

Abstract
Mitochondrial production of reactive oxygen species is often considered an unavoidable consequence of aerobic metabolism and currently cannot be manipulated without perturbing oxidative phosphorylation. Antioxidants are widely used to suppress effects of reactive oxygen species after formation, but they can never fully prevent immediate effects at the sites of production. To identify site-selective inhibitors of mitochondrial superoxide/H2O2 production that do not interfere with mitochondrial energy metabolism, we developed a robust small-molecule screen and secondary profiling strategy. We describe the discovery and characterization of a compound (N-cyclohexyl-4-(4-nitrophenoxy)benzenesulfonamide; CN-POBS) that selectively inhibits superoxide/H2O2 production from the ubiquinone-binding site of complex I (site IQ) with no effects on superoxide/H2O2 production from other sites or on oxidative phosphorylation. Structure/activity studies identified a core structure that is important for potency and selectivity for site IQ. By employing CN-POBS in mitochondria respiring on NADH-generating substrates, we show that site IQ does not produce significant amounts of superoxide/H2O2 during forward electron transport on glutamate plus malate. Our screening platform promises to facilitate further discovery of direct modulators of mitochondrially-derived oxidative damage and advance our ability to understand and manipulate mitochondrial reactive oxygen species production in both normal and pathological conditions.
Keywords: superoxide, hydrogen peroxide, antioxidant, glycerol 3-phosphate dehydrogenase, NADH:Q oxidoreductase, complex II, complex III, energy metabolism, respiratory complexes
INTRODUCTION
Mitochondrial production of reactive oxygen species (ROS) is central to the free radical theory of aging [1] and implicated in the pathogenesis of virtually all age-associated diseases including cardiovascular disease, neurodegeneration, cancer and diabetes [2–6]. Most research on the roles of ROS in aging and disease has focused on two areas: defining the mechanisms and sites of ROS production under normal and pathogenic conditions, and developing broad-acting antioxidants to decrease damage caused by ROS. Considerable progress has been made in defining sites of production and it is generally accepted that at least ten distinct sites exist within mitochondria [7]. Although mitochondrial production of superoxide and/or H2O2 has been implicated in a wide range of pathological events, the current inability to dissociate it from energy metabolism has made attribution to any site equivocal at best. Further, the contribution of each (or any) site under normal circumstances remains completely unknown.
Complex I and complex III have high capacities for production of superoxide/H2O2 and are commonly stated to be the sites most relevant to disease [8], although there is scant evidence for this view. In complex I, there is evidence for the existence of two sites of superoxide/H2O2 production, the flavin binding site (site IF) and the ubiquinone-binding site (site IQ) [9]. Site IQ generates superoxide/H2O2 at high rates during reverse electron flow from a reduced ubiquinone pool and is dependent upon a high protonmotive force across the mitochondrial inner membrane. In contrast, site IF is prominent during forward electron flow from a reduced NADH pool in the mitochondrial matrix and has a much lower maximum rate of production than site IQ in intact mitochondria. Blockade of site IQ by rotenone potently inhibits superoxide/H2O2 production from site IQ by preventing oxidation of ubiquinone and reverse electron transport into the site. In contrast, rotenone potentiates superoxide production from site IF by increasing its reduction state [9]. Inhibition of complex I activity by rotenone or the neurotoxin MPP+ has been linked to parkinsonism in both rodents and humans suggesting a link between dysfunctional complex I, ROS production, and neurodegeneration [10, 11]. In contrast, comparative analyses show an inverse relationship between maximal superoxide/H2O2 production from site IQ, but not site IF, and maximum life span across diverse vertebrate species [12, 13]. Therefore, selective modulators of superoxide/H2O2 production from site IQ or site IF would offer unique opportunities to probe the putative role of mitochondrial ROS production in normal and pathological processes.
The outer ubiquinone-binding site of complex III (site IIIQo) has the highest absolute capacity for superoxide production of all sites identified [14], partly because of the relative abundance of complex III. Although these high rates occur in the presence of the complex III inhibitor antimycin A, this site is active even in mitochondria respiring on different substrates in the absence of inhibitors [15, 16]. Site IIIQo and mitochondrial glycerol 3-phosphate dehydrogenase (mGPDH) are distinguished from the other sites by their production of superoxide to both the matrix and cytosolic sides of the mitochondrial inner membrane [17, 18]. This fact, combined with the reported high rates of production from site IIIQo and genetic manipulations of complex III activity, has led to speculation that superoxide production from site IIIQo plays an important role in cellular signaling during hypoxia and other events [19–21]. However, this conclusion remains controversial [22]. Unfortunately, because of the intimate links between ROS production, redox signaling and broader metabolism there is no definitive evidence for a direct link between any of these sites of superoxide/H2O2 production and a particular physiologically-relevant condition.
Antioxidants have been instrumental in the development of theories regarding the physiological roles of ROS. Antioxidant research has yielded thousands of natural and man-made chemicals and numerous gene products that broadly modulate ROS with differing selectivity and potency and within different cellular compartments. While antioxidants generally do not interfere with electron transport or oxidative phosphorylation, they scavenge ROS downstream from production and therefore can never fully suppress the effects of ROS. In addition, although ROS have been implicated in numerous disease states and antioxidants have shown promise in many pre-clinical experiments, virtually all clinical trials of antioxidant-based therapeutics have shown limited efficacy [23–25]. Therefore, from a fundamental understanding of the mechanisms of ROS production to the development of effective therapeutics, there remains a pressing need to identify the ways in which mitochondrial superoxide/H2O2 production is involved in (patho)physiological processes as well as the means by which this production can be modulated selectively.
Toward both of these ends, we developed a panel of high-throughput assays for the identification of compounds that modulate superoxide/H2O2 production at defined sites in isolated mitochondria without concomitant changes in energy metabolism. Our unbiased approach efficiently identifies site-specific modulators of superoxide/H2O2 production while also revealing less-specific effectors such as broad-acting antioxidants and novel inhibitors of mitochondrial function. We describe, for the first time, a site-selective inhibitor of mitochondrial superoxide/H2O2 production that does not compromise normal mitochondrial function. This novel inhibitor, CN-POBS, suppresses the majority of superoxide/H2O2 production from site IQ without altering superoxide/H2O2 production at other sites and without affecting mitochondrial energetic status. Our screening strategy represents a significant advance in the ability to discover small molecules with potent and selective effects on mitochondrial electron flow while our novel inhibitors promise to advance our understanding of the mechanisms of mitochondrial ROS production in more complex systems.
Experimental procedures
Reagents, animals, mitochondrial isolation, and standard buffers
CaCl2 standard was from Thermo Scientific. Amplex UltraRed and tetramethylrhodamine methyl ester (TMRM) were from Invitrogen. Compounds for screening were from a diverse, non-combinatorial library obtained from ChemBridge. New stocks of primary hits and their structural analogs were also from ChemBridge. The biological activity of N-cyclohexyl-4-(4-nitrophenoxy)benzenesulfonamide, CN-POBS, obtained from ChemBridge (ID 5229982) was validated using compound obtained from ChemDiv (ID 0668-0074) and found to be essentially identical. All other reagents were from Sigma-Aldrich.
Skeletal muscle mitochondria were isolated from hindlimbs of 5–8 week old female Wistar rats (Harlan Laboratories) as previously described [26]. The animal protocol was approved by the Buck Institute Animal Care and Use Committee, in accordance with IACUC standards.
Unless stated otherwise, freshly isolated muscle mitochondria were assayed in KHEB medium containing 120 mM KCl, 5 mM HEPES, 1 mM EGTA and 0.3% (w/v) bovine serum albumin. Where indicated, total and free calcium concentrations in assay buffers were calculated using the Extended MaxChelator program available at http://maxchelator.stanford.edu.
Screen for site-selective inhibitors of mitochondrial H2O2 production
Screening
To screen for site-selective small-molecule inhibitors of mitochondrial H2O2 production, we designed a core set of five assays using distinct combinations of substrates and inhibitors of electron transport (Fig. 1). Assays were performed in parallel and differed only in the amount of mitochondria and the substrates and inhibitors used. Superoxide and H2O2 were measured fluorometrically as H2O2 without distinguishing between them, in the presence of exogenous superoxide dismutase (25 U • mL−1), horseradish peroxidase (1 U • mL−1), and Amplex UltraRed (25 µM) as described [26] except assays were performed in 96-well format at room temperature (about 21 °C). Compounds that generally inhibited mitochondrial function were filtered with a sixth assay, of mitochondrial membrane potential (ΔΨm) using the potentiometric dye TMRM.
Figure 1. Six assays for the identification of site-selective inhibitors of mitochondrial superoxide/H2O2 production.

