

Abstract
Microphthalmia-associated transcription factor (MiTF) is a key transcription factor for melanocyte lineage survival. Most previous work on this gene has been focused on its role in development. A role in carcinogenesis has emerged recently, but the mechanism is unclear. We classified melanoma cells into MiTF-positive and -negative groups and explored the function of MiTF in regulating cellular responses to reactive oxygen species (ROS). The MiTF-positive melanoma cell lines accumulated high levels of apurinic/apyrimidinic endonuclease (APE-1/Ref-1, redox effector-1), a key redox sensor and DNA endonuclease critical for oxidative DNA damage repair. We demonstrate that APE-1 is a transcriptional target for MiTF. Knocking down MiTF led to reduced APE-1 protein accumulation, as well as abolished induction of APE-1 by ROS. MiTF-negative melanoma cells survived more poorly under ROS stress than the MiTF-positive cells based on 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay and Trypan blue staining. Overexpression of APE-1 partially rescued ROS-induced cell death when MiTF was depleted. We conclude that MiTF regulates cellular response to ROS by regulation of APE-1, and this may provide a mechanism of how MiTF is involved in melanoma carcinogenesis.
INTRODUCTION
Much evidence supports the notion that MiTF is the master control gene for melanocyte lineage survival. Mutations of the MiTF gene in mice cause a microphthalmia phenotype, deafness, and lack of pigmentation (Steingrimsson et al., 2004). Mutations of MiTF in humans cause Waardenburg syndrome type IIA in which patients exhibit deafness and pigment disturbances because of a lack of melanocytes (Tassabehji et al., 1994). The MiTF melanocytes survival pathway involves a number of genes including Pax3, c-Kit, Sox9, Sox10, LEF1, and Bcl2 (Levy et al., 2006), among which Bcl2 is a downstream transcriptional target of MiTF, whereas others are upstream regulatory factors.
The role of MiTF in differentiation and development is well established, but evidence of its role in tumorigenesis remains contradictory. In human melanocytes transfected with mutant BRAF, overexpression of MiTF leads to transformation (Garraway et al., 2005). As the MiTF gene is amplified in some melanomas, it has been suggested that MiTF can function as a melanoma oncogene (Levy et al., 2006). In another report, however, MiTF was downregulated in Braf-transformed murine melanocytes and BRAF-expressing human melanocytes; reexpression of MiTF in these cells inhibited cell proliferation (Wellbrock and Marais, 2005). This is consistent with the role of MiTF in cell-cycle exit by activating p21 and p16ink4A (Carreira et al., 2005; Loercher et al., 2005). These contradictions have been elegantly reconciled in a recent review that has pointed out that an optimal amount of MiTF is required for maintaining proliferation and differentiation, whereas MiTF levels that are too low or too high may cause cell-cycle arrest and apoptosis (Gray-Schopfer et al., 2007).
It is unknown whether MiTF is directly involved in response to elevated ROS in melanocytes or melanoma cells. As a transcription factor, a number of downstream targets of MiTF have been identified. Most of these targets are melanogenesis proteins such as tyrosinase, TYRP-1, Dct, Silver, Mc1R, MLANA, and Aim1 (Vance and Goding, 2004). Recently Hif1α, a well-studied hypoxia response factor, was identified as a MiTF transcriptional target (Busca et al., 2005).
It is well known that persistent oxidative stress has a key role in many cancer types, including melanoma (Meyskens et al., 2001; Sander et al., 2004; Gidanian et al., 2008). Several factors contribute to elevated ROS level in melanoma cells. During melanogenesis, hydrogen peroxide is generated by autooxidation of eumelanin precursors, 6-dihydroxyindole and 5,6-dihydroxyindole-2-carboxylic acid (Nappi and Vass, 1996). Melanoma cells also contain strikingly malformed melanosomes (Farmer et al., 2003; Vachtenheim and Borovansky, 2004), which leads to leaking of oxidative quinone and semiquinone intermediates required for melanin synthesis. Under the oxidative environment, the normal antioxidative melanin can be oxidized and becomes a prooxidative ROS generator (Meyskens et al., 2001; Farmer et al., 2003). Overall, melanoma cells exhibit aberrant redox regulation as compared to normal human melanocytes (Meyskens et al., 1997, 2007).
