

Introduction
The contemporary development of drugs that modulate G protein-coupled receptors (GPCRs) is based not only on organic synthesis, but also on structural studies of the protein targets. GPCRs contain seven membrane-spanning helical domains (TMs). The structural characterization of ligand interactions with specific regions of GPCRs presently requires mutagenesis (van Rhee and Jacobson, 1996) in conjunction with molecular modeling (Moro et al., 1998), since direct analysis has not yet been achieved. As molecular modeling of cloned GPCR sequences using a rhodopsin template has been refined, it has become possible to generate hypotheses for location of the binding sites that are consistent with mutagenesis results and ligand specificities. To obtain an energetically refined 3D structure of the ligand–receptor complex, we have introduced a new computational approach, a “cross docking” procedure, which simulates the reorganization of the native receptor induced by the ligand (Moro et al., 1998).
Mutagenesis and model for binding of agonists at human P2Y1 receptors
Extracellular nucleotides may act in cellular signaling through two families of membrane-bound P2 receptors: P2Y subtypes, G protein-coupled receptors (GPCRs) activated by both adenine and uracil nucleotides; and P2X subtypes, ligand-gated ion channels activated principally by adenine nucleotides (Fredholm et al, 1994). As many as seven subtypes have been cloned within each family. We have selected the human P2Y1 receptor as a model system for development of ligands with the aid of molecular modeling and mutagenesis (Jiang et al., 1997; Moro et al., 1998). The P2Y1 receptor is a membrane bound G protein-coupled receptor of the rhodopsin family and is stimulated by extracellular adenine nucleotides.
The P2Y1 receptor subtype is a phospholipase C-activating receptor (Schachter et al, 1996) present in heart, skeletal muscle, and various smooth muscles. At this receptor, the potency order for activation is 2-methylthioadenosine 5′-diphosphate (2-MeSADP) > 2-(hexylthio)adenosine 5′-monophosphate (HT-AMP) > ADP > ATP, while AMP and UTP are inactive (Boyer et al., 1996a).
Within the TM domains
In order to ascertain which residues of the human P2Y1 receptor are involved in ligand recognition (Fig. 1), we have mutated (Jiang et al., 1997; Hoffmann et al., 1999) the transmembrane helical domains (TM 3, 5, 6, and 7) and the extracellular loops (EL). The mutant receptors expressed in COS-7 cells were measured for stimulation of phospholipase C (PLC) in the presence of 2-MeSADP, an agonist which activates the wild-type receptor with an EC50 value of 2 nM, and other adenine nucleotides. A cluster of positively charged amino acids, Lys and Arg residues near the exofacial side of TMs 3 and 7 and to a lesser extent TM6, predicted by molecular modeling to coordinate the phosphate moieties of nucleotide ligands in the human P2Y, receptor, were replaced with alanine and, in some cases, by other amino acids (Jiang et al., 1997). Agonists had no activity at R128A (TM3) and R310A and S314A (TM7) mutant receptors and a markedly reduced potency at K280A (TM6) and Q307A (TM7) mutant receptors. Previously, positively charged residues of the human P2Y2 receptor (H262, R265, and R292 in TM6 and TM7) were similarly found to be critical for activation (Erb et al., 1995). These results suggest that residues on the exofacial side of TM3 and TM7 are critical determinants of the ATP binding pocket. In contrast, there was no change in the potency or maximal effect of agonists with the S317A mutant receptor, and alanine replacement of F131, H132, Y136, F226, or H277 resulted in mutant receptors that exhibited a seven- to 18-fold reduction in potency compared to that observed with the wild-type receptor. These residues thus appear to subserve a less important modulatory role in ligand binding to the P2Y1 receptor.
Fig. 1.

Human P2Y1 receptor topology showing transmembrane helical (TM) regions, extracellular loops (EL), and intracellular loops. Single amino acid replacements were made in TM3, 5, 6, and 7, in EL2 and EL3, and in the N-terminal segment. Residues which when mutated to either Ala or the indicated amino acid (brackets), decreased the potency of 2-MeSADP by factors of one-to-five-fold (shaded), five-to-20-fold (shaded and underlined), or >20-fold (heavy circles).
Since changes in the potency of 2-MeSADP and HT-AMP paralleled the changes in potency of 2-MeSATP at the various mutant receptors, the β- and γ-phosphates of the adenine nucleotides appear to be less important than the α-phosphate in ligand P2Y1 receptor interactions (Jiang et al., 1997). However, T221A and T222A mutant receptors exhibited much larger reductions in triphosphate (89- and 33-fold vs. wild-type receptors, respectively), rather than di- or monophosphate potency. This result may be indicative of a greater role of these TM5 residues in γ-phosphate recognition. Taken together the results suggest that the adenosine and a-phosphate moieties of ATP bind to critical residues in TM3 and TM7 on the exofacial side of the receptor.
