

Abstract
Recent work has identified nucleotide agonists selective for P2Y1, P2Y2 and P2Y6 receptors and nucleotide antagonists selective for P2Y1, P2Y12 and P2X1 receptors. Selective non-nucleotide antagonists have been reported for P2Y1, P2Y2, P2Y6, P2Y12, P2Y13, P2X2/3/P2X3 and P2X7 receptors. For example, the dinucleotide INS 37217 (Up4dC) potently activates the P2Y2 receptor, and the non-nucleotide antagonist A-317491 is selective for P2X2/3/P2X3 receptors. Nucleotide analogues in which the ribose moiety is substituted by a variety of novel ring systems, including conformation-ally locked moieties, have been synthesized as ligands for P2Y receptors. The focus on conformational factors of the ribose-like moiety allows the inclusion of general modifications that lead to enhanced potency and selectivity. At P2Y1,2,4,11 receptors, there is a preference for the North conformation as indicated with (N)-methanocarba analogues. The P2Y1 antagonist MRS2500 inhibited ADP-induced human platelet aggregation with an IC50 of 0.95 nM. MRS2365, an (N)-methanocarba analogue of 2-MeSADP, displayed potency (EC50) of 0.4 nM at the P2Y1 receptor, with >10 000-fold selectivity in comparison to P2Y12 and P2Y13 receptors. At P2Y6 receptors there is a dramatic preference for the South conformation. Three-dimensional structures of P2Y receptors have been deduced from structure activity relationships (SAR), mutagenesis and modelling studies. Detailed three-dimensional structures of P2X receptors have not yet been proposed.
Extracellular purine and pyrimidine nucleotides act as neurotransmitters/modulators (Jacobson et al 2002). These ubiquitous signalling molecules modulate the function of diverse mammalian cell types and tissues under both normal and pathophysiological conditions. Receptors for extracellular nucleotides have been characterized through medicinal chemical, molecular biological, and pharmacological approaches. The eight subtypes of P2Y receptors, denoted P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, P2Y13 and P2Y14, are all seven transmembrane-spanning (7TM) receptors, which couple to G proteins. The seven P2X receptor subunits (P2X1–P2X7) form multimeric ligand-gated ion channels. The distribution of P2Y receptors is broad, and the relevant therapeutic interests include antithrombotic therapy, modulation of the immune system and cardiovascular system, and treatment of cystic fibrosis and other pulmonary diseases (Yerxa et al 2002). Several of the P2Y receptors have been linked to either induction/suppression of apoptosis or hypoxic stress and proliferation (Kim et al 2003a, Yitzhaki et al 2005, Coutinho-Silva et al 2005).
Ligand development at the P2 receptors has, in general, proceeded more slowly than at other 7TM receptors. Nevertheless, there are now definitive pharmacological probes available for characterizing some of the P2 receptor subtypes (Jacobson et al 2004). Decision trees for use of selective or partially selective agonists and antagonists for the initial pharmacological characterization of P2Y receptors (Fig. 1) and homomultimeric P2X receptors (Fig. 2) are presented. Most of the non-selective ligand probes must be used cautiously given the limitations of low potency and stability, mixed selectivity, and the tendency of P2 antagonists to inhibit ecto-nucleotidases, thus increasing the concentration of available nucleotide agonist. The presence of heteromultimeric P2X receptors that display unique pharmacology complicates ligand development (Khakh et al 2001). Recent work has identified agonists selective for P2Y1, P2Y2 and P2Y6 receptors and antagonists selective for P2Y1, P2Y2, P2Y12, P2Y13, P2X1, P2X2/3/P2X3 and P2X7 receptors based on studies of structure–activity relationships (SARs).
FIG. 1.

Progressive use of agonist (grey ovals) and antagonist (black rectangles) ligands for defining subtypes of human (or rat, when indicated) P2Y receptors in pharmacological experiments, as condensed from recent literature. The most general set of agonist ligands (corresponding to the endogenous agonists) appears in the upper row, and below are more specialized receptor probes. Each among the agonists ADP, ATP and UTP activates multiple P2Y receptor subtypes (open ovals). Antagonists of the P2Y14 receptor have not yet been reported. Abbreviations: AR-C67085MX, 2-(propylthio)-β,γ-dichloromethylene-ATP; MRS2179, N6-methyl-2′-deoxyadenosine-3′,5′-bisphosphate; RB2, Reactive blue 2. Note: Ap4A also activates various P2X receptors and the rat P2Y4 receptor (Wildman et al 2003), but not the human P2Y4 receptor (Shaver et al 2005). Suramin is known to inhibit G proteins and other intracellular targets. The selectivity of AZD6140 in comparison to the P2Y13 receptor remains to be established. MRS 2578 is an insurmountable antagonist, which is of limited use due to its reactivity and hydrophobicity (Mamedova et al 2004).
FIG. 2.