ROS can be detoxified by a number of mechanisms, including the cellular antioxidative system involving glutathione, thioredoxin, catalase, superoxide dismutase, and glutathione peroxidase (McEligot et al., 2005). These factors and proteins are regulated by redox sensors and transcription factors, among which a multifunctional protein, apurinic/apyrimidinic endonuclease (APE-1/Ref-1), has an important role (Evans and Smith, 1997). APE-1 has two essential well-characterized functions: a DNA endonuclease that can recognize and cut DNA at the abasic sites and therefore has a key role in the base excision DNA repair pathway (Demple and Sung, 2005), and a redox sensor that enhances the DNA binding activities of a large number of transcription factors, including p53, Hif1α, AP-1, and nuclear factor-κB upon an increase of intracellular ROS (Evans and Smith, 1997; Yang et al., 2005). Depletion of APE-1 by siRNA leads to apoptosis in human cells due to accumulation of damaged DNA, especially when ROS increases (Fung and Demple, 2005; Yang et al., 2007). APE-1 expression is upregulated at both mRNA and protein level with increased ROS (Ramana et al., 1998), but the mechanism of this upregulation is not fully understood (Evans and Smith, 1997).
In the current study, we explored the function of MiTF in regulating cellular ROS in normal human melanocytes and human melanoma cell lines. We demonstrated that APE-1 is a transcriptional target for MiTF, which activates its basal level transcription as well as enhances its stimulation by ROS. As a consequence, MiTF-negative melanoma cells accumulated less APE-1 and lacked an induction of this protein by ROS, leading to poorer survival as compared to MiTF-positive cells.
RESULTS
APE-1 is a transcriptional target for MiTF
To search for additional MiTF transcriptional targets, a genome-wide promoter screening was performed using E-boxes (CANNTG) and M-box sequences (Aksan and Goding, 1998) as the MITF binding motif according to Xie et al. (2005). Three E-boxes were found on the APE-1 promoter region, located at −10, +52, and +73, respectively (Figure 1a). The numbers of the nucleotide sequence are based on APE-1 cDNA (GenBank accession no. NM_001641) and its genomic DNA, chromosome 14 genomic contig (GenBank accession no. NT_026437). To confirm MiTF can bind to APE-1 promoter, a chromatin immunoprecipitation analysis was performed using MiTF antibody. MiTF protein was found on the APE-1 promoter in SK-Mel-28 cells, whereas MEK1, an MAPK pathway kinase, which served as a negative control, did not bind to the APE-1 promoter. MiTF did not bind to the α-tubulin coding sequence, suggesting that the MiTF binding to APE-1 promoter is specific (Figure 1b). When a reporter construct of pAPE-Luc was co-transfected into MiTF-negative Lu1205 cells with MiTF expression construct pcDNA-MiTF (Molina et al., 2005), the APE-1 promoter activity increased 3- to 4-fold in the presence of MiTF as compared to pcDNA3.1 empty vector (Figure 1c). When MiTF was knocked down in MiTF-positive SK-Mel-28 cells by DNA-based short hairpin RNA (shRNA) constructs pTER-mi3 or pTER-mi5 (two different shRNAs, both targeting all isoforms of MiTF), the APE-1 promoter activity decreased about 50% (Figure 1d, lanes 3 and 4), which did not occur in cells co-transfected with pTER-GFPi control construct (Figure 1d, lane 2). As shown in Figure 1d top right corner, pTER-mi3 and pTER-mi5 knocked down MiTF efficiently, whereas pTER-GFPi did not alter MiTF accumulation. Furthermore, a dominant-negative MiTF (Miller et al., 2004) repressed APE-1 promoter activity in SK-Mel-28 cells (Figure 1e).
Figure 1. APE-1 is a transcriptional target for MiTF.