Within the extracellular domains
We have investigated the role in receptor activation of all charged amino acids (D, E, K and R) and cysteines in the extracellular loops (EL) 2 and 3 of the human P2Y1 receptor by alanine scanning mutagenesis (Hoffmann et al., 1999, Fig. 1).
Surface detection of most of the mutant receptors by ELISA showed at least 10% expression at the surface of the plasma membrane compared to the wild-type receptor. Control experiments in which COS-7 cells were transfected with lower amounts of P2Y1 wild-type DNA showed full stimulation and no shift for EC50 values but surface expression rates dropped to approximately 10%. Therefore, assuming ≥10% receptor expression, shifts in the potency of 2-MeSADP and other agonists upon single amino acid replacement directly reflect the structural perturbations of the receptor, rather than insufficient receptor protein reaching the surface. In a few cases (see below), the receptor protein was undetectable at the cell surface. In those cases, the mutation was assumed to interfere with trafficking, and the effect on ligand recognition/activation could not be determined.
Two essential disulfide bridges in the extracellular domains have been identified, and several charged residues in the EL 2 (E209) and 3 (R287) have been found to be critical for receptor activation (Hoffmann et al., 1999). The C124A and C202A mutation, located in the upper part of transmembrane helix (TM) 3 and EL 2, prevented PLC stimulation by up to 100 μM 2-MeSADP. These data indicate a disulfide bridge in the P2Y1 receptor between loop 2 and the upper part of TM 3, as found in many other G protein-coupled receptors. This disulfide bridge seems to be critical for the proper receptor trafficking to the cell surface. Presently it is unknown whether it is also critical for receptor activation. In contrast, the C42A and C296A mutant receptors (located in the N-terminal domain and EL 3) were activated by 2-MeSADP, but the EC50 values for stimulation were over 1000-fold greater than for the wild-type receptor. The double mutant receptor C42A/C296A exhibited no additive shift in the dose–response curve for 2-MeSADP. These data indicate that C42 and C296 form another disulfide bridge in the extracellular region which is critical for activation processes.
Upon replacement of charged amino acids in EL 2 and 3, we found only minor deviations from the agonist affinity at wild type receptors, with two remarkable exceptions (Hoffmann et al., 1999). E209 in the EL 2 exhibits a > 1000-fold shift in EC50 value when substituted with alanine, while it responds like wild-type if substituted with amino acids capable of H-bonding (D, Q or R). Thus, E209 appears to be required for its ability to form H-bonding alone.
R287 in EL 3 was impaired similarly to E209 when substituted by alanine, i.e. dose–response curves where shifted by > 1000-fold, and the curve shape was identical. Substitution of R287 by lysine, another positively charged residue, only partially restored the potency of 2-MeSADP as a P2Y1 receptor agonist, with an EC50 value of 75 nM. The possibility of a required ionic interaction between essential residues R287 and E209, which may be in physical proximity according to preliminary molecular modeling of the loop regions, was considered. This possibility was ruled out since the double mutant E209A/R287A receptor demonstrates a shift that is additive relative to the single mutations. Refined molecular modeling indicates a direct interaction of the negatively charged phosphate chain of the ligand with R287. Moreover, the selective reduction in potency of 3′NH2–ATP in activating the E209R mutant receptor is consistent with the hypothesis of direct contact between EL2 and nucleotide ligands. Our findings support ATP binding to at least two distinct domains of the P2Y1 receptor, both outside the TM region (“meta-binding” sites) and within the TM core (Moro et al., 1999).
Novel P2X receptor antagonists
We have developed selective agonists and antagonists for both P2Y and P2X receptors (Table 1). The P2X ligands were developed through an empirical rather than computational approach. Molecular modeling of inotropic P2X receptors is limited by the lack of a template protein, as well as specific knowledge of loop conformation (van Rhee et al., 1997).
TABLE 1.
