

Abstract
JAK/STAT3 is one of the major signaling pathways that is aberrantly activated in ovarian cancer and associated with tumor progression and poor prognosis in ovarian cancer patients. In this study, we evaluated the therapeutic potential of targeting JAK/STAT3 signaling in ovarian cancer using a peritoneal dissemination mouse model. We developed this mouse model by injecting a metastatic human ovarian cancer cell line, SKOV3-M-Luc, into the peritoneal cavity of immunodeficient mice. This model displayed a phenotype similar to late stage ovarian cancer, including extensive peritoneal metastasis and ascites production. The constitutive activation of STAT3 in human ovarian cancer cells appeared to be mediated by an autocrine-cytokine loop involving the IL-6 family of cytokines and JAK1 kinase. shRNA-mediated knockdown of JAK1 or STAT3 in ovarian cancer cells led to reduced tumor growth, decreased peritoneal dissemination and diminished ascites production, suggesting a critical role of STAT3 in ovarian cancer progression. Similar results were obtained when a small-molecule inhibitor (JAKi) of the JAK1 kinase was used to treat ovarian cancer in this model. In addition, we found that the expression level of IL-6 was correlated with activation of STAT3 in ovarian cancer cells both in vitro and in vivo, suggesting a potential application of IL-6 as a biomarker. Altogether, our results demonstrate that targeting JAK1/STAT3, using shRNA knockdown or a small molecule inhibitor, effectively suppressed ovarian tumor progression and, therefore, could be a potential novel therapeutic approach for treating advanced ovarian cancer.
Keywords: JAK1/STAT3, shRNA, peritoneal metastasis, ascites, late stage ovarian cancer
Introduction
Ovarian cancer is the most deadly gynecological malignancy in women. Unfortunately, it is often not diagnosed until it has reached an advanced stage (1-3). Unlike other malignancies, ovarian cancer metastasizes less frequently to distant sites; it disseminates within the peritoneal cavity, and is associated with ascites formation (4-6). The extent of peritoneal carcinomatosis and ascites production are important prognostic factors for survival (5,7,8). Therefore, identifying approaches that inhibit peritoneal dissemination would be an important advance for treating ovarian cancer.
Standard management for advanced ovarian cancer is combination of cytoreductive surgery and paclitaxel-platinum treatment (3,9,10). Maximum cytoreductive surgery is associated with the longest patient survival, especially when combined with intraperitoneal chemotherapy (11). Managing large volumes of ascites requires repeated paracenteses, which only temporarily relieves the symptoms, and so remains a clinical challenge. The initial response rate to standard therapy is over 70%, however, most patients who have advanced tumors will eventually experience a recurrence. In spite of the improvements in surgical methods and chemotherapy, the mortality rates in women with both advanced and recurrent ovarian cancer remain high (1,12), highlighting the critical need to develop novel strategies for treating advanced and recurrent ovarian cancer.
Identifying the molecular events that regulate ovarian cancer metastasis holds promise for developing more effective therapeutic agents to treat human ovarian cancer (12-16). One of the major signaling pathways that is aberrantly activated and is critical for ovarian tumor growth is the JAK/STAT3 pathway (17-24). STAT3, which is a member of the STAT family of transcription factors, mediates cellular responses to growth factors and cytokines, including IL-6, IL-10, and VEGF. STAT3 is predominantly located in the cytoplasm in an inactive form, but is activated by phosphorylation at Tyr705 by the Janus family kinases (JAK) (25). The phosphorylated STAT3 protein can translocate into the nucleus, where it binds to DNA and activates the transcription of various genes that regulate vital cellular functions, including cell survival, proliferation, angiogenesis and tumor evasion (26). In normal tissues, the activation of STAT3 by the JAK kinase occurs transiently and is tightly regulated. In contrast, STAT3 is constitutively activated in cancer cells (19,26-29) and its continued activation is associated with a poor prognosis for ovarian cancer patients (30). Elevated levels of IL-6 in serum, ascites and malignant cancer cells also correlate with poor overall patient survival (30-34). These data suggest a relationship between STAT3 activation and ovarian cancer progression, but it remains to be determined if the STAT3 pathway is required for ovarian cancer progression.
Here, we have developed a peritoneal mouse model of human ovarian cancer to investigate the therapeutic potential of targeting the STAT3 pathway. This model displays extensive peritoneal dissemination and ascites formation, which resembles late stage human ovarian cancer. Our studies revealed that inhibiting the JAK/STAT3 signaling pathway, either by genetic manipulation through knocking down JAK1 or STAT3 expression or by pharmacological intervention using a small molecule JAK inhibitor, significantly suppressed both the dissemination of tumor cells into the peritoneal cavity and the production of ascites in vivo.
Materials and methods
Reagent
JAKi, AZD1480, was kindly provided by AstraZeneca. Antibodies against p-STAT3, STAT3, JAK1, P-JAK2, JAK2 and GAPDH were obtained from Cell Signaling.
Cell culture
SKOV3 and MDAH2774 were obtained from ATCC in 2010, SKOV3-Luc-D3 was obtained from Perkin Elmer in 2011, OVCAR 8 was from National Cancer Institute in 2013 and Kuramochi was from National Institute of Biomedical Innovation JCRB Cell Bank in 2014. No authorization of these cell lines was performed by the authors. SKOV3 and MDAH cells were cultured in DMEM medium containing 10% FBS. OVCAR 8 and Kuramochi cells were cultured in RPMI1640 medium containing 10% FBS.
Primary ovarian cancer cells were isolated from ascites fluid. The ascites was taken from primary serous ovarian cancer debulking case where advanced (Stage III/IV) disease was present. As previously described (35), 25ml ascites fluid was transferred to a culture flask and mixed with equal volume of MCDB105/M199 medium containing 10% FBS. The flask was placed in the incubator undisturbed for 3-4 days and then replaced with fresh MCDB105/M199 medium every 2-3 days. The human ascites fluid was used under a research protocol approved by the Institutional Review Board.
SKOV3-M-Luc cell line was derived from SKOV3-Luc-D3 cell line by selecting for a peritoneal metastatic phenotype in the mice. SKOV3-Luc-D3 cells were inoculated into peritoneal cavity of immunodeficient mice (athymic nude mice) and mice that produced the most ascites and had extensive peritoneal metastasis were used to isolate the tumor cells from the ascites. The isolated tumor cells produced ascites and numerous tumor nodules throughout the peritoneal cavity when inoculated into the peritoneal cavity of immunodeficient mice that were either athymic nude (National Cancer Institute) or NOD/SCID/IL2Rgamma null (NSG) mice. This cell line, named SKOV3-M-Luc, was used throughout this study.
Preparation of lysates and immunoblotting
To prepare cell lysates, cells treated with either DMSO or drugs in the complete growth medium were washed in cold PBS and lysed in lysis buffer (Thermo Scientific) containing protease inhibitor and phosphatase inhibitor (Thermo Scientific). To prepare tumor lysates, tumor tissues were homogenized in lysis buffer with a Bullet BlenderTM (Next Advance) following the manufacturer’s instructions. The lysate samples were centrifuged at 13000 rpm and the supernatants were transferred to a new tube and protein concentration was determined using BCA protein assay reagent (Thermo Scientific). Equal amounts of protein from each lysate were separated by electrophoresis on SDS gels, transferred to PVDF (Polyvinylidene Fluoride) membranes and incubated with total and phosphorylated antibodies. Binding of the primary antibody was detected using a horseradish peroxidase (HRP)-conjugated secondary antibody and chemiluminescent substrate (Thermo Scientific).