(a) The APE-1 promoter and gene structure. nCaRE, binding site for negative calcium response elements; Sox site, SOX transcription factor binding site; cAMP response element-binding protein, cAMP response element binding site; E2F site, E2F binding site; E-box, consensus MiTF binding site; ATG, translation initiation site; 3′ UTR, 3′ untranslated region. (b) MiTF binds to APE-1 promoter. Chromatin immunoprecipitation was performed using SK-Mel-28 cells and anti-Mek1 or anti-MiTF antibodies. The precipitated chromatin was PCR amplified with either APE-1 promoter-specific primers (top) or α-tubulin primers (bottom). (c) MiTF activates APE-1 promoter activity. pAPE1-Luc reporter construct was co-transfected with MiTF expression plasmid into Lu1205 cells, luciferase activities were assayed 48 hours after transfection and normalized by β-galatosidase activity (pCH110) that is also co-transfected. (d) knockdown of MiTF in Sk-Mel-28 cells decreased APE-1 promoter activity. pAPE-Luc reporter construct was co-transfected with MiTF shRNA plasmids pTER-mi3 or pTER-mi5 or control plasmid pTER-GFPi that carries shRNA targeting GFP. Luciferase activity was assayed 48 hours after transfection. Western blot in the top right corner indicates the MiTF level (top) and α-tubulin level from cells in lanes 2, 3, and 4. (e) A dominant-negative MiTF protein inhibited APE-1 promoter activity in SK-Mel-28 cells. At 12 hours after transfection with the APE-1 wild-type promoter, cells were infected with adenovirus expressing a control nonsense polypeptide (PolyP), wild-type MiTF (MiTF-WT) or dominant-negative MiTF (MiTF-DN) (Miller et al., 2004). Luciferase activities were measured 48 hours after transfection. Cells transfected but not infected with any virus were used as a control (uninfected). (f) MiTF regulates APE-1 promoter activity through E-boxes. Mutant APE-1 promoters lacking the first E box (pΔ1E-Luc) or all three E boxes (pΔ3E-Luc) were analyzed as above along with the wild-type APE-1 promoter in A375 cells. (g) The first E box is critical for MiTF regulation. Site mutagenesis was performed to make point-mutated promoters, which were used for activity analysis in the presence or absence of MiTF construct. The data were normalized to co-transfected β-galactosidase activity and the induction fold of each promoter was calculated by comparing activity in the presence of MiTF to that in the absence of MiTF.
MiTF regulates APE-1 promoter activity through E-boxes
To investigate whether the MiTF regulation of APE-1 is directly through E-boxes on APE-1 promoter, the first E box (at −10 position) was deleted to generate pΔ1E-Luc construct. The mutant Δ1E APE-1 promoter activity decreased to about half that of wild-type promoter (Figure 1f, lanes 1 and 3). More importantly, this mutation abolished most of the induction of APE-1 promoter activity by MiTF (Figure 1f, lanes 1 though 4). The wild-type APE-1 promoter increased 3- to 4-fold in the presence of MiTF, whereas the Δ1E promoter increased only about 20%. Deletion of all three E boxes reduced the promoter activity to only about 10% of the wild type, and completely eliminated the activation by MiTF (Figure 1f, lanes 5 and 6). As deletion of all three E boxes also deleted transcription initiation sites and part of the 5′ untranslated region, the decrease of APE-1 promoter activity may not be the direct effect of E box mutation. To further investigate which E box is critical for MiTF regulation, we made point mutations of each E boxes, designated as E1, E2, and E3 promoters corresponding to the mutated first, second, and third E box. These promoters were transfected into A375 melanoma cells with MiTF construct or the empty vector as control. Again we observed nearly four-fold of induction by MiTF in wild-type promoter, no induction at all on E1 promoter, about 1.4-fold of induction of E2 promoter, and about 2.3-fold of induction of E3 promoter (Figure 1g). These results suggest that the first E box (E1) is critical for MiTF regulation of APE-1 promoter. The second E box also participates in this regulation, and the third E box is less critical.