Estimates for antagonist potencies (IC50 values, nM) in functional assays at P2 receptorsa
| Compound | P2X1 | P2X2 | P2X3 | P2X4 | P2Y1 | P2Y2 | P2Y4 | P2Y6 | P2YAC |
|---|---|---|---|---|---|---|---|---|---|
| PPADS | 98.5± 5.5e |
1200± 200e |
239.5 ± 38.0e, 1700 ± 200(h) |
b | 18,200 ± 1700e. ~4000 (h) |
c | c | 69%f(h) | b |
| isoPPADS | 43±18 | 398± 129 |
84±4 | ND | 28.1 ± 8.0 | ND | ND | ND | ND |
| pyridoxal-5-phosphate | ~3000 | 39,500± 19,000 |
ND | 219,000± 2400 |
~50,000(h) | b | ND | ND | ND |
| suramin | 4900 ± 1000 | 10,400± 2000e |
~3000, 14,900± 1900(h) |
b | d | 48,000± 17,000 |
b | 27%f(h) | 4000± 2300 |
| reactive blue 2 | ND | 360±80 | ND | 128,000± 11,800 |
d | b | 33%f(h) | 87%f(h) | 25±7 |
| MRS 2179 | 1150±200 | c | ~10,000 | c | 330±59 | b | b | b | b |
| MRS 2270 | 10,200± 2600 |
b | 58,300± 100 |
b | b | b | b | b | b |
Effects of antagonists on inward current induced by activation by ATP, at the indicated concentrations, of recombinant rat P2X1 (3 μM), P2X2 (10 μM), P2X3 (1 μM), and P2X4 (30 μM) receptors, expressed in Xenopus oocytes, using the twin electrode voltage clamping technique; or at phospholipase C-coupled P2Y1 receptor of turkey erythrocytes, at recombinant human P2Y2 and P2Y1 receptors, and at recombinant rat P2Y6 receptors, unless noted. (h) indicates human clone.
inactive as antagonist at 100 μM.
inactive as antagonist at 10 μM.
right shift at 30 μM and decrease in maximal effect (Boyer et al., 1994).
Similar experiments gave IC50 ~ 1 μM (Charlton et al., 1996; North and Barnard, 1997).
Percent inhibition at 100 μM (Communi et al., 1996; Robaye et al, 1997)
ND not determined.
Structural modifications of the non-selective P2 antagonist pyridoxal-5′-phosphate-6-azophenyl-2′,4′-disulfonate (PPADS, Lambrecht et al., 1992, Fig. 2), have been made through functional group substitution on the sulfophenyl ring and at the phosphate moiety through the inclusion of phosphonates, demonstrating that a phosphate linkage is not required for P2 receptor antagonism (Kim et al., 1998). Phosphonates are not hydrolyzable and thus may be more stable than phosphate analogues in pharmacological studies. Substituted 6-azophenyl and 6-azonaphthyl derivatives were evaluated (Fig. 2). Among the 6-azophenyl derivatives, 5′-methyl, ethyl, propyl, vinyl, and allyl phosphonates were included. The compounds have been tested as antagonists at P2Y receptors in the turkey erythrocyte (Harden et al, 1988) and in the guinea-pig taenia coli and at P2X1 receptors in guinea-pig vas deferens and bladder and at recombinant rat P2X2 receptors expressed in Xenopus oocytes. Competitive binding assay at human P2X1 receptors in differentiated HL-60 cell membranes was carried out using [35S]ATP-γ-S. A 2′-chloro analogue of isoPPADS (MRS 2157, Fig. 2), a vinyl phosphonate derivative, MRS 2206, and an azonaphthyl derivative, MRS 2166, were particularly potent in binding at human P2X1 receptors. Potencies of phosphate derivatives at turkey erythrocyte P2Y receptors were generally similar to PPADS itself (IC50 18.2 ± 1.7 μM; Fig. 3), except for the p-carboxyphenylazo phosphate derivative, MRS 2159, and its m-chloro analogue, MRS 2160, which were selective for P2X vs. P2Y1 (IC50 > 100 μM) receptors. At 30 μM, both MRS 2159 and 2160 significantly antagonized P2X receptor-induced contraction of guinea pig urinary bladder and vas deferens. MRS 2160 was very potent at rat P2X2 receptors expressed in Xenopus oocytes with an IC50 value of 0.82 ± 0.28 μM, while MRS 2159 was less potent with an IC50 value of 11.9 ± 1.4 μM.
Fig. 2.

Structures of pyridoxal-5′-phosphate (P-5-P) and other related P2 receptor antagonists, including those recently synthesized in the molecular recognition section (MRS).
Fig. 3.

Stimulation of phospholipase C in turkey erythrocyte membranes (Harden et al., 1988). Membranes were incubated for 30 min at 37°C. Data are presented as percent of maximum accumulation of tritiated inositol phosphates above basal levels in the presence of the antagonist and 10 nM 2-MeSATP, average of two experiments. EC50 values were 4.35 μM for MRS 2192 and 18.2 μM for PPADS.
