase (TH) in the striatum 1 week after saline or METH administration. Photomicrographs show representative coronal sections of the striatum of rats treated with 1×107 (a,b) or 2×107 (c,d) TUs of AAV2/6-parkin 3 weeks before saline (a,c) or METH (b,d) treatment. (B) Quantification of the TH-immunoreactive fibers in the striatum. The average optical density of TH-positive fibers in parkin-overexpressing striatum, as compared to the contralateral striatum, was significantly higher in the 1×107 parkin group (−37% vs. −23%) and 2×107 parkin group (−42% vs. −19%), suggesting that parkin overexpression in the left striatum protected 14% and 23% of total DAergic terminals (***p<0.001, **p<0.01, significant difference between saline and METH; #p<0.05, significant difference between injected and non-injected; mean ± SEM; n=5-6 rats/group; one-way ANOVA followed by the Student-Newman-Keuls post-hoc test: 1×107, F(3,18)=19.48, p<0.001; 2×107, F(3,18)=6.03, p<0.01). There was a significant main effect of treatment condition (saline vs. METH) (F(1,18)=59.70, p<0.001) but not of pre-treatment condition (AAV2/6-parkin injection vs. no injection) in 1×107 parkin group; however, there was a trend for treatment × pre-treatment interaction (F(1,18)=3.68, p=0.071) (two-way ANOVA with the Bonferroni post-hoc test). In 2×107 parkin group, there was a significant main effect of treatment condition (saline vs. METH) (F(1,18) = 12.50, p<0.01) and a trend for an effect in pre-treatment condition (AAV2/6-parkin injection vs. no injection) (F(1,18)=3.48, p=0.078) (two-way ANOVA with the Bonferroni post-hoc test). There was a significant main effect of pre-treatment as well as treatment condition when 1×107 and 2×107 parkin groups were compared: pre-treatment (1×107 vs. 2×107 TUs of AAV2/6-parkin, F(1,18)=5.94, p<0.05); treatment (saline vs. METH, F(1,18)=22.08, p<0.001) (two-way ANOVA with Bonferroni post-hoc test). Abbreviations: METH, methamphetamine; TUs, transducing units.
Core body temperature in parkin neuroprotection
METH causes hyperthermia which influences the degree of METH neurotoxicity with higher core body temperatures increasing the toxicity of the drug and vice versa (Bowyer, et al., 1994). On the other hand, parkin might play a role in thermoregulation in rodents (Itier, et al., 2003, Takamatsu, et al., 2011). To examine whether parkin overexpression influences basal body temperature or METH-induced hyperthermia, parkin overexpressing rats were treated with saline or METH together with AAV2/6-injected controls. Core body temperatures were taken at 1 hour after each saline and METH injection. As expected, binge METH administration caused significant hyperthermia. Parkin overexpression did not change core body temperature profile during saline or METH administration (n=6 rats/group) (Fig.9A).
Figure 9.

The effect of parkin overexpression on core body temperature and chymotrypsin-like activity of 20S proteasome in the striatum. (A) The effect of parkin overexpression on core body temperature. Rats were microinjected with the non-coding AAV2/6 (1×107 or 2×107 TUs) or AAV2/6-parkin (1×107 or 2×107 TUs) into the left SNc were treated with binge METH or saline (arrows). Core body temperatures were taken one hour after each injection. AAV2/6-parkin microinjections did not significantly reduce core body temperature either in METH-treated or saline-treated rats, as compared to corresponding AAV2/6-injected controls. *p<0.05; significant difference between METH-treated rats and corresponding saline controls (two-way ANOVA with repeated measures followed by Student-Newman-Keuls post-hoc test; F(5,30) =21.27, p<0.0001; mean ± SEM; n=6 rats/group). Arrows indicate times of METH injections. (B) The effect of parkin overexpression on chymotrypsin-like activity of the 20S proteasome. Rats were microinjected with 2×107 TUs of AAV2/6-parkin and sacrificed 3 weeks later. There was no difference in Vmax of chymotrypsin-like activity of 20S proteasome between synaptosomes from left and right striatum of parkin-overexpressing rats (p>0.05, unpaired two-tailed Student’s t-test, t=1.696, df=8, n=5). Data are expressed as mean ± SEM. Abbreviations: AMC, amido-4-methylcoumarin; AAV, adeno-associated virus; SAL, saline; METH, methamphetamine; TUs, transducing units.
The 20S proteasome in parkin neuroprotection
In vivo administration of binge METH causes progressive increase in the levels of oxidized proteins (Gluck, et al., 2001). Degradation of oxidized proteins requires increased levels of free 20S core particles of the proteasome (Wang, et al., 2010). To determine whether parkin neuroprotection might be exerted via increasing basal activity of 20S proteasome, we examined chymotrypsin-like activity of the 20S catalytic core in striatal synaptosomes from rats injected with 2×107 TUs parkin and sacrificed without the drug treatment. There was no difference in Vmax of chymotrypsin-like activity of 20S proteasome between synaptosomes from left and right striatum in parkin-overexpressing rats (n=5) (Fig.9B).