Oxidizable substrates are highlighted by yellow boxes. Chemicals added to produce more specific signals are highlighted by orange boxes. Additional modulators of electron and proton flux used as positive controls for each assay are highlighted by green boxes. Important redox centers within the electron transport chain complexes are shown 1) as white circles when uninhibited but not contributing predominantly to superoxide (O2•−)/H2O2 production, 2) as red circles when uninhibited and acting as the predominant source of O2•−/H2O2 production, and 3) as black circles when blocked by potent inhibitors and not contributing to O2•−/H2O2 production. (A) Site IQ with 5 mM succinate. Electrons from succinate reduce the mobile ubiquinone pool (Q-pool) before ultimately reducing O2 at complex IV (solid line with arrowheads). Electron transfer through complex III and complex IV drives the pumping of protons to the intermembrane space resulting in an electrochemical gradient of protons across the inner membrane (proton motive force, PMF, composed of a pH gradient, ΔpH, and a membrane potential, ΔΨm). The combination of a reduced Q-pool and high PMF drives the reverse transport of electrons from the Q-pool to NAD+ in the matrix via complex I (dashed line with arrowheads). Under this condition, most of the O2•−/H2O2 production is from site IQ although a minor portion comes from site IF/DH and site IIIQo [16]. Site IIF O2•−/H2O2 production is inhibited by high succinate and mGPDH is substrate-limited. The protonophore FCCP dissipates PMF causing an oxidation of all redox centers and acts as a positive control for this assay. An alternative assay utilizing subsaturating succinate was also used during compound retesting. In this condition, site IQ remains active but contributes proportionally less O2•−/H2O2 due to lower PMF and increased activity from site IIF. (B) Site IF/DH with 5 mM malate, 5 mM glutamate and 4 µM rotenone. Malate is oxidized to oxaloacetate by malate dehydrogenase (MDH) to generate NADH that is oxidized by site IF. Glutamate is added to convert oxaloacetate to 2-oxoglutarate and aspartate by aspartate aminotransferase (AAT) and facilitate the continual uptake and oxidation of malate. Rotenone prevents oxidation of redox centers upstream of site IQ. This increases the matrix NADH/NAD+ ratio to induce O2•− production from site IF while oxidizing redox centers downstream of complex I. The formation of 2-oxoglutarate in the presence of a high NADH/NAD+ ratio also induces significant O2•− /H2O2 production from 2-oxoglutarate dehydrogenase (OGDH). The addition of 20 mM aspartate disfavors the transamination of oxaloacetate to 2-oxoglutarate resulting in lower O2•−/H2O2 production from both site IF and OGDH and is used as a positive control for this assay. (C) Site IIF with 15 µM palmitoylcarnitine, 2 µM myxothiazol and 2.5 µM antimycin A. After reaction with coenzyme A, palmitoylcarnitine is metabolized by enzymes of the β-oxidation pathway to yield acetyl-CoA and NADH as well as reducing equivalents that enter the Q-pool via electron transferring flavoprotein (ETF) and ETF:ubiquinone oxidoreductase (ETFQOR). Oxidation of the Q-pool is prevented by myxothiazol and antimycin A, facilitating the backward entry of electrons into complex II and the production of O2•−/H2O2 from site IIF (dashed line with arrowheads). Site IIF predominates heavily in this condition, although low levels of production from site IF/DH are also observed due to the NADH generated during β-oxidation and TCA cycle activity. Malonate potently inhibits this production from site IIF and is used as a positive control in this assay. (D) Site IIIQo with 5 mM succinate, 4 µM rotenone and 2.5 µM antimycin A. In the presence of rotenone and antimycin A, the oxidation of succinate strongly reduces the Q-pool. Blockade of site IIIQi by antimycin A promotes O2•− production from site IIIQo to both sides of the inner membrane. The saturating levels of succinate prevent O2•−/H2O2 production from site IIF while rotenone and a lack of PMF prevent production from site IQ. Site IF/DH has a minor contribution due to a small increase in matrix NADH due to Krebs cycle activity. Myxothiazol potently inhibits site IIIQo and is used as a positive control in this assay. An alternative assay utilizing subsaturating succinate was also used during compound retesting. In this condition, partial reduction of the Q-pool results in maximal O2•−/H2O2 production from site IIIQo. However, unlike with saturating succinate, putative inhibitors may cause a lower signal simply through partial inhibition of succinate oxidation. (E) mGPDH with 25 mM glycerol phosphate, 4 µM rotenone, 2.5 µM antimycin A, 1 mM malonate and 2 µM myxothiazol. Oxidation of glycerol 3-phosphate causes an over-reduction of redox centers in mGPDH when the oxidation of the Qpool is prevented by myxothiazol and antimycin A. This results in O2•−/H2O2 production from mGPDH towards both sides of the inner membrane. O2•−/H2O2 production is prevented at site IQ, site IIF and site IIIQo by rotenone, malonate and myxothiazol, respectively. Site IF/DH is fully oxidized under this condition. There were no positive controls used for this assay; instead, signals were scaled to the signals with FCCP in the site IQ assay. (F) ΔΨm with 5 mM malate, 5 mM glutamate and 80 ng • mL−1 nigericin. Malate and glutamate are metabolized as described in the site IF/DH assay. However, in the absence of rotenone, electrons are transported to reduce O2 at complex IV with proton pumping by complexes I, III and IV generating a PMF. The ΔpH component of PMF is converted to ΔΨm by the addition of the K+/H+ exchanger nigericin. The uptake of the fluorescent cation TMRM into the mitochondrial matrix reports ΔΨm. The protonophore FCCP dissipates PMF and acts as a positive control for this assay. O2•−, superoxide; PMF, proton motive force across the mitochondrial inner membrane; Q-pool, mobile ubiquinone pool; MDH, malate dehydrogenase; OGDH, 2-oxoglutarate dehydrogenase; AAT, asparate aminotransferase; ETF, electron transferring flavoprotein; ETFQOR, ETF:ubiquinone oxidoreductase; GF/Q, FAD and ubiquinone-binding sites in mGPDH.
The screening workflow is shown in Fig. 2A. 3,200 compounds (~10 mM in DMSO) were arbitrarily selected from a library of 24,000 compounds obtained from ChemBridge. Master screening plates each with 80 compounds were prepared and diluted with KHEB medium using a Biomek FX liquid handling workstation (Beckman) just prior to screening (final testing concentration ~2.5 µM). On each master plate, eight DMSO wells (negative controls) were evenly distributed in the first and last columns of wells. Eight wells on each plate were reserved for duplicates of four known mitochondrial inhibitors (positive controls; their effect is reflective of a “positive” hit in certain designated assays), prepared fresh in KHEB. Final concentrations were: 1 µM carbonylcyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), a chemical uncoupler used as a positive control for site IQ superoxide production and the assay of ΔΨm; 2 µM myxothiazol, a potent inhibitor of site IIIQo and positive control for site IIIQo superoxide production; 1 mM malonate, a competitive inhibitor of complex II and positive control for the superoxide/H2O2 production by the flavin binding site of complex II (site IIF); 20 mM aspartate, a substrate for aspartate aminotransferase that removes matrix 2-oxoglutarate to act as a positive control for superoxide/H2O2 production from site IF plus NADH-linked matrix dehydrogenases including 2-oxoglutarate dehydrogenase and pyruvate dehydrogenase (collectively site IF/DH) [16].3
Figure 2. Multiple-parallel screening identifies site-specific inhibitors of H2O2 production that do not impair mitochondrial function.

(A) Screening workflow. Compounds in 96-well plates were screened in duplicate against the six assays in Fig. 1. Five assays each targeted a distinct site of superoxide/H2O2 production using different substrates without or with inhibitors. The sites assayed were site IQ, site IF/DH, site IIF, site IIIQo, and mGPDH. A sixth parallel assay of ΔΨm counterscreened for general inhibitors of mitochondrial function. All assays were initiated by the addition of Start Solutions containing the substrates and inhibitors listed in parentheses. Details are in "Experimental procedures" and Fig. 1. Endpoint fluorescence was measured and the effect of compounds was scaled to positive and negative controls included on each assay plate. (B – G) Average normalized effects of 3200 compounds screened against all six assays in duplicate (gray circles). 180 compounds gave >20% inhibition of site IQ superoxide/H2O2 production (dashed line in B). However, after evaluating their effects in the other screens, only 13 were selective for superoxide/H2O2 production at site IQ and did not alter ΔΨm (red circles in B – G). AUR, resorufin product of Amplex UltraRed oxidation; Suc, succinate; Glu, glutamate; Mal, malate; PalmCarn, palmitoylcarnitine; GP, glycerol 3-phosphate; Rot, rotenone; Ant A, antimycin A; Myx, myxothiazol; Malon, malonate; Nig, nigericin.
The mitochondrial suspension and a solution of Amplex UltraRed with superoxide dismutase and horseradish peroxidase were diluted in KHEB just prior to use. Sites of mitochondrial superoxide/H2O2 production were targeted individually using different combinations of mitochondrial substrates and inhibitors (Fig. 2A). These different “Start Solutions” were designed to generate maximal rates of H2O2 production predominantly from a single site within the chain. The sites of production targeted [15, 16] and the Start Solutions used to stimulate H2O2 production during screening were: site IQ with 5 mM succinate; site IF/DH with 5 mM glutamate, 5 mM malate and 4 µM rotenone; site IIIQo with 5 mM succinate, 4 µM rotenone and 2.5 µM antimycin A; site IIF with 15 µM palmitoylcarnitine, 2.5 µM antimycin A and 2 µM myxothiazol; mGPDH with 25 mM glycerol phosphate, 4 µM rotenone, 2.5 µM antimycin A, 1 mM malonate and 2 µM myxothiazol (Fig. 1). A Biomek FX workstation was used to dispense all components into black 96-well assay plates. The addition of the Start Solutions initiated each assay. Plates were incubated in the dark at room temperature (about 21 °C) for 30 – 40 min and endpoint fluorescence was measured on a Victor 3V plate reader (Perkin Elmer) equipped with a λex 550 nm/λem 590 nm filter cube.