Progressive use of agonist (grey ovals) and antagonist (black rectangles) ligands for defining homomultimeric subtypes of P2X ion channels (except the P2X6 receptor, which functions as a heteromultimer) in pharmacological experiments, as condensed from recent literature. A potentiating ligand is also shown for the P2X4 receptor. The four most generally useful agonist ligands appear in the upper row, and below are more specialized receptor probes. Agonists other than ATP only partially activate the P2X4 receptor. The pEC50 or pIC50 value for human (or rat, when indicated) is provided. Heteromultimeric P2X ion channels may display unique pharmacology, thus this scheme is to be used cautiously. NE, not effective; BBG, Brilliant blue G; RB2, Reactive blue 2; PAPET-ATP, 2-[2-(p-aminophenyl)ethylthio]-ATP; TNP-ATP, 2′,3′-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate. Note: Bz-ATP (2′,3′-O-[4-benzoyl-benzoyl]-ATP) is more potent at the P2X1 than at the P2X7 receptor. Suramin is known to inhibit G-proteins and other intracellular targets. At P2X1 and P2X3 receptors desensitization is rapid, thus, an agonist may appear to act as an antagonist. Agonist SARs at the P2X2 receptor (Spelta et al 2003), P2X1 and P2X3 receptors (Bianchi et al 1999), P2X4 receptor (Garcia-Guzman et al 1997, Jarvis et al 2004), P2X5 receptor (Wildman et al 2002, Bo et al 2003), P2X6 receptor (Jones et al 2003) and P2X7 receptor (Baraldi et al 2004) have been characterized. An uncharged, selective P2X1 antagonist was recently reported (Jaime-Figueroa et al 2005).
A variety of analogues of native P2 agonists (ATP, ADP, UTP, UDP and UDP-glucose) have provided subtype selectivity. For example, UTP-γ-S and UDP-β-S are selective agonists for P2Y2/P2Y4 and P2Y6 receptors, respectively (Malmsjö et al 2000). These thiophosphate derivatives may be prepared using enzymatic methods (Lazarowski et al 1996). The dinucleotide INS 37217 (Up4dC) potently activates the P2Y2 receptor, and is less prone to enzymatic hydrolysis than naturally-occurring dinucleotide agonists (Yerxa et al 2002).
Use of ring constraints to define the conformational preferences of nucleotides at P2Y receptors
We have synthesized nucleotide analogues containing novel ring systems as ligands for P2 receptors (Ohno et al 2004). The focus on conformational factors of the ribose or ribose-like moiety allows introduction of general modifications that lead to enhanced potency and selectivity at certain P2Y subtypes. We combine these ribose modifications with exploration of structure–activity relationships merging known enhancing modifications at other sites on the molecules.
In solution, the ribose ring of an unbound nucleotide may exist in a dynamic equilibrium between (N) (North; 2′-exo/3′-endo) and (S) (South; 2′-endo/3′-exo) conformations, and X-ray crystallographic structures of diverse nucleotide complexes through nature indicate a clustering around these conformations. It is possible to stabilize each of these conformational clusters by chemical bridging within a ring. Replacement of the ribose moiety of ATP with a ‘methanocarba’ ring system, i.e. fused cyclopropane and cyclopentane rings, locks the analogue in either a (N) or (S) conformation, depending on the position of the –CH2–bridge. These isomeric variants based on the bicyclo[3.1.0]hexane ring system produce agonists having widely differing activities at P2 receptors (Kim et al 2002). In the (N) conformation (e.g. ATP analogue MRS2340) there was a dramatic increase in potency at the P2Y1 receptor. Curiously, at the hP2Y4 receptor this compound had weak agonist activity, in contrast to ATP, which is an antagonist at this receptor. Constraining the pseudoribose ring in the (S) conformation in the racemic ATP analogue MRS2312 resulted in a decrease in potency at the P2Y2 and P2Y11 receptors. At the P2Y1 receptor the (S)-methanocarba analogue had similar potency to ATP. Applying the (N)-methanocarbo modification to 2-methylthio-ADP, itself a potent agonist of P2Y1, P2Y12 and P2Y13 receptors, resulted in MRS2365 (Chhatriwala et al 2004). MRS2365 proved to be the most potent agonist reported for the P2Y1 receptor (EC50 0.4 nM) with high selectivity (>10 000-fold) in comparison to the P2Y12 and P2Y13 receptors. Exposure of platelets to MRS2365 induced the characteristic shape change without proceeding to aggregation, which requires co-activation of the P2Y12 receptor.
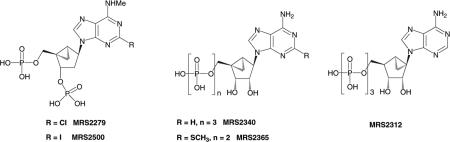
At four different P2X receptors (P2X1, P2X2, P2X3 and P2X2/3), characterized using whole cell patch clamp recording, MRS2340 and MRS 2312 were compared (Dunn et al 2004). The (N) analogue was roughly as potent as ATP and the (S) analogue was inactive. Thus, the receptors at which the (N) conformation of nucleotide derivatives is known to be preferred over the (S) are: P2Y1, P2Y2, P2Y4, P2Y11, P2X1, P2X2, P2X2/3 and P2X3.
However, the conformational requirements of the ribose moiety in binding to the P2Y6 receptor are very different from those of the above-mentioned P2 receptors. Dramatically, a uridine 5′-diphosphate analogue locked in the (N) envelope conformation was inactive (Kim et al 2002). Based on a prediction from docking of nucleotide derivatives to the P2Y6 receptor model, (S)-mc-dUDP was synthesized and found to be more potent than the corresponding riboside, dUDP, indicating a preference for the South conformation (Costanzi et al 2005). Thus, there is a fundamental conformational difference between the binding sites of P2Y6 and various other P2Y receptors.
Nucleotide derivatives have also been developed as P2Y1 and P2Y12 receptor antagonists. At the P2Y1 receptor, antagonists include both riboside (e.g. MRS2179) and acyclic nucleotide (e.g. bisphosphate MRS2298 or bisphosphonate MRS2496) structures (Cattaneo et al 2004). MRS2496 (with a binding Ki at the human P2Y1 receptor of 76 nM), by virtue of being a phosphonate rather than phosphate, is not subject to hydrolysis by nucleotidases. The nucleotide/nucleoside derivatives AR-C69931X, AZD6140, INS49266 and INS 50589 are selective P2Y12 receptor antagonists that do not require metabolic activation in vivo (Ingall et al 1999, van Giezen & Humphries 2005, Douglas et al 2002).