APE-1 protein levels are correlated with MiTF-M protein levels
To examine whether the accumulation levels of APE-1 protein in melanoma cells are correlated with MiTF protein levels, we knocked down MiTF in melanoma MNT1 cells by infection of adenovirus carrying shRNA (Ad-shMiTF). Adenovirus is used because MNT1 is very difficult to transfect, various transfection methods were tested and no more than 10% of transfection efficiency was achieved. As shown in Figure 2a, MiTF protein levels decreased 48 hours after infection, and more dramatically after 72 and 96 hours. Both MiTF bands were diminished simultaneously. The shRNA degrades mRNA and therefore inhibits newly synthesized nonphosphorylated MiTF. When this MiTF form decreases, the corresponding phosphorylated MiTF may also decrease due to less substrate for its kinase(s). The APE-1 protein level decreased concomitantly. As a control, MiTF and APE-1 protein level remained unchanged in cells infected with adenovirus carrying luciferase shRNA (Ad-shLuc) (Figure 2a). We further examined the protein accumulation of MiTF and APE-1 levels in normal human melanocytes and melanoma cell lines. Of the eight melanoma cell lines examined, five were MiTF negative, in which all isoforms of MiTF protein were undetectable using 80 μg of input total protein. These were cell lines c8161, c81-46A, WM3211, Lu1205, and A375 (Figure 2b). Three melanoma cell lines, SK-Mel-28, MNT1, and c83-2C, were MiTF positive. In addition to the MiTF-M isoforms, all these latter cells also accumulated MiTF-A isoform (Figure 2b). Normal human melanocytes accumulate mainly the MiTF-M isoform, the MiTF-A isoform is barely detectable (Figure 2b). Hereafter MiTF is referred to as MiTF-M isoform in this paper because this is the main isoform expressed and observed in melanoma and melanocytes. APE-1 accumulation level is correlated to that of MiTF protein level in normal melanocytes and melanoma cell lines. APE-1 accumulation from the western blot was quantitated by a densitometer and normalized to that of α-tubulin (Figure 2c). The APE-1 accumulation in normal melanocytes is defined as 1. The average APE-1 of five MiTF-negative cells was 0.74, and the average APE-1 level of three MiTF-positive melanoma cells was 1.37 (P=0.046, <0.05). This comparative data suggest that MiTF levels in melanoma cells are correlated with APE-1 levels. To further demonstrate that APE-1 level is correlated to MiTF level, normal human melanocytes were precultured in media with no 12-O-tetradecanoylphorbol- 13-acetate and low bovine pituitary extract (0.03%) for 24 hours, cells were then treated with α-MSH. Addition of α-MSH induced MiTF accumulation after 6 and 12 hours, which was also correlated to an increase of APE-1 accumulation under such condition (Figure 2d).
Figure 2. APE-1 protein level is correlated with that of MiTF.
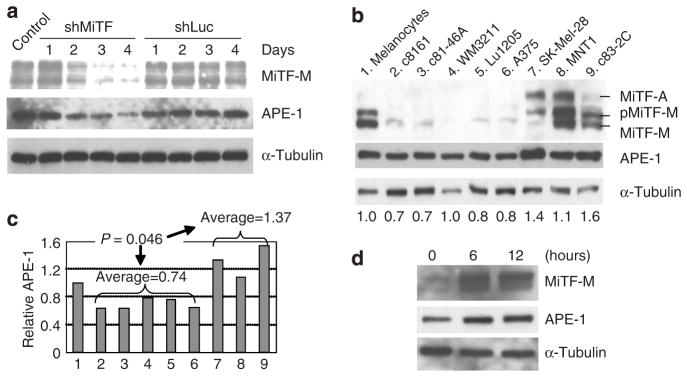
(a) Knockdown of MiTF led to a decrease in APE-1 protein accumulation. MNT1 cells were infected with Ad-shMiTF or Ad-shLuc and collected for western blot analysis at the indicated days. (b) Western blot analysis of MiTF and APE-1 accumulation in normal human melanocytes and melanoma cell lines: (1) Normal human melanocytes, (2) c8161, (3) c81-46A, (4) WM3211, (5) Lu1205, (6) A375, (7) SK-Mel-28, (8) MNT1, and (9) c83-2C. α-Tubulin serves as a loading control. Numbers below the western blot indicate the quantitated relative APE-1 levels (see c). (c) Quantitative analysis of APE-1 accumulation levels based on (b). The western results were quantitated by a densitometer and APE-1 level is normalized to α-tubulin. Normalized APE-1 accumulation in normal human melanocytes was defined as 1.0 and its levels in other cell lines were relative to normal human melanocytes. (d) APE-1 protein level is correlated to MiTF upregulation by α-MSH. Normal human melanocytes were cultured in media without 12-O-tetradecanoylphorbol-13-acetate and reduced bovine pituitary extract levels for 24 hours (lane 1), α-MSH was then added and cells were collected at 6 and 12 hours (lanes 2 and 3) for western blot analysis.