Discussion
The present study demonstrates that overexpression of rat parkin in the rat nigrostriatal DA system partially protects against METH-induced neurotoxicity as indicated by the attenuated loss of TH immunoreactivity in the parkin-overexpressing striatum. The neuroprotective effect of parkin does not involve decreasing METH-induced hyperthermia or increasing basal chymotrypsin-like activity of the 20S proteasome.
Parkin overexpression in the nigrostriatal system
In agreement with the previous studies (D’Agata, et al., 2000, D’Agata, et al., 2002, Horowitz, et al., 1999, Vercammen, et al., 2006) we detected very low levels of endogenous parkin in the SNc and striatum. For in vivo parkin overexpression we used pseudotype transfer vector AAV2/6 (the AAV serotype 2 genome packaged in the serotype 6 capsid). The AAV2/6 was shown to transduce the majority of nigral neurons in rats, resulting in a robust overexpression of the protein of interest (Azeredo da Silveira, et al., 2009, Ciron, et al., 2012, Dusonchet, et al., 2009). The transduction efficiency of AAV2/6 vector and levels of parkin overexpression in the present investigation were relatively high (Fig.2 and 3). Parkin-overexpressing nigral cells appeared in medium-to-dark brown as compared to light brown endogenous parkin-positive cells in the contralateral SN, suggesting that an uneven number of viral genomes actively expressed parkin in these cells. Variations between neurons in terms of transgene expression levels are very common. Parkin overexpression in the SNc was dose-dependent due to an increase in both the number of parkin-overexpressing cells and the levels of parkin expression in these cells (Fig.3). Co-localization of immunofluorescence for parkin and TH, a DAergic marker, demonstrated that DA neurons in the SNc, albeit not all, overexpressed parkin (Fig.4). Number of parkin overexpressing DA neurons decreased with the distance from the injection site (Fig.2D and 4C). Immunofluorescence and western blotting experiments showed that parkin was dose-dependently expressed also in striatal DAergic neuronal terminals (Fig.5), indicating that overexpressed parkin travelled by the anterograde transport to the striatum.
The levels of TH were not changed by the injection of 2×107 TUs of non-coding AAV2/6 vector when assessed by western blotting, whereas they were decreased when assessed by immunohistochemistry (Fig.6 and 7). This discrepancy can be explained by the fact that western blotting measured TH levels in the whole striatum, whereas immunohistochemistry measured TH levels in a number of collected striatal slices.
When assessed by immunohistochemistry, the injection of 2×107 TUs, but not 1×107 TUs, of non-coding AAV2/6 caused a mild decrease in TH immunoreactivity in the striatum and SNc (Fig.7A,B). Possibly, viral particles and protein contaminants in vector preparations caused a mild transient inflammation in the nigrostriatal system, resulting in decreased TH synthesis (Lang, et al., 2007, Mak and Cheung, 2007) but not neuronal cell death as a loss of TH-positive neurons in the SNc was not observed (Fig.7C). AAV vectors offer stable gene expression with little negative side effects such as inflammation (McCown, 2011). They can cause cell death only at very high concentrations, four orders of magnitude higher than concentrations used in the present study (Royo, et al., 2008). The decrease in TH levels triggered in the SNc by the higher dose of non-coding AAV2/6 vector is a likely reason for the decreases in striatal TH levels observed in saline and METH-treated rats. The finding of no difference in TH levels between non-injected and 2×107 AAV2/6-parkin-microinjected striatum (Fig.8) suggests that parkin overexpression slightly increases striatal TH levels. This agrees with a previous finding (Manfredsson, et al., 2007). Potentially, parkin also decreases contaminant-induced inflammation (Khandelwal, et al., 2010). Since striatal TH levels did not differ between hemispheres in parkin overexpressing rats prior to METH administration (Fig.6A), TH was used as a marker of DAergic terminal integrity and, therefore, as a marker of METH toxicity to these terminals.