Among the phosphonate derivatives (Fig. 2), [4-formyl-3-hydroxy-2-methyl-6-azo-(2′-chloro-5′-sulfonylphenyl)-5-pyridyl]-methylphosphonic acid (MRS 2192) showed high potency at P2Y1 receptors with an IC50 of 4.35 ± 0.36 μM (Fig. 3). The corresponding 2′,5′-disulfonylphenyl derivative, MRS 2191, was nearly inactive at turkey erythrocyte P2Y1 receptors. Thus a single ring substitution, sulfo instead of chloro, has a major effect on the selectivity of these methylphosphonates as P2Y receptor antagonists. MRS 2191 was relatively potent at recombinant P2X1 receptors with an IC50 value of 1.1 ± 0.2 μM. Also, an ethyl phosphonate derivative, MRS 2142, while inactive at turkey P2Y1 receptors, was particularly potent at recombinant P2X2 receptors (IC50 1.5 ± 0.1 μM).
A pyridoxine cyclic phosphate (cyclic pyridoxine-α4,5-monophosphate, MRS 2219; Fig. 2) and its 6-azoaryl derivative (cyclic pyridoxine-α4,5-monophosphate-6-azophenyl-2′,5′-disulfonic acid, MRS 2220) selectively potentiate and antagonize, respectively, P2X1 receptors expressed in Xenopus oocytes (Jacobson et al., 1998). These derivatives are novel analogues of the P2 receptor antagonists pyridoxal-5′-phosphate and the 6-azophenyl-2′,4′-disulfonate derivative (Lambrecht et al., 1992; PPADS), in which the phosphate group was cyclized by esterification to a CH2OH group at the 4-position. The cyclic pyridoxine-α4,5-monophosphate, MRS 2219, was found to be a selective potentiator of ATP-evoked responses at rat P2X1 receptors with an EC50 value of 5.9 ± 1.8 μM, while the corresponding 6-azophenyl-2′,5′-disulfonate derivative, MRS 2220, was a selective antagonist (Fig. 4). The potency of compound MRS 2220 at the recombinant P2X1 receptor (IC50 10.2 ± 2.6 μM) was lower than PPADS (IC50 98.5 ± 5.5 nM) or iso-PPADS (IC50 42.5 ± 17.5 nM), although unlike PPADS its effect was reversible with washout and surmountable. Compound MRS 2220 also showed weak antagonistic activity at the rat P2X1 receptor (IC50 58.3 ± 0.1 μM), while at recombinant rat P2X2 and P2X4 receptors no enhancing or antagonistic properties were evident. MRS 2219 and MRS 2220 were found to be inactive as either agonists or antagonists at the phospholipase C-coupled P2Y1 receptor of turkey erythrocytes, at recombinant human P2Y2 and P2Y4 receptors, and at recombinant rat P2Y6 receptors. Similarly, neither MRS 2219 nor MRS 2220 at a concentration of 100 μM had measurable affinity at adenosine rat A1, rat A2A, or human A3 receptors. The lack of an aldehyde group in these derivatives indicates that Schif’s base formation with the P2X1 receptor is not necessarily required for recognition of pyridoxal phosphate derivatives. Thus, MRS 2219 and MRS 2220 are relatively selective pharmacological probes of P2X1 receptors, filling a long-standing need in the P2 receptor field, and may also be important lead compounds for future studies.
Fig.4.

Effects of MRS 2219 (A) and MRS 2220 (B) on inward current induced by ATP, at the indicated concentrations, of recombinant rat P2X1 (3 μM), P2X2 (10 μM), P2X3 (1 μM), and P2X4 (30 μM) receptors, expressed in Xenopus oocytes, using the twin electrode voltage clamping technique. The agonist concentrations correspond approximately to the EC70 values. IC50 values for MRS 2220 (n = 4) at P2X1 and P2X3 receptors were 10.2 ± 2.6 μM and 58.3 ± 0.1 μM, respectively.