As for agonists, the (N)-methanocarba ring-constrained analogues provided the P2Y1 antagonists endowed with the highest potency, including the first general-use radioligand for the P2Y1 receptor (Waldo et al 2002), [3H]MRS2279 ((1′R,2′S,4′S,5′S)-4-(2-chloro-6-methylamino-purin-9-yl)-1-[(phosphato)-methyl]-2-(phosphato)-bicyclo[3.1.0]-hexane), and its more potent 2-iodo analogue MRS2500 with a binding Ki of 0.78 nM (Kim et al 2003b). These antagonists are highly selective for P2Y1 versus other P2Y receptor-subtypes. MRS2500 was shown to be a potent inhibitor of ADP-induced platelet aggregation (Cattaneo et al 2004). [32P]MRS2500 has been synthesized and studied as a radioligand having a Kd value of <1 nM at the P2Y1 receptor (Houston et al 2006). Other constrained ring systems, such as a carbocyclic LNA (oxabicyclo[2.2.1]heptane), which maintains a different form of the (N) conformation, have been applied to P2Y receptor agonists and antagonists, but none have been found to be as enhancing of potency as the (N)-methanocarba at the P2Y1 receptor (Ohno et al 2004).
Novel non-nucleotide antagonist of P2 receptors
Non-nucleotide antagonists have been reported for P2Y1, P2Y2, P2Y12, P2Y2/3, P2X3, and P2X7 receptors. Uracil-derived P2Y2 receptor antagonists such as ARC126313 have been reported (Meghani 2002). Various directly-acting P2Y12 receptor antagonists, including CT50547 and novel pyrazolidine-3,5-dione derivatives, are under development as antithrombotic agents (Scarborough et al 2001, Fretz et al 2005). The suramin derivative NF449 is selective for the P2X1 receptor (Rettinger et al 2005). The selective P2X2,3/P2X3 receptor antagonist A-317491 has antinociceptive properties and has been developed as a radioligand, but suffers from low bioavailability ( Jarvis et al 2002). Derivatives of the isoquino-line KN-62, such as MRS2427 (Chen et al 2002) and a high affinity radioligand (Romagnoli et al 2004), and novel 4,5-diarylimidazolines and other antagonists identified through screening of chemical libraries are selective for the P2X7 receptor (Merriman et al 2003, Baxter et al 2003).
We have synthesized and characterized insurmountable antagonists of P2Y receptors, consisting of symmetric aryl diisothiocyanate derivatives that are selective for P2Y6 or other subtypes within the P2Y family (Mamedova et al 2004). We examined the ability of these compounds to inhibit agonist-induced activation of five subtypes of recombinant P2Y receptors. A 1,4-di-(phenylthioureido) butane derivative (MRS2578) was more potent at inhibiting UDP-induced phospholipase C (PLC) activity through both human (IC50 37 nM) and rat (IC50 98 nM) P2Y6 receptors expressed in 1321N1 human astrocytes in comparison to human P2Y1, P2Y2, P2Y4 and P2Y11 receptors. MRS2578 (1 μM) completely blocked the protection by UDP of cells undergoing tumour necrosis factor (TNF)a-induced apoptosis. A related derivative of 1,4-phenylendiisothiocyanate (MRS2575) inhibited only human but not rat P2Y6 receptor activity. Limitations of using these isothiocyanate derivatives as P2Y antagonists include the pharmacological irreversibility, relative instability of the compounds in aqueous medium, and hydrophobicity and consequent low aqueous solubility.
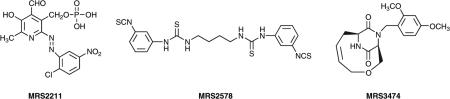
Derivatives of the known P2 receptor antagonist PPADS (pyridoxal-5′-phosphate-6-azo-phenyl-2,4-disulfonate) and isoPPADS (its 2,5-disulfonate isomer) have been examined at various P2 receptors. Recently, a selective antagonist was found to inhibit functional activity of the recombinant human P2Y13 nucleotide receptor expressed in 1321N1 human astrocytoma cells co-expressing a promiscuous Gα16 protein (Kim et al 2005). The highest antagonistic potency was observed for 6-(3-nitrophenylazo) derivatives of pyridoxal-5′-phosphate. The 2-chloro-5-nitro analogue (MRS2211) inhibited ADP (100 nM)-induced inositol trisphosphate (IP3) formation with a pIC50 value of 5.97, being 45-fold more potent than PPADS. The antagonism of MRS2211 was competitive with a pA2 value of 6.3. MRS2211 displayed >20-fold selectivity as an antagonist of the P2Y13 receptor in comparison to P2Y1 and P2Y12 receptors.
Diketopiperazines (DKPs) are a common motif in various biologically active natural products, and hence they may be useful scaffolds for the rational design of receptor probes and therapeutic agents. We constructed a new bicyclic scaffold that combines a DKP bridged with a 10-membered ring and tested for activity in astrocytoma cells expressing receptors coupled to phospholipase C (Besada et al 2005). One member of this series MRS3474 selectively inhibited calcium mobilization (IC50 value of 486±16 nM) and phosphoinositide turnover elicited by the P2Y1 receptor agonist 2-MeSADP, but not by the muscarinic receptor agonist carbachol. However, this compound did not compete for binding of a radiolabelled nucleotide-competitive receptor antagonist. Therefore, the new class of DKP derivatives shows utility as pharmacological tools for P2Y receptors.