Parkin neuroprotection against METH neurotoxicity
A deficit in parkin function in DA neurons leads to their neurodegeneration (Buneeva and Medvedev, 2006, Schiesling, et al., 2008). Conversely, overexpression of parkin protects DA neurons against a variety of cellular insults, including factors that mediate METH toxicity such as DA-induced oxidative stress (Jiang, et al., 2004), proteasome inhibiton-induced apoptosis (Yang, et al., 2005), excitotoxicity (Staropoli, et al., 2003), and agents affecting mitochondrial function (Darios, et al., 2003). In vivo, parkin protects DA neuronal cell bodies in the SNc of rodents from a variety of insults including 6-hydroxydopamine (Manfredsson, et al., 2007, Vercammen, et al., 2006), MPTP (Paterna, et al., 2007), and overexpression of proteins that self-aggregates in specific neurodegenerative disease, such as alpha-synuclein (Lo Bianco, et al., 2004, Yamada, et al., 2005, Yang, et al., 2003) and tau (Klein, et al., 2006). The protection of DAergic terminals by parkin overexpression in the SNc was demonstrated after administration of 6-hydroxydopamine (Vercammen, et al., 2006) and in α-synuclein-induced neuropathology (Lo Bianco, et al., 2004). The present study demonstrated that overexpression of parkin attenuated METH-induced decreases in striatal TH immunoreactivity (Fig.8) thus indicating that parkin overexpression can protect DAergic terminals against METH neurotoxicity in rat striatum. This finding not only supports existing evidence for parkin neuroprotective qualities but also provides evidence that parkin has an ability to protect DAergic terminals in the absence of damage to the SNc, thus suggesting that parkin protective activity might have taken place in these terminals. Parkin-mediated neuroprotection observed in the present study was partial (Fig.8), which was an expected result. METH neurotoxicity is mediated by a variety of molecular mechanisms, some of which likely do not involve parkin (Yamamoto, et al., 2010). The partial protection could be also due to the fact that the extent of parkin-mediated neuroprotection in the striatum differed from slice to slice, resulting in partial average protection. The striatal regions without protection tended to be at the far rostral or caudal regions of the striatum, i.e. regions harboring terminals of nigral DA cell bodies which the transgene did not reach (neurons away from the microinjection site) (Fig.2 and 4). Recombinant AAV2 and AAV6 vectors primarily transduce neurons and are very effective in the SN (Ciron, et al., 2012, Dusonchet, et al., 2009, Kaplitt, et al., 1994, Klein, et al., 1998, Wang, et al., 2011); however, their limitation is a relatively small volume of transduction (Markakis, et al., 2010, Nguyen, et al., 2001). As aforementioned, variations between neurons in terms of transgene expression levels are very common. Variations in parkin overexpression between nigral neurons observed in our study and lack of parkin expression in some of them (Fig. 3 and 4) both could contribute to partial parkin protection against METH in the striatum. One more confounding factor to be considered is the TH-decreasing effect of the 2×107 TUs dose of the vector. With this dose, but not with 1×107 TUs of AAV2/6-parkin, parkin might protect against both METH toxic effects and contaminant-triggered inflammation. The increase in TH immunoreactivity observed with the lower parkin overexpression was solely in response to METH.
Our present finding of parkin protection from METH together with our previous finding of METH-induced parkin deficit (Moszczynska and Yamamoto, 2011) suggest that parkin deficit mediates, in part, toxicity of METH to striatal DAergic terminals. In contrast, the study of Perez et al. (Perez, et al., 2005) found no difference between wt and parkin KO mice in terms of sensitivity to METH toxicity, suggesting no role for parkin in mediating toxicity of the drug. It is possible that one of many other RING E3 ligases functioning in neurons takes over parkin functions in case of its deficit. This notion is supported by findings from parkin knock-out mice of nigrostriatal deficits without a loss of DA neurons (Goldberg, et al., 2003, Itier, et al., 2003). Alternatively, the results are not in agreement due to different METH regimens. The study of Perez et al. used single medium dose of METH that cause moderate toxicity to DAergic terminals, whereas our study used binge high-dose METH administration.
Hyperthermia is an important factor in neurotoxicity of amphetamines (Bowyer, et al., 1994, Yamamoto, et al., 2010). Parkin may play a role in temperature regulation in amphetamine-exposed rodents. Firstly, parkin KO mice have decreased body temperature (Itier, et al., 2003), whereas 3,4-Methylenedioxymethamphetamine (MDMA)-exposed parkin KO mice show enhanced hyperthermia (Takamatsu, et al., 2011). Secondly, both temperature and parkin function are regulated by DA D1 receptor (Ares-Santos, et al., 2012, Berthet, et al., 2012) and, therefore, they might be functionally connected. Overexpression of parkin in the nigrostriatal system did not lower either basal core body temperature or METH-induced hyperthermia (Fig.9A), thus eliminating temperature regulation as parkin-mediated protective mechanism in METH neurotoxicity.