ΔΨm was assayed manually by transferring diluted compounds to black 96-well assay plates. A bulk solution was prepared containing the following components and dispensed into two assay plates (all concentrations final): 0.2 mg protein • mL−1 mitochondria, 5 µM TMRM, 5 mM glutamate, 5 mM malate and 80 ng • mL−1 of the K+/H+ exchanger nigericin to collapse the pH gradient across the inner membrane (ΔpH) and maximize ΔΨm. Assay plates were incubated in the dark at room temperature (about 21 °C) for 10 – 15 min and endpoint fluorescence was measured on a Victor 3V plate reader equipped with a λex 550 nm/λem 590 nm filter cube.
Hit selection
Endpoint fluorescence values were scaled as % change from the average of eight intra-plate DMSO negative control wells, with the duplicate wells containing the relevant positive control inhibitor defined as −100% (see “Screening” for the assignment of inhibitors to each assay). Values for the mGPDH superoxide/H2O2 assay were arbitrarily scaled to the values for FCCP wells in the parallel site IQ H2O2 assay since no potent and selective inhibitors of mGPDH superoxide/H2O2 production existed at the start of this project [27]. Subsequently, this decision to use the FCCP values from the site IQ superoxide/H2O2 assay was found to underestimate the potency of compounds against mGPDH-specific H2O2 production. However, we maintained this procedure throughout the screen for consistency and lowered the threshold for hit identification in the mGPDH assay accordingly.
Following initial scaling, normalized intra-plate values were subjected to Tukey’s two-way median polish to remove row- and column-dependent positional effects [28, 29]. Initially, hits in each H2O2 assay were identified by applying a threshold of at least a 20% decrease in that assay. A critical component of the procedure was the use of two-tiered filtering for non-specific hits. First, each H2O2 assay was used to counterscreen each of the other H2O2 assays. This parallel counterscreening eliminated the vast majority of non-selective modulators of H2O2 production or consumption as well as compounds that produced artifacts in the detection system such as fluorescence quenching or interference with the HRP-Amplex UltraRed reaction. A smaller set of non-selective modulators or general inhibitors of mitochondrial function was flagged at the second step using the ΔΨm counterscreen. Most frequently, the ΔΨm assay eliminated marginal compounds or those structurally related to more potent inhibitors of ΔΨm. Hits in a single assay at the −20% threshold were subsequently eliminated if they altered the other H2O2 or ΔΨm assays by more than 10% or 4%, respectively. Based on the final number of hits in each assay, consideration of structural similarities among hits, and consideration of the underestimation in the mGPDH assay mentioned above, the thresholds for inclusion for the site IF/DH and mGPDH assays were adjusted to −18% and -10%, respectively, to generate additional hits for further testing. 2 – 6% of tested compounds surpassed the threshold in a given assay, but, after the two-tiered elimination process, only 0.1 – 0.5% were retained as potent and selective hits.
Retesting against an expanded panel of superoxide/H2O2 and ΔΨm assays
Hits from the primary screen and selected structural analogs were sourced from ChemBridge and retested against an expanded set of site-specific H2O2 and ΔΨm assays. Important changes from the original screen were as follows: four compounds were titrated in duplicate between 0.08 – 80 µM on each 96-well plate and a total of 32 DMSO controls were interspersed across each plate to serve as local normalization controls in place of two-way median polishing. Two additional H2O2 assays were included for all retesting: site IQ with 0.5 mM succinate, and site IIIQo with 0.5 mM succinate, 4 µM rotenone and 2.5 µM antimycin A. For retesting certain inhibitors of site IQ H2O2 production, an assay containing 5 mM succinate and 0.5 mM glutamate was also included. On select plates, FCCP (0.0025 – 2.5 µM) or rotenone (0.02 – 20 µM) was included for comparison to candidate inhibitors of site IQ H2O2 production. ΔΨm was assayed manually in four different conditions: 5 mM glutamate and 5 mM malate; 5 mM glutamate, 5 mM malate and 80 ng • mL−1 nigericin; 5 mM succinate and 4 µM rotenone; 5 mM succinate, 4 µM rotenone and 80 ng • mL−1 nigericin.
Hit validation
Endpoint fluorescence values were scaled to intra-plate DMSO and positive control inhibitor wells as described in “Hit Selection” except that the median values for interspersed DMSO wells (3 – 6 nearest DMSO wells for each compound well) were used as negative control values rather than only the eight DMSO wells in columns 1 and 12. This method replaced the two-way median polish used during high-throughput screening but had the similar effect of minimizing positional row and column effects in low signal:noise assays. Hits were validated if they caused progressive, dose-dependent inhibition of H2O2 production from a single site over at least a 5 – 10 fold concentration range without strong effects on other sites of H2O2 production or the maintenance of ΔΨm. At this stage, strong candidates were not necessarily excluded from further testing if subtle off-target effects were observed (e.g. loss of specificity or signs of general inhibition at the highest concentrations). Often, these candidates were pursued for more detailed mechanistic studies with the expectation that subsequent structure-activity analysis would yield structurally-related compounds with better selectivity.
Steady-state measurements of NAD(P)H, cytochrome b566, H2O2 production and ΔΨm
Steady-state % reduction levels of NAD(P)H and cytochrome b566 were determined as described in [15]. Steady-state rates of H2O2 production were measured using Amplex UltraRed in a Varian Cary Eclipse fluorimeter as described in [26]. Although no effect of CN-POBS on the HRP-Amplex UltraRed detection system was observed, H2O2 calibration curves were always generated in the presence and absence of CN-POBS and applied as appropriate. Steady-state ΔΨm was calibrated using an electrode sensitive to the cation methyltriphenylphosphonium (TPMP) in the absence or presence of 80 ng • ml−1 nigericin as described in [15, 26]. The effect of CN-POBS on steady-state variables was tested at 37 °C with continuous stirring in either KHEB or KHEB plus 5 mM KH2PO4 and 2.5 mM MgCl2.
Measurement of the rate of reverse electron transport
Dose-dependent effects of CN-POBS, FCCP or rotenone on the rate of reverse electron transport from succinate to NAD(P)+ was measured using a Varian Cary Eclipse fluorimeter with minor changes to previous procedures [30]. Briefly, to fully activate complex II and obtain maximal rates of NAD+ reduction, mitochondria (0.3 mg protein • mL−1) were equilibrated in KHEB medium for 15 s at 37 °C with continuous stirring then 0.5 mM glutamate was added to facilitate conversion of oxaloacetate to aspartate by aspartate aminotransferase. After another 15 s, 5 mM succinate was added and the immediate, rapid rate of NAD(P)+ reduction was measured. Rates of NAD(P)+ reduction were calculated as % of vehicle control (DMSO for CN-POBS and rotenone, ethanol for FCCP).
Measurement of respiration
The effect of candidate compounds on basal state 2 (substrate only), phosphorylating state 3 (5 mM ADP) and non-phosphorylating state 4o (0.5 ug • mL−1 oligomycin) were measured in plate-attached skeletal muscle mitochondria using a Seahorse XF24 Analyzer according to [27, 31]. Compounds were added just prior to loading the assay plate into the instrument. Each concentration of compound was tested in at least two wells on each plate. At least three biological replicates of each titration in each substrate condition were performed. Measurements were performed in a mannitol- and sucrose-based medium (Seahorse MAS buffer [31]) containing 0.3% (w/v) bovine serum albumin and 1 mM EGTA without or with 250 nM free calcium at pH 7.0.
Data presentation and statistical analysis
Data are presented as mean ± SE except screening data where error bars represent ranges of duplicate values. Statistical differences between conditions were determined with GraphPad Prism software using Student’s t-test, one-way ANOVA with Newman-Keuls post-test, or two-way ANOVA with Bonferroni post-test as specified in the Figure legends. p values < 0.05 were considered significant.
Results and Discussion
Unbiased profiling for site-selective inhibitors of mitochondrial H2O2 production
Our goal was to discover compounds that suppress the leak of electrons onto oxygen that occurs from multiple sites within mitochondria. Importantly, we desired compounds that act in a site-selective manner and without altering the normal electron and proton fluxes that drive mitochondrial oxidative phosphorylation. To accomplish this goal we designed a set of microplate-based assays to monitor H2O2 production from five distinct sites along with an assay to monitor ΔΨm. Five sites of H2O2 production were targeted separately by adding to a common assay mixture different substrates without or with selected inhibitors (Fig. 2A). In parallel, a distinct counterscreen to monitor ΔΨm was used to eliminate compounds that were likely general inhibitors of the electron transport chain or uncouplers of mitochondrial ATP production (rightmost assay, Fig. 2A). Each assay was robust, with Z-factors [32] above 0.5, and all but one assay had a coefficient of variation below 5% (Table 1). The combination of this robustness and our use of five separate counterscreens for each assay of H2O2 production resulted in an efficient platform for identifying site-selective inhibitors of superoxide/H2O2 production. Of 3200 compounds tested in our primary screening, approximately 2 – 6% had a strong effect on a given assay. For example, for the assay of superoxide/H2O2 production at site IQ, 180 compounds (5.6% of total) surpassed the threshold of −20% designated for this assay (gray circles below dashed line in Fig. 2B). However, when each of these compounds was crosschecked for effects on any of the other four sites of superoxide/H2O2 production or in the ΔΨm assay, only 13 compounds remained (red circles in Fig. 2B – 2G; 0.4% of total). These 13 compounds represented the initial leads in our search for site-selective inhibitors of superoxide/H2O2 production from site IQ. In comparison, between four and 17 site-selective hits were identified for the other four sites of superoxide/H2O2 production (Table 1).
Table 1. Summary statistics for the screen.