Use of homology modelling to study receptor structure
The experimental knowledge of the structure of a receptor and its binding site recognition elements greatly facilitates the process of ligand design.
Crystallographic structural determination has not yet been accomplished for P2X and P2Y receptors. While the P2X receptors are not yet amenable to modelling at atomic detail, we have approached the structure–functional analysis of the P2Y receptors by indirect means, using a multidisciplinary combination of muta-genesis, chemical modification of the ligands, and homology modelling (Erb et al 1995, Moro & Jacobson 2002, Costanzi et al 2004, 2005), most recently based on a high resolution rhodopsin template. The structural insights gained provided by this approach assisted in the design of novel ligands.
In order to ascertain which residues were involved in ligand recognition and activation, individual residues of the P2Y1 receptor located in the transmembrane domains (TMs 3, 5, 6, and 7), as well as in the extracellular loops (ELs 2 and 3) were mutated to Ala and various charged residues. A cluster of positively charged Lys and Arg residues near the exofacial side of TMs 3, 6 and 7, putatively coordinated the phosphate moieties of nucleotide agonists and antagonists (7, 8). Two subclasses of P2Y receptors have been defined based on receptor sequence analysis, mechanism of ligand recognition and second messengers. The clusters of cationic residues involved in the coordination of the phosphate groups are different in the two subclasses (Costanzi et al 2004). In the P2Y1 receptor, these residues are Arg128 (3.29), Lys280 (6.55) and Arg310 (7.39), while in the P2Y12 receptor, the phosphate-coordinating residues are proposed to be Lys174 (EL2), Arg256 (6.55) and Lys280 (7.35). Both studies of site-directed mutagenesis of P2Y1 receptors (Moro et al 1999) and of chimeric P2Y1/6 receptors (Hoffmann et al 2004) emphasize the importance of extracellular loops in recognition of small molecule (nucleotide) ligands. Upon replacement of extracellular domains of the P2Y1 receptor with the corresponding domains of the P2Y6 receptor we observed a trend toward gain of receptor-induced PLC activation by UDP. This effect was particularly pronounced in the P2Y1/6 chimera containing replacements of both the N-terminus and EL1.
Conclusion
In conclusion, recent developments have greatly expanded the armamentarium of ligand tools for defining P2Y and P2X receptor subtypes pharmacologically. Attention to conformational factors of nucleotides has enabled the design of highly selective ligands for P2Y1 and other receptors. Screening of diverse chemical libraries and optimization of non-nucleotide antagonists has provided novel selective ligands for P2X3 receptors and other subtypes. Molecular modelling of P2Y receptors has aided ligand development, while the ability to model P2X receptors trails due to the lack of a suitable protein template.
DISCUSSION
Fields: Is the amount of phenol red that is in a normal culture medium sufficient to block P2X1?
Burnstock: It is: this is a serious problem (see King et al 2005).
Neary: This is not all bad because it gives us another tool for looking at P2X receptor function. If we want to stimulate P2X7 receptors we add Bz-ATP, but this is also an agonist for P2X1. In cell culture we don't have to worry about this if the culture medium contains phenol red. Conversely, if we want to stimulate P2X1 and P2X3 receptors, we can prepare medium without phenol red and test for the effects of a,b-methylene ATP.
Zalc: Have you used any of these ligands for PET studies?
Jacobson: Not yet. PET ligands are being developed for two of the adenosine receptors. It would be nice to have such ligands for the P2 receptors.
Zalc: You showed some ligands with a Kd of 0.3 or 0.5 nM. I would assume these would be suitable for a PET study.
Jacobson: I agree. It would be worth going in this direction.
Zalc: It would seem to be easy to substitute them.
Jacobson: That class of N-methanocarba compounds requires 14 synthetic steps, so it is very complicated.
Chao: The adenosine receptors are quite different from the P2Y receptors. Would any of the agonists or antagonists cross-react with the adenosine receptors?
Jacobson: There is little cross-reactivity.
Chao: Structurally, there is very little similarity among the G protein-coupled receptors in these two families.
Jacobson: That is correct. Even the position of the adenine ring docked in the putative binding site is flipped.
Burnstock: We ought to mention that recently a P2Y15 receptor was proposed (Inbe et al 2004), which was said to be activated by adenosine and AMP. Papers were published subsequently that negated this claim (e.g. Qi et al 2004, Abbracchio et al 2005). So please don't refer to the P2Y15 receptor—the sooner it is eliminated from the literature the better.
Fields: What about interactions with ectonucleotidases? Do these compounds have any actions there?
Jacobson: This has been a serious problem throughout the development of P2 receptor antagonists. Most of these compounds have either not been checked adequately, or in some cases are known to have blocking effects. There is evidence that N-methaonocarba nucleotides such as MRS2500, the high-affinity P2Y1 antagonist, have reduced interaction with ectonucleotidases, in comparison with native nucleotides.
Haydon: You mentioned separating the two phosphate groups is more likely to make a compound an antagonist. Is it known why this is?
Jacobson: We don't yet have a good template for modelling the activated form of the receptor, so we can't adequately answer that question.
Di Virgilio: Are there any studies on allosteric modulators of the P2 receptors? Jacobson: There are some candidates. Pyridyl isatogen is not a competitive antagonist, but may be an allosteric antagonist to the P2Y1 receptor. We also recently reported on some diketopiperazines such as MRS3474 that block P2Y1 signalling, these may be allosteric antagonists.
