

Summary
DNA assembler enables rapid construction and engineering of biochemical pathways in a one-step fashion by exploitation of the in vivo homologous recombination mechanism in Saccharomyces cerevisiae. It has many applications in pathway engineering, metabolic engineering, combinatorial biology, and synthetic biology. Here we use two examples including the zeaxanthin biosynthetic pathway and the aureothin biosynthetic gene cluster to describe the key steps in the construction of pathways containing multiple genes using the DNA assembler approach. Methods for construct design, pathway assembly, pathway confirmation, and functional analysis are shown. The protocol for fine genetic modifications such as site-directed mutagenesis for engineering the aureothin gene cluster is also illustrated.
Keywords: DNA assembler, In vivo homologous recombination, Pathway engineering, Synthetic biology, Metabolic engineering, Gene cluster characterization and engineering
1. Introduction
Methods that enable rapid construction and engineering of biochemical pathways are invaluable in pathway engineering, metabolic engineering, combinatorial biology and synthetic biology (1-6). In all these studies, the conventional multi-step sequential cloning method is typically used, which includes primer design, PCR amplification, restriction digestion, in vitro ligation and transformation (7, 8). Moreover, multiple plasmids are often required (7, 8). This method is not only time-consuming and inefficient but also relies on unique restriction sites that become limited for large recombinant DNA molecules.
Thanks to its high efficiency and ease to work with, in vivo homologous recombination in yeast has been widely used for gene cloning, plasmid construction and library creation (9-12). Recently, we developed a new method, called “DNA assembler,” which enables design and rapid construction of large biochemical pathways in a one-step fashion by exploitation of the in vivo homologous recombination mechanism in S. cerevisiae (13). This method is highly efficient and circumvents many potential problems associated with conventional cloning methods such as requirement of time-consuming, multi-step procedures, low efficiency, and dependence on unique restriction sites. Therefore, it represents a versatile approach for construction of biochemical pathways for synthetic biology, metabolic engineering, and functional genomics studies.
In addition to assembling biochemical pathways, DNA assembler also offers unprecedented flexibility and versatility in characterizing and engineering natural product gene clusters (14). Microorganisms and plants have evolved to produce a myriad array of complex molecules known as natural products or secondary metabolites that are of biomedical and biotechnological importance (15-17). Sequenced genomes and metagenomes represent a tremendously rich source for discovery of novel pathways involved in natural product biosynthesis (18, 19). Over the last two decades, the complete genome sequences of more than 1900 organisms have been determined, with more than 10,000 organisms in the pipeline (http://www.genomesonline.org/cgi-bin/GOLD/bin/gold.cgi). However, only a tiny fraction of the biosynthetic pathways from these organisms have been characterized, and discovery and sustainable production of natural products are often hampered by our limited ability to manipulate the corresponding biosynthetic pathways. Using the DNA assembler approach, we developed a genomics-driven, synthetic biology-based strategy for rapid characterization and engineering of natural product biosynthetic pathways (14).
Here we use the zeaxanthin biosynthetic pathway and the aureothin biosynthetic cluster as two examples to illustrate the experimental procedures. Fig. 1 shows the scheme for constructing the zeaxanthin biosynthetic pathway. Briefly, for each individual gene in the zeaxanthin pathway, an expression cassette including a promoter, a structural gene, and a terminator is PCR-amplified and assembled using overlap extension PCR (OE-PCR) (20). The 5′ end of the first gene expression cassette is designed to overlap with a vector, while the 3′ end is designed to overlap with the second cassette. Each successive cassette is designed to overlap with the two flanking ones, and the 3′ end of the last cassette overlaps with the vector. All overlaps are designed to be at least 50 bp for efficient in vivo homologous recombination (see Note 1). The resulting multiple expression cassettes are co-transformed into S. cerevisiae with the linearized vector through electroporation, which allows the entire pathway to be assembled into a vector. Restriction digestion or DNA sequencing is subsequently used to verify the correctly assembled pathway, after which the cells carrying the correct construct are checked for zeaxanthin production.
Figure 1.

(a) Preparation of each gene expression cassette using OE-PCR. Promoters (Px, Px+1), genes (Gx, Gx+1), and terminators (Tx, Tx+1) are individually PCR-amplified and joined together by OE-PCR. The resulting two cassettes are fused through the in vivo homologous recombination (HR) process. To generate an overlap of approximately 50 bp, the reverse primer used to amplify Tx contains a sequence of the first 20-25 nucleotides of Px+1, and the forward primer used to amplify Px+1 contains a sequence of the last 20-25 nucleotides of Tx. (b) One-step method for assembly of a biochemical pathway using in vivo homologous recombination (HR) in S. cerevisiae.
For engineering the aureothin gene cluster, as shown in Fig. 2, pathway fragments encoding the aureothin biosynthetic pathway are amplified from the isolated genomic DNA of the native aureothin producer Streptomyces thioluteus. The helper fragments carrying the genetic elements needed for DNA maintenance and replication in S. cerevisiae, E. coli, and the target heterologous expression host Streptomyces lividans (see Note 2) are amplified from the corresponding vectors. Since PCR primers are designed to generate an overlap region between two adjacent fragments (see Note 3), these fragments will be assembled into a single DNA molecule in S. cerevisiae through homologous recombination after co-transformation. The isolated plasmids are transformed into E. coli for plasmid enrichment and verification, and the correct construct is transformed into S. lividans for heterologous expression of the aureothin biosynthetic pathway. Of special note, because the pathway fragments can be readily modified by PCR, various sophisticated genetic manipulations such as point mutagenesis and scarless gene substitution and deletion can be easily performed to confirm gene function, locate key amino acid residues, study biosynthetic mechanisms, express biosynthetic pathways heterologously, and generate new derivatives (see Note 4).
Figure 2.
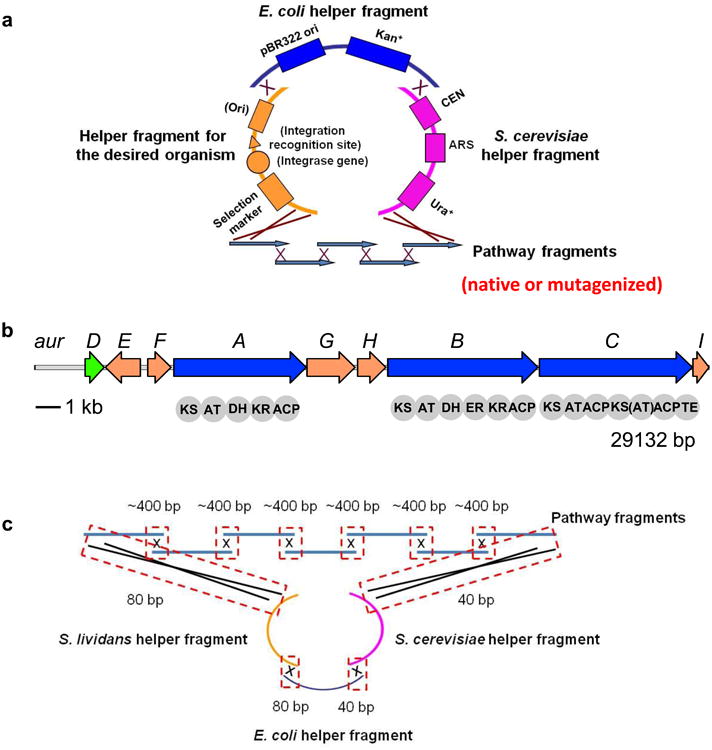
(a) The DNA assembler-based strategy for efficient manipulation of natural product biosynthetic pathways. Various genetic modifications are introduced in the pathway fragments to be assembled. (b) The aureothin biosynthetic gene cluster from S. thioluteus. (c) The overlap lengths between adjacent fragments in assembly.