Cell viability assays
Cells (7000 per well for MDAH2774, 4000 per well for other cells,) were plated in 96-well plate in 100μl growth medium. Cells were treated with DMSO or JAKi next day at indicated concentration and incubated for additional 3 days. Viable cells were determined either by MTS assay (Promega, Madison, WI, USA) or acid phosphotase assay (36). For MTS assay, 25μl MTS solution was added directly into each well according to the manufacturer’s instructions. For acid phophotase asaay, all the media was removed and paranitrophenol phosphotase substrate (10mM 100μl) was added into each well and incubated at 37°C for 1hr. NaOH was added to stop the reaction and the absorbance was read at 415nM. IC50 was determined using Calcusyn software (Biosoft, ferguson, MO).
Transfection with siRNA and shRNA
To knock-down STAT3, JAK1 and JAK2 kinase, we transfect SKOV3-M-Luc cells with synthetic siRNA (Santa Cruz) using RNAiMAX (Invitrogen) according to the manufacturer’s instruction. For long term knockdown of STAT3 and JAK1, lentiviral vectors expressing STAT3, JAK1 or a non-targeting control shRNA, were produced as previously described using 293T as packaging cells (37). Subconfluent SKOV3-M-Luc cells were incubated with viral supernatants in the presence of polybrene for 48 hours. The medium was replaced with fresh medium containing puromycin (1 μg/mL) to select transduced cells expressing STAT3 shRNA, or JAK1 shRNA or NT shRNA. The stable cells lines were maintained in media containing puromycin.
Peritoneal tumor formation in mice
All procedures involving animals and their care were approved by the Institutional Animal Care and Use Committee according to the guidelines of the association for Assessment and Accreditation of Laboratory Animal Care.
Human ovarian cancer cells (5×106) were inoculated into the peritoneal cavity of either athymic nude (National Cancer Institute) or NOD/SCID/IL2Rgamma null (NSG) mice. Six to 12 mice were used for each group. To assess the effect of small molecule inhibitor on ovarian tumor growth, mice were individually given JAKi at 20 mg/kg based on previous studies (37-39) or vehicle (30% Solutol HS15) by daily gavage starting one week after inoculation. The mice were monitored for ascites production and any adverse effect, then euthanized 3-5 weeks after cell inoculation. Visible tumor nodules were excised and weighed and the ascites fluid was collected and measured for the volume.
Luciferase imaging
To monitor tumor growth in the peritoneal cavity, tumor bearing mice were injected intraperitoneally with 150μL of D-Luciferin (Perkin Elmer) at 4.29 mg/mouse and imaged by a Xenogen IVIX system (Caliper Life Science) once a week.
Luminex immunoassay
Ascites and plasma were collected from each mouse and centrifuged. Plasma, ascites and tumor lysates from the respective groups (4-8) were pooled and used for the experiments. The expression level of IL-6 and other cytokines in the pooled samples, as well as in each individual sample, was determined with an ELISA- based bead multiplex assay (Luminex Corp, Austin, TX, USA) that used a human 30-plex or 5-plex cytokine kit from Invitrogen.
Statistical analysis
Data are presented as mean ± S.D. Comparisons between two groups were determined by student’s t-test. Each assay was repeated 2-4 times; P < 0.05 was considered statistically significant.
Results
Development of a mouse model for advanced ovarian cancer
To study the role of JAK/STAT3 signaling pathway in ovarian tumor progression, a mouse tumor model that represents late stage ovarian cancer with peritoneal metastasis and ascites formation was developed by inoculating a highly metastatic human ovarian cancer cell line, SKOV3-M-Luc, into peritoneal cavity of immunodeficient mice. We derived SKOV3-M-Luc cell from SKOV3-Luc cells by selecting for a peritoneal metastatic phenotype in the mice (Materials and Methods). STAT3 is constitutively activated in these cells. Four weeks following i.p. injection of SKOV3-M-Luc cells, we found that hundreds of tumor nodules, together with large primary tumors, had developed and attached themselves to the peritoneal surface, gastrointestinal tract, omentum, and diaphragm throughout the peritoneal cavity in these mice (Figure 1A). Disseminated tumor nodules were excised from the cavity for analysis, and some weighed up to 2 grams in total in an individual mouse. The average weight of large primary tumor was about 0.91 g and the average volume of ascites was about 6 mL per mouse (Table 1a). Our model, therefore, closely resembled late stage ovarian cancer.
Figure 1.
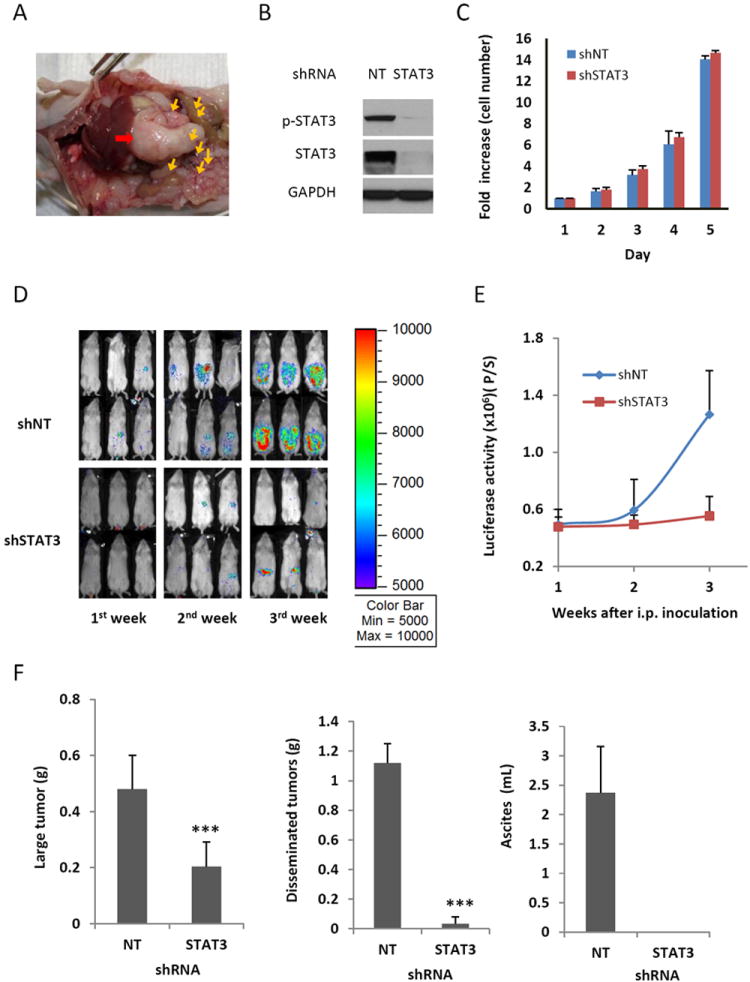
Knocking down STAT3 expression using RNA interference suppresses peritoneal metastasis and ascites production in NSG mice. (A) Representative view of the peritoneal cavity of an athymic nude mouse from 8 mice inoculated with SKOV3-M-Luc cells. Red arrow: large primary tumor; yellow arrows: small tumor nodules. (B) STAT3 expression was blocked in SKOV3-M-Luc cells with shRNA targeted against STAT3. Cells were transduced with STAT3 shRNA or non-targeted (NT) shRNA, and expression of total STAT3 and phosphorylated STAT3 was determined by Western blot analysis. Results are representative of 3 experiments. (C) In vitro proliferation assay. Both shNT cells and shSTAT3 cells were plated and counted each day. (D-E) Luciferase images show cancer progression in mice. STAT3 deficient cells (shSTAT3) and STAT3 active cells (shNT) were inoculated into the peritoneal cavity of NSG mice. Luciferase activities were measured each week after initial cell inoculation (D) and quantified (E). (F) Effect of STAT3 knockdown on tumor burden and ascites volume. At the end of experiment, mice were euthanized. Large primary tumors and small tumor nodules were excised and weighed. Ascites was collected and the volume was measured. n=5-8, ***, P<0.0005, vs. shNT control.