In vivo administration of binge METH causes progressive increase in the levels of oxidized proteins (Gluck, et al., 2001). Degradation of oxidized proteins requires increased activity of 20S proteasome (Grune, et al., 1997, Wang, et al., 2010). We have previously found that binge METH causes deficit in parkin and 26S proteasome function, accompanied by increased activity of 20S proteasome (Moszczynska and Yamamoto, 2011), likely an adaptational response to the METH-induced increase in oxidized proteins. Parkin deficit can decrease 20S activity whereas parkin overexpression can augment it (Hyun, et al., 2002, Wang, et al., 2005). Therefore, the increase in 20S activity observed in our previous study could have been, potentially, insufficient to dispose of the neurotoxic surplus of oxidized proteins. We found no difference in activity of 20S proteasome between METH-exposed synaptosomes from left and right striata of parkin-overexpressing rats (Fig.9B), suggesting that parkin overexpression does not protect DAergic terminals via increasing 20S function. Whether or not the parkin protection is mediated via increasing function of 26S proteasome is currently under investigation.
Stereology techniques were not employed in the quantifications as the goal of our study was to qualitatively, not quantitatively, relate the level parkin overexpression to the level of protection against METH. We asked a general question whether parkin has a potential to protect DAergic terminals from METH by comparing experimental groups. We intentionally chose for some analyses slices close to the injection site where parkin overexpression was the highest. Otherwise, tissue sampling and processing was aimed to reduced bias. Consequently even though the results obtained in the present study are qualitative, they came close to an unbiased estimate and answered our research question.
In conclusion, the presented results suggest that parkin overexpression attenuates METH-induced neurotoxicity in vivo. These results together with our previous findings of decreased striatal parkin shortly after METH administration suggest that a deficit in parkin function mediates, in part, the toxicity of METH to striatal DAergic terminals and that parkin has the potential to be a target in therapeutic strategies to mitigate the harmful effects of acute METH overdose as well as the effects of chronic METH on the CNS in human users of the drug.
Highlights.
Rat parkin was successfully overexpressed in the nigrostriatal dopamine system in adult rats using adeno-associated viral transfer vector.
Parkin overexpression protected dopaminergic terminals in the nigrostriatal system from neurodegeneration triggered by high doses of binge methamphetamine
Parkin neuroprotection did not involve decreasing core body temperature or methamphetamine-induced hyperthermia
Parkin protection did not involve upregulation of the 20S proteasome.
The results from the study indicate that parkin might be a drug target in protection from methamphetamine overdose and abuse.
Acknowledgements
This work was supported by an NIH grant DA023085. We are grateful to Dr. Patrick Aebischer (Brain Mind Institute, Ecole Polytechnique Federale de Lausanne, Lausanne, Switzerland) for generously providing non-codining AAV2/6 and AAV2/6-parkin vectors. Thanks to Dr. Bryan K. Yamamoto (University of Toledo School of Medicine, Toledo, OH, USA ) for initial consultation on the project and Derek McCrory for technical assistance.
Abbreviations used
- AAV
adeno-associated virus
- AMC
amido-4-methylcoumarin
- AP
anterior-posterior
- CNS
central nervous system
- DA
dopamine
- DAB
diaminobenzidine
- METH
methamphetamine
- ML
medio-lateral
- SAL
saline
- SNc
substantia nigra pars compacta
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- Suc-LLVY-AMC
Suc-Leu-Leu-Val-Tyr-7-amido-4-methylcoumarin
- TH
tyrosine hydroxylase
- TU
transducing unit
- V
ventral
- Vmax
maximal velocity