Novel P2Y receptor antagonists
Adenosine 3′3′- and 2′,5′-bisphosphates were previously demonstrated to act as competitive antagonists at the P2Y1 receptor (Boyer et al., 1996b). 2′- and 3′-Deoxyadenosine bisphosphate analogues containing various structural modifications at the 2- and 6-position of the adenine ring, on the ribose moiety, and on the phosphate groups have been synthesized with the goal of developing more potent and selective P2Y1 antagonists (Camaioni et al., 1998). Single-step phosphorylation reactions of adenosine nucleoside precursors were carried out. The activity of each analogue at P2Y1 receptors (Table 2) was determined by measuring its capacity to stimulate phospholipase C in turkey erythrocyte membranes (agonist effect) and to inhibit phospholipase C stimulation elicited by 10 nM 2-MeSATP (antagonist effect). Both 2′- and 3′-deoxy modifications were well tolerated. The carbocyclic analogue of compound 3 proved to be a partial agonist of moderate potency at P2Y1 receptors (Nandanan et al., 1999). The N6-methyl modification (2′-deoxy-N6-methyladenosine-3′,5′-bisphosphate, MRS 2179) both enhanced antagonistic potency of 2′-deoxyadenosine 3′,5′-bisphosphate by 17-fold and eliminated residual agonist properties observed with the lead compounds (Boyer et al., 1998). In a Schild analysis MRS 2179 was found to be a competitive antagonist with a KB value 104 nM at P2Y1 receptors in turkey erythrocyte membranes (Fig. 5, Boyer et al., 1998). MRS 2179 did not partially activate either the turkey or human P2Y1 receptor, thus it is a pure antagonist. MRS 2179 was inactive at the P2Y1 receptor in C6 glioma cells which is coupled to inhibition of adenylate cyclase (J. Boyer et al., unpublished). The N6-ethyl modification provided intermediate potency as an antagonist, while the N6-propyl group completely abolished both agonist and antagonist properties (Camaioni et al., 1998). 2-Methylthio and 2-chloro analogues were partial agonists of intermediate potency. A 2′-methoxy group provided intermediate potency as an antagonist while enhancing agonist activity. An N1-methyl analogue was a weak antagonist with no agonist activity. An 8-bromo substitution and replacement of the N6-amino group with methylthio, chloro, or hydroxy groups, greatly reduced the ability to interact with P2Y1 receptors. Benzoylation or dimethy lation of the N6-amino group also abolished the antagonist activity. In summary, our results further define the structure activity of adenosine bisphosphates as P2Y1 receptor antagonists and have led to the identification of MRS 2179 as the most potent antagonist reported to date for this receptor. Thus, MRS 2179 was a potent, competitive antagonist, selective for the P2Y1 receptor vs. four other P2Y subtypes. However MRS 2179 has not yet been evaluated at the recently cloned P2Y11 receptor (Communi et al., 1997).
TABLE 2.
Effects of 3′,5′-bisphosphate derivatives on stimulation of PLC in turkey erythrocytes and inhibition of effects of 10 nM 2-MeSATP
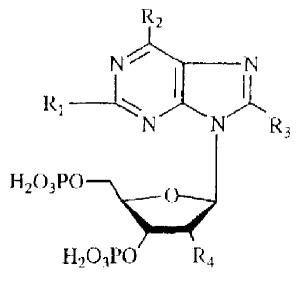
|
||||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Compound | R1 | R2 | R3 | R4 | Agonist Effect (% maximum) |
EC50 (μM) |
Antagonist Effect (%maximum) |
IC50 (μM) |
| 1 | H | NH2 | H | OH | 21% | 1.28 | 77% | 4.19 |
| 2 | H | NH2 | H | OCH3 | 35% | 12.9 | 65% | 12.4 |
| 3 | H | NH2 | H | H | 12% | 6.26 | 87% | 5.76 |
| 4 | Cl | NH2 | H | H | 19% | 0.651 | 80% | 2.01 |
| 5 | SCH3 | NH2 | H | H | 22% | 0.550 | 77% | 2.11 |
| 6 | H | NH2 | Br | H | 0% | 100% | 36.7 | |
| 7 | H | NHCH3 | H | H | 0% | 99% | 0.330 | |
| (MRS 219) | ||||||||
| 8 | H | NHCH2CH3 | H | H | 0% | 100% | 1.08 | |
| 9 | H | NH(CH2)2CH3 | H | H | 0% | <20% | ||
| 10 | H | NHCOC6H6 | H | H | 0% | 0% | ||
| 11 | H | N(CH3)2 | H | H | 0% | 70% | 46.7 | |
| 12 | H | Cl | H | H | <10% | <10% | ||
| 13 | H | OH | H | H | 0% | <20% | ||
| 14 | H | SCH3 | H | H | 0% | 78% | 29.1 | |
Fig. 5.

Competitive inhibition by MRS 2179 of phopholipase C activation (by 10 nM 2-MeSATP) in turkey erythrocyte membranes (Boyer et al., 1998).