Raff: Are there studies comparing the effects of chronic treatment with P2Y1 antagonists with the knockout mice?
Jacobson: The best support for this is in the case of P2Y1 receptors, where there are well characterized effects on thrombus formation, based on the absence of P2Y1 receptors. In our collaborative studies with Christian Gachet (Hechler et al 2006) we found that potent P2Y1 antagonists have similar effects to genetic deletion of the receptors. It is consistent. However, concerning the use of knockouts, nature has already knocked out one of the P2Y receptors: the mouse has no P2Y11 sequence. One might conclude from this that P2Y11 receptors are totally super-fluous, which is certainly not true in humans.
Raff: Are there non-platelet effects with either the P2Y1 antagonists or the knockouts?
Jacobson: The P2Y1 receptor knockout mice have no apparent abnormality in their development, survival or reproductive function. I am not aware of other phenotypes.
Raff: Have people looked for phenotypes in other cell types that express the receptors to see whether they can find defects there?
Jacobson: No, I think almost all the work has been done with platelets.
Raff: Do you think that the receptors are only on platelets? Stojilkovic: The pituitary lactotrophs express them (He et al 2003).
Raff: Does the knockout have an effect on lactation?
Stojilkovic: I do not know.
Burnstock: P2Y1 receptors are strongly expressed on endothelial cells, and in the brain.
Raff: Do these drugs cross the blood–brain barrier (BBB)?
Jacobson: Most do not. They are negatively charged.
Abbracchio: Does the new compound (AZD6140) cross the BBB? At a certain point you mentioned that it may be useful for behavioural studies.
Jacobson: Being a carbocyclic nucleoside it is a good candidate for crossing the BBB, since we know that many adenosine agonists that are similar nucleosides do cross to a limited extent.
Di Virgilio: I want to comment on the number of knockouts that nature has made. The P2X7 receptor is highly polymorphic. Some of these polymorphisms are loss-of-function. As well as in humans, they are also common in mice. The original P2X7 knockout was made in a loss-of-function P2X7 polymorphism background.
Illes: The blood–brain permeability question is an important one. Is there any chance of getting P2 agonists or antagonists which pass the BBB? As an example, take opioid peptides. In the first run you had opioid agonists, such as morphine itself which only slowly permeates the BBB. However, the diacetyl derivative of morphine, heroin has a much improved permeability. Finally, in contrast to exogenous alkaloids or their structural analogues, the endogenous opioid peptides enkephalin or dynorphin fail to enter the brain on systemic application. Also there is a range of antagonists that is BBB permeable. Is a comparable (although reverse) development for P2 receptor agonists and antagonists possible, and might we expect sometime in the future non-natural ligands with a BBB-permeable structure?
Jacobson: There are efforts to examine chemical libraries for interactions with P2Y or P2X receptors. This promises to provide some novel uncharged structures that might cross the BBB.
Fields: I imagine that BBB permeability and stability are the big issues here. Illes: Is the Abbott P2X3 antagonist A-317491 BBB permeable?
Jacobson: No, because it contains three carboxylate groups.
Salter: It has to be given intrathecally.
Neary: Are you planning on producing selective antagonists for the P2Y2s and Y4s? It would be a really big help in the rat, where UTP activates Y2 and Y4. Geoff Burnstock mentioned that it is possible to distinguish them to some degree with suramin and RB2, but in astrocytes where there are other receptors besides the Y2 and Y4 that are impacted by suramin and RB2, it makes it a little harder. It would be a nice advance if we had something more selective.
Jacobson: We are working on that. We would also like to be able to convert agonists into antagonists as we did for the P2Y1 receptor, but so far our efforts haven't been successful. Our study of structure activity relationships at P2Y2 and P2Y4 receptors was recently published (Jacobson et al 2006).
Fields: Ken, is your lab at all interested in taking the approach of synthesizing fluorescent compounds that would allow us to localize receptors?
Jacobson: We are potentially interested, but I assume that the availability of antibodies would fill some of that need. We have already applied that approach successfully to adenosine receptors (McCabe et al 1992).
Salter: A lot of GPCRs undergo ligand-dependent internalization. Do any of these ligands for the P2Ys cause internalization?
Jacobson: Yes. That is being studied in G. Reiser’s lab (Tulapurkar et al 2004). Salter: Most ligand-gated ion channels exist in large complexes. What is known about the complexes for P2X or P2Y receptors?
Burnstock: Are you talking about heteromultimers and heterodimers?
Salter: No. Presumably the receptors don't just sit their by themselves. They have trafficking, scaffolding and signalling proteins nearby.
Burnstock: As far as I am aware, little is known about this.
Inoue: Dr H. Nakata reported that P2Y1 makes a heteromultimer and the properties of the complex are completely different from those of the parent receptors. In this case will your modelling software make a good ligand?
Jacobson: We are currently modelling dimerization of GPCRs, specifically the A3 adenosine receptor. However, it is premature to expect this to lead to selective ligands. It may be possible to bridge the dimers with bivalent ligands.
Fields: I gather the P2X molecular structure is not quite as well worked out as the P2Y. From your point of view, what is the major direction you are planning to take in synthesis?
Jacobson: With the exception of P2X7 receptors, our efforts are mostly concentrated on the P2Y receptors. We'd like to make progress with the uracil nucleotide receptors. These have been somewhat neglected, except P2Y2. A lot of work is needed here.