2. Materials
Prepare all solutions using ultrapure water, prepared by purifying deionized water to attain a resistivity of 18.2 mΩ cm at 25 °C. Prepare and store all reagents at room temperature unless indicated otherwise.
2.1. DNA preparation
pRS416: Obtained from New England Biolabs (Beverly, MA, USA) and serves as the template for amplifying the S. cerevisiae helper fragment for assembling the aureothin gene cluster variants.
pRS416m: pRS416 with a hisG sequence and a delta2 (21) sequence that flank the multiple cloning site and serves as the vector for assembly of the zeaxanthin pathway (Fig. 3a) (see Note 5).
pCAR-ΔCrtX: It contains the genes crtE, crtB, crtI, crtY and crtZ from Erwinia uredovora for zeaxanthin biosynthesis (Prof. E.T. Wurtzel, City University of New York, NY, USA) (22-24).
S. thioluteus strain: Obtained from the American Tissue Culture Collection (ATCC 12310, Manassas, VA)
The genomic DNA of S. thioluteus: Isolated from S. thioluteus using Wizard Genomic DNA Isolation Kit (Promega, Madison, WI, USA).
pAE4: A Streptomyces-E. coli shuttle vector obtained from Professor William Metcalf (University of Illinois, Urbana, IL, USA) and serves as the template for amplifying the S. lividans helper fragment and the E. coli helper fragment for assembly of the aureothin gene cluster (the complete sequence of the plasmid can be obtained by request from the authors).
E. coli strain WM6026: Obtained from Professor William Metcalf and serves as the donor strain for conjugating plasmids to S. lividans (the details of strain construction can be obtained by request from the authors).
0.5 M ethylenediamine tetraacetic acid (EDTA) solution (pH 8.0): For a 500 mL of stock solution of 0.5 M EDTA, weigh out 93.05 g of EDTA disodium salt (MW = 372.2) and dissolve it in 400 mL of deionized water. Adjust to pH 8.0 with NaOH and correct the final volume to 500 mL. EDTA will not be dissolved completely in water unless the pH is adjusted to about 8.0.
Concentrated stock solution of TAE (50×): Weigh 242 g of Tris base (MW = 121.14) and dissolve it in approximately 750 mL of deionized water. Carefully add 57.1 mL of glacial acid and 100 mL of 0.5 M EDTA, and adjust the solution to a final volume of 1 L. This stock solution can be stored at room temperature. The pH of this buffer is not adjusted and should be about 8.5.
Working solution of TAE buffer (1×): Dilute the stock solution by 50 fold with deionized water. Final solute concentrations are 40 mM Tris acetate and 1 mM EDTA.
0.7% Agarose gel in 1× TAE buffer: Add 0.7 g of agarose into 100 mL of 1× TAE buffer and microwave until agarose is completely melted. Cool the solution to approximately 70-80 °C. Add 5 μL of ethidium bromide into the solution and mix well. Pour 25-30 mL of solution onto an agarose gel rack with appropriate 2-well or 8-well combs.
Wizard Genomic DNA Isolation Kit (Promega, Madison, WI, USA).
QIAquick Gel Extraction Kit (QIAGEN, Valencia, CA, USA).
QIAprep Miniprep Kit (QIAGEN, Valencia, CA, USA).
DNA polymerase: Any polymerase with high fidelity can be used.
Failsafe PCR 2× Premix G: Containing dNTPs and PCR reaction buffer (EPICENTRE Biotechnologies, Madison, WI, USA).
5× Phusion GC Reaction Buffer (New England Biolabs, Beverly, MA, USA).
40× dNTPs premix: 10 mM each nucleotide.
Dimethyl sulfoxide (DMSO).
BamHI restriction enzyme (New England Biolabs, Ipswich, MA, USA).
3 M sodium acetate pH 5.0: Weigh 12.3 g of sodium acetate (MW=82.03) and dissolve it into 50 mL of deionized water. Adjust to pH 5.0 by HCl.
10 mg/mL glycogen: Dissolve 10 mg of glycogen in 1 mL of deionized water.
NanoDrop2000c: Used to measure the concentration of DNA and check the OD600 of the cells (Thermo Scientific, Wilmington, DE, USA).
Benchtop centrifuges to separate cells and supernatant.
Molecular imager gel doc: Used to check DNA on the agarose gel (Bio-Rad, Hercules, CA, USA).
Figure 3.
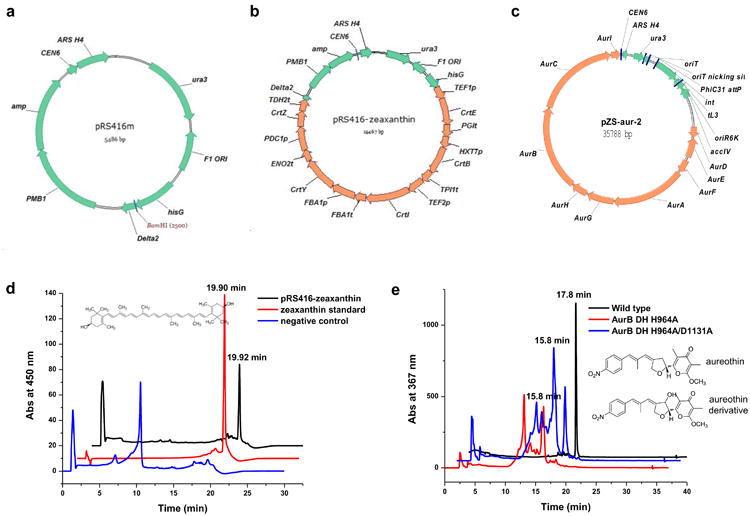
(a) The vector map of pRS416m. (b) The vector map of the construct pRS416m-zeaxanthin. CEN6: centromere; ARS H4: automatic replication sequence; ura3: selection marker in S. cerevisiae; amp: selection marker in E. coli; hisG and delta2: two regions flanking the zeaxanthin biosynthetic pathway; PMB1: E. coli origin of replication. F1 ORI: this region is contained in the original pRS416 vector, but is not required for the construction of the plasmid containing the zeaxanthin biosynthetic pathway. (c) The vector map of the construct pZS-aur-2. oriR6K: E. coli origin of replication; accIV: apramycin resistance gene (it can be used as the selection marker in both Streptomyces and E. coli); oriT: conjugal transfer ori; PhiC31 attP: the ϕC31 recognition site; int: the ϕC31 integrase; tL3: a terminator.
2.2. Transformation
S. cerevisiae HZ848 (MATα, ade2-1, Δura3, his3-11, 15, trp1-1, leu2-3, 112, can1-100): Used as the host for DNA assembly (see Note 6).
YPAD medium: Dissolve 6 g of yeast extract, 12 g of peptone, 12 g of dextrose, and 60 mg of adenine hemisulphate in 600 mL of deionized water. Autoclave at 121 °C for 15 min.