Table 1.
a: Tumor burden and ascites volume in the mice inoculated with human ovarian cancer cells.
| Ovarian cancer xenograph model (i.p.) (SKOV3-M-Luc) | Value (n=7) |
|---|---|
| Primary tumor (g) | 0.91±0.08 |
| Disseminated tumor (g) | 2.06±0.29 |
| Ascites (ml) | 6.14±1.34 |
| b: Expression of IL-6 family of cytokines | ||
|---|---|---|
| Concentration (pg/ml) | ||
| IL-6 family of cytokines | Conditioned medium (SKOV3-M-Luc cells) | Ascites (bearing SKOV3-M-Luc tumor) |
| IL-6 | 21921 ±6231 | 663 ±283 |
| LIF | 510±45 | 978±20 |
| OSM | ND | ND |
| IL-10 | ND | ND |
| IL-27 | ND | ND |
Effect of STAT3 knockdown on peritoneal tumor growth and ascites formation
We next investigated whether blocking STAT3 expression had any effect on cancer progression in this model. We first used a genetic approach to silence STAT3 expression via RNA interference (shRNA). The constitutively activated STAT3 in SKOV3-M-Luc cells was blocked when cells were transduced with a lentiviral vector expressing shRNA against STAT3, but not non-targeting control shRNA (Figure 1B). There was no significant difference observed in the in vitro proliferation between STAT3 shRNA knockdown cells (shSTAT3) and non-targeted shRNA control cells (shNT), which had active STAT3 (Figure 1C). However, the ability of these two cell lines to disseminate, form tumors and produce ascites in the peritoneal cavities of mice was strikingly different. Tumor growth in the peritoneal cavity was monitored weekly by luciferase imaging after inoculation of tumor cells into the peritoneal cavity of immunodeficient mice (NSG). Luciferase activity was significantly reduced in the mice inoculated with the shSTAT3 cells compared to mice inoculated with shNT cells (Figures 1D and 1E). Four weeks after injection, mice inoculated with shNT cells displayed signs of severe ascites and all mice were euthanized at that time point. Large amounts of ascites fluid (mean volume 2.4 mL) had accumulated, and hundreds of tumor nodules had developed on the peritoneal wall, gastrointestinal tract, diaphragm in the peritoneal cavities of mice inoculated with shNT cells expressing activated STAT3. In contrast, no measurable amount of ascites was produced and there were fewer small tumor nodules found in the peritoneal cavity of mice inoculated with the shSTAT3 cells, in which STAT3 expression was blocked. The total weight of all disseminated small tumor nodules was decreased by ~ 25-fold in mice inoculated with shSTAT3 knockdown cells (0.045 g) compared to the shNT controls (1.12 g). The weight of the large primary tumors was reduced by ~60 % (0.48 g vs 0.20 g) (Figure 1F). These results indicate that knocking down the expression of STAT3 in ovarian cancer cells decreased their ability to metastasize and produce ascites.
Activation of STAT3 mediated by an autocrine cytokine loop
The constitutive activation of STAT3 in ovarian cancer cells could be mediated by an autocrine cytokine loop through JAK kinases, or by the activation of oncogenes, such as EGFR and Src. To understand the mechanism by which STAT3 is activated in ovarian cancers, we first determined if cytokines secreted into the medium were responsible for activating STAT3. Human ovarian cancer cells, SKOV3 and MDAH2774, were grown in culture medium for two days, and then medium was replaced with fresh medium for 30 mins. Phosphorylation of STAT3 was lost when the old medium was replaced with fresh medium (Figure 2A), but could be restored by replacing with old medium (Figures 2B and 2C), suggesting cytokines secreted by the cancer cells into the medium might be critical in mediating the phosphorylation of STAT3 (Figures 2A to 2C). Furthermore, STAT3 phosphorylation was suppressed by adding a neutralizing antibody against gp130, a co-receptor for the IL-6 family of cytokines, suggesting that IL-6 family of cytokines was involved in the activation of STAT3 (Figures 2B and 2C). To determine what are the IL-6 family cytokines that are produced by ovarian cancer cells, we measured protein level of IL-6, leukemia inhibitory factor (LIF), IL-10, IL-27 and oncostatin M (OSM) in the conditioned media using an ELISA based multiplex assay. As shown in Table 1b, the expression level of IL-10, IL-27 and OSM was very low, out of detection range. However the expression of IL-6 and LIF was high and may contribute to the activation of STAT3. Taken together, these results suggest that autocrine production of cytokines, involving members of IL-6 family, mediates STAT3 phosphorylation in ovarian cancer cells.
Figure 2.
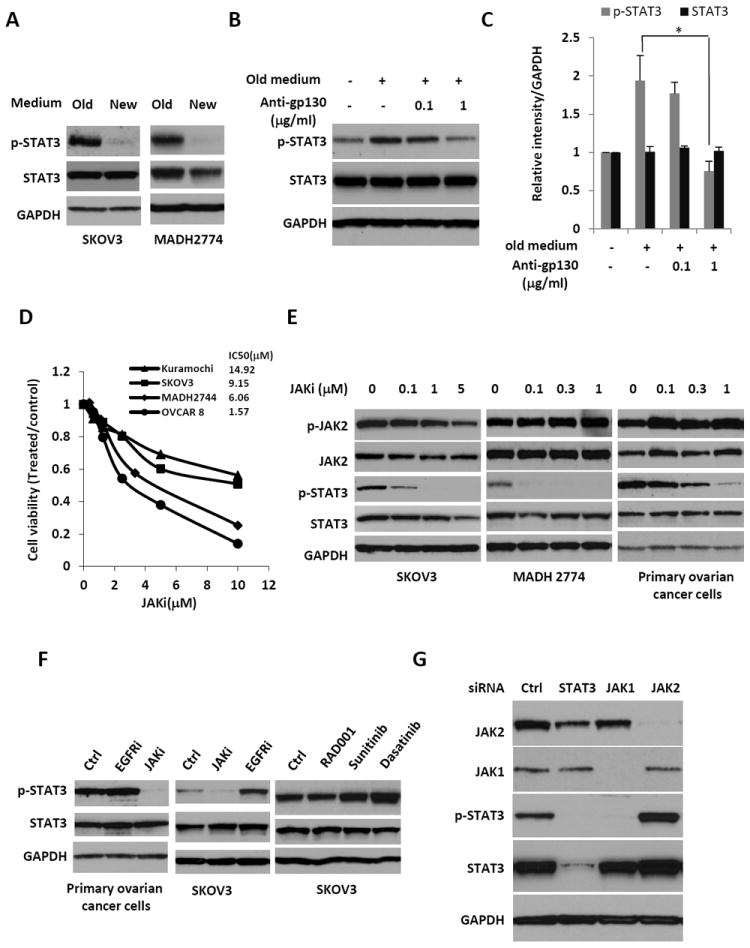
Suppressing the STAT3 pathway by inhibiting JAK1 kinase activity. (A) SKOV3 and MDAH2774 cells were first grown in regular culture medium for two days, then the medium was replaced with fresh medium for 30 mins. Cells, both before and after replacing the medium, were harvested and analyzed for STAT3 phosphorylation. (B) Old medium was added back to the SKOV3 cells, which had been in the fresh medium for 30 mins, either in the presence or absence of antibodies against the gp130 protein, and incubated for an additional 30 mins. Cells were harvested and analyzed for the phosphorylation of STAT3. (C) Relative levels of p-STAT3 and STAT3 were determined by measuring the density of each band and normalized to that of GAPDH. Densitometry data were relative changes in protein expression and were mean ± SD of 2-3 preparations. *, P<0.05 vs. control without anti-gp130 in old medium. (D) Effect of JAKi on cell viability of human ovarian cancer cells. Cells were plated and treated with various concentration of JAKi, cell viability was determined 72 hrs later. (E) Dose-dependent inhibition of STAT3 phosphorylation in SKOV3, MDAH2774 and primary human ovarian cancer cells by the JAK inhibitor (JAKi), AZD1480. Cells were treated with JAKi at various concentrations for 24 hours and the phosphorylation of STAT3 and JAK2 were analyzed on Western blots. (F) Effect of other kinase inhibitors on phosphorylation of STAT3. SKOV3 cells were treated with EGFRi (gefitinib), SRC inhibitor (dasatinib), multiple kinase inhibitor (sunitinib) and mTOR inhibitor (RAD001) for 24hrs. Phosphophrylation of STAT3 was determined by Western Blot. (G) STAT3 phosphorylation is inhibited by treating with an siRNA against JAK1. SKOV3-M-Luc cells were transfected with siRNA against STAT3, JAK1 or JAK2. Cells were analyzed for the expression levels of JAK1, JAK2 or the phosphorylation of STAT3 by Western blot.