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ares-Santos S, Granado N, Oliva I, O’Shea E, Martin ED, Colado MI, Moratalla R. Dopamine D(1) receptor deletion strongly reduces neurotoxic effects of methamphetamine. Neurobiol Dis. 2012;45:810–820. doi: 10.1016/j.nbd.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 2.Azeredo da Silveira S, Schneider BL, Cifuentes-Diaz C, Sage D, Abbas-Terki T, Iwatsubo T, Unser M, Aebischer P. Phosphorylation does not prompt, nor prevent, the formation of alpha-synuclein toxic species in a rat model of Parkinson’s disease. Hum Mol Genet. 2009;18:872–887. doi: 10.1093/hmg/ddn417. [DOI] [PubMed] [Google Scholar]
- 3.Berthet A, Bezard E, Porras G, Fasano S, Barroso-Chinea P, Dehay B, Martinez A, Thiolat ML, Nosten-Bertrand M, Giros B, Baufreton J, Li Q, Bloch B, Martin-Negrier ML. L-DOPA impairs proteasome activity in parkinsonism through D1 dopamine receptor. J Neurosci. 2012;32:681–691. doi: 10.1523/JNEUROSCI.1541-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bowyer JF, Davies DL, Schmued L, Broening HW, Newport GD, Slikker W, Jr., Holson RR. Further studies of the role of hyperthermia in methamphetamine neurotoxicity. J Pharmacol Exp Ther. 1994;268:1571–1580. [PubMed] [Google Scholar]
- 5.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 6.Broening HW, Pu C, Vorhees CV. Methamphetamine selectively damages dopaminergic innervation to the nucleus accumbens core while sparing the shell. Synapse. 1997;27:153–160. doi: 10.1002/(SICI)1098-2396(199710)27:2<153::AID-SYN6>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 7.Bulteau AL, Lundberg KC, Humphries KM, Sadek HA, Szweda PA, Friguet B, Szweda LI. Oxidative modification and inactivation of the proteasome during coronary occlusion/reperfusion. J Biol Chem. 2001;276:30057–30063. doi: 10.1074/jbc.M100142200. [DOI] [PubMed] [Google Scholar]
- 8.Buneeva OA, Medvedev AE. Ubiquitin-protein ligase parkin and its role in the development of Parkinson’s disease. Biochemistry (Mosc) 2006;71:851–860. doi: 10.1134/s0006297906080050. [DOI] [PubMed] [Google Scholar]
- 9.Callaghan RC, Cunningham JK, Sajeev G, Kish SJ. Incidence of Parkinson’s disease among hospital patients with methamphetamine-use disorders. Mov Disord. 2010;25:2333–2339. doi: 10.1002/mds.23263. [DOI] [PubMed] [Google Scholar]
- 10.Callaghan RC, Cunningham JK, Sykes J, Kish SJ. Increased risk of Parkinson’s disease in individuals hospitalized with conditions related to the use of methamphetamine or other amphetamine-type drugs. Drug Alcohol Depend. 2011 doi: 10.1016/j.drugalcdep.2011.06.013. [DOI] [PubMed] [Google Scholar]
- 11.Cass WA. Decreases in evoked overflow of dopamine in rat striatum after neurotoxic doses of methamphetamine. J Pharmacol Exp Ther. 1997;280:105–113. [PubMed] [Google Scholar]
- 12.Ciron C, Lengacher S, Dusonchet J, Aebischer P, Schneider BL. Sustained expression of PGC-1alpha in the rat nigrostriatal system selectively impairs dopaminergic function. Hum Mol Genet. 2012;21:1861–1876. doi: 10.1093/hmg/ddr618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D’Agata V, Zhao W, Cavallaro S. Cloning and distribution of the rat parkin mRNA. Brain Res Mol Brain Res. 2000;75:345–349. doi: 10.1016/s0169-328x(99)00286-7. [DOI] [PubMed] [Google Scholar]
- 14.D’Agata V, Zhao W, Pascale A, Zohar O, Scapagnini G, Cavallaro S. Distribution of parkin in the adult rat brain. Prog Neuropsychopharmacol Biol Psychiatry. 2002;26:519–527. doi: 10.1016/s0278-5846(01)00301-3. [DOI] [PubMed] [Google Scholar]
- 15.Darios F, Corti O, Lucking CB, Hampe C, Muriel MP, Abbas N, Gu WJ, Hirsch EC, Rooney T, Ruberg M, Brice A. Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Hum Mol Genet. 2003;12:517–526. doi: 10.1093/hmg/ddg044. [DOI] [PubMed] [Google Scholar]
- 16.Dusonchet J, Bensadoun JC, Schneider BL, Aebischer P. Targeted overexpression of the parkin substrate Pael-R in the nigrostriatal system of adult rats to model Parkinson’s disease. Neurobiol Dis. 2009;35:32–41. doi: 10.1016/j.nbd.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 17.Gluck MR, Moy LY, Jayatilleke E, Hogan KA, Manzino L, Sonsalla PK. Parallel increases in lipid and protein oxidative markers in several mouse brain regions after methamphetamine treatment. J Neurochem. 2001;79:152–160. doi: 10.1046/j.1471-4159.2001.00549.x. [DOI] [PubMed] [Google Scholar]