Synthetic complete drop-out medium lacking uracil (SC-Ura): Dissolve 3 g of ammonium sulfate, 1 g of yeast nitrogen source without ammonium sulfate and amino acids, 0.5 g of complete synthetic medium minus uracil (CSM-Ura; MP Biomedicals, Solon, OH, USA), 26 mg of adenine hemisulphate, and 12 g of dextrose in 600 mL of deionized water, and adjust the pH to 5.6 by NaOH. Autoclave at 121 °C for 15 min.
SC-Ura-agar: SC-Ura and 20 g/L of agar.
1 M sorbitol solution: Dissolve 91.1 g of sorbitol (MW = 182.17) in 400 mL of deionized water and adjust to a final volume of 500 mL. Sterilize the solution by filtering it through a filter with a pore size of 0.22 μm.
Gene Pulser II and Pulse controller plus: used to transform plasmids into S. cerevisiae and E. coli through electroporation (Bio-Rad, Hercules, CA, USA).
2.3. Verification of the clones
Zymoprep II yeast plasmid miniprep (Zymo Research, Orange, CA, USA).
E. coli strain BW25141 (F-, Δ(araD-araB)567, ΔlacZ4787(∷rrnB-3), Δ(phoB-phoR)580, λ-, galU95, ΔuidA3∷pir+, recA1, endA9(del-ins)∷FRT, rph-1, Δ(rhaD-rhaB)568, hsdR514): Work as a standard cloning strain (obtained from Professor William Metcalf at the University of Illinois, Urbana, IL).
ScaI, PsiI, SacI and AscI restriction enzymes (New England Biolabs, Ipswich, MA, USA) (see Note 7).
1 M glucose solution: Dissolve 90 g of D-glucose in 400 mL of deionized water and adjust to a final volume of 500 mL. Filter-sterilize it.
SOC medium: Add 20 g of Bacto-tryptone, 5 g of yeast extract, 0.5 g of NaCl, 186.4 mg of KCl into 980 mL of deionized water. Adjust the pH to 7.0 with NaOH. Autoclave at 121 °C for 15 min. After the solution cools down to 70-80 °C, add 20 mL of sterile 1 M glucose.
100 mg/mL ampicillin stock solution: Dissolve 1 g of ampicillin powder in 10 mL of deionized water and filter-sterilize it.
50 mg/mL apramycin stock solution: Dissolve 0.5 g of apramycin powder in 10 mL of deionized water and filter-sterilize it.
LB broth: Add 10 g of bacto-tryptone, 5 g of yeast extract, 10 g of NaCl into 1 L of deionized water. Autoclave at 121 °C for 15 min.
LB agar: LB broth and 20 g/L agar.
LB-Amp+ agar plates: Autoclave LB-agar and when the solution cools down to 70-80 °C, add 1 mL of 100 mg/mL ampicillin to 1 L of LB-agar. Pour 20-25 mL into each Petri dish.
LB-Apr+ agar plates: Autoclave LB-agar and when the solution cools down to 70-80 °C, add 1 mL of 50 mg/mL apramycin to 1 L of LB-agar. Pour 20-25 mL into each Petri dish.
2.4. Detection of zeaxanthin
0.1% trifluoroacetic acid (TFA) buffer: Add 1 mL of TFA into 999 mL of deionized water.
10 μg/mL zeaxanthin standard solution: Dissolve 1 mg of zeaxanthin (Sigma, St Louis, MO, USA) in 100 mL of methanol.
French pressure cell press: Used to lyse the yeast cells.
Rotary evaporator R-205: Used to evaporate the solvent.
High performance liquid chromatography (HPLC) equipment.
ZORBAX SB-C18 column (Agilent Technologies, Palo Alto, CA, USA).
2.5. Conjugation of plasmids into S. lividans
3.8 mg/mL 2,6-diaminopimelic acid (DAP) stock solution: Dissolve 38 mg of DAP powder in 10 mL of deionized water and filter-sterilize it.
LB-Apr+-DAP plates: LB-Apr+ plates and 19 μg/mL DAP.
A mixture of nalidixic acid and apramycin each at the concentration of 1 mg/mL: Dissolve 20 mg of nalidixic acid powder in 15 mL of deionized water. Nalidixic acid will not be dissolved completely in water unless the pH is adjusted to approximately 12.0. Slowly adjust pH to 9.0∼9.5 with 1 M HCl and add 400 μL of 50 mg/mL apramycin. The final pH after supplementing apramycin should drop to approximately 7.8-8.2. Correct the final volume to 20 mL and filter-sterilize it.
S. lividans spores: Prepared according to the protocol in the book “Practical Streptomyces Genetics”, second edition, page 44-47 (25).
R2 no-sucrose agar plates: Add 0.25 g of K2SO4, 10.12 g of MgCl2·6H2O, 10 g of glucose, 0.1 g of Difco casamino acids, 5.73 g of TES into 990 mL of deionized water. Autoclave at 121 °C for 15 min. After the solution cools down to 70-80 °C, add 1 mL of 5 mg/mL KH2PO4, 8 mL of 36.8 mg/mL CaCl2·2H2O, 1.5 mL of 0.2 g/mL L-proline and 0.5 mL of 1M NaOH (All these solutions need to be filter-sterilized in advance). Pour 20-25 mL into each Petri dish. Please refer to the recipe in the book “Practical Streptomyces Genetics”, page 408 (25).
ISP2-Apr+ agar plates: Add 10 g of malt extract, 4 g of yeast extract, 4 g of glucose, and 20 g of agar into 1 L of deionized water. Adjust pH to 7.2 and autoclave at 121 °C for 15 min. When the solution cools down to 70-80 °C, add 1 mL of 50 mg/mL apramycin. Mix well and pour 20-25 mL into each Petri dish.
2.6. Culturing of the aureothin pathway variants and detection of aureothin and its derivatives
MYG medium: Add 10 g of malt extract, 4 g of yeast extract, and 4 g of glucose into 1 L of deionized water. Adjust pH to 7.2 and autoclave at 121 °C for 15 min.
Ethyl acetate
100 μg/mL aureothin standard solution: Dissolve 1 mg of aureothin (BioAustralis, Smithfield, Australia) in 10 mL of methanol.
Agilent 1100 series LC/MSD XCT plus ion trap mass spectrometer (Agilent, Palo Alto, CA)
3. Methods
3.1. DNA preparation for assembling the zeaxanthin biosynthetic pathway
Amplify the genes crtE, crtB, crtI, crtY and crtZ from the plasmid pCAR-ΔCrtX and amplify the corresponding promoter and terminator from the genomic DNA of S. cerevisiae using the primers listed in Table 1. Set up the reaction mixtures as follows: 50 μL of FailSafe PCR 2×PreMix G, 2.5 μL of forward primer (20 pmol/μL), 2.5 μL of reverse primer (20 pmol/μL), 1 μL of template (10-50 ng of S. cerevisiae genomic DNA or the plasmid pCAR-ΔCrtX), 1 μL of DNA polymerase, and 43 μL of ddH2O in a total volume of 100 μL.
PCR condition: Fully denature at 98 °C for 30 s, followed by 25 cycles of 98 °C for 10 s, 58 °C for 30 s and 72 °C for 1 min, with a final extension at 72 °C for 10 minutes.
Load the 100 μL of PCR products onto 0.7% agarose gels and perform electrophoresis at 120 V for 20 minutes.