Effect of JAK1 knockdown on peritoneal tumor growth and ascites formation
To understand the role of the JAK kinases in activating STAT3, we studied the effect of JAK kinase inhibitor (JAKi) (37), AZD1480, on cell proliferation and phosphorylation of STAT3 in SKOV3 cells, MDAH2774 cells and primary human ovarian cancer cells isolated from ascites of ovarian cancer patients. As shown in Figure 2D, the effect of JAKi on cell viability was weak, which could be due to the activation and compensation of multiple survival pathways in vitro. However, the inhibitory effect of JAKi on phosphorylation of STAT3 was strong in a dose-dependent manner (Figure 2E). In contrast, the phosphorylation of STAT3 was not inhibited by other kinase inhibitors, such as EGFR inhibitor (gefitinib), SRC inhibitor (dasatinib), multiple kinase inhibitor (sunitinib) and mTOR inhibitor (RAD001)(Figure 2F). Taken together, these results suggested that JAK kinase is required for the persistent activation of STAT3. To determine whether JAK1, JAK2 or both activate STAT3, we knocked down the expression of each individual JAK kinase with siRNA. Knockdown of JAK1, but not JAK2, blocked the phosphorylation of STAT3 (Figure 2G), suggesting that JAK1 is the critical kinase that phosphorylates and activates STAT3 in human ovarian cancer cells.
To investigate the effect of suppressing JAK1 expression on the progression of ovarian cancer in mice, we knocked down expression of JAK1 in SKOV3-M-luc cells with a lentiviral vector expressing shRNA against JAK1 (shJAK1). STAT3 activation was only suppressed in cells stably expressing the JAK1 shRNA, but not in cells expressing the control shRNA (shNT). Total STAT3 appeared to be reduced in cells expressing shJAK1 (Figures 3A and 3B). We inoculated the cells into immunodeficient mice to determine if JAK1 knockdown cells (shJAK1) could metastasize and produce ascites in the peritoneal cavity. Consistent with the results in mice inoculated with STAT3 knockdown cells (shSTAT3), peritoneal disseminated tumor nodules were reduced by ~7 fold (0.18 g) in mice inoculated with shJAK1 cells, compared to 1.25 grams in mice inoculated with the shNT tumor cells (Figures 3C-3E). The weight of large primary tumors was reduced only by ~30% in mice bearing shJAK1 cells compared to mice bearing shNT cells. The ascites volume was decreased by ~12 fold (0.475 mL) in the mice bearing shJAK1 cells, compared to 5.55 mL in mice bearing STAT3 active cells (shNT) (Figure 3E). Western blot analysis showed that phosphorylation of STAT3 was inhibited in the tumor lysates of both shSTAT3 and shJAK1 tumors (Figures 3F and 3G). Altogether, the results indicate that targeting the JAK1/STAT3 pathway effectively inhibited peritoneal metastasis and ascites production by ovarian cancer cells.
Figure 3.
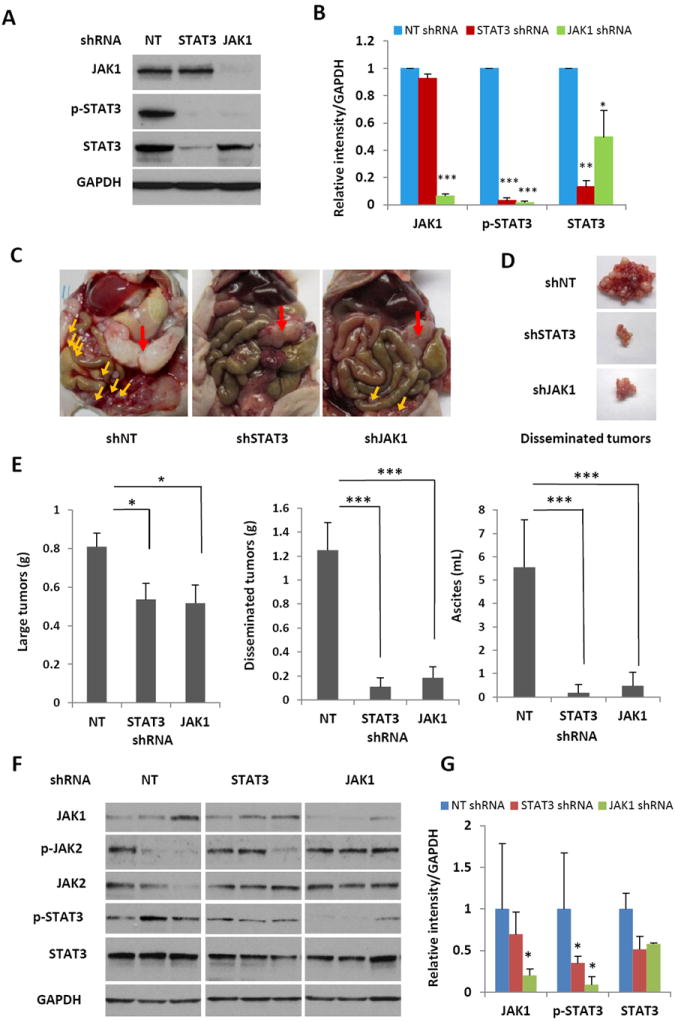
Knocking down JAK1 expression using RNA interference suppresses peritoneal dissemination and ascites formation in nude mice. (A) SKOV3-M-Luc cells were infected with shRNA against JAK1, STAT3 and control non-targeted shRNA (NT). The expression of JAK1, STAT3 as well as p-STAT3 was detected on Western blots. (B) Relative levels of p-STAT3, STAT3 and JAK1 were determined by measuring the density of each band and normalized to that of GAPDH. Densitometry data were relative changes in protein expression and were mean ± SD of 2-3 preparations. *, P<0.05, **, P<0.005, ***, P<0.0005, vs. shNT control. (C) Representative photographs of the peritoneal cavities of athymic nude mice 30 days after peritoneal inoculation of STAT3 active cells (shNT) or STAT3 inactive cells (shSTAT3 or shJAK1). Red arrow: large tumor; yellow arrows: small tumor nodules. (D) Representative photographs of disseminated peritoneal tumors. (E) Tumor burden and ascites production 30 days after tumor inoculation into the peritoneal cavity of athymic nude mice. Small tumor nodules, as well as the large primary tumor, were excised and weighed. Ascites was collected and the volume was measured. n=4-8, *, P<0.05, ***, P<0.0005, vs. shNT control. (F) Expression of STAT3 in xenograft tumors. Protein extracts were isolated from xenograft tumors and analyzed for the expression of p-STAT3, STAT3, p-JAK2, JAK2, and JAK1 on Western blots. (G) Relative level of JAK1, p-STAT3 and STAT3 were determined by measuring the density of each band and normalized to that of GAPDH. Densitometry data were relative changes in protein expression and were mean ± SD of 2-3 preparations. *, P<0.05, vs. shNT control.