- 18.Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, Gajendiran M, Roth BL, Chesselet MF, Maidment NT, Levine MS, Shen J. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem. 2003;278:43628–43635. doi: 10.1074/jbc.M308947200. [DOI] [PubMed] [Google Scholar]
- 19.Grune T, Reinheckel T, Davies KJ. Degradation of oxidized proteins in mammalian cells. FASEB J. 1997;11:526–534. [PubMed] [Google Scholar]
- 20.Harvey BK, Chou J, Shen H, Hoffer BJ, Wang Y. Diadenosine tetraphosphate reduces toxicity caused by high-dose methamphetamine administration. Neurotoxicology. 2009;30:436–444. doi: 10.1016/j.neuro.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haughey HM, Fleckenstein AE, Hanson GR. Differential regional effects of methamphetamine on the activities of tryptophan and tyrosine hydroxylase. J Neurochem. 1999;72:661–668. doi: 10.1046/j.1471-4159.1999.0720661.x. [DOI] [PubMed] [Google Scholar]
- 22.Horowitz JM, Myers J, Stachowiak MK, Torres G. Identification and distribution of Parkin in rat brain. Neuroreport. 1999;10:3393–3397. doi: 10.1097/00001756-199911080-00025. [DOI] [PubMed] [Google Scholar]
- 23.Hotchkiss AJ, Gibb JW. Long-term effects of multiple doses of methamphetamine on tryptophan hydroxylase and tyrosine hydroxylase activity in rat brain. J Pharmacol Exp Ther. 1980;214:257–262. [PubMed] [Google Scholar]
- 24.Hyun DH, Lee M, Hattori N, Kubo S, Mizuno Y, Halliwell B, Jenner P. Effect of wild-type or mutant Parkin on oxidative damage, nitric oxide, antioxidant defenses, and the proteasome. J Biol Chem. 2002;277:28572–28577. doi: 10.1074/jbc.M200666200. [DOI] [PubMed] [Google Scholar]
- 25.Itier JM, Ibanez P, Mena MA, Abbas N, Cohen-Salmon C, Bohme GA, Laville M, Pratt J, Corti O, Pradier L, Ret G, Joubert C, Periquet M, Araujo F, Negroni J, Casarejos MJ, Canals S, Solano R, Serrano A, Gallego E, Sanchez M, Denefle P, Benavides J, Tremp G, Rooney TA, Brice A, Garcia de Yebenes J. Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum Mol Genet. 2003;12:2277–2291. doi: 10.1093/hmg/ddg239. [DOI] [PubMed] [Google Scholar]
- 26.Jiang H, Ren Y, Yuen EY, Zhong P, Ghaedi M, Hu Z, Azabdaftari G, Nakaso K, Yan Z, Feng J. Parkin controls dopamine utilization in human midbrain dopaminergic neurons derived from induced pluripotent stem cells. Nat Commun. 2012;3:668. doi: 10.1038/ncomms1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang H, Ren Y, Zhao J, Feng J. Parkin protects human dopaminergic neuroblastoma cells against dopamine-induced apoptosis. Hum Mol Genet. 2004;13:1745–1754. doi: 10.1093/hmg/ddh180. [DOI] [PubMed] [Google Scholar]
- 28.Kaplitt MG, Leone P, Samulski RJ, Xiao X, Pfaff DW, O’Malley KL, During MJ. Long-term gene expression and phenotypic correction using adeno-associated virus vectors in the mammalian brain. Nat Genet. 1994;8:148–154. doi: 10.1038/ng1094-148. [DOI] [PubMed] [Google Scholar]
- 29.Khandelwal PJ, Dumanis SB, Feng LR, Maguire-Zeiss K, Rebeck G, Lashuel HA, Moussa CE. Parkinson-related parkin reduces alpha-Synuclein phosphorylation in a gene transfer model. Mol Neurodegener. 2010;5:47. doi: 10.1186/1750-1326-5-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kirik D, Bjorklund A. Parkinson’s disease: viral vector delivery of parkin generates model results in rats. Gene Ther. 2005;12:727–729. doi: 10.1038/sj.gt.3302497. [DOI] [PubMed] [Google Scholar]
- 31.Klein RL, Dayton RD, Henderson KM, Petrucelli L. Parkin is protective for substantia nigra dopamine neurons in a tau gene transfer neurodegeneration model. Neurosci Lett. 2006;401:130–135. doi: 10.1016/j.neulet.2006.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klein RL, Meyer EM, Peel AL, Zolotukhin S, Meyers C, Muzyczka N, King MA. Neuron-specific transduction in the rat septohippocampal or nigrostriatal pathway by recombinant adeno-associated virus vectors. Exp Neurol. 1998;150:183–194. doi: 10.1006/exnr.1997.6736. [DOI] [PubMed] [Google Scholar]
- 33.Lang CH, Frost RA, Vary TC. Regulation of muscle protein synthesis during sepsis and inflammation. Am J Physiol Endocrinol Metab. 2007;293:E453–459. doi: 10.1152/ajpendo.00204.2007. [DOI] [PubMed] [Google Scholar]