Gel-purify PCR products using the QIAquick Gel Extraction Kit.
Check the concentrations of the purified products using NanoDrop.
Perform OE-PCR to generate each gene expression cassette (see Note 8). Set up the first-step reaction mixture as follows: 10 μL of FailSafe PCR 2×PreMix G, 100 ng of promoter fragment, 100 ng of crt gene fragment, 100 ng of terminator fragment, and 0.2 μL of DNA polymerase. Add ddH2O to a final volume of 20 μL.
Reaction condition: Fully denature at 98 °C for 30 s, followed by 10 cycles of 98 °C for 10 s, 58 °C for 30 s and 72 °C for 1 min, with a final extension at 72 °C for 10 minutes.
Set up the second-step reaction mixture as follows: 50 μL of FailSafe PCR 2× PreMix G, 10 μL of first-step reaction mixture, 1 μL of DNA polymerase, 2.5 μL of forward primer (20 pmol/μL), 2.5 μL of reverse primer (20 pmol/μL), and 34 μL ddH2O in a total volume of 100 μL.
Reaction condition: Fully denature at 98 °C for 30 s, followed by 25 cycles of 98 °C for 10 s, 58 °C for 30 s and 72 °C for 1 min, with a final extension at 72 °C for 10 minutes.
Digest pRS416m by BamHI at 37 °C for 3 hours. Digestion condition: 5 μL of 10× buffer, 0.5 μL of 100× BSA, 3 μg of pRS416m, and 30 units of BamHI. Add ddH2O to a final volume of 50 μL.
Load the PCR and digestion products onto 0.7% agarose gels and perform electrophoresis at 120 V for 20-30 minutes.
Gel-purify the PCR and digestion products using the QIAquick Gel Extraction Kit.
Check the concentrations of the purified products using NanoDrop.
Take 200-300 ng of each fragment, mix in a tube and calculate the final volume.
Add 10% v/v 3 M sodium acetate and 2% v/v 10 mg/mL glycogen (e.g. if there is 100 μL of mixture, add 10 μL of sodium acetate and 2 μL of glycogen) and mix well.
Add 2× v/v 100% ethanol (e.g. if the final volume is about 110 μL, add 220 μL ethanol) and mix well.
Store the DNA mixture at -80 °C for at least an hour.
Centrifuge at 4 °C, 13,200 rpm for 20 minutes. Usually the precipitated DNA can be seen at the bottom of the tube.
Remove the supernatant completely (do not touch the DNA).
Add 500 μL of 70% ethanol to wash the DNA pellet, and centrifuge at room temperature, 13,200 rpm for 3 minutes.
Remove the ethanol completely and air dry the pellet for 1-2 minutes (do not over-dry it).
Resuspend the DNA pellet in 4 μL of ddH2O. Now the DNA is ready for transformation (see Note 9).
Table 1.
The primers used in assembling the zeaxanthin pathway in pRS416m.
| Name | Sequence |
|---|---|
| hisG-f | GGCCAGTGAGCGCGCGTAATACGACTCACTATAGGCGCGCCTGCGTGAAGTCGAAG |
| hisG-r | GGAGTAGAAACATTTTGAAGCTATTTCCAGTCAATCAGGGTATTG |
| TEF1p-f | CTTCAATACCCTGATTGACTGGAAATAGCTTCAAAATGTTTCTACTC |
| TEF1p-r | GAACGTGTTTTTTTGCGCAGACCGTCATTTTGTAATTAAAACTTAGATTAG |
| CrtE-f | CTAATCTAAGTTTTAATTACAAAATGACGGTCTGCGCAAAAAAACACGTTC |
| CrtE-r | GGTATATATTTAAGAGCGATTTGTTTAACTGACGGCAGCG |
| PGIt-f | CGCTGCCGTCAGTTAAACAAATCGCTCTTAAATATATACC |
| PGIt-r | CCGAAATTGTTCCTACGAGAAGTGGTATACTGGAGGCTTCATGAGTTATG |
| HXT7p-f | CATAACTCATGAAGCCTCCAGTATACCACTTCTCGTAGGAACAATTTCGG |
| HXT7p-r | GAGTAACGACGGATTATTCATTTTTTGATTAAAATTAAAAAAAC |
| CrtB-f | GTTTTTTTAATTTTAATCAAAAAATGAATAATCCGTCGTTACTC |
| CrtB-r | GATAATATTTTTATATAATTATATTAATCCTAGAGCGGGCGCTGCCAGAG |
| TPI1t-f | CTCTGGCAGCGCCCGCTCTAGGATTAATATAATTATATAAAAATATTATC |
| TPI1t-r | CTATATGTAAGTATACGGCCCTATATAACAGTTGAAATTTGG |
| TEF2p-f | CCAAATTTCAACTGTTATATAGGGCCGTATACTTACATATAG |
| TEF2p-r | CACCAATTACCGTAGTTGGTTTCATGTTTAGTTAATTATAGTTCGTTG |
| CrtI-f | CAACGAACTATAATTAACTAAACATGAAACCAACTACGGTAATTGGTG |
| CrtI-r | CTCATTAAAAAACTATATCAATTAATTTGAATTAACTCATATCAGATCCTCCAGCATC |
| FBA1t-f | GATGCTGGAGGATCTGATATGAGTTAATTCAAATTAATTGATATAGTTTTTTAATGAG |
| FBA1t-r | GTTCAAGCCAGCGGTGCCAGTTGGAGTAAGCTACTATGAAAGACTTTAC |
| FBA1p-f | GTAAAGTCTTTCATAGTAGCTTACTCCAACTGGCACCGCTGGCTTGAAC |
| FBA1p-r | CAGATCATAATGCGGTTGCATTTTGAATATGTATTACTTGGTTATGG |
| CrtY-f | CCATAACCAAGTAATACATATTCAAAATGCAACCGCATTATGATCTG |
| CrtY-r | CTAATAATTCTTAGTTAAAAGCACTTTAACGATGAGTCGTCATAATGG |
| ENO2t-f | CCATTATGACGACTCATCGTTAAAGTGCTTTTAACTAAGAATTATTAG |
| ENO2t-r | GGAACATATGCTCACCCAGTCGCATGAGGTATCATCTCCATCTCCCATATG |
| PDC1p-f | CATATGGGAGATGGAGATGATACCTCATGCGACTGGGTGAGCATATGTTCC |
| PDC1p-r | GGCATTCCAAATCCACAACATTTTGATTGATTTGACTGTG |
| CrtZ-f | CACAGTCAAATCAATCAAAATGTTGTGGATTTGGAATGCC |
| CrtZ-r | CATTAAAGTAACTTAAGGAGTTAAATTTACTTCCCGGATGCGGGCTC |
| TDH2t-f | GAGCCCGCATCCGGGAAGTAAATTTAACTCCTTAAGTTACTTTAATG |
| TDH2t-r | GATCCGTTAGACGTTTCAGCTTCCAGCGAAAAGCCAATTAGTGTGATAC |
| Delta2-f | GTATCACACTAATTGGCTTTTCGCTGGAAGCTGAAACGTCTAACGGATC |
| Delta2-r | TTACGCCAAGCGCGCAATTAACCCTCACTAAAGGCGCGCCGAGAACTTCTAGTATATTC |
3.2. DNA preparation for assembling the aureothin gene cluster variants
Amplify the aureothin biosynthetic pathway fragments from the genomic DNA of S. thioluteus and amplify the S. cerevisiae, E. coli and S. lividans helper fragments from the corresponding vectors using the primers listed in Table 2. For constructing the mutant pathways, mutations are added into the corresponding primers. Set up the reaction for amplifying the S. cerevisiae helper fragment as follows: 20 μL of Buffer GC, 2.5 μL of dNTP premix, 2.5 μL of forward primer (20 pmol/μL), 2.5 μL of reverse primer (20 pmol/μL), 1 μL of template pRS416 at the concentration of 50-100 ng/μL, 1 μL of DNA polymerase, and 70.5 μL of ddH2O in a total volume of 100 μL (see Note 10). Set up all the other reaction mixtures as follows: 50 μL of FailSafe PCR 2× PreMix G, 2.5 μL of forward primer (20 pmol/μL), 2.5 μL of reverse primer (20 pmol/μL), 1 μL of template (10-50 ng of S. thioluteus genomic DNA or the plasmid pAE4), 1 μL of DNA polymerase, 5 μL of DMSO (see Note 11) and 38 μL of ddH2O in a total volume of 100 μL.