References
- Baxter A, Bent J, Boweres K, et al. Hit-to-Lead studies: the discovery of potent adamantane amide P2X7 receptor antagonists. Bioorg Med Chem Lett. 2003;13:4047–4050. doi: 10.1016/j.bmcl.2003.08.034. [DOI] [PubMed] [Google Scholar]
- Besada P, Mamedova L, Thomas CJ, Costanzi S, Jacobson KA. Design and synthesis of new bicyclic diketopiperazines as scaffolds for receptor probes of structurally diverse functionality. Org Biomol Chem. 2005;3:2016–2025. doi: 10.1039/b416349d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bo X, Jiang LH, Wilson HL, et al. Pharmacological and biophysical properties of the human P2X5 receptor. Mol Pharmacol. 2003;63:1407–1416. doi: 10.1124/mol.63.6.1407. [DOI] [PubMed] [Google Scholar]
- Bianchi BR, Lynch KJ, Touma E, et al. Pharmacological characterization of recombinant human and rat P2X receptor subtypes. Eur J Pharmacol. 1999;376:127–138. doi: 10.1016/s0014-2999(99)00350-7. [DOI] [PubMed] [Google Scholar]
- Cattaneo M, Lecchi A, Ohno M, et al. Antiaggregatory activity in human platelets of potent antagonists of the P2Y1 receptor. Biochem Pharmacol. 2004;68:1995–2002. doi: 10.1016/j.bcp.2004.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Ravi RG, Kertesy SB, Dubyak GR, Jacobson KA. Functionalized congeners of tyrosine-based P2X7 receptor antagonists: probing multiple sites for linking and dimerization. Bioconj Chem. 2002;13:1100–1111. doi: 10.1021/bc020025i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhatriwala M, Ravi RG, Patel RI, Boyer JL, Harden TK, Jacobson KA. Induction of novel agonist selectivity for the ADP-activated P2Y1 receptor versus the ADP-activated P2Y12 and P2Y13 receptors by conformational constraint of an ADP analogue. J Pharm Exp Therap. 2004;311:1038–1043. doi: 10.1124/jpet.104.068650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzi S, Mamedova L, Gao ZG, Jacobson KA. Architecture of P2Y nucleotide receptors: Structural comparison based on sequence analysis, mutagenesis, and homology modeling. J Med Chem. 2004;47:5393–5404. doi: 10.1021/jm049914c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzi S, Joshi BV, Maddileti S, et al. Human P2Y6 receptor: Molecular modeling leads to the rational design of a novel agonist based on a unique conformational preference. J Med Chem. 2005;48:8108–8111. doi: 10.1021/jm050911p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho-Silva R, Stahl L, Cheung KK, et al. P2X and P2Y purinergic receptors on human intestinal epithelial carcinoma cells: effects of extracellular nucleotides on apoptosis and cell proliferation. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1024–1035. doi: 10.1152/ajpgi.00211.2004. [DOI] [PubMed] [Google Scholar]
- Douglass J, Patel RI, Redick C, et al. Ribose and nucleobase modifications to nucleotides that confer antagonist properties against the P2Y12 platelet receptor. Haematologica. 2002;87S1:22. [Google Scholar]
- Dunn PM, Kim HS, Jacobson KA, Burnstock G. Northern ring conformation of methanocarba-adenosine 5′-triphosphate required for activation of P2X receptors. Drug Devel Res. 2004;61:227–232. doi: 10.1002/ddr.10381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erb L, Garrad R, Wang Y, Quinn T, Turner JT, Weisman GA. Site-directed mutagenesis of P2U purinoceptors. Positively charged amino acids in transmembrane helices 6 and 7 affect agonist potency and specificity. J Biol Chem. 1995;270:4185–4188. doi: 10.1074/jbc.270.9.4185. [DOI] [PubMed] [Google Scholar]
- Fretz H, Houille O, Hillpert K, et al. Novel pyrazolidine-3, 5-dione derivatives are P2Y12 receptor antagonists and inhibit ADP-triggered blood platelet aggregation.. 229th National Meeting of the American Chemical Soc; San Diego, CA. March 11, 2005; 2005. Abstract MEDI 80. [Google Scholar]
- Garcia-Guzman M, Soto F, Gomez-Hernandez JM, Lund PE, Stuhmer W. Characterization of recombinant human P2X4 receptor reveals pharmacological differences to the rat homologue. Mol Pharmacol. 1997;51:109–118. doi: 10.1124/mol.51.1.109. [DOI] [PubMed] [Google Scholar]
- Hoffmann CA, Soltysiak K, West PL, Jacobson KA. Shift in purine/pyrimidine base recognition upon exchanging extracellular domains in P2Y1/6 chimeric receptors. Biochem Pharmacol. 2004;68:2075–2086. doi: 10.1016/j.bcp.2004.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houston D, Ohno M, Nicholas RA, Jacobson KA, Harden TK. [2P]2-iodo-N6-methyl-(N)-methanocarba-2′-deoxyadenosine-3′,5′-bisphosphate ([32P]MRS2500), a novel radioligand for quantification of native P2Y1 receptors. Brit J Pharmacol. 2006 doi: 10.1038/sj.bjp.0706453. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingall AH, Dixon J, Bailey A, et al. Antagonists of the platelet P2T receptor: A novel approach to antithrombotic therapy. J Med Chem. 1999;42:213–220. doi: 10.1021/jm981072s. [DOI] [PubMed] [Google Scholar]
- Jacobson KA, Jarvis MF, Williams M. Purine and pyrimidine (P2) receptors as drug targets. J Med Chem. 2002;45:4057–4093. doi: 10.1021/jm020046y. [DOI] [PubMed] [Google Scholar]