Coefficients of variation (%CV) were determined from eight DMSO control wells included on each assay plate (n ≥ 36 plates). Z-factors [32] were calculated using raw average fluorescence values and their standard deviations for DMSO controls and selected inhibitor controls (Z-factor comparisons shown) included on each assay plate (n ≥ 36 plates). Total Hits are the numbers of compounds that surpassed the threshold set for each individual assay, whereas Selective Hits are the numbers of compounds remaining after excluding those that also surpassed a minimum threshold in at least one of the other five assays (see “Hit Selection” in "Experimental procedures" for threshold cutoffs).
| Assay | %CV | Z-Factor | Z-Factor Comparison | Total Hits | Selective Hits |
|---|---|---|---|---|---|
| IQ | 3.4 ± 0.3 | 0.88 ± 0.01 | DMSO vs FCCP | 180 | 13 |
| IF/DH | 4.5 ± 0.4 | 0.62 ± 0.02 | DMSO vs Aspartate | 199 | 17 |
| IIF | 6.3±0.6 | 0.54 ± 0.04 | DMSO vs Malonate | 80 | 4 |
| IIIQo | 4.8 ± 0.4 | 0.77 ± 0.02 | DMSO vs Myxothiazol | 72 | 13 |
| mGFDH | 3.4 ± 0.2 | n.d. | n.a. | 87 | 8 |
| ΔΨm | 2.2 ± 0.4 | 0.87 ± 0.03 | DMSO vs FCCP | n.a. | n.a. |
Next, we retested many of the 55 site-selective compounds against an expanded panel of H2O2 and ΔΨm assays (see "Experimental procedures" for specific conditions) to verify their selectivity and to begin to probe for mechanisms of action. The IF/DH, IIIQo, and mGPDH assays all yielded one or more structural classes of novel inhibitors of superoxide/H2O2 production from these sites to be described in detail elsewhere. Briefly, the majority of these compounds validated the results of our primary screen under the conditions tested. However, the hits in these assays were ultimately found to have condition-dependent effects on H2O2 production or substrate oxidation. For example, inhibitors in the assay of mGPDH superoxide production were found to inhibit mGPDH enzymatic activity in more detailed follow-up experiments [27].
The assay for IQ superoxide/H2O2 production had the highest Z-factor and lowest CV of the five H2O2 assays used in the primary screen and yielded 13 compounds that were selective inhibitors for this site of superoxide/H2O2 production (Table 1). Among these 13 were several structurally-similar compounds, the most potent of which was CN-POBS.
Validating inhibitors of site IQ superoxide/H2O2 production
To verify the activity of these compounds, we generated dose-response curves between 0.08 and 80 µM against our panel of H2O2 and ΔΨm assays for several of the 13 original hits and over 20 structurallyrelated compounds available through ChemBridge. To better compare the selectivity of these IQ superoxide/H2O2 inhibitors, we first estimated their IC50 against superoxide/H2O2 production from site IQ with 5 mM succinate. We then determined the % change in H2O2 or ΔΨm signal in the other assays at these concentrations (Table 2).
Table 2. Partial list of selective suppressors of superoxide/H2O2 production from site IQ, select structural analogs, and their effects on H2O2 production, ΔΨm and the rate of NAD(P)+ reduction from succinate via complex I.
Summary of effects on H2O2 production and ΔΨm of five compounds identified in the primary screen and ten structural analogs of one of these (CN-POBS). Dose-response curves between 0.08 and 80 µM were generated for each compound against the five sites of superoxide/H2O2 production (sites IQ, IF/DH, IIF, IIIQo and mGPDH) and ΔΨm assayed in the primary screen (data not shown; see Experimental procedures). IC50 values in the site IQ assay were estimated to determine relative potency against this site and relative selectivity in the other five assays. Estimated IC50 values were defined as the tested concentration, or the average of two tested concentrations (gray boxes), that yielded a normalized decrease of ~50% in site IQ superoxide/H2O2 production. The relative selectivity of inhibitors or analogs against the other assays was then compared at these defined IC50 values (or at 80 µM for compound 15). The five most potent and selective compounds were tested further for effects on the rate of NAD(P)+ reduction by succinate via complex I (rightmost column). LogP values are from the NCBI PubChem compound database. Values for all assays are the average of the normalized percent changes from DMSO for two duplicate runs, n.d., not determined.
| Compound | ChemBridge ID |
Structure | LogP | IC50(µM) | IQ | IF/DH | IIF | IIIQo | mGPDH | ΔΨm | Rate of NAD(P)+ Reduction |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 5212116 | 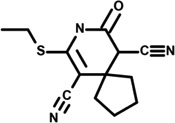 |
2.2 | 2.5 | −52 | −7 | 3 | 2 | 0 | −3 | n.d. |
| 2 | 5192566 | 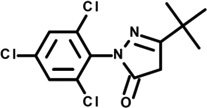 |
4.6 | 2.5 | −57 | 5 | −10 | −6 | 0 | −1 | n.d. |
| 3 | 5619111 | 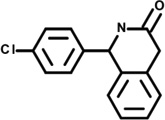 |
3.0 | 25 | −56 | −6 | −30 | −5 | −3 | −1 | n.d. |
| 4 | 5154627 | 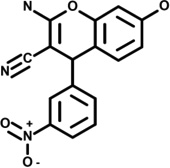 |
2.8 | 25 | −49 | 8 | −21 | 3 | −5 | −2 | n.d. |
| CN-POBS | 5229982 | 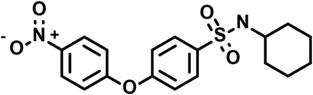 |
4.1 | 4.5 | −56 | 7 | 4 | 0 | −2 | −1 | −1 |
| 6 | 5918711 | 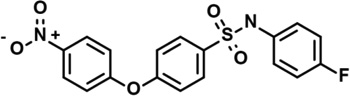 |
4.5 | 0.45 | −48 | −1 | 1 | 1 | 0 | −1 | −14 |
| 7 | 6669125 | 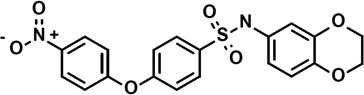 |
4.8 | 0.8 | −51 | 0 | 4 | −1 | −3 | −1 | −6 |
| 8 | 6491212 | 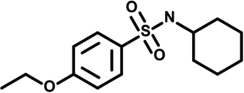 |
3.0 | 8 | −57 | 2 | −3 | 1 | 1 | −2 | 6 |
| 9 | 5315316 | 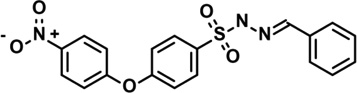 |
3.9 | 16 | −54 | −7 | −6 | 0 | −2 | −1 | n.d. |
| 10 | 5229983 | 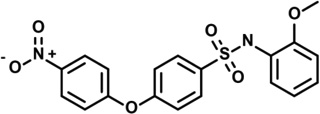 |
3.6 | 0.8 | −51 | 1 | −10 | 2 | 1 | 0 | 2 |
| 11 | 5229985 | 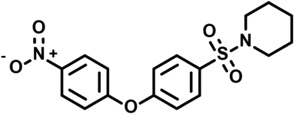 |
3.3 | 25 | −47 | 8 | −43 | −16 | −7 | 0 | n.d. |
| 12 | 5109687 | 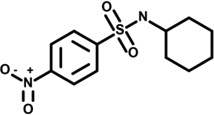 |
2.5 | 45 | −60 | −30 | −5 | 45 | 4 | −5 | n.d. |
| 13 | 9040813 | 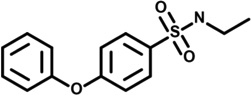 |
2.8 | 80 | −56 | 30 | −11 | 38 | 14 | −2 | n.d. |
| 14 | 7954145 | 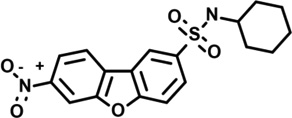 |
4.3 | −25 | −57 | −17 | 8 | 89 | 14 | −12 | n.d. |
| 15 | 7965586 | 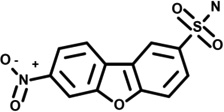 |
2.1 | »80 | −10 | 20 | −1 | 2 | 2 | 0 | n.d. |
Two of the most potent hits in the original screen, compounds 1 and 2, remained potent inhibitors of site IQ superoxide/H2O2 production with 5 mM succinate. However, 1 gave slight inhibition of site IF/DH H2O2 production at 2.5 µM as well as a subtle inhibition of succinate oxidation revealed by differing effects on site IIIQo superoxide production that were dependent upon the concentration of succinate (data not shown). Specifically, it caused a dose-dependent decrease in superoxide production from site IIIQo driven by 0.5 mM succinate, but increased superoxide production from the same site when driven by 5 mM succinate. In the absence of ΔΨm, superoxide production from site IIIQo peaks at an intermediate reduction state of the ubiquinone pool [33]. Our assays for superoxide production from site IIIQo were designed to target both the peak rate of production with 0.5 mM succinate and a lower, but significant rate near complete reduction of the ubiquinone pool with 5 mM succinate. The effects of 1 were best explained by an inhibition of succinate oxidation that progressively lowered the reduction state of the ubiquinone pool causing a drop from the peak of the curve with 0.5 mM succinate and an increase toward the top of the peak with 5 mM succinate. Low concentrations of malonate can cause a similar effect [33].