PCR condition for amplifying the S. cerevisiae helper fragment: Fully denature at 98 °C for 30 s, followed by 25 cycles of 98 °C for 10 s, 45 °C for 30 s and 72 °C for 1 min, with a final extension at 72 °C for 10 minutes (see Note 10). PCR condition for all the other fragments: Fully denature at 98 °C for 30 s, followed by 25 cycles of 98 °C for 10 s, 58 °C for 30 s and 72 °C for 1-3 min, with a final extension at 72 °C for 10 minutes (see Note 11).
Load the 100 μL of PCR products onto 0.7% agarose gels and perform electrophoresis at 120 V for 20 minutes.
Gel-purify PCR products using the QIAquick Gel Extraction Kit.
Check the concentrations of the purified products using NanoDrop.
Take 200-300 ng of each fragment, mix in a tube, and calculate the final volume.
Add 10% v/v 3 M sodium acetate and 2% v/v 10 mg/mL glycogen (e.g. if there is 100 μL of mixture, add 10 μL of sodium acetate and 2 μL of glycogen) and mix well.
Add 2× v/v 100% ethanol (e.g. if the final volume is about 110 μL, add 220 μL ethanol) and mix well.
Store the DNA mixture at -80 °C for at least an hour.
Centrifuge at 4 °C, 13,200 rpm for 20 minutes. Usually the precipitated DNA can be seen at the bottom of the tube.
Remove the supernatant completely (do not touch the DNA).
Add 500 μL of 70% ethanol to wash the DNA pellet, and centrifuge at room temperature, 13,200 rpm for 3 minutes.
Remove the ethanol completely and air dry the pellet for 1-2 minutes (do not over-dry it).
Resuspend the DNA pellet by 4 μL of ddH2O. Now the DNA is ready for transformation (see Note 9).
Table 2.
The primers used in assembling the aureothin gene cluster variants. The mutated codons are underlined.
| Name | Sequence |
|---|---|
| Aur-1-for | TGGTACTGCAAATACGGCATCAGTTACCGTGAGCAGATCGGATCAGCTCGTCCCGTTCGG |
| Aur-1-rev | GCTGCTCTTCTCGCATCGTC |
| Aur-2-for | CGTAGAGGAGCTCCAGCAGC |
| Aur-2-rev | CTCCTCCAGCACCTCGCAGC |
| Aur-3-for | TCCTGACCTTCGACTCGCTG |
| Aur-3-rev | CATGTTCGATCCTTCCGTTG |
| Aur-4-for | GACGGTGCACCAGCTGGTCA |
| Aur-4-rev | GTTGCCGGTCATGTGGTAGC |
| Aur-5-for | ATGACCAATGACGCCAAGAC |
| Aur-5-rev | CCGTCCATCAGGTCGAACGC |
| Aur-6-for | CCTACTACGGCCTGGTGGAC |
| Aur-6-rev | CATCGCCGTCATCGAGACGA |
| Aur-7-for | GCAACGAAGGACATGTCCAG |
| Aur-7-rev | TTATAGCACGTGATGAAAAGGACCCAGGTGGCACTTTTCGTCAGTCAGTCGTCCAGGCGC |
| Yeast-for | CGAAAAGTGCCACCTGGGTC |
| Yeast-rev | AATATTGTGAGTTTAGTATACATGCA |
| Strep-for | GTATTATAAGTAAATGCATGTATACTAAACTCACAATATT ATGGCGCGCCGACGTGCTCA |
| Strep-rev | ATTAGCCATGGCATCACAGTATCGTGATGACATTAATTAACGCAATCCAGTGCAAAGCTA |
| E. coli-for | AGACGAAGAAGCTAGCTTTGCACTGGATTGCGTTAATTAATGTCATCACGATACTGTGAT |
| E. coli-rev | GCCCATGACCACCGTCGTCTCCGAACGGGACGAGCTGATCCGATCTGCTCACGGTAACTG |
| AurB-DH-H964A-for | TGTCGGCGCGGACCGAGTCCTGGCTGGCCGACGCCGTCGTGCTCGGCTCCACGCTCGTCC |
| AurB-DH-H964A-rev | GGACGAGCGTGGAGCCGAGCACGACGGCGTCGGCCAGCCAGGACTCGGTCCGCGCCGACA |
| AurB-DH-D1131A-for | GCTACGGCGTCCACCCCGCGCTCCTCGCCGCCGCACTGCACACCGCCCTCCTGAAGGAGG |
| AurB-DH-D1131A-rev | CCTCCTTCAGGAGGGCGGTGTGCAGTGCGGCGGCGAGGAGCGCGGGGTGGACGCCGTAGC |
3.3. Transformation
Inoculate a single colony of HZ848 into 3 mL of YPAD medium and grow overnight in a shaker at 30 °C and 250 rpm.
Measure the OD600 of the seed culture and inoculate the appropriate amount to 50 mL of fresh YPAD medium to obtain an OD600 of 0.2 (e.g. if the overnight culture has an OD600 of 10, then add 1 mL into 50 mL of fresh YPAD medium).
Continue growing the 50 mL of culture for approximately 4 hours to obtain an OD600 of 0.8 (see Note 12).
Spin down the yeast cells at 4 °C, 4,000 rpm for 10 minutes and remove the spent medium.
Use 50 mL of ice cold ddH2O to wash the cells once and centrifuge again.
Discard water, add 1 mL of ice-cold ddH2O to resuspend the cells and move them to a sterile Eppendorf tube.
Spin down the cells using a bench top centrifuge for 30 s at 4 °C, 7,000 rpm.
Remove water and use 1 mL of 1 M ice-cold sorbitol to wash the cells once (now the cells look slightly yellow). Centrifuge again and remove the sorbitol.
Resuspend the cells in 250-300 μL of chilled 1 M sorbitol and distribute them into 50 μL aliquots.
Now each 50 μL of cells is ready for electroporation (see Note 13). Mix the 4 μL of DNA with 50 μL of yeast cells and put the mixture into a chilled electroporation cuvette.
Electroporate the cells at 1.5 kV, and quickly add 1 mL of pre-warmed (30 °C) YPAD medium to resuspend cells (see Note 14).