- Jacobson KA, Costanzi S, Ohno M, et al. Molecular recognition at purine and pyrimidine nucleotide (P2) receptors. Current Topics Med Chem. 2004;4:805–819. doi: 10.2174/1568026043450961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaime-Figueroa S, Greenhouse R, Padilla F, Dillon MP, Gever JR, Ford APDW. Discovery and synthesis of a novel and selective drug-like P2X1 antagonist. 2005;15:3292–3295. doi: 10.1016/j.bmcl.2005.04.049. [DOI] [PubMed] [Google Scholar]
- Jarvis MF, Burgard EC, McGaraughty S, et al. A-317491, a novel potent and selective non-nucleotide antagonist of P2X3 and P2X2/3 receptors, reduces chronic inflammatory and neuropathic pain in the rat. Proc Natl Acad Sci USA. 2002;99:17179–17184. doi: 10.1073/pnas.252537299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CA, Vial C, Sellers LA, Humphrey PP, Evans RJ, Chessell IP. Functional regulation of P2X6 receptors by N-linked glycosylation: identification of a novel alpha beta-methylene ATP-sensitive phenotype. Mol Pharmacol. 2004;65:979–985. doi: 10.1124/mol.65.4.979. [DOI] [PubMed] [Google Scholar]
- Khakh BS, Burnstock G, Kennedy C, et al. International union of pharmacology. XXIV. Current status of the nomenclature and properties of P2X receptors and their subunits. Pharmacol Rev. 2001;53:107–118. [PubMed] [Google Scholar]
- Kim HS, Ravi RG, Marquez VE, et al. Methanocarba modification of uracil and adenine nucleotides: High potency of Northern ring conformation at P2Y1, P2Y2, or P2Y4 and P2Y11, but not P2Y6 receptors. J Med Chem. 2002;45:208–218. doi: 10.1021/jm010369e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SG, Gao ZG, Soltysiak KA, Chang TS, Brodie C, Jacobson KA. P2Y6 nucleotide receptor activates PKC to protect 1321N1 astrocytoma cells against tumor necrosis factor-induced apoptosis. Cell Mol Neurobiol. 2003a;23:401–418. doi: 10.1023/A:1023696806609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Ohno M, Xu B, et al. 2-Substitution of adenine nucleotide analogues containing a bicyclo[3.1.0]hexane ring system locked in a Northern conformation: Enhanced potency as P2Y1 receptor antagonists. J Med Chem. 2003b;46:4974–4987. doi: 10.1021/jm030127+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y-C, Lee J-S, Sak K, et al. Synthesis of pyridoxal phosphate derivatives with antagonist activity at the P2Y13 receptor. Biochem Pharmacol. 2005;70:266–274. doi: 10.1016/j.bcp.2005.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarowski ER, Watt WC, Stutts MJ, Brown HA, Boucher RC, Harden TK. Enzymatic synthesis of UTPgS, a potent hydrolysis resistant agonist of P2U-purinoreceptors. Br J Pharmacol. 1996;117:203–209. doi: 10.1111/j.1476-5381.1996.tb15175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmsjö M, Adner M, Harden TK, Pendergast W, Edvinsson L, Erlinge D. The stable pyrimidines UDPbS and UTPgS discriminate between the P2 receptors that mediate vascular contraction and relaxation of the rat mesenteric artery. Br J Pharmacol. 2000;131:51–56. doi: 10.1038/sj.bjp.0703536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamedova L, Joshi BV, Gao ZG, von Kügelgen I, Jacobson KA. Diisothiocyanate derivatives as potent, insurmountable antagonists of P2Y6 nucleotide receptors. Biochem Pharmacol. 2004;67:1763–1770. doi: 10.1016/j.bcp.2004.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meghani P. The design of P2Y2 antagonists for the treatment of inflammatory diseases.. 224th ACS National meeting, Abstracts, Division of Medicinal Chemistry.2002. p. 12. [Google Scholar]
- Merriman GH, Ma L, Shum P, et al. Synthesis and SAR of novel 4,5-diarylimidazolines as potent P2X7 receptor antagonists. Bioorg Med Chem Lett. 2003;15:435–438. doi: 10.1016/j.bmcl.2004.10.052. [DOI] [PubMed] [Google Scholar]
- Moro S, Hoffmann C, Jacobson KA. Role of the extracellular loops of G protein-coupled receptors in ligand recognition: a molecular modeling study of the human P2Y1 receptor. Biochemistry. 1999;38:3498–3507. doi: 10.1021/bi982369v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro S, Jacobson KA. Molecular modeling as a tool to investigate molecular recognition in P2Y receptors. Curr Pharmaceut Design. 2002;8:99–110. doi: 10.2174/1381612023392892. [DOI] [PubMed] [Google Scholar]
- Ohno M, Costanzi S, Kim HS, et al. Nucleotide analogues containing 2-oxabicyclo[2.2.1]heptane and L-a-threofuranosyl ring systems: Interactions with P2Y receptors. Bioorganic Med Chem. 2004;12:5619–5630. doi: 10.1016/j.bmc.2004.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettinger J, Braun K, Hochmann H, et al. Profiling at recombinant homomeric and heteromeric rat P2X receptors identifies the suramin analogue NF449 as a highly potent P2X1 receptor antagonist. Neuropharmacology. 2005;48:461–468. doi: 10.1016/j.neuropharm.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Romagnoli R, Baraldi PG, Pavani MG, et al. Synthesis, radiolabeling, and preliminary biological evaluation of [3H]-1-[(S)-N,O-bis-(isoquinolinesulfonyl)-N-methyl-tyrosyl]-4-(otolyl)-piperazine, a potent antagonist radioligand for the P2X7 receptor. Bioorg Med Chem Lett. 2004;14:5709–5712. doi: 10.1016/j.bmcl.2004.07.095. [DOI] [PubMed] [Google Scholar]