Compound 2 likely also inhibited succinate oxidation or uptake. Its effects on site IIIQo superoxide production were more potent than 1 in that it fully inhibited IIIQo superoxide production driven by low succinate below 8 µM whereas 1 inhibited only ~50% at 80 µM. Between 0.08 – 0.8 µM, 2 increased site IIIQo superoxide production with high succinate but progressively and completely inhibited this production by 80 µM. Again, these effects were best explained by a progressive inhibition of succinate uptake or oxidation that lowered the reduction state of the ubiquinone pool driven by succinate. Further support for this explanation came from the observations that superoxide/H2O2 production from site IQ was more sensitive to 2 when driven by 0.5 mM succinate than with 5 mM succinate and that ΔΨm driven with succinate was more sensitive than ΔΨm driven with glutamate and malate (data not shown). Evidence to support an effect on succinate uptake rather than a direct effect on oxidation at complex II was the fact that site IIF superoxide/H2O2 production driven by palmitoylcarnitine was inhibited only ~30% at 80 µM 2 (data not shown). Interestingly, the effect of 2 on site IIIQo superoxide production with 5 mM succinate was not detected in the original screen because the inflection point caused by 2 in the signal of this assay occurred near the screening concentration of 2.5 µM. Therefore, it showed no change in H2O2 signal relative to DMSO controls at this concentration but greatly increased or decreased H2O2 signal at lower or higher concentrations, respectively. Together, these results emphasize the effectiveness of the primary screen to reveal novel modulators of mitochondrial function and the efficiency with which our retesting strategy could both eliminate false positives and provide useful information on mechanisms of action of novel modulators.
Two compounds, 3 and 4, did not reach the threshold of −20% set for the IQ H2O2 assay, but shared structural similarity and were therefore selected for further testing (Table 2). As in the primary screen, these compounds were less potent against IQ superoxide/H2O2 production (IC50 ~25 µM). However, at this concentration both compounds inhibited site IIF superoxide/H2O2 production driven with palmitoylcarnitine, although they had no significant effects on site IIIQo superoxide production with either low or high succinate (data not shown and Table 2). Therefore, these compounds appeared to have off-target effects involving fatty acid oxidation. Further evaluation of structurally similar compounds may reveal if the dual effects of this class of compound are separable.
Most of the other compounds tested for selectivity against IQ superoxide/H2O2 production were members of a structural class exemplified by the most potent and selective hit identified in the primary screen, CN-POBS (Table 2). This class was characterized by a core 4-phenoxy-benzenesulfonamide "POBS” structure. A comparison of multiple members of this class revealed structural features that were important for selectivity against IQ superoxide/H2O2 production. Three compounds (6, 7 and 10) were more potent against IQ superoxide/H2O2 production than CN-POBS, with IC50 < 1 µM, and differed structurally only in substitutions at the terminal nitrogen in the sulfonamide group. Specifically, while CN-POBS has a cyclohexyl group on the terminal nitrogen, these three compounds have benzene rings with additional sizable or electron-withdrawing groups. Several other compounds with similar substitutions were also tested (data not shown). Commonly, these compounds had a greater effect on site IIF superoxide/H2O2 production at or just above their IC50 (Table 2 and data not shown) than did CNPOBS, which affected site IIF superoxide/H2O2 production driven by palmitoylcarnitine only at the highest concentrations tested (Fig. 3). Notably, as observed for 3 and 4, this effect was likely due to inhibition of fatty acid oxidation and not complex II because CN-POBS did not inhibit superoxide/H2O2 production from site IIF when driven by succinate (data not shown). It is possible that these compounds interfered with palmitoylcarnitine transport or metabolism and that the terminal group off the sulfonamide modulated this off-target effect. In addition, some compounds more strongly inhibited the reverse flux of electrons from succinate to NAD(P)+ via complex I at their IC50 than did CN-POBS (rightmost column in Table 2). Other structural modifications decreased potency, selectivity, or both. Selected examples of these modifications and their effects on site-specific superoxide/H2O2 production and ΔΨm are provided in Table 2 (compounds 6 – 15).
Figure 3. CN-POBS is a selective inhibitor of superoxide/H2O2 production from site IQ.

Effect of CN-POBS on normalized H2O2 production from site IF/DH (black diamonds), site IQ (with 0.5 or 5 mM succinate; white and black squares, respectively), site IIF (black circles), mGPDH (black stars), and site IIIQo (with 0.5 or 5 mM succinate; white and black triangles, respectively). Details of the conditions to induce H2O2 production from each site are given in "Experimental procedures" and Fig. 1. Data are normalized means ± S.E. (n = 3). AUR, resorufin product of Amplex UltraRed oxidation; Suc, succinate.
Importantly, our retesting strategy identified CN-POBS as the most potent and selective inhibitor of site IQ superoxide/H2O2 production among the compounds tested. All further experiments focused on characterizing the effects of this novel inhibitor in more detail.
CN-POBS is a novel site-selective inhibitor of superoxide/H2O2 production from site IQ
Dose-response curves for CN-POBS against the expanded panel of site-specific superoxide/H2O2 assays are shown in Fig. 3. CN-POBS showed remarkable selectivity for superoxide/H2O2 production from site IQ. No significant effects on H2O2 production were observed below 25 µM for any of the other four sites tested.
CN-POBS slightly, but consistently, inhibited H2O2 production driven by saturating succinate more potently than H2O2 production driven by subsaturating succinate (compare white and black squares in Fig. 3). This difference was likely due to superoxide/H2O2 production from sites other than IQ during oxidation of succinate alone [16]. Succinate oxidation increases the reduction state of both the NADH pool in the matrix and the ubiquinone pool of the inner membrane and induces superoxide/H2O2 production from site IF/DH and site IIIQo, respectively [9, 15, 33]. In the presence of subsaturating succinate, site IIF may also be active [15]. Depending on the relative protonmotive forces, relative reduction states of the redox centers, and the contribution from site IIF with either high or low succinate, it is possible that the relative contribution of these sites to the total signal versus the contribution from site IQ may be greater with subsaturating succinate. Such a shift in the sources of production would lower the relative effect of CN-POBS on total H2O2 production under this condition. Importantly, this relative effect is the opposite of what was observed for novel and known inhibitors of succinate oxidation as described in “Validating inhibitors of site IQ superoxide/H2O2 production” above. For these compounds, superoxide/H2O2 production with low succinate was more sensitive than with high succinate alone. This distinction argues against inhibition of succinate oxidation as a likely mechanism of action for CN-POBS.
To further evaluate the selectivity of CN-POBS for effects on electron leak to oxygen at site IQ and not on electron flow through the electron transport chain, we measured ΔΨm (Fig. 4A) and respiration (Fig. 4B) with different substrates and energetic conditions. CN-POBS had no effect on succinate or glycerol 3-phosphate oxidation below the highest concentration tested, 80 µM. However, there was a significant effect of CN-POBS on ΔΨm (at 8 µM) and respiration (at 80 µM) powered by the complex I substrates glutamate plus malate. The greater potency of CN-POBS in the TMRM-based ΔΨm assays versus the respiration measurements driven with glutamate and malate was subsequently identified as a sensitizing effect of 5 µM TMRM specifically during oxidation of glutamate and malate (data not shown). Similar inhibitory effects of TMRM on respiration driven by glutamate plus malate have been shown previously [34]. To confirm that CN-POBS had no off-target effects on substrate oxidation at concentrations near its IC50, we also measured ΔΨm using an electrode sensitive to the potentiometric cation TPMP. We observed no effect of 2.5 µM CN-POBS on ΔΨm powered by either glutamate plus malate or succinate either in the presence or absence of the K+/H+-exchanger nigericin (Table 3).
Figure 4. CN-POBS does not inhibit ΔΨm or respiration below 8 µM.

(A) Effect of CN-POBS on normalized ΔΨm generated by oxidation of 5 mM glutamate plus 5 mM malate without or with 80 ng • mL−1 nigericin (white or black circles), or 5 mM succinate and 4 µM rotenone without or with nigericin (white or black triangles). ΔΨm with glutamate plus malate in the absence of nigericin was significantly decreased by 8, 25 and 80 µM CN-POBS and in the presence of nigericin only by 25 and 80 µM CNPOBS. ΔΨm with succinate was significantly decreased by 80 µM CN-POBS. (*p < 0.05 versus vehicle control; one-way ANOVA with Newman-Keuls post-test). Data are normalized means ± S.E. (n = 3). Glu, glutamate; Mal, malate; Nig, nigericin; Suc, succinate; Rot, rotenone. (B) Effect of CN-POBS on the rates of mitochondrial respiration driven by 5 mM glutamate plus 5 mM malate, 5 mM succinate with 4 µM rotenone, or 16.7 mM glycerol phosphate with 4 µM rotenone and 250 nM free calcium. Respiratory states 2, 3, and 4o were defined by the sequential additions of substrate, 5 mM ADP, and 0.5 µg • mL−1 oligomycin, respectively. CN-POBS at 80 µM significantly decreased state 3 respiration with all substrates and increased state 2 respiration with succinate. (*p < 0.05 versus vehicle control or #p < 0.05 versus 2.5 and 8 µM CN-POBS; one-way ANOVA with Newman-Keuls post-test). Data are means ± S.E. (n = 3).
Table 3. Effect of CN-POBS on ΔΨm and the reduction levels of NAD(P)H and cytochrome b566 in mitochondria oxidizing succinate or glutamate plus malate.