Grow in a shaker at 30 °C, 250 rpm for 1 hour.
Spin down the cells in a sterile tube at 13,200 rpm for 30 s and remove the YPAD medium.
Use 1 mL of room temperature sorbitol solution to wash the cells two to three times and finally resuspend the cells in 1 mL sorbitol.
Spread 100 μL of resuspended cells onto SC-Ura plates.
Incubate the plates at 30 °C for 2-3 days until colonies appear.
3.4. Verification of the correctly assembled pathways
Randomly pick 10 colonies from the SC-Ura plate and inoculate each colony into 1.5 mL of SC-Ura liquid medium. Grow at 30 °C for 1.5 days (see Note 15).
Purify yeast plasmid DNA from each 1.5 mL of culture using the Zymoprep II kit.
Mix 2 μL of isolated plasmid with 50 μL of E. coli BW25141 cells and put the mixture into a chilled electroporation cuvette (see Note 16).
Electroporate the cells at 2.5 kV, and quickly add 1 mL of SOC medium to resuspend the cells (see Note 14).
Grow in a shaker at 37 °C, 250 rpm for 1 hour.
Spin down the cells, remove 800 μL of SOC medium, resuspend the pellet with the remaining 200 μL of SOC medium and spread the cells on a LB-Amp+ plate for the zeaxanthin construct or on a LB-Apr+ plate for the aureothin constructs.
Incubate the plates at 37 °C for 16-18 hours until colonies appear (see Note 17).
Inoculate a single colony from each plate to 5 mL of LB supplemented with 100 μg/mL ampicillin (the zeaxanthin construct) or 50 μg/mL apramycin (the aureothin constructs), and grow at 37 °C for 12-16 hours.
Purify E. coli plasmids from each 5 mL of culture using the QIAgen Miniprep kit.
Check the plasmid concentrations by NanoDrop.
Verify the correctly assembled pathway through two separate restriction digestion reactions for each construct (see Note 7).
Digestion condition by ScaI at 37 °C for 3 hours: 1.5 μL of 10× buffer, 0.15 μL of 100× BSA, 300 ng of plasmid, and 5 units of ScaI. Add ddH2O to a final volume of 15 μL. Expected bands for the assembled zeaxanthin pathway (Fig. 3b): 1750 bp, 2131 bp, 2628 bp, 3223 bp, 5735 bp.
Digestion condition by PsiI at 37 °C for 3 hours: 1.5 μL of 10× buffer, 0.15 μL of 100× BSA, 300 ng of plasmid, and 5 units of PsiI. Add ddH2O to a final volume of 15 μL. Expected bands for the assembled zeaxanthin pathway (Fig. 3b): 215 bp, 1389 bp, 1689 bp, 1782 bp, 2425 bp, 2752 bp, 5215 bp.
Digestion condition by SacI at 37 °C for 3 hours: 1.5 μL of 10× buffer, 0.15 μL of 100× BSA, 300 ng of plasmid, and 5 units of SacI. Add ddH2O to a final volume of 15 μL. Expected bands for the assembled aureothin gene cluster variants (Fig. 3c): 163 bp, 696 bp, 752 bp, 839 bp, 1207 bp, 1965 bp, 2370 bp, 2956 bp, 3074 bp, 3612 bp, 3693 bp, 5250 bp, 9211 bp.
Digestion condition by AscI at 37 °C for 3 hours: 1.5 μL of 10× buffer, 0.15 μL of 100× BSA, 300 ng of plasmid, and 5 units of AscI. Add ddH2O to a final volume of 15 μL. Expected bands for the assembled aureothin gene cluster variants (Fig. 3c): 487 bp, 1890 bp, 2927 bp, 3154 bp, 3716 bp, 5419 bp, 5433 bp, 5810 bp, and 6952 bp.
3.4. Detection of zeaxanthin
Inoculate a single colony carrying the zeaxanthin biosynthetic pathway into 3 mL of SC-Ura liquid medium and grow at 30 °C, 250 rpm for 1.5 days.
Inoculate 2.5 mL of seed culture into 250 mL of fresh SC-Ura medium and continue to grow at 30 °C, 250 rpm for 4 days.
Cells are collected by centrifugation at 4,000 rpm, resuspended with 5 mL of acetone and lysed by French pressure cell at 10,000 psi.
Supernatants are collected after centrifugation at 13,200 rpm for 3 minutes and evaporated to dryness using rotary evaporator.
After resuspension in 0.5-1 mL of methanol, 100 μL of sample is loaded onto the Agilent ZORBAX SB-C18 column and analyzed at 450 nm by HPLC with a 0.5 mL/min flow rate as follows: buffer A: H2O with 0.1% TFA, buffer B: 100% CH3OH; 0-3 minutes, 60% CH3OH; 3-15 minutes, linear gradient from 60% CH3OH to 100% CH3OH; 15-17 minutes, 100% CH3OH; 17-20 minutes, linear gradient from 100% CH3OH to 60% CH3OH. Authentic zeaxanthin is used as standard, which was eluted at 19.9 minutes.
3.5. Conjugation and heterologous expression of the aureothin pathway variants in S. lividans
Mix 50-100 ng of each verified plasmid with 50 μL of E. coli WM6026 cells and put the mixture into a chilled electroporation cuvette.
Electroporate the cells at 2.5 kV, and quickly add 1 mL of SOC medium and 5 μL of 38 mg/mL DAP to resuspend cells (see Note 14 and 18).
Grow in a shaker at 37 °C, 250 rpm for 1 hour.
Spread 100 μL of each culture on a LB-Apr+-DAP plate.
Incubate the plates at 37 °C for 16 hours until colonies appear.
Inoculate a single colony from each plate to 2 mL of LB supplemented with 50 μg/mL apramycin and 10 μL of 38 mg/mL DAP, and grow at 37 °C for approximately 2 hours until OD600 reaches 0.6∼0.8.
Spin down 100 μL of cell cultures in an Eppendorf tubes and wash the cell pellets each with 1 mL of fresh LB medium.
Spin down the cells, and wash one more time.
Resuspend the cell pellets each with 1 mL of LB.
Mix 2 μL of the resuspended cells with 25 μL of S. lividans spores by pipetting and spot 2 μL of aliquots onto R2 no-sucrose plates. Wait until all the spotted drops are absorbed into the plates.
Incubate the plates at 30 °C for 16-18 hours.
Flood the plates with 2 mL of a mixture of nalidixic acid and apramycin each at a concentration of 1 mg/mL (see Note 19).
Incubate the plates at 30 °C for additional 3-5 days until exconjugants appear, at which point exconjugants are picked and restreaked on ISP2-Apr+ plates and allowed to grow for 2 days.
Inoculate single colonies from the ISP2-Apr+ plates into 10 mL of MYG supplemented with 50 μg/mL apramycin and grow the cultures at 30 °C for 2 days as seed cultures, of which 2.5 mL is subsequently inoculated to 250 mL of fresh MYG supplemented with 50 μg/mL apramycin and grown for another 84 hours.
3.6. Detection of aureothin and its derivatives
Centrifuge the cultures at 4,000 rpm for 10 minutes to remove the cells.
Extract the supernatants with an equal volume of ethyl acetate and evaporate to dryness using rotary evaporator.