Effect of suppressing the JAK1/STAT3 signaling pathway with small molecule inhibitor on peritoneal tumor growth and ascites formation
We next investigated if targeting JAK1/STAT3 signaling with a small molecule inhibitor also suppressed ovarian cancer growth and progression. We used a small molecule inhibitor of JAK kinase (JAKi) (37-39), AZD1480, to block STAT3 activation both in vitro and in vivo. We showed that JAKi blocked STAT3 phosphorylation but not cell proliferation in human ovarian cancer cells in vitro (Figures 2D & 2E), To determine anti-tumor activity of JAKi in vivo, nude mice were inoculated with SKOV3-M-Luc cells, allowed to grow for seven days, and then treated with JAKi at a daily dose of 20 mg/kg (37-39). The weight of the small tumor nodules in the peritoneal cavity was significantly reduced (5-fold) in treated mice (0.4 g), compared to the control mice (2.0g). The weight of large primary tumors was reduced by 40%, 0.58 g compared to 0.98 g. The ascites volume decreased 10-fold, 0.67 mL in JAKi treated mice compared to 6.4mL in mice treated with vehicle (Figures 4A and 4B). To determine if the anti-tumor activity of JAKi in mice was due to reduced phosphorylation of STAT3, whole tumor lysates were prepared and analyzed by Western blot. The phosphorylation of STAT3 was decreased in the tumors from mice treated with JAKi, but not in the tumors from vehicle treated mice (Figures 4C and 4D). The results indicate that targeting JAK1 with a small molecule inhibitor was able to effectively inhibit ovarian tumor progression and ascites production, providing the basis for a new therapeutic approach for the treatment of advanced human ovarian cancer patients.
Figure 4.
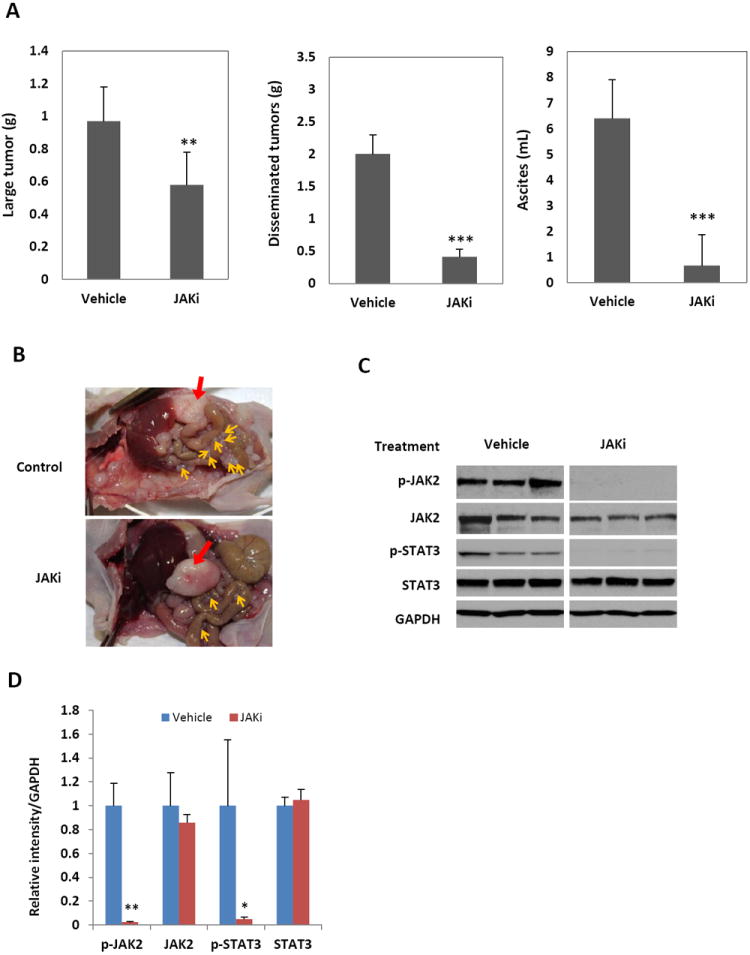
Inhibition of the JAK/STAT3 pathway with a small molecule inhibitor of JAK kinases blocks peritoneal metastasis and ascites production in nude mice. (A) SKOV3-M-Luc cells were inoculated into the peritoneal cavity of nude mice. Mice were treated with vehicle control or JAKi (20 mg/kg) each day. Mice were euthanized four weeks later. The large tumor and the small tumor nodules throughout the peritoneal cavity were excised and weighed. Ascites was collected and the volumes were measured. n=4-8, **, P<0.005, ***, P<0.0005, vs. vehicle control. (B) Representative photographs of peritoneal cavities of mice. Red arrow: large tumor; yellow arrows: small tumor nodules. (C) JAKi inhibited the activation of STAT3 in the tumor. Whole tumor lysates were prepared and analyzed for expression of p-STAT3, p-JAK2, total STAT3 and total JAK2 by Western blot. (D) Relative level of JAK1, p-STAT3 and STAT3 were determined by measuring the density of each band and normalized to that of GAPDH. Densitometry data were relative changes in protein expression and were mean ± SD of 2-3 preparations. *, P<0.05, **, P<0.005, vs. shNT control.