- 34.Lim KL. Ubiquitin-proteasome system dysfunction in Parkinson’s disease: current evidence and controversies. Expert Rev Proteomics. 2007;4:769–781. doi: 10.1586/14789450.4.6.769. [DOI] [PubMed] [Google Scholar]
- 35.Lo Bianco C, Schneider BL, Bauer M, Sajadi A, Brice A, Iwatsubo T, Aebischer P. Lentiviral vector delivery of parkin prevents dopaminergic degeneration in an alpha-synuclein rat model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2004;101:17510–17515. doi: 10.1073/pnas.0405313101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mak RH, Cheung W. Cachexia in chronic kidney disease: role of inflammation and neuropeptide signaling. Curr Opin Nephrol Hypertens. 2007;16:27–31. doi: 10.1097/MNH.0b013e3280117ce7. [DOI] [PubMed] [Google Scholar]
- 37.Manfredsson FP, Burger C, Sullivan LF, Muzyczka N, Lewin AS, Mandel RJ. rAAV-mediated nigral human parkin over-expression partially ameliorates motor deficits via enhanced dopamine neurotransmission in a rat model of Parkinson’s disease. Exp Neurol. 2007;207:289–301. doi: 10.1016/j.expneurol.2007.06.019. [DOI] [PubMed] [Google Scholar]
- 38.Markakis EA, Vives KP, Bober J, Leichtle S, Leranth C, Beecham J, Elsworth JD, Roth RH, Samulski RJ, Redmond DE., Jr. Comparative transduction efficiency of AAV vector serotypes 1-6 in the substantia nigra and striatum of the primate brain. Mol Ther. 2010;18:588–593. doi: 10.1038/mt.2009.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCown TJ. Adeno-Associated Virus (AAV) Vectors in the CNS. Curr Gene Ther. 2011;11:181–188. doi: 10.2174/156652311795684759. [DOI] [PubMed] [Google Scholar]
- 40.Moore DJ. Parkin: a multifaceted ubiquitin ligase. Biochem Soc Trans. 2006;34:749–753. doi: 10.1042/BST0340749. [DOI] [PubMed] [Google Scholar]
- 41.Morgan ME, Gibb JW. Short-term and long-term effects of methamphetamine on biogenic amine metabolism in extra-striatal dopaminergic nuclei. Neuropharmacology. 1980;19:989–995. doi: 10.1016/0028-3908(80)90010-6. [DOI] [PubMed] [Google Scholar]
- 42.Moszczynska A, Yamamoto BK. Methamphetamine oxidatively damages parkin and decreases the activity of 26S proteasome in vivo. J Neurochem. 2011;116:1005–1017. doi: 10.1111/j.1471-4159.2010.07147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nguyen JB, Sanchez-Pernaute R, Cunningham J, Bankiewicz KS. Convection-enhanced delivery of AAV-2 combined with heparin increases TK gene transfer in the rat brain. Neuroreport. 2001;12:1961–1964. doi: 10.1097/00001756-200107030-00037. [DOI] [PubMed] [Google Scholar]
- 44.Oluwatosin-Chigbu Y, Robbins A, Scott CW, Arriza JL, Reid JD, Zysk JR. Parkin suppresses wild-type alpha-synuclein-induced toxicity in SHSY-5Y cells. Biochem Biophys Res Commun. 2003;309:679–684. doi: 10.1016/j.bbrc.2003.08.059. [DOI] [PubMed] [Google Scholar]
- 45.Olzmann JA, Li L, Chin LS. Aggresome formation and neurodegenerative diseases: therapeutic implications. Curr Med Chem. 2008;15:47–60. doi: 10.2174/092986708783330692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paterna JC, Leng A, Weber E, Feldon J, Bueler H. DJ-1 and Parkin modulate dopamine-dependent behavior and inhibit MPTP-induced nigral dopamine neuron loss in mice. Mol Ther. 2007;15:698–704. doi: 10.1038/sj.mt.6300067. [DOI] [PubMed] [Google Scholar]
- 47.Perez FA, Curtis WR, Palmiter RD. Parkin-deficient mice are not more sensitive to 6-hydroxydopamine or methamphetamine neurotoxicity. BMC Neurosci. 2005;6:71. doi: 10.1186/1471-2202-6-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petrucelli L, O’Farrell C, Lockhart PJ, Baptista M, Kehoe K, Vink L, Choi P, Wolozin B, Farrer M, Hardy J, Cookson MR. Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron. 2002;36:1007–1019. doi: 10.1016/s0896-6273(02)01125-x. [DOI] [PubMed] [Google Scholar]
- 49.Ricaurte GA, Guillery RW, Seiden LS, Schuster CR, Moore RY. Dopamine nerve terminal degeneration produced by high doses of methylamphetamine in the rat brain. Brain Res. 1982;235:93–103. doi: 10.1016/0006-8993(82)90198-6. [DOI] [PubMed] [Google Scholar]
- 50.Riddle EL, Topham MK, Haycock JW, Hanson GR, Fleckenstein AE. Differential trafficking of the vesicular monoamine transporter-2 by methamphetamine and cocaine. Eur J Pharmacol. 2002;449:71–74. doi: 10.1016/s0014-2999(02)01985-4. [DOI] [PubMed] [Google Scholar]