ΔΨm and the redox states of NAD(P)H and cytochrome b566 were determined under the conditions listed in parallel to measurements of H2O2 production in Fig. 6 and Fig. 7. In addition, 80 ng • ml−1 nigericin was added to measurements of ΔΨm to determine if CN-POBS altered ΔpH. There were no significant differences between DMSO vehicle and 2.5 μM CN-POBS for any measurements, or in % reduced NAD(P)H with 5 mM succinate versus succinate plus 4 μM rotenone for either DMSO vehicle or CN-POBS. Data are means ± S.E. (n ≥ 3). The reduction state of cytochrome b566 in mitochondria oxidizing succinate in the presence of rotenone was 58.8 ± 1.2% (mean ± SE, n = 14) and was not significantly different from that observed with succinate alone (p = 0.18, Student’s t-test). This measurement was not performed in parallel with the others and therefore was not included in the table.
| ΔΨm(mV) | % reduced NAD(P)H | % reduced cytochrome b566 | ||||
|---|---|---|---|---|---|---|
| Condition CN-POBS (µM) | 0 | 2.5 | 0 | 2.5 | 0 | 2.5 |
| Succinate | 147.0 ± 3.1 | 150.8 ± 2.3 | 86.0 ± 3.1 | 85.8 ± 2.5 | 55.0 ± 0.6 | 57.1 ± 0.5 |
| Succinate + Nigericin | 186.0 ± 4.8 | 186.9 ± 3.3 | ||||
| Succinate + Rotenone | 75.7 ± 3.3 | 77.0 ± 3.7 | ||||
| Glutamate + Malate + Oligomycin | 143.8 ± 4.7 | 144.8 ± 0.7 | 91.7 ±0.6 | 92.5 ± 0.8 | 41.7 ± 1.6 | 39.8 ± 2.1 |
| Glutamate + Malate + Oligomycin + Nigericin | 166.2 ± 3.6 | 164.8 ± 0.6 | ||||
Together, these data indicate that CN-POBS can suppress at least 50% of superoxide/H2O2 production from site IQ without any effects at other sites of H2O2 production, on generation of protonmotive force, or on oxidative phosphorylation.
CN-POBS lowers superoxide/H2O2 production at site IQ without inhibiting electron flux through complex I
The superoxide/H2O2 production that we attribute to site IQ is best associated with reverse electron transport and is dependent on the normal kinetic mechanisms of electron flux and proton pumping of complex I [35]. Reverse electron transport will also cause the FMN at site IF to become reduced and generate superoxide [35, 36]. Therefore, it is possible that the suppression of superoxide/H2O2 associated with reverse electron transport observed with CN-POBS is due to an impairment in flux through complex I. Such an impairment would decrease superoxide/H2O2 production at both sites. To determine if the inhibition of superoxide/H2O2 production associated with reverse electron transport by CN-POBS was due to altered kinetics of complex I, we measured the rate of NAD(P)+ reduction from succinate via complex I. A dose-response curve showed that CN-POBS had no significant effect on reverse electron flux through complex I below 25 µM (Fig. 5A) although it significantly decreased H2O2 production at every concentration tested between 0.25 and 80 µM (Fig. 5B). A correlation plot of the relative effects on the rate of NAD(P)+ reduction versus the rate of H2O2 production confirmed that at least 50% of superoxide/H2O2 production was suppressed by CN-POBS with no effect on the flux through complex I (Fig. 5C).
Figure 5. CN-POBS significantly lowers H2O2 production at site IQ without altering electron flux through complex I.

(A) Effect of FCCP (white circles), rotenone (gray circles), and CN-POBS (black circles), on normalized rates of NAD(P)+ reduction driven by 5 mM succinate plus 0.5 mM glutamate. FCCP significantly lowered the rate of NAD(P)+ reduction at all concentrations except 0.0025 µM. Rotenone significantly lowered the rate of NAD(P)+ reduction at all concentrations except 0.02 µM. CNPOBS lowered the rate of NAD(P)+ reduction at 25 and 80 µM. (ns = not significant. All other points significant at p < 0.05; one-way ANOVA with Newman-Keuls post-test). Data are means ± S.E. (n = 3 – 5). (B) Effect of FCCP (white circles), rotenone (gray circles), and CN-POBS (black circles), on normalized rates of H2O2 production driven by 5 mM succinate plus 0.5 mM glutamate. All three compounds significantly decreased the rate of H2O2 production at all concentrations except FCCP at 0.0025 µM. (ns = not significant. All other points significant at p < 0.05; one-way ANOVA with Newman-Keuls post-test). Data are means ± S.E. (n = 4). (C) Replot of the data in A and B to reveal the effect of FCCP (white circles), rotenone (gray circles), and CN-POBS (black circles) on the rates of NAD(P)+ reduction relative to their effects on the normalized rates of H2O2 production. CN-POBS was more effective than either FCCP or rotenone at decreasing the rate of H2O2 production without inhibiting the rate of NAD(P)+ reduction.
Other known inhibitors of superoxide/H2O2 production during reverse electron transport act by inhibiting electron transport through site IQ (e.g. rotenone), by dissipating the protonmotive force that drives reverse electron transport (e.g. FCCP), or by inhibiting oxidation of the substrate providing electrons to site IQ (e.g. malonate during succinate oxidation). Our data ruled out a direct effect on succinate oxidation by CN-POBS below 80 µM (see above and Figs. 3, 4A, 4B). To test the other two mechanisms, we compared the rates of NAD(P)+ reduction and H2O2 production during succinate oxidation in the presence of differing amounts of rotenone or FCCP. As expected, each potently inhibited both the rate of NAD(P)+ reduction via complex I (Fig. 5A) and the rate of H2O2 production (Fig. 5B). However, FCCP was not able to dissociate these effects, and only the lowest concentration of rotenone (20 nM) lowered H2O2 production with a small but not significant decrease in the rate of NAD(P)+ reduction (Fig. 5C). In comparison, CN-POBS had a much broader window of differential effects.
We conclude that CN-POBS suppresses superoxide/H2O2 production by a unique mechanism that is independent of altered flux through complex I and is therefore best described as acting at site IQ. Because CN-POBS is lipophilic (logP = 4.1), we speculate that it may interact with specificity near site IQ in the hinge region connecting the membrane-associated domains to the soluble subunits in the matrix. At high concentrations, the lipophilicity of CN-POBS may promote non-specific interactions with other membrane-associated domains like the proton pumping channels to fully inhibit the complex.
CN-POBS suppresses two-thirds of superoxide/H2O2 production at site IQ without changes in other mitochondrial functions
As mentioned above, multiple sites of superoxide/H2O2 production are active during oxidation of succinate alone [16]. Because other sites of H2O2 production are unaffected by CN-POBS below 25 µM (Fig. 3), their contribution to the total H2O2 production may cause an underestimation of the effect of CNPOBS on site IQ superoxide/H2O2 production with succinate alone. To more accurately determine the potency of CN-POBS against superoxide/H2O2 production at site IQ, we determined the effect of CNPOBS on calibrated steady-state rates of H2O2 production driven by succinate alone without and with complex I activity fully inhibited by rotenone. First, to verify that CN-POBS was without effect on various aspects of oxidative phosphorylation, we measured the reduction states of NAD(P)H and ubiquinone and the value of ΔΨm after addition of succinate. CN-POBS had no effect on any of these variables (Table 3). However, it significantly lowered the rate of H2O2 production measured under the same conditions by 50% (Fig. 6A). Using rotenone to fully block site IQ resulted in a significantly larger decrease in the total rate of H2O2 production from succinate than observed with CN-POBS and succinate alone. However, CN-POBS had no additional effect in the presence of rotenone (Fig. 6A). The addition of rotenone to mitochondria oxidizing succinate can change the distribution of electrons at superoxide/H2O2- producing redox centers such as site IF/DH and site IIIQo [16]. Therefore, it is important to account for changes in superoxide/H2O2 production at these sites prior to assignment of the entire rotenone-sensitive component to site IQ. This correction is made using the reduction states of the matrix NAD(P)H signal and cytochrome b566 signal in complex III to determine the relative rates of superoxide/H2O2 production that are produced by site IF/DH and site IIIQo, respectively [15, 16]. We observed no significant difference in either the % reduction of NAD(P)H or % reduction of cytochrome b566 with the addition of rotenone to succinate (Table 3). Therefore, in this particular case, the rotenone-sensitive component of the H2O2 production accurately described the rate attributable to site IQ. The site IQ-specific rate of H2O2 production with succinate alone was 1144 ± 86 pmol • min−1 • mg protein−1 (n = 3). In comparison, the amount sensitive to 2.5 µM CN-POBS was 762 ± 69 pmol • min−1 • mg protein−1 (n = 3). Therefore, CN-POBS selectively suppressed 67% of the H2O2 production from site IQ without altering H2O2 production from at least five other major sites of mitochondrial superoxide/H2O2 production (sites IF/DH, IIF, IIIQo, and mGPDH) and without affecting any measured aspect of mitochondrial activity including oxidation of fatty-acyl carnitine, basal and phosphorylating respiration, protonmotive force (ΔΨm or ΔpH), electron flux through complex I, and the reduction states of the NAD(P)H and ubiquinone pools. This represents the first identification of such a site-selective inhibitor of mitochondrial superoxide/H2O2 production without effects on oxidative phosphorylation. Previous studies [30] identified diphenyleneiodonium as an inhibitor of superoxide/H2O2 production from site IQ, but not site IF/DH or site IIIQo. However, this compound has strong off-target effects on oxidative phosphorylation, greatly limiting its utility.
Figure 6. CN-POBS suppresses the majority of the superoxide/H2O2 production from site IQ without inhibiting superoxide/H2O2 production from site IF.