- Scarborough RM, Laibelman AM, Clizbe LA, et al. Novel tricyclic benzothiazolo[2,3-c]thiadiazine antagonists of the platelet ADP receptor (P2Y12). Bioorg Med Chem Lett. 2001;11:1805–1808. doi: 10.1016/s0960-894x(01)00313-4. [DOI] [PubMed] [Google Scholar]
- Spelta V, Mekhalfia A, Rejman D, Thompson M, Blackburn GM, North RA. ATP analogues with modified phosphate chains and their selectivity for rat P2X2 and P2X2/3 receptors. Br J Pharmacol. 2003;140:1027–1034. doi: 10.1038/sj.bjp.0705531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaver SR, Rideout JL, Pendergast W. Structure–activity relationships of dinucleotides: potent and selective agonists of P2Y receptors. Purinergic Signalling. 2005;1:183–191. doi: 10.1007/s11302-005-0648-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Giezen JJ, Humphries RG. Preclinical and clinical studies with selective reversible direct P2Y12 antagonists. Semin Thromb Hemost. 2005;31:195–204. doi: 10.1055/s-2005-869525. [DOI] [PubMed] [Google Scholar]
- Waldo GL, Corbitt J, Boyer JL, et al. Quantitation of the P2Y1 receptor with a high affinity radiolabeled antagonist. Mol Pharmacol. 2002;62:1249–1257. doi: 10.1124/mol.62.5.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildman SS, Brown SG, Rahman M, et al. Sensitization by extracellular Ca2+ of rat P2X5 receptor and its pharmacological properties compared with rat P2X1. Mol Pharmacol. 2002;62:957–966. doi: 10.1124/mol.62.4.957. [DOI] [PubMed] [Google Scholar]
- Wildman SS, Unwin RJ, King BF. Extended pharmacological profiles of rat P2Y2 and rat P2Y4 receptors and their sensitivity to extracellular H+ and Zn2+ ions. Br J Pharmacol. 2003;140:1177–1186. doi: 10.1038/sj.bjp.0705544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yerxa BR, Sabater JR, Davis CW, et al. Pharmacology of INS37217 [P(1)-(uridine 5′)-P4- (2′-deoxycytidine 5′) tetraphosphate, tetrasodium salt], a next-generation P2Y2 receptor agonist for the treatment of cystic fibrosis. J Pharmacol Exp Ther. 2002;302:871–880. doi: 10.1124/jpet.102.035485. [DOI] [PubMed] [Google Scholar]
- Yitzhaki S, Shneyvais V, Jacobson KA, Shainberg A. Involvement of uracil nucleotides in protection of cardiomyocytes from hypoxic stress. Biochem Pharmacol. 2005;69:1215–1223. doi: 10.1016/j.bcp.2005.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbracchio MP, Burnstock G, Boeynaems JM, et al. The recently deorphanized GPR80 (GPR99) proposed to be the P2Y15 receptor is not a genuine P2Y receptor. Trends Pharmacol Sci. 2005;26:8–9. doi: 10.1016/j.tips.2004.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He ML, Gonzalez-Iglesias AE, Stojilkovic SS. Role of nucleotide P2 receptors in calcium signaling and prolactin release in pituitary lactotrophs. J Biol Chem. 2003;278:46270–46277. doi: 10.1074/jbc.M309005200. [DOI] [PubMed] [Google Scholar]
- Hechler B, Nonne C, Roh EJ, et al. MRS2500 [2-iodo-N6-methyl-(N)-methanocarba- 2′-deoxyadenosine-3′,5′-bisphosphate], a potent, selective, and stable antagonist of the platelet P2Y1 receptor with strong antithrombotic activity in mice. J Pharmacol Exp Ther. 2006;316:556–563. doi: 10.1124/jpet.105.094037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inbe H, Watanabe S, Miyawaki M, Tanabe E, Encinas JA. Identification and characterization of a cell-surface receptor, P2Y15, for AMP and adenosine. J Biol Chem. 2004;279:19790–19799. doi: 10.1074/jbc.M400360200. [DOI] [PubMed] [Google Scholar]
- Jacobson KA, Costanzi S, Ivanov AA, et al. Structure activity and molecular modeling analyses of ribose- and base-modified uridine 5′-triphosphate analogues at the human P2Y2 and P2Y4 receptors. Biochem Pharmacol. 2006;71:540–549. doi: 10.1016/j.bcp.2005.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King BF, Liu M, Townsend-Nicholson A, et al. Antagonism of ATP responses at P2X receptor subtypes by the pH indicator dye, Phenol red. Br J Pharmacol. 2005;145:313–322. doi: 10.1038/sj.bjp.0706187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe RT, Skolnick P, Jacobson KA. FITC-APEC: A fluorescent ligand for A2-adenosine receptors. J Fluoresc. 1992;2:217–223. doi: 10.1007/BF00865279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulapurkar ME, Laubinger W, Nahum V, Fischer B, Reiser G. Subtype specific internalization of P2Y1 and P2Y2 receptors induced by novel adenosine 5′-O-(1-boranotriphosphate) derivatives. Br J Pharmacol. 2004;142:869–878. doi: 10.1038/sj.bjp.0705859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi A-D, Harden TK, Nicholas RA. GPR80/99, proposed to be the P2Y15 receptor activated by adenosine and AMP, is not a P2Y receptor. Purinergic Signalling. 2004;1:67–74. doi: 10.1007/s11302-004-5069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]