- 51.Royo NC, Vandenberghe LH, Ma JY, Hauspurg A, Yu L, Maronski M, Johnston J, Dichter MA, Wilson JM, Watson DJ. Specific AAV serotypes stably transduce primary hippocampal and cortical cultures with high efficiency and low toxicity. Brain Res. 2008;1190:15–22. doi: 10.1016/j.brainres.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schiesling C, Kieper N, Seidel K, Kruger R. Review: Familial Parkinson’s disease--genetics, clinical phenotype and neuropathology in relation to the common sporadic form of the disease. Neuropathol Appl Neurobiol. 2008;34:255–271. doi: 10.1111/j.1365-2990.2008.00952.x. [DOI] [PubMed] [Google Scholar]
- 53.Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- 54.Staropoli JF, McDermott C, Martinat C, Schulman B, Demireva E, Abeliovich A. Parkin is a component of an SCF-like ubiquitin ligase complex and protects postmitotic neurons from kainate excitotoxicity. Neuron. 2003;37:735–749. doi: 10.1016/s0896-6273(03)00084-9. [DOI] [PubMed] [Google Scholar]
- 55.Takamatsu Y, Shiotsuki H, Kasai S, Sato S, Iwamura T, Hattori N, Ikeda K. Enhanced Hyperthermia Induced by MDMA in Parkin Knockout Mice. Curr Neuropharmacol. 2011;9:96–99. doi: 10.2174/157015911795016985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tata DA, Raudensky J, Yamamoto BK. Augmentation of methamphetamine-induced toxicity in the rat striatum by unpredictable stress: contribution of enhanced hyperthermia. Eur J Neurosci. 2007;26:739–748. doi: 10.1111/j.1460-9568.2007.05688.x. [DOI] [PubMed] [Google Scholar]
- 57.Vercammen L, Van der Perren A, Vaudano E, Gijsbers R, Debyser Z, Van den Haute C, Baekelandt V. Parkin protects against neurotoxicity in the 6-hydroxydopamine rat model for Parkinson’s disease. Mol Ther. 2006;14:716–723. doi: 10.1016/j.ymthe.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 58.Wang C, Ko HS, Thomas B, Tsang F, Chew KC, Tay SP, Ho MW, Lim TM, Soong TW, Pletnikova O, Troncoso J, Dawson VL, Dawson TM, Lim KL. Stress-induced alterations in parkin solubility promote parkin aggregation and compromise parkin’s protective function. Hum Mol Genet. 2005;14:3885–3897. doi: 10.1093/hmg/ddi413. [DOI] [PubMed] [Google Scholar]
- 59.Wang J, Faust SM, Rabinowitz JE. The next step in gene delivery: molecular engineering of adeno-associated virus serotypes. J Mol Cell Cardiol. 2011;50:793–802. doi: 10.1016/j.yjmcc.2010.10.017. [DOI] [PubMed] [Google Scholar]
- 60.Wang X, Yen J, Kaiser P, Huang L. Regulation of the 26S proteasome complex during oxidative stress. Sci Signal. 2010;3:ra88. doi: 10.1126/scisignal.2001232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yamada M, Mizuno Y, Mochizuki H. Parkin gene therapy for alpha-synucleinopathy: a rat model of Parkinson’s disease. Hum Gene Ther. 2005;16:262–270. doi: 10.1089/hum.2005.16.262. [DOI] [PubMed] [Google Scholar]
- 62.Yamamoto BK, Moszczynska A, Gudelsky GA. Amphetamine toxicities: classical and emerging mechanisms. Ann N Y Acad Sci. 2010;1187:101–121. doi: 10.1111/j.1749-6632.2009.05141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang H, Zhou HY, Li B, Chen SD. Neuroprotection of Parkin against apoptosis is independent of inclusion body formation. Neuroreport. 2005;16:1117–1121. doi: 10.1097/00001756-200507130-00017. [DOI] [PubMed] [Google Scholar]
- 64.Yang H, Zhou X, Liu X, Yang L, Chen Q, Zhao D, Zuo J, Liu W. Mitochondrial dysfunction induced by knockdown of mortalin is rescued by Parkin. Biochem Biophys Res Commun. 2011;410:114–120. doi: 10.1016/j.bbrc.2011.05.116. [DOI] [PubMed] [Google Scholar]
- 65.Yang Y, Nishimura I, Imai Y, Takahashi R, Lu B. Parkin suppresses dopaminergic neuron-selective neurotoxicity induced by Pael-R in Drosophila. Neuron. 2003;37:911–924. doi: 10.1016/s0896-6273(03)00143-0. [DOI] [PubMed] [Google Scholar]
- 66.Zhang Y, Gao J, Chung KK, Huang H, Dawson VL, Dawson TM. Parkin functions as an E2-dependent ubiquitin-protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci U S A. 2000;97:13354–13359. doi: 10.1073/pnas.240347797. [DOI] [PMC free article] [PubMed] [Google Scholar]