Although MRS 2179 is selective for P2Y1 from among five different metabotropic P2 receptors, caution is advised when using this agent when inotropic P2 receptors are present, since it was found to antagonize one subtype, i.e. the rat P2X1 receptor, expressed in Xenopus oocytes (Fig. 6, King et al., unpublished). Ion current induced by 3 μM ATP acting at P2X1 receptors was blocked by MRS 2179 with an IC50 value of 1.2 ± 0.2 μM. At the rat P2X3 receptor, MRS 2179 is a much weaker antagonist with an IC50 value of ~10 μM. The compound at a concentration of 10 μM was inactive at the rat P2X2 receptor, while at the rat P2X4 receptor, a potentiation of ion current by 25% was observed.
Fig. 6.

Effect of 10 μM MRS 2179 on inward current induced by activation by ATP, at the indicated concentrations, of recombinant rat P2X1 (3 μM), P2X3 (10 μM), P2X3 (1 μM), and P2X4 (30 μM) receptors, expressed in Xenopus oocytes, using the twin electrode voltage clamping technique (n = 4). The agonist concentrations correspond approximately to the EC50 values. IC50 values for MRS 2179 at P2Xl and P2X3 receptors were 1.2±0.2 μM and ~10 μM, respectively. Potentiation of 25% was observed at P2X4 receptors.
Binding model for antagonists at human PZY1 receptors
An antagonist P2Y1 receptor binding model has been developed (Moro et al., 1998). The structural similarity between the potent antagonist MRS 2179 and nucleotide agonists, which bind to a single putative binding region within the TM domains, suggests that receptor activation resulting in a specific conformational change may depend on subtle differences between ligands.
The molecular basis for recognition by human P2Y1 receptors of the selective, competitive antagonist MRS 2179 was probed using site-directed mutagenesis and molecular modeling. The potency of this antagonist was measured in mutant receptors in which key residues in TMs 3, 5, 6, and 7 were replaced by Ala or other amino acids. The capacity of MRS 2179 to block stimulation of phospholipase C promoted by 2-MeSADP was lost in P2Y1 receptors having F226A, K280A, or Q307A mutations, indicating that these residues are critical for the binding of the antagonist molecule. Mutation of the residues His132, Thr222, and Tyr136 had an intermediate effect on the capacity of MRS 2179 to block the P2Y1 receptor. These positions therefore appear to have a modulatory role in recognition of this antagonist. F131A, H277A, T221A, R310K, or S317A mutant receptors exhibited an apparent affinity for MRS 2179 that was similar to that observed with the wild-type receptor. Thus, Phel31, Thr221, His277, and Ser317 are not essential for recognition of the nucIeotide antagonist. A computer-generated model of the human P2Y1 receptor was built and analyzed to help interpret these results. The model was derived from using primary sequence comparison, secondary structure predictions, and three-dimensional homology building, using rhodopsin as a template, and was consistent with data obtained from mutagenesis studies. A putative nucleotide binding site was localized, following a cross docking procedure to obtain energetically refined 3D structures of the ligand-receptor complexes, and used to predict which residues are likely to be in proximity to agonists and antagonists. According to our computational model TM6 and TM7 are close to the adenine ring, TM3 and TM6 are close to the ribose moiety, and TM3, TM6, and TM7 are near the triphosphate chain.
Initial results indicated that both suramin and PPADS retained antagonist properties at the P2Y, mutant receptors examined (Jiang et al, 1997). However, recently we have determined with full dose response curves that K280A and Q307A mutations greatly diminish the potency of the pyridoxal phosphate-related antagonists, i.e. 20 μM PPADS is ineffective (Guo et al., 1998). Thus, as for adenosine receptor antagonists, there appears to be a spatial overlap between the binding regions of the receptor for agonist and antagonist ligands, even those of highly divergent structure. Molecular modeling (Moro et al., 1998) using PowerFit (Molecular Simulations, Mahwah NJ) has suggested a possible model of superimposition of two classes of antagonists, nucleotides related to MRS 2179 and non-nucleotides related to pyridoxal phosphate (Fig. 7). In these energetically minimized conformations, it is possible to demonstrate overlap of negatively charged moieties, two monophosphate groups for MRS 2179 and 5′-phosphate and phenyl-4-sulfonate groups for PPADS. Thus, a preliminary pharmacophore model features a hydrogen bond donor (e.g. the 6-NH2 of MRS 2179 and 3-OH of PPADS), which is proposed to bind in vicinity of Glu307 of the P2Y1 receptor (Moro et al., 1998), and two anionic groups. The sulfonate group of PPADS (corresponding to 5′-phosphate of MRS 2179) would interact directly with Lys280. By analogy (Moro et al., 1998), this also allows prediction of overlap between PPADS and ATP-related agonists. In conclusion, pyridoxal phosphate antagonists appear to bind in the TM region of the human P2Y receptor, since the capacity of PPADS to block stimulation of phospholipase C was lost in receptors having K280A or Q307A mutations (TM6 and 7).