(A) Effect of 2.5 µM CN-POBS on the rate of H2O2 production driven by 5 mM succinate only (left hand set of columns) or by 5 mM succinate with 4 µM rotenone (right hand set of columns). CN-POBS significantly inhibited 67% of the rotenone-sensitive H2O2 production (from site IQ) driven by succinate alone but had no significant effect on H2O2 production by succinate in the presence of rotenone (from site IF/DH). (p < 0.01 between conditions indicated by different letters; two-way ANOVA with Bonferroni post-test). Data are means ± S.E. (n = 3). Suc, succinate; Rot, Rotenone. (B) Effect of 2.5 µM CN-POBS on the rate of H2O2 production driven by 5 mM malate in the presence of 2.5 mM ATP, 1.5 mM aspartate, 4 µM rotenone and 1 µg • mL−1 oligomycin. CN-POBS had no effect on H2O2 production (specifically from site IF) in this condition. Data are means ± S.E. (n = 3).
The hypothesis that complex I has two distinct sites of superoxide/H2O2 production (site IF and site IQ) rather than a single site (IF) activated under different conditions, remains a subject of debate [9, 36]. Although our data indicates no effect of CN-POBS on superoxide production by site IF/DH, our screening assays were not designed to distinguish between superoxide/H2O2 production from site IF and superoxide/H2O2 production from matrix dehydrogenases, and may not have been sensitive enough to detect subtle effects specifically on site IF. Therefore, we tested the effect of CN-POBS under conditions in which site IF is the sole producer of superoxide.3 In agreement with our screening data, CN-POBS had no effect on superoxide/H2O2 production specifically from site IF (Fig. 6B). Therefore, our discovery of CN-POBS provides strong support for the existence of two sites of superoxide/H2O2 production in complex I, as it significantly inhibits superoxide/H2O2 production associated with reverse electron transport into site IQ but has no effect on reverse electron transport to site IF, on steady-state NAD(P)H reduction, or superoxide/H2O2 production under any conditions in which site IF is expected to be active.
Site IQ does not produce significant amounts of superoxide/H2O2 during forward flux of electrons through complex I
In isolated mitochondria, site IQ has a high capacity for superoxide/H2O2 production during the oxidation of substrates such as succinate and glycerol 3-phosphate that directly reduce ubiquinone and generate a large enough protonmotive force to drive electron transport in reverse into complex I [16, 18]. In intact cells, it remains unclear how much site IQ contributes to superoxide/H2O2 production under conditions that strongly favor forward electron flux through complex I. Recently, through the use of endogenous reporters of site IF/DH and site IIIQo, we demonstrated that these sites account for all of the measured H2O2 production under several conditions [15]. However, during non-phosphorylating oxidation of glutamate plus malate, a significant portion of the measured H2O2 production could not be attributed to site IF/DH and site IIIQo. Of the conditions tested, this had the highest membrane potential and reduction state of the ubiquinone pool, conditions that favor superoxide/H2O2 production from site IQ. Therefore, to determine if site IQ contributed significantly to the total rate of H2O2 production under this condition, we repeated these measurements of calibrated rates of H2O2 production, ΔΨm, % reduction of NAD(P)H, and % reduction of cytochrome b566 in both the absence and presence of CN-POBS. As observed under other conditions, 2.5 µM CN-POBS did not alter ΔΨm or the reduction state of either redox pool (Table 3). In calibrated measurements of H2O2 production, CN-POBS had no significant effect on the total rate of production (Fig. 7). Although CN-POBS caused small (2 – 7%) decreases in total H2O2 production in three out of four trials, the average effect was non-significant at −2.5 ± 2.0% (n = 4). With the additional consideration that CN-POBS suppresses only 67% of site IQ superoxide/H2O2 production at 2.5 µM (see previous section), the potential maximal contribution of site IQ likely was still less than 4%. Therefore, we conclude that site IQ is not a significant source of superoxide/H2O2 production under these conditions. This exclusion leaves open the idea that the source of the unattributed H2O2 production is likely 2-oxoglutarate dehydrogenase [16]3.
Figure 7. Site IQ is not active during forward electron transport driven by glutamate plus malate.

CN-POBS had no effect on the rate of H2O2 production driven by 5 mM glutamate plus 5 mM malate under non-phosphorylating conditions. Data are means ± S.E. (n = 4).
It is important to note that the absence of superoxide/H2O2 production from site IQ during oxidation of glutamate plus malate in isolated mitochondria does not rule out a role for site IQ superoxide/H2O2 production under other conditions in which the matrix NADH pool is reduced and there is forward electron flux through complex I (e.g. in intact cells respiring on glucose or fatty acids). Indeed, the rate of H2O2 production in isolated mitochondria metabolizing succinate alone is stimulated by additional substrates that reduce matrix NAD+, such as glutamate, 2-oxoglutarate, pyruvate and palmitoylcarnitine [37]. In uninhibited systems, the rate of H2O2 production and the participation of different sites depend on the substrates being oxidized [16]. Therefore, context-dependent shifts in the sizes of different substrate pools along with changes in energetic demand will determine the contribution of site IQ in more complex systems. For example, there is evidence that increased flux through mGPDH drives superoxide/H2O2 production at site IQ during T-cell activation [38]. In a pathological context, a role for site IQ superoxide/H2O2 production may be most likely during reperfusion after ischemic events. During ischemia, succinate levels rise significantly and remain elevated after reperfusion [39, 40]. Therefore, it is possible that site IQ contributes to the burst of superoxide/H2O2 production on reperfusion [41]. A potential role for site IQ superoxide/H2O2 production during reperfusion is supported by recent observations that a cysteine residue adjacent to site IQ is selectively nitrosylated during ischemia and that this modification protects against ROS production and tissue damage induced by subsequent reperfusion [42]. Interestingly, inhibition of complex I with pyridaben significantly attenuated, and inhibition of complex II with 3-nitropropionic acid significantly exacerbated, tissue damage caused by ischemia/reperfusion in neonatal mice [43]. Because these inhibitors may alter ΔΨm, rates of ATP synthesis, and electron distribution at multiple sites including site IQ, it would be interesting to repeat these effects in the presence of CN-POBS to determine more directly whether site IQ superoxide/H2O2 production plays a significant role in ischemia/reperfusion injury.
Conclusions
Previous strategies to address questions of mitochondrial superoxide/H2O2 production involved acute pharmacological intervention with potent inhibitors of mitochondrial activity, longer-term molecular suppression of gene products that invariably alter normal mitochondrial metabolism, or chemical or enzymatic antioxidants that act downstream of production in a relatively non-specific manner and often indirectly influence cellular redox balance. The use of site-selective inhibitors of superoxide/H2O2 production that do not impair mitochondrial function allows more direct testing of hypotheses regarding the mechanisms of superoxide/H2O2 production, the contributions of distinct sites to overall production in complex systems, and the (patho)physiological consequences of production from specific sites in intact systems. As the first known site-selective inhibitor of superoxide/H2O2 production that does not interfere with oxidative phosphorylation, CN-POBS provides a proof-of-concept that such molecules exist. Despite the observed lack of a significant contribution from site IQ to the total H2O2 production during oxidation of glutamate plus malate, our ability to address this hypothesis using a site-selective inhibitor of superoxide/H2O2 production that does not interfere with normal mitochondrial energetics represents a fundamental shift in the approach to questions of mitochondrial ROS production. In addition, our efficient and adaptable high-throughput screening platform shows great promise for further identification of novel modulators of mitochondrial function, including selective inhibitors of other sites of superoxide/H2O2 production. As more potent and selective inhibitors of the numerous sites of superoxide/H2O2 production are identified, we can begin to address longstanding questions regarding the role of mitochondrial ROS production in health and disease.
Acknowledgements
Supported by The Ellison Medical Foundation grant AG-SS-2288-09 (MDB) and National Institutes of Health grants R01 AG033542 (MDB), RL1 GM084432 (REH), and TL1 AG032116 (ALO).
Abbreviations
- mGPDH
mitochondrial sn-glycerol 3-phosphate dehydrogenase
- TPMP
methyltriphenylphosphonium
- TMRM
tetramethylrhodamine methyl ester
- ΔΨm
potential difference across the mitochondrial inner membrane
- ΔpH
pH difference across the mitochondrial inner membrane
- site IF
flavin mononucleotide site of complex I
- site IF/DH
site IF plus matrix NAD-linked dehydrogenases
- site IQ
ubiquinone-binding site of complex I
- site IIF
flavin site of complex II
- site IIIQo
outer ubiquinone-binding site of complex III
- FCCP
carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone
- CN-POBS
N-cyclohexyl-4-(4-nitrophenoxy)benzenesulfonamide
- DMSO
dimethylsulfoxide
- ROS
reactive oxygen species.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
C. L. Quinlan, M. H. Mogensen, R. L. S. Goncalves, V. I. Bunik, M. D. Brand, Manuscript in preparation.
Author contributions
Adam Orr designed, performed, and analyzed all experiments. Deepthi Ashok assisted with screening, mitochondrial respiration, and TPMP measurements. Melissa Sarantos and Tong Shi assisted with screening. Robert Hughes provided the chemical library and advised in the design, execution, and analysis of the screen. Martin Brand conceived and oversaw the project. Adam Orr and Martin Brand wrote the paper.