Effect of suppressing JAK1/STAT3 signaling on the expression of IL-6
Increased IL-6 levels in serum, ascites and tumor are associated with poor prognosis (30-34). High levels of phosphorylated STAT3 (p-STAT3) are also linked to poor prognosis (31,33,34). However, it remains to be determined if IL-6 levels are directly correlated with STAT3 activation in human ovarian cancer cells. To address this question, we investigated if the activation of STAT3 had any effect on IL-6 expression in ovarian cancer cells. We found that IL-6 levels were dramatically reduced in the supernatant of ovarian STAT3 knockdown cancer cells as compared to control cells (Figure 5A), suggesting the STAT3 pathway could mediate the production of IL-6. To determine if IL-6 level was associated with STAT3 activation in tumor, we measured the expression level of IL-6 in tumor lysates and found that IL-6 level was higher in the tumor with active STAT3 (shNT) than in the tumor with inactive STAT3 (shSTAT3 and shJAK1) (Figure 5B). To further understand whether circulating IL-6 level was also associated with STAT3 activation in tumor, ascites and plasma were collected from mice inoculated with STAT3 knockdown cells and control cells, and the levels of IL-6 was measured using a multiplex assay. We found that IL-6 levels in the plasma were very low in the nude mice without tumor (None), but they were remarkably elevated 34-fold in mice inoculated with tumor cells expressing activated STAT3 (shNT). However, IL-6 levels were almost as low as in the mice without tumor when mice were inoculated with STAT3 shRNA-expressing cancer cells (Figure 5C). To investigate whether activation of STAT3 also increased expression of other cytokines, we measured the levels of IL-2 and interferon gamma (IFN-γ), two cytokines that are distinct from the IL-6 family, in the same samples. In contrast to IL-6, circulating IFN-γ levels, although fivefold higher compared to levels in normal non-inoculated mice, were similar in both the shSTAT3 mice and shNT mice. The circulating IL-2 levels were similar in all mice irrespective of their inoculation or tumor status (Figure 5C). Overall the results suggest that circulating IL-6 levels were correlated with the levels of activated STAT3 in the tumors. Consistent with these results, the IL-6 levels in ascites were also correlated with the levels of activated STAT3 in the tumor. IL-6 level was decreased by 67% in shSTAT3 mice and by 85% in shJAK1 mice. The expression of IFN-γ and IL-2 was similar in both STAT3 active tumor mice and shSTAT3 mice (Figure 5D). We further investigated if there was a similar correlation between IL-6 levels when JAKi was used to block STAT3 activation. We found that IL-6 levels were significantly lower in plasma and ascites of mice treated with JAKi (Figures 5E and 5F).
Figure 5.
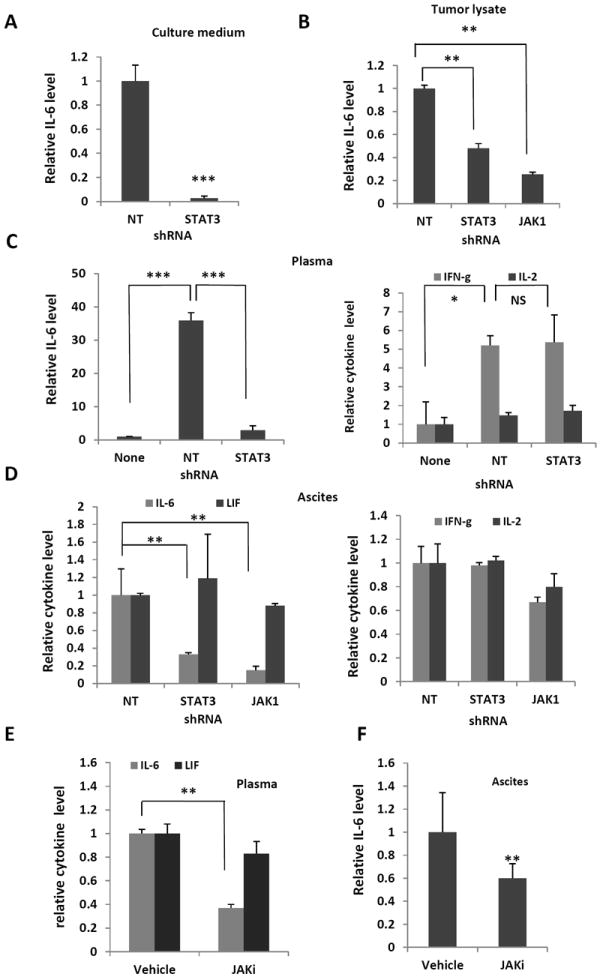
Loss of IL-6 expression by cells in conditioned medium (A), tumor lysate (B), plasma (C), and ascites (D), induced by the knock-down of STAT3 and JAK1 expression. The expression level of IL-6 in these samples was measured using a multiplex assay. Results are presented as the ratio of IL-6 from samples to the active STAT3 group (shNT). n=4-8, *, P<0.05, **, P<0.005, vs. shNT control. NS, not significant. (E)-(F) The small molecule inhibitor of JAK, JAKi, significantly reduced IL-6 expression in plasma (E), and ascites (F). Data are expressed as the ratio of treated samples to vehicle control. n=4-8, **, P<0.005, vs. vehicle control.
To determine whether the expression of other members of IL-6 family of cytokine was also correlated with activation of STAT3 in tumor, we measured expression level of LIF, OSM, IL-10, IL-27, in ascites and plasma. While the expression level of IL-10, IL-27 and OSM was too low to be detected, the expression of LIF was high. However, the LIF level was similar in mice bearing either STAT3 active tumor or STAT3 inactive tumor (Figures 5D and 5E). Although it has been well documented that expression of IL-6 is regulated by STAT3 pathway, it remained to be investigated whether the expression of other members of IL-6 family of cytokines can be regulated by STAT3.
Altogether, our results indicate that the JAK1/STAT3 pathway plays a critical role in the autocrine production of IL-6 by human ovarian cancer, and there is a direct link between IL-6 levels and activated STAT3 both in vitro and in vivo. We propose that IL-6 could be a useful marker for monitoring STAT3 activation during ovarian cancer development and treatment.
Discussion
Unlike other solid tumors, human ovarian cancer infrequently spreads to distant sites. Peritoneal dissemination is the most common pathway for human ovarian cancer to metastasize (6,40). Although the role of STAT3 in promoting metastasis through the blood stream to remote sites has been reported for other solid tumors, including melanoma and breast cancer, the role of STAT3 in ovarian cancer peritoneal metastasis and ascites production was unknown (39,41-44). We show here that by targeting JAK1/STAT3 signaling using genetic or pharmacological methods, we could significantly inhibit peritoneal metastasis, ascites production and autocrine production of IL-6 family cytokine in our peritoneal human ovarian cancer model.
Although most solid tumors are often surrounded by stroma, human ovarian cancer is surrounded by ascites, a very unique tumor-friendly microenvironment (8). Ascites is an abnormal accumulation of fluid in the peritoneal cavity due to lymphatic obstruction and increased vessel permeability (45). Ascites is enriched with chemokines, cytokines and growth factors, which promote inflammation, tumor growth and chemoresistance (33). Increased production of ascites is often associated with poor prognosis and poor quality of life. Managing ascites has remained a clinical challenge, given its limited and short-lived treatment options. Here we show that suppressing STAT3 phosphorylation inhibited the production of ascites, suggesting a novel strategy for controlling ascites production.