Fig. 7.

Lower left: possible superposition of MRS 2179, PPADS, and MRS 2192 using steric and electrostatic alignment methodology. Lower right: scheme of the hypothetical pharmacophore map extrapolated using the superposition of MRS 2179, PPADS, and MRS 2192 structures. At top: Structures of MRS 2179, PPADS, and MRS 2192 showing positions corresponding to the general pharmacophore.
Conclusions
The cloning of at least 13 subtypes of P2 receptors has presented a unique challenge to medicinal chemists: the design of selective agonists and antagonists for this multiplicity of receptors with few existing leads. The human P2Y1 receptor as representative of the P2Y family of metabotropic purine and pyrimidine nucleotide receptors may be modeled based on a rhodopsin template, and the resulting model is highly consistent with pharmacological and mutagenesis results. Charged residues in both the transmembrane and extracellular domains and two disulfide bridges essential for receptor activation have been identified. Selective P2Y1 receptor antagonists such as MRS 2179 are under development. Modeling of P2X receptors has not been achieved, since no template for the extracellular nucleotide binding region exists. Nevertheless, a selective antagonist, MRS 2220, and a potentiator, MRS 2219, of this subtype have been identified. Both are based on pyridoxal-5′-phosphate antagonists (such as PPADS), for which the SAR is being examined at all of the P2 receptor subtypes.
References
- Boyer JL, Zohn IE, Jacobson KA, Harden TK. Differential effects of P2-purinergic receptor antagonists on phospholipase C- and adenylyl cyclase-coupled P2Y purinergic receptors. Br. J. Phannacol. 1994;113:614–620. doi: 10.1111/j.1476-5381.1994.tb17034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer JL, Siddiqi S, Fischer B, Romera-Avila T, Jacobson KA, Harden TK. Identification of potent P2Y purinoceptor agonists that are derivatives of adenosine 5′-monophosphate. Br. J. Pharmacol. 1996a;118:1959–1964. doi: 10.1111/j.1476-5381.1996.tb15630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer JL, Romero-Avila T, Schachter JB, Harden TK. Identification of competitive antagonists of the P2Y1 receptor. Mol. Pharmacol. 1996b;50:1323–1329. [PubMed] [Google Scholar]
- Boyer JL, Mohanram A, Camaioni E, Jacobson KA, Harden TK. Competitive and selective antagonism of P2Y1 receptors by N6-methyl 2′-deoxyadenosine 3′,5′-bisphosphate. Br. J. Pharmacol. 1998;124:1–3. doi: 10.1038/sj.bjp.0701837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camaioni E, Boyer JL, Mohanram A, Harden TK, Jacobson KA. Deoxyadenosine-bisphosphate derivatives as potent antagonists at P2Y1 receptors. J. Med. Chem. 1998;41:183–190. doi: 10.1021/jm970433l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlton SJ, Brown CA, Weisman GA, Turner JT, Erb L, Boarder MR. PPADS and suramin as antagonists at cloned P2Y- and P2U-purinoceptors. Br. J. Pharmacol. 1996;118:704–710. doi: 10.1111/j.1476-5381.1996.tb15457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Communi D, Motte S, Boeynaems J-M, Pirroton S. Pharmacological characterization of the human P2Y4 receptor. Eur. J. Pharmacol. 1996;317:383–389. doi: 10.1016/s0014-2999(96)00740-6. [DOI] [PubMed] [Google Scholar]
- Communi D, Govaerts C, Parmentier M, Boeynaems J-M. Cloning of a Human P2 Receptor Coupled to Phospholipase C and Adenylyl Cyclase. J. Biol. Chem. 1997;272:31969–31973. doi: 10.1074/jbc.272.51.31969. [DOI] [PubMed] [Google Scholar]
- Erb L, Garrad R, Wang YJ, Quinn T, Turner JT, Weisman GA. Site-directed mutagenesis of P2U purinoceptors – positively charged amino-acids in transmebrane helix-6 and helix-7 affect agonist potency and specificity. J. Biol. Chem. 1995;270:4185–4188. doi: 10.1074/jbc.270.9.4185. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, Abbracchio MP, Burnstock G, Daly JW, Harden TK, Jacobson KA, Leff P, Williams M. Nomenclature and classification of purinoceptors: A report from the IUPHAR subcommittee. Pharmacol. Rev. 1994;46:143–156. [PMC free article] [PubMed] [Google Scholar]