Perform LC-MS on an Agilent 1100 series LC/MSD XCT plus ion trap mass spectrometer with an Agilent SB-C18 reverse-phase column. HPLC parameters for detection of aureothin and its derivatives are as follows: solvent A, 1% acetic acid in water; solvent B, acetonitrile; gradient, 10% B for 5 min, to 100% B in 10 min, maintain at 100% B for 5 min, return to 10% B in 10 min and finally maintain at 10% B for 7 min; flow rate 0.3 mL/min; detection by UV spectroscopy at 367 nm. Under such conditions, aureothin and its derivative are eluted at 17.8 min and 15.8 min, respectively. Mass spectra are acquired in ultra-scan mode using electrospray ionization (ESI) with positive polarity. The MS system is operated using a drying temperature of 350 °C, a nebulizer pressure of 35 psi, a drying gas flow of 8.5 L min-1, and a capillary voltage of 4500 V (see Note 20).
Footnotes
If a larger biochemical pathway needs to be assembled, increasing the length of the overlaps between the adjacent fragments is necessary. For example, to assemble a pathway with a size of ∼25 kb, a longer overlap (e.g. 125 bp) could ensure high assembly efficiency (> 50%), while low efficiency (10-20%) is obtained if the length of the overlaps is only 50 bp (13).
S. cerevisiae is used as the assembly host, E. coli serves as the DNA enrichment host, and S. lividans is a widely used heterologous host for studying gene clusters from other Streptomyces species.
As shown in Fig. 2c, to ensure high assembly efficiency, overlaps of 400 bp between internal adjacent pathway fragments are generated. For example, the forward primer for amplifying the second pathway fragment can be located at ∼400 bp upstream of the annealing position of the reverse primer for amplifying the first pathway fragment. Overlaps between other fragments are generated by adding tails to primers, thus they could not be very long. We encountered difficulties in amplifying the S. cerevisaie helper fragment, and in the end, the correct product was only obtained by using the pair of primers without any tails. As a result, the S. cerevisaie helper fragment only overlaps with the last pathway fragment and the adjacent helper fragment with 40 bp. Overlaps of 80 bp are generated between the S. lividans helper fragment and its neighbors.
Site-directed mutagenesis can be easily performed using DNA assembler. Unlike the labor-intensive and time-consuming procedures used in the conventional methods (26-28), DNA assembler only requires adding site-specific mutation(s) into the PCR primers used to generate pathway fragments. As an example, the active sites of the dehydratase domains (DH) of AurB are targeted (Fig. 2b). The motifs HXXXGXXXXP and DXXX(Q/H) were found to be conserved among the DH domains of polyketide synthases and the histidine and the aspartic acid were identified as the catalytic residues (29-31). Based on this information, AurB DH H964A mutant and AurB DH H964A/D1131A double mutant will be generated. To confirm the necessity of gene(s), gene disruption can be carried out by two strategies: (1) a stop codon can be introduced into the early portion of the gene, resulting in in situ gene inactivation; (2) the complete gene can be omitted in pathway assembly by redesigning the overlaps. Refer to the reference (14) for more details.
The hisG and delta2 sequences are not essential for assembling a pathway. As long as the 5′ end of the first promoter and the 3′ end of the last terminator contain overlaps (at least 50 bp) with the vector backbone, the pathway can be assembled on a plasmid. Similarly, any linearized S. cerevisiae-E. coli shuttle vector containing an ura3 gene as a selection marker can be used as the vector backbone.
S. cerevisiae HZ848 is used as the host for DNA assembly. However, any S. cerevisiae strain with a non-functional ura3 gene can be used as a host.
In order to verify the correctly assembled constructs through restriction digestion, a set of digestion consisting of one or two enzymes that cut the expected construct multiple times are chosen. Usually, two to three sets of digestions need to be set up in order to ensure the correct assembly. For a plasmid with a size of 15-20 kb, such as pRS416-zeaxanthin, find one or two enzymes which cut the DNA molecule 5-9 times. For a plasmid with a size of 20-30 kb, such as pZS-aur-2, find one or two enzymes which cut the DNA molecule 9-13 times. Try to avoid using enzyme digestion that will result in multiple fragments with similar sizes. Besides restriction digestion, the correctly assembled constructs can be confirmed by DNA sequencing.
In the construction of the first gene expression cassette and the last gene cassette, the hisG sequence and the delta2 sequence are also included. Therefore, for these two reactions, four fragments are spliced together.
The fragment mixture can be maintained at -20 °C for several months.
We encountered difficulties in amplifying the S. cerevisaie helper fragment, and in the end, the correct product was obtained by using the designated reagents and PCR program mentioned above.
58 °C is used as a standard annealing temperature. In some cases, especially for amplifying certain fragments from Streptomyces, fine-tuning annealing temperature is often necessary for obtaining the correct amplicons or improving PCR yields. Due to the high GC content of Streptomyces genome (>70%), including 5% DMSO in the reaction mixture will reduce the chance of forming secondary structures in general, resulting in better amplification efficiency. However, in some cases, we did encounter lower amplification efficiencies or even failures when DMSO was used. Generally, when difficulties are encountered to amplify a certain gene, a set of PCR conditions with various annealing temperatures and inclusion or exclusion of 5% DMSO need to be tested.
Normally, the doubling time for a S. cerevisiae laboratory strain is approximately two hours.
Unlike E. coli, yeast competent cells need to be freshly prepared each time.
For an efficient electroporation, a time constant of 5.0-5.2 ms should be obtained.
Assembly efficiency is defined as the percentage of the correct clones among the transformants appearing on the plate. Usually, ten colonies are picked, and an average efficiency of 60-80% can be obtained for assembly of the two target pathways.
E. coli strain BW25141 was used for plasmid enrichment and verification. However, any E. coli strain suitable for DNA cloning, such as DH5α and JM109, can be used.
The number of obtained E. coli transformants could vary from a few to several thousands. This is mainly due to the low quality of the isolated yeast plasmids. However, as long as colonies appear, experiments can proceed.
WM6026 is an auxotrophic E. coli strain whose growth relies on exogenously supplemented DAP.
Nalidixic acid is used to kill E. coli after it donates the plasmid into S. lividans, and apramycin is used to select the successful S. lividans exconjugants.
Aureothin has a molecular weight of 398.4 and the aureothin derivative generated by the AurB DH H964A mutant and the AurB DH H964A/D1131A double mutant has a molecular weight of 414.4. The MS2 fragmentation patterns of the produced compounds are compared with that of authentic aureothin. The products generated by the AurB DH H964A mutant and the AurB DH H964A/D1131A mutant exhibit the expected +16 patterns in the MS and MS2 profiles.