Although suppression of the STAT3 pathway remarkably reduced the tumor burden and accumulation of ascites fluid, the suppression of STAT3 phosphorylation by shRNA knockdown or JAKi had no striking effect on cell viability in vitro. One possible explanation for this is that multiple survival pathways are activated in ovarian cancer cells, and therefore, suppressing a single pathway may not be sufficient to suppress cell growth in vitro due to compensation by other survival pathways. It’s also possible that STAT3 signal is redundant in the presence of the multiple growth factors in serum in cell culture, but it becomes essential under physiological conditions where cells are not bathed 24/7 in growth factors and IL-6/JAK1/STAT3 becomes a key driver. More studies are needed to address whether other pathways are involved in ovarian cancer cell proliferation and survival, and whether anti-tumor activity of inhibiting STAT3 can be enhanced when other survival pathways are blocked simultaneously either in the presence of inhibitors or in the absence of growth factors under serum free culture conditions. The inhibitory effect of targeting the STAT3 pathway on tumor growth and progression in vivo could also be mediated by regulating the tumor microenvironment in human ovarian cancer. The tumor microenvironment is complex; it is composed of a number of cell types, many of which are critical for tumor growth and progression (26,46). For example, myeloid-derived suppressor cells (MDSCs) are one of the most important cell populations in tumor microenvironment. Specific targeting of STAT3 in MDSC cells decreases tumor growth and metastasis (26). Endothelial cells are another important cell type in the microenvironment. Angiogenesis, a process by which new blood vessels form, is critical for tumors to grow and progress (47). It has been reported that activated STAT3 up-regulates VEGF levels in human cancer cells (19). Further studies are underway to address whether the critical role played by the JAK1/STAT3 pathway in promoting peritoneal metastasis and ascites production is mediated by regulating the ovarian tumor microenvironment.
Many studies have shown that IL-6 is one of the critical regulators that mediates crosstalk between tumor cells and their microenviroment (48). The importance of IL-6 autocrine production in tumor growth and progression has been demonstrated in many tumors (48-50). Activated STAT3 has been implicated in modulating the production of cytokines, including IL-6. Here we show that suppressing the STAT3 pathway reduced IL-6 expression both in vitro and in vivo, suggesting that the STAT3 pathway mediates the autocrine production of IL-6 in ovarian tumor cells. The increased IL-6 levels could activate STAT3 in surrounding cells, which would increase production of IL-6 and other cytokines by these cells. This, in turn, would activate tumor cells to produce even more IL-6, promoting an inflammatory tumor environment that supports tumor growth and metastasis (26,48). Suppressing STAT3 phosphorylation may put a break on this feed-forward loop by blocking STAT3 activation and IL-6 production, and thus blocking ovarian tumor growth and metastasis.
Following inhibition of JAK1/STAT3 signaling in ovarian cancer cells, we saw decreased levels of IL-6 in the ascites and tumor cells. Consistent with these results, we showed that circulating IL-6 was significantly decreased in the plasma of mice inoculated with shSTAT3 tumor cells, and IL-6 levels were comparable to non-inoculated mice.
Circulating IL-6 levels were also reduced in the plasma of mice treated with the JAK inhibitor. These results indicate that IL-6 levels are closely correlated with the activation of STAT3 in the tumor cells, suggesting that IL-6 is a potential biomarker that could be used to optimize patient selection and to guide treatment.
Overall, our results indicate that targeting the JAK1/STAT3 pathway is an effective method for inhibiting peritoneal metastasis and ascites production in ovarian cancer in our mouse model, and may provide a new therapeutic avenue for treating these events in human ovarian cancer patients.
Acknowledgments
We thank our lab members for their valuable suggestions and discussions, Dr. Dennis Huszar at AstraZeneca for providing AZD1480, Dr. Paul Lin for human ascites, Mrs. Yan Wang for her assistance in the animal studies and Dr. Margaret Morgan for critical reading of this manuscript. We thank Dr. Mi Shu and Clinical Immunobiology Correlative Studies laboratory Core, and Animal Facility for their technical assistance.
Grant Support:
This work was supported by NIH grant R01 CA 115674-05 to R. Jove. The core facilities used in this study were supported by NCI p30 CA033572.
Footnotes
Disclosure of Potential Conflicts of Interest:
No potential conflicts of interest were disclosed.
References
- 1.Cannistra SA. Cancer of the ovary. N Engl J Med. 2004;351(24):2519–29. doi: 10.1056/NEJMra041842. [DOI] [PubMed] [Google Scholar]
- 2.Romero I, Bast RC., Jr Minireview: human ovarian cancer: biology, current management, and paths to personalizing therapy. Endocrinology. 2012;153(4):1593–602. doi: 10.1210/en.2011-2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vaughan S, Coward JI, Bast RC, Jr, Berchuck A, Berek JS, Brenton JD, et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer. 2011;11(10):719–25. doi: 10.1038/nrc3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tan DS, Agarwal R, Kaye SB. Mechanisms of transcoelomic metastasis in ovarian cancer. The lancet oncology. 2006;7(11):925–34. doi: 10.1016/S1470-2045(06)70939-1. [DOI] [PubMed] [Google Scholar]
- 5.Naora H, Montell DJ. Ovarian cancer metastasis: integrating insights from disparate model organisms. Nature reviews Cancer. 2005;5(5):355–66. doi: 10.1038/nrc1611. [DOI] [PubMed] [Google Scholar]
- 6.Masoumi Moghaddam S, Amini A, Morris DL, Pourgholami MH. Significance of vascular endothelial growth factor in growth and peritoneal dissemination of ovarian cancer. Cancer metastasis reviews. 2012;31(1-2):143–62. doi: 10.1007/s10555-011-9337-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lengyel E. Ovarian cancer development and metastasis. The American journal of pathology. 2010;177(3):1053–64. doi: 10.2353/ajpath.2010.100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahmed N, Stenvers K. Getting to know ovarian cancer ascites: opportunities for targeted therapy- based translational research. Frontiers in Oncology. 2013;3 doi: 10.3389/fonc.2013.00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morgan RJ, Jr, Alvarez RD, Armstrong DK, Boston B, Burger RA, Chen LM, et al. Epithelial ovarian cancer. Journal of the National Comprehensive Cancer Network : JNCCN. 2011;9(1):82–113. doi: 10.6004/jnccn.2011.0008. [DOI] [PubMed] [Google Scholar]
- 10.Cristea M, Han E, Salmon L, Morgan RJ. Practical considerations in ovarian cancer chemotherapy. Therapeutic advances in medical oncology. 2010;2(3):175–87. doi: 10.1177/1758834010361333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Landrum LM, Java J, Mathews CA, Lanneau GS, Jr, Copeland LJ, Armstrong DK, et al. Prognostic factors for stage III epithelial ovarian cancer treated with intraperitoneal chemotherapy: a Gynecologic Oncology Group study. Gynecol Oncol. 2013;130(1):12–8. doi: 10.1016/j.ygyno.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yap TA, Carden CP, Kaye SB. Beyond chemotherapy: targeted therapies in ovarian cancer. Nat Rev Cancer. 2009;9(3):167–81. doi: 10.1038/nrc2583. [DOI] [PubMed] [Google Scholar]
- 13.Blagden S, Gabra H. Promising molecular targets in ovarian cancer. Current opinion in oncology. 2009;21(5):412–9. doi: 10.1097/CCO.0b013e32832eab1f. [DOI] [PubMed] [Google Scholar]