- Guo D, Moro S, Von Kügelgen I, Kim Y-C, Jacobson KA. Recognition of pyridoxal phosphate-related antagonists occurs within transmembrane domains of the human P2Y1 receptor. Faseb J. 1999;12 A465, Abstract 390.9. [Google Scholar]
- Harden TK, Hawkins PT, Stephens L, Boyer JL, Downes P. Phosphoinositide hydrolysis by guanosine 5′-(γ-thio)triphosphate-activated phospholipase C of turkey erythrocyte membranes. Biochem. J. 1988;252:583–593. doi: 10.1042/bj2520583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann C, Moro S, Nicholas RA, Harden TK, Jacobson KA. The role of amino acids of the extracellular loops of the human P2Y1 receptor in surface expression and activation processes. J. Biol. Chem. 1999 doi: 10.1074/jbc.274.21.14639. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Kim Y-C, Wildman SS, Mohanram A, Harden TK, Boyer JL, King BF, Bumstock G. A pyridoxine cyclic-phosphate and its 6-arylazo-derivative selectively potentiate and antagonize activation of P2X1 receptors. J. Med. Chem. 1998;41:2201–2206. doi: 10.1021/jm980183o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Guo D, Lee BX, van Rhee AM, Kim YC, Nicholas R, Schachter J, Harden TK, Jacobson KA. Mutational analysis of residues essential for ligand recognition at the human P2Y1 receptor. Mol. Pharmacol. 1997;52:499–507. doi: 10.1124/mol.52.3.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y-C, Camaioni E, Ziganshin AU, Ji X-J, King BF, Wildman SS, Rychkov A, Yobum J, Kim H, Mohanram A, Harden TK, Boyer JL, Burnstock G, Jacobson KA. Synthesis and structure activity relationships of pyridoxal-6-azoaryl-5′-phosphate and phosphonate derivatives as P2 receptor antagonists. Drug Devel. Res. 1998;45:52–66. doi: 10.1002/(SICI)1098-2299(199810)45:2<52::AID-DDR2>3.0.CO;2-V. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrecht G, Friebe T, Grimm U, Windscheif U, Bungardt E, Hildebrandt C, Baumert HG, Spatzkumbel G, Mutschler E. PPADS, a novel functionally selective antagonist of P2 purinoceptor-mediated responses. Eur J. Pharmacol. 1992;217:217–219. doi: 10.1016/0014-2999(92)90877-7. [DOI] [PubMed] [Google Scholar]
- Moro S, Guo D, Camaioni E, Boyer JL, Harden TK, Jacobson KA. Human P2Y1 receptor: Molecular modeling and site-directed mutagenesis as tools to identify agonist and antagonist recognition sites. J. Med. Chem. 1998;41:1456–1466. doi: 10.1021/jm970684u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro S, Hoffmann C, Jacobson KA. Role of the extracellular loops of G protein-coupled receptors in ligand recognition: A molecular modeling study of the human P2Y1 receptor. Biochemistry. 1999;38:3498–3507. doi: 10.1021/bi982369v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandanan E, Camaioni E, Jang SY, Kim Y-C, Cristalli G, Herdewijn P, Secrist JA, Tiwari KN, Mohanram A, Harden TK, Boyer JL, Jacobson KA. Structure activity relationships of bisphosphate nucleotide derivatives as P2Y1 receptor antagonists and partial agonists. J. Med. Chem. 1999 doi: 10.1021/jm980657j. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RA, Barnard EA. Nucleotide receptors. Current Opinion in Neurobiol. 1997;7:346–357. doi: 10.1016/s0959-4388(97)80062-1. [DOI] [PubMed] [Google Scholar]
- Robaye B, Boeynaems J-M, Communi D. Slow desensitization of the human P2Y6 receptor. Eur J. Pharmacol. 1997;329:231–236. [PubMed] [Google Scholar]
- Schachter JB, Li Q, Boyer JL, Nicholas RA, Harden TK. Second messenger cascade specificity and pharmacological selectivity of the human P2Y1-purinoceptor. Br. J. Pharmacol. 1996;118:167–173. doi: 10.1111/j.1476-5381.1996.tb15381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rhee AM, Jacobson KA. Molecular architecture of G protein-coupled receptors. Drug Devel. Res. 1996;37:1–38. doi: 10.1002/(SICI)1098-2299(199601)37:1<1::AID-DDR1>3.0.CO;2-S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rhee AM, Jacobson KA, Garrad R, Weisman GA, Erb L. P2 receptor modeling and identification of ligand binding sites. In: Turner JT, Weisman G, Fedan J, editors. The P2 Nucleotide Receptors, in the series “The Receptors”. Humana Press; Clifton, New Jersey: 1997. pp. 135–166. [Google Scholar]