References
- 1.Harman D. The free radical theory of aging: Effect of age on serum copper levels. J. Gerontol. 1965;20:151–153. doi: 10.1093/geronj/20.2.151. [DOI] [PubMed] [Google Scholar]
- 2.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 3.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 4.Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444:875–880. doi: 10.1038/nature05487. [DOI] [PubMed] [Google Scholar]
- 5.Muller FL, Lustgarten MS, Jang Y, Richardson A, Van Remmen H. Trends in oxidative aging theories. Free Rad. Biol. Med. 2007;43:477–503. doi: 10.1016/j.freeradbiomed.2007.03.034. [DOI] [PubMed] [Google Scholar]
- 6.Nunnari J, Suomalainen A. Mitochondria: In sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brand MD, Orr AL, Perevoshchikova IV, Quinlan CL. The role of mitochondrial function and cellular bioenergetics in ageing and disease. Br. J. Dermatol. 2013;169(Suppl 2):1–8. doi: 10.1111/bjd.12208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brand MD. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010;45:466–472. doi: 10.1016/j.exger.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Treberg JR, Quinlan CL, Brand MD. Evidence for two sites of superoxide production by mitochondrial NADH-ubiquinone oxidoreductase (complex I) J. Biol. Chem. 2011;286:27103–27110. doi: 10.1074/jbc.M111.252502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat. Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 11.Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- 12.Lambert AJ, Boysen HM, Buckingham JA, Yang T, Podlutsky A, Austad SN, Kunz TH, Buffenstein R, Brand MD. Low rates of hydrogen peroxide production by isolated heart mitochondria associate with long maximum lifespan in vertebrate homeotherms. Aging Cell. 2007;6:607–618. doi: 10.1111/j.1474-9726.2007.00312.x. [DOI] [PubMed] [Google Scholar]
- 13.Lambert AJ, Buckingham JA, Boysen HM, Brand MD. Low complex I content explains the low hydrogen peroxide production rate of heart mitochondria from the long-lived pigeon, columba livia. Aging Cell. 2010;9:78–91. doi: 10.1111/j.1474-9726.2009.00538.x. [DOI] [PubMed] [Google Scholar]
- 14.Perevoshchikova IV, Quinlan CL, Orr AL, Gerencser AA, Brand MD. Sites of superoxide and hydrogen peroxide production during fatty acid oxidation in rat skeletal muscle mitochondria. Free Rad. Biol. Med. 2013;61:298–309. doi: 10.1016/j.freeradbiomed.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quinlan CL, Treberg JR, Perevoshchikova IV, Orr AL, Brand MD. Native rates of superoxide production from multiple sites in isolated mitochondria measured using endogenous reporters. Free Rad. Biol. Med. 2012;53:1807–1817. doi: 10.1016/j.freeradbiomed.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Orr AL, Brand MD. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013;1:304–312. doi: 10.1016/j.redox.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002;277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 18.Orr AL, Quinlan CL, Perevoshchikova IV, Brand MD. A refined analysis of superoxide production by mitochondrial sn-glycerol 3-phosphate dehydrogenase. J. Biol. Chem. 2012;287:42921–42935. doi: 10.1074/jbc.M112.397828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bell EL, Klimova TA, Eisenbart J, Moraes CT, Murphy MP, Budinger GRS, Chandel NS. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 2007;177:1029–1036. doi: 10.1083/jcb.200609074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tormos KV, Anso E, Hamanaka RB, Eisenbart J, Joseph J, Kalyanaraman B, Chandel NS. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab. 2011;14:537–544. doi: 10.1016/j.cmet.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GRS, Chandel NS. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Nat. Acad. Sci. 2010;107:8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoffman DL, Salter JD, Brookes PS. Response of mitochondrial reactive oxygen species generation to steady-state oxygen tension: Implications for hypoxic cell signaling. Am. J. Physio. – Heart Circ. Physiol. 2007;292:H101–H108. doi: 10.1152/ajpheart.00699.2006. [DOI] [PubMed] [Google Scholar]
- 23.Kerr DS. Treatment of mitochondrial electron transport chain disorders: A review of clinical trials over the past decade. Mol. Genet. Metab. 2010;99:246–255. doi: 10.1016/j.ymgme.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 24.Gane EJ, Weilert F, Orr DW, Keogh GF, Gibson M, Lockhart MM, Frampton CM, Taylor KM, Smith RAJ, Murphy MP. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int. 2010;30:1019–1026. doi: 10.1111/j.1478-3231.2010.02250.x. [DOI] [PubMed] [Google Scholar]
- 25.Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O'Sullivan JD, Fung V, Smith RAJ, Murphy MP, Taylor KM. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant mitoQ as a disease-modifying therapy in Parkinson's disease. Mov. Disord. 2010;25:1670–1674. doi: 10.1002/mds.23148. [DOI] [PubMed] [Google Scholar]
- 26.Affourtit C, Quinlan CL, Brand MD. Measurement of proton leak and electron leak in isolated mitochondria. Methods Mol. Biol. 2012;810:165–182. doi: 10.1007/978-1-61779-382-0_11. [DOI] [PubMed] [Google Scholar]
- 27.Orr AL, Ashok D, Sarantos MR, Ng R, Shi T, Gerencser AA, Hughes RE, Brand MD. Novel inhibitors of mitochondrial sn-glycerol 3-phosphate dehydrogenase. doi: 10.1371/journal.pone.0089938. Manuscript in submission. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brideau C, Gunter B, Pikounis B, Liaw A. Improved statistical methods for hit selection in high-throughput screening. J. Biomol. Screen. 2003;8:634–647. doi: 10.1177/1087057103258285. [DOI] [PubMed] [Google Scholar]
- 29.Malo N, Hanley JA, Cerquozzi S, Pelletier J, Nadon R. Statistical practice in highthroughput screening data analysis. Nat. Biotech. 2006;24:167–175. doi: 10.1038/nbt1186. [DOI] [PubMed] [Google Scholar]
- 30.Lambert AJ, Buckingham JA, Boysen HM, Brand MD. Diphenyleneiodonium acutely inhibits reactive oxygen species production by mitochondrial complex I during reverse, but not forward electron transport. Biochim. Biophys. Acta. 2008;1777:397–403. doi: 10.1016/j.bbabio.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 31.Rogers GW, Brand MD, Petrosyan S, Ashok D, Elorza AA, Ferrick DA, Murphy AN. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS One. 2011;6:e21746. doi: 10.1371/journal.pone.0021746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang JH, Chung TDY, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 33.Quinlan CL, Gerencser AA, Treberg JR, Brand MD. The mechanism of superoxide production by the antimycin-inhibited mitochondrial Q-cycle. J. Biol. Chem. 2011;286:31361–31372. doi: 10.1074/jbc.M111.267898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scaduto RC, Grotyohann LW. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys. J. 1999;76:469–477. doi: 10.1016/S0006-3495(99)77214-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Treberg JR, Brand MD. A model of the proton translocation mechanism of complex I. J. Biol. Chem. 2011;286:17579–17584. doi: 10.1074/jbc.M111.227751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pryde KR, Hirst J. Superoxide is produced by the reduced flavin in mitochondrial complex I: a single, unified mechanism that applies during both forward and reverse electron transfer. J. Biol. Chem. 2011;286:18056–18065. doi: 10.1074/jbc.M110.186841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muller FL, Liu Y, Abdul-Ghani MA, Lustgarten MS, Bhattacharya A, Jang YC, Van Remmen H. High rates of superoxide production in skeletal-muscle mitochondria respiring on both complex I- and complex II-linked substrates. Biochem. J. 2008;409:491–499. doi: 10.1042/BJ20071162. [DOI] [PubMed] [Google Scholar]
- 38.Kaminski MM, Sauer SW, Kaminski M, Opp S, Ruppert T, Grigaravicius P, Grudnik P, Gröne HJ, Krammer PH, Gülow K. T cell activation is driven by an ADP-dependent glucokinase linking enhanced glycolysis with mitochondrial reactive oxygen species generation. Cell Rep. 2012;2:1300–1315. doi: 10.1016/j.celrep.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 39.Folbergrova J, Ljunggren B, Norberg K, Siesjo BK. Influence of complete ischemia on glycolytic metabolites, citric acid cycle intermediates, and associated amino acids in the rat cerebral cortex. Brain Res. 1974;80:265–279. doi: 10.1016/0006-8993(74)90690-8. [DOI] [PubMed] [Google Scholar]
- 40.Benzi G, Arrigoni E, Marzatico F, Villa RF. Influence of some biological pyrimidines on the succinate cycle during and after cerebral ischemia. Biochem. Pharm. 1979;28:2545–2550. doi: 10.1016/0006-2952(79)90024-8. [DOI] [PubMed] [Google Scholar]
- 41.Sanderson T, Reynolds C, Kumar R, Przyklenk K, Hüttemann M. Molecular mechanisms of ischemia–reperfusion injury in brain: Pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol. Neurobiol. 2013;47:9–23. doi: 10.1007/s12035-012-8344-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chouchani ET, Methner C, Nadtochiy SM, Logan A, Pell VR, Ding S, James AM, Cocheme HM, Reinhold J, Lilley KS, Partridge L, Fearnley IM, Robinson AJ, Hartley RC, Smith RAJ, Krieg T, Brookes PS, Murphy MP. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 2013;19:753–759. doi: 10.1038/nm.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Niatsetskaya ZV, Sosunov SA, Matsiukevich D, Utkina-Sosunova IV, Ratner VI, Starkov AA, Ten VS. The oxygen free radicals originating from mitochondrial complex I contribute to oxidative brain injury following hypoxia–ischemia in neonatal mice. J. Neurosci. 2012;32:3235–3244. doi: 10.1523/JNEUROSCI.6303-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]