- 14.Tagawa T, Morgan R, Yen Y, Mortimer J. Ovarian cancer: opportunity for targeted therapy. Journal of oncology. 2012;2012 doi: 10.1155/2012/682480. 682480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han ES, Wakabayashi M, Leong L. Angiogenesis inhibitors in the treatment of epithelial ovarian cancer. Current treatment options in oncology. 2013;14(1):22–33. doi: 10.1007/s11864-012-0220-6. [DOI] [PubMed] [Google Scholar]
- 16.Bast RC, Jr, Hennessy B, Mills GB. The biology of ovarian cancer: new opportunities for translation. Nature reviews Cancer. 2009;9(6):415–28. doi: 10.1038/nrc2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miklossy G, Hilliard TS, Turkson J. Therapeutic modulators of STAT signalling for human diseases. Nat Rev Drug Discov. 2013;12(8):611–29. doi: 10.1038/nrd4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duan Z, Foster R, Bell DA, Mahoney J, Wolak K, Vaidya A, et al. Signal transducers and activators of transcription 3 pathway activation in drug-resistant ovarian cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12(17):5055–63. doi: 10.1158/1078-0432.CCR-06-0861. [DOI] [PubMed] [Google Scholar]
- 19.Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer. 2004;4(2):97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 20.Buettner R, Mora LB, Jove R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clinical cancer research : an official journal of the American Association for Cancer Research. 2002;8(4):945–54. [PubMed] [Google Scholar]
- 21.Silver DL, Naora H, Liu J, Cheng W, Montell DJ. Activated signal transducer and activator of transcription (STAT) 3: localization in focal adhesions and function in ovarian cancer cell motility. Cancer research. 2004;64(10):3550–8. doi: 10.1158/0008-5472.CAN-03-3959. [DOI] [PubMed] [Google Scholar]
- 22.Sansone P, Bromberg J. Targeting the Interleukin-6/Jak/Stat Pathway in Human Malignancies. Journal of Clinical Oncology. 2012;30(9):1005–14. doi: 10.1200/JCO.2010.31.8907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miklossy G, Hilliard TS, Turkson J. Therapeutic modulators of STAT signalling for human diseases. Nat Rev Drug Discov. 2013;12(8):611–29. doi: 10.1038/nrd4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang M, Page C, Reynolds RK, Lin J. Constitutive activation of stat 3 oncogene product in human ovarian carcinoma cells. Gynecologic oncology. 2000;79(1):67–73. doi: 10.1006/gyno.2000.5931. [DOI] [PubMed] [Google Scholar]
- 25.Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264(5164):1415–21. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 26.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell. 1999;98(3):295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 28.Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10(1):105–15. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- 29.Darnell JE. Validating Stat3 in cancer therapy. Nat Med. 2005;11(6):595–6. doi: 10.1038/nm0605-595. [DOI] [PubMed] [Google Scholar]
- 30.Rosen DG, Mercado-Uribe I, Yang G, Bast RC, Jr, Amin HM, Lai R, et al. The role of constitutively active signal transducer and activator of transcription 3 in ovarian tumorigenesis and prognosis. Cancer. 2006;107(11):2730–40. doi: 10.1002/cncr.22293. [DOI] [PubMed] [Google Scholar]
- 31.Scambia G, Testa U, Benedetti Panici P, Foti E, Martucci R, Gadducci A, et al. Prognostic significance of interleukin 6 serum levels in patients with ovarian cancer. British journal of cancer. 1995;71(2):354–6. doi: 10.1038/bjc.1995.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tempfer C, Zeisler H, Sliutz G, Haeusler G, Hanzal E, Kainz C. Serum evaluation of interleukin 6 in ovarian cancer patients. Gynecol Oncol. 1997;66(1):27–30. doi: 10.1006/gyno.1997.4726. [DOI] [PubMed] [Google Scholar]
- 33.Lane D, Matte I, Rancourt C, Piche A. Prognostic significance of IL-6 and IL-8 ascites levels in ovarian cancer patients. BMC Cancer. 2011;11:210. doi: 10.1186/1471-2407-11-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yigit R, Figdor CG, Zusterzeel PL, Pots JM, Torensma R, Massuger LF. Cytokine analysis as a tool to understand tumour-host interaction in ovarian cancer. Eur J Cancer. 2011;47(12):1883–9. doi: 10.1016/j.ejca.2011.03.026. [DOI] [PubMed] [Google Scholar]
- 35.Shepherd TG, Theriault BL, Campbell EJ, Nachtigal MW. Primary culture of ovarian surface epithelial cells and ascites-derived ovarian cancer cells from patients. Nature protocols. 2006;1(6):2643–9. doi: 10.1038/nprot.2006.328. [DOI] [PubMed] [Google Scholar]
- 36.Yang TT, Sinai P, Kain SR. An acid phosphatase assay for quantifying the growth of adherent and nonadherent cells. Analytical biochemistry. 1996;241(1):103–8. doi: 10.1006/abio.1996.0383. [DOI] [PubMed] [Google Scholar]
- 37.Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, et al. The JAK2 Inhibitor AZD1480 Potently Blocks Stat3 Signaling and Oncogenesis in Solid Tumors. Cancer Cell. 2009;16(6):487–97. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scuto A, Krejci P, Popplewell L, Wu J, Wang Y, Kujawski M, et al. The novel JAK inhibitor AZD1480 blocks STAT3 and FGFR3 signaling, resulting in suppression of human myeloma cell growth and survival. Leukemia. 2011;25(3):538–50. doi: 10.1038/leu.2010.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xin H, Herrmann A, Reckamp K, Zhang W, Pal S, Hedvat M, et al. Antiangiogenic and antimetastatic activity of JAK inhibitor AZD1480. Cancer Res. 2011;71(21):6601–10. doi: 10.1158/0008-5472.CAN-11-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim MY, Oskarsson T, Acharyya S, Nguyen DX, Zhang XH, Norton L, et al. Tumor self-seeding by circulating cancer cells. Cell. 2009;139(7):1315–26. doi: 10.1016/j.cell.2009.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ranger JJ, Levy DE, Shahalizadeh S, Hallett M, Muller WJ. Identification of a Stat3-dependent transcription regulatory network involved in metastatic progression. Cancer research. 2009;69(17):6823–30. doi: 10.1158/0008-5472.CAN-09-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barbieri I, Quaglino E, Maritano D, Pannellini T, Riera L, Cavallo F, et al. Stat3 is required for anchorage-independent growth and metastasis but not for mammary tumor development downstream of the ErbB-2 oncogene. Molecular carcinogenesis. 2010;49(2):114–20. doi: 10.1002/mc.20605. [DOI] [PubMed] [Google Scholar]
- 43.Chang Q, Bournazou E, Sansone P, Berishaj M, Gao SP, Daly L, et al. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia. 2013;15(7):848–62. doi: 10.1593/neo.13706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sahai E. Illuminating the metastatic process. Nature reviews Cancer. 2007;7(10):737–49. doi: 10.1038/nrc2229. [DOI] [PubMed] [Google Scholar]
- 45.Nagy JA, Masse EM, Herzberg KT, Meyers MS, Yeo KT, Yeo TK, et al. Pathogenesis of ascites tumor growth: vascular permeability factor, vascular hyperpermeability, and ascites fluid accumulation. Cancer research. 1995;55(2):360–8. [PubMed] [Google Scholar]
- 46.Bournazou E, Bromberg J. Targeting the tumor microenvironment: JAK-STAT3 signaling. Jak-Stat. 2013;2(2):e23828. doi: 10.4161/jkst.23828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 48.Bromberg J, Wang TC. Inflammation and Cancer: IL-6 and STAT3 Complete the Link. Cancer cell. 2009;15(2):79–80. doi: 10.1016/j.ccr.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, et al. gp130-Mediated Stat3 Activation in Enterocytes Regulates Cell Survival and Cell-Cycle Progression during Colitis-Associated Tumorigenesis. Cancer cell. 2009;15(2):91–102. doi: 10.1016/j.ccr.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 50.Grivennikov S, Karin E, Terzic J, Mucida D, Yu G-Y, Vallabhapurapu S, et al. IL-6 and Stat3 Are Required for Survival of Intestinal Epithelial Cells and Development of Colitis-Associated Cancer. Cancer cell. 2009;15(2):103–13. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]