References
- 1.Hjersted JL, Henson MA, Mahadevan R. Genome-scale analysis of Saccharomyces cerevisiae metabolism and ethanol production in fed-batch culture. Biotechnol Bioeng. 2007;97:1190–1204. doi: 10.1002/bit.21332. [DOI] [PubMed] [Google Scholar]
- 2.Keasling JD. Synthetic biology for synthetic chemistry. ACS Chem Biol. 2008;3:64–76. doi: 10.1021/cb7002434. [DOI] [PubMed] [Google Scholar]
- 3.Menzella HG, Reid R, Carney JR, Chandran SS, Reisinger SJ, Patel KG, Hopwood DA, Santi DV. Combinatorial polyketide biosynthesis by de novo design and rearrangement of modular polyketide synthase genes. Nat Biotechnol. 2005;23:1171–1176. doi: 10.1038/nbt1128. [DOI] [PubMed] [Google Scholar]
- 4.Pitera DJ, Paddon CJ, Newman JD, Keasling JD. Balancing a heterologous mevalonate pathway for improved isoprenoid production in Escherichia coli. Metab Eng. 2007;9:193–207. doi: 10.1016/j.ymben.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 5.Ro DK, Paradise EM, Ouellet M, Fisher KJ, Newman KL, Ndungu JM, Ho KA, Eachus RA, Ham TS, Kirby J, Chang MC, Withers ST, Shiba Y, Sarpong R, Keasling JD. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature. 2006;440:940–943. doi: 10.1038/nature04640. [DOI] [PubMed] [Google Scholar]
- 6.Szczebara FM, Chandelier C, Villeret C, Masurel A, Bourot S, Duport C, Blanchard S, Groisillier A, Testet E, Costaglioli P, Cauet G, Degryse E, Balbuena D, Winter J, Achstetter T, Spagnoli R, Pompon D, Dumas B. Total biosynthesis of hydrocortisone from a simple carbon source in yeast. Nat Biotechnol. 2003;21:143–149. doi: 10.1038/nbt775. [DOI] [PubMed] [Google Scholar]
- 7.Dejong JM, Liu Y, Bollon AP, Long RM, Jennewein S, Williams D, Croteau RB. Genetic engineering of taxol biosynthetic genes in Saccharomyces cerevisiae. Biotechnol Bioeng. 2006;93:212–224. doi: 10.1002/bit.20694. [DOI] [PubMed] [Google Scholar]
- 8.Yan Y, Kohli A, Koffas MA. Biosynthesis of natural flavanones in Saccharomyces cerevisiae. Appl Environ Microbiol. 2005;71:5610–5613. doi: 10.1128/AEM.71.9.5610-5613.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gunyuzlu PL, Hollis GF, Toyn JH. Plasmid construction by linker-assisted homologous recombination in yeast. Biotechniques. 2001;31:1246, 1248, 1250. doi: 10.2144/01316bm03. [DOI] [PubMed] [Google Scholar]
- 10.Ma H, Kunes S, Schatz PJ, Botstein D. Plasmid construction by homologous recombination in yeast. Gene. 1987;58:201–216. doi: 10.1016/0378-1119(87)90376-3. [DOI] [PubMed] [Google Scholar]
- 11.Oldenburg KR, Vo KT, Michaelis S, Paddon C. Recombination-mediated PCR-directed plasmid construction in vivo in yeast. Nucleic Acids Res. 1997;25:451–452. doi: 10.1093/nar/25.2.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raymond CK, Pownder TA, Sexson SL. General method for plasmid construction using homologous recombination. Biotechniques. 1999;26:134–138. 140–131. doi: 10.2144/99261rr02. [DOI] [PubMed] [Google Scholar]
- 13.Shao Z, Zhao H, Zhao H. DNA assembler, an in vivo genetic method for rapid construction of biochemical pathways. Nucleic Acids Res. 2009;37:e16. doi: 10.1093/nar/gkn991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shao Z, Luo Y, Zhao H. Rapid characterization and engineering of natural product biosynthetic pathways via DNA assembler. Mol Biosyst. 2011;7:1056–1059. doi: 10.1039/c0mb00338g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dewick PM. Medical natural products A biosynthetic approach. 2nd. John Wiley and Sons; Chichester, UK: 2002. [Google Scholar]
- 16.Herbert RB. The biosynthesis of secondary metabolites. 2nd. Chapman and Hall; London, UK: 1989. [Google Scholar]
- 17.Li JW, Vederas JC. Drug discovery and natural products: end of an era or an endless frontier? Science. 2009;325:161–165. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- 18.Challis GL. Mining microbial genomes for new natural products and biosynthetic pathways. Microbiology. 2008;154:1555–1569. doi: 10.1099/mic.0.2008/018523-0. [DOI] [PubMed] [Google Scholar]
- 19.Zerikly M, Challis GL. Strategies for the discovery of new natural products by genome mining. Chembiochem. 2009;10:625–633. doi: 10.1002/cbic.200800389. [DOI] [PubMed] [Google Scholar]
- 20.Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes - gene-splicing by overlap extension. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 21.Lee FW, Da Silva NA. Sequential delta-integration for the regulated insertion of cloned genes in Saccharomyces cerevisiae. Biotechnol Prog. 1997;13:368–373. doi: 10.1021/bp970055d. [DOI] [PubMed] [Google Scholar]
- 22.Chemler JA, Yan Y, Koffas MA. Biosynthesis of isoprenoids, polyunsaturated fatty acids and flavonoids in Saccharomyces cerevisiae. Microb Cell Fact. 2006;5:20. doi: 10.1186/1475-2859-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Misawa N, Nakagawa M, Kobayashi K, Yamano S, Izawa Y, Nakamura K, Harashima K. Elucidation of the Erwinia uredovora carotenoid biosynthetic pathway by functional analysis of gene products expressed in Escherichia coli. J Bacteriol. 1990;172:6704–6712. doi: 10.1128/jb.172.12.6704-6712.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Misawa N, Shimada H. Metabolic engineering for the production of carotenoids in non-carotenogenic bacteria and yeasts. J Biotechnol. 1997;59:169–181. doi: 10.1016/s0168-1656(97)00154-5. [DOI] [PubMed] [Google Scholar]
- 25.Kieser T, Bibb JM, Buttner JM, Chater FK, Hopwood AD. Practical Streptomyces Genetics. The John Innes Foundation; Norwich: 2000. [Google Scholar]
- 26.Blodgett JA, Thomas PM, Li G, Velasquez JE, van der Donk WA, Kelleher NL, Metcalf WW. Unusual transformations in the biosynthesis of the antibiotic phosphinothricin tripeptide. Nat Chem Biol. 2007;3:480–485. doi: 10.1038/nchembio.2007.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ito T, Roongsawang N, Shirasaka N, Lu W, Flatt PM, Kasanah N, Miranda C, Mahmud T. Deciphering pactamycin biosynthesis and engineered production of new pactamycin analogues. Chembiochem. 2009;10:2253–2265. doi: 10.1002/cbic.200900339. [DOI] [PubMed] [Google Scholar]
- 28.Karray F, Darbon E, Nguyen HC, Gagnat J, Pernodet JL. Regulation of the biosynthesis of the macrolide antibiotic spiramycin in Streptomyces ambofaciens. J Bacteriol. 2010;192:5813–5821. doi: 10.1128/JB.00712-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keatinge-Clay A. Crystal structure of the erythromycin polyketide synthase dehydratase. J Mol Biol. 2008;384:941–953. doi: 10.1016/j.jmb.2008.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moriguchi T, Kezuka Y, Nonaka T, Ebizuka Y, Fujii I. Hidden function of catalytic domain in 6-methylsalicylic acid synthase for product release. J Biol Chem. 2010;285:15637–15643. doi: 10.1074/jbc.M110.107391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pawlik K, Kotowska M, Chater KF, Kuczek K, Takano E. A cryptic type I polyketide synthase (cpk) gene cluster in Streptomyces coelicolor A3(2) Arch Microbiol. 2007;187:87–99. doi: 10.1007/s00203-006-0176-7. [DOI] [PubMed] [Google Scholar]