MiTF phosphorylation after ROS elevation is correlated to increased APE-1 protein level
APE-1 is a key protein for redox sensing, and its association with MiTF suggests that MiTF itself may be involved in regulating cellular ROS response. Next, we treated SK-Mel-28 cells with 0.1mM H2O2 to elevate cellular ROS levels. The MiTF protein was phosphorylated 1 hour after treatment and increased slightly from 1 to 4 hours, then at 9 to 12 hours degraded almost completely (Figure 3a). These results are consistent with a previously published data, which indicated that MiTF degrades with an increase of cellular ROS level (Jimenez-Cervantes et al., 2001). However, the status of phosphorylation of MiTF was not previously reported. When cells were treated with U0126, an Mek1/2-Erk1/2 kinase inhibitor, before exposure to ROS, the upper band was not enhanced as compared to cells without the inhibitor (Figure S1), suggesting this band represents phosphor-MiTF, and the phosphorylation is via Erk1/2 mitogen-activated protein kinase pathway. To confirm this phosphorylation of MiTF is a particular response to elevated cellular ROS levels, we then treated c83-2C cells with glucose oxidase (GOX, 100 mUml−1), which reacts with glucose and oxygen to produce constant low level of H2O2, and examined the response of MiTF and APE-1. As shown in Figure 3b, at time 0 when cells were treated with GOX for 1 hour MiTF accumulation level increased slightly and the phosphorylated form also increased as compared to the untreated cells (lanes 1 and 2). Again the MiTF protein degraded 9–12 hours after GOX treatment, and then recovered to its original untreated level 16 hours after treatment. Interestingly, protein accumulation of APE-1 increased after GOX treatment and remained elevated during the time course as compared to that in the untreated cells. It seemed that APE-1 levels reached its peak 3–6 hours after recovering from GOX treatment, following the elevated phosphor-MiTF protein level (Figure 3b, bottom). The kinetics of phosphor-MiTF and APE-1 was similar during the first 9 hours of recovering. A similar response of MiTF and APE-1 accumulation was also observed in normal human melanocytes after 0.1mM of H2O2 treatment (Figure 3c), with a slightly different kinetics over the time course. MiTF-M was phosphorylated 1 hour after H2O2 treatment, decreased after 4–6 hours, and recovered 9–24 hours after the treatment (Figure 3c). The APE-1 protein accumulation increased 2–4 hours after treatment and remained elevated until 16 hours, then returned to its pretreatment level (Figure 3c).
Figure 3. MiTF is phosphorylated and required for APE-1 induction after ROS treatment.

(a) Western blot analysis of MiTF in SK-Mel-28 cells after H2O2 treatment. The Sk-Mel-28 cells were treated with 0.1mM H2O2 and collected for western blot analysis at the indicated time points. p84 serves as a loading control. (b) MiTF and APE-1 accumulation in c83-2C cells after GOX (150mUml−1) treatment. Cells were incubated with GOX for 1 hour, washed with 1 × phosphate-buffered saline, and then returned to incubator with fresh media (time 0). The bottom graph is quantiated MiTF (total) and APE-1 protein levels based on the western blot on top. (c) Western blot analysis of MiTF and APE-1 accumulation in normal human melanocytes after treatment with 0.1mM H2O2. (d) Western blot analysis of APE-1 accumulation in two MiTF-negative melanoma cells A375 (top) and c81-46A (bottom) after 0.15mM H2O2 treatment. (e) Induction of APE-1 promoter activity requires MiTF. SK-Mel-28 cells were transfected with pAPE-Luc and one of the following plasmids: pcDNA3.1, pcDNA-MiTF, pTER-mi3, or pTER-GFPi, together with pCH110. Cells were treated with 0.1mM H2O2 36 hours after transfection and luciferase activity was assayed 48 hours after transfection.
MiTF is required for the upregulation of APE-1 after elevated ROS
As seen in Figure 2b, although the APE-1 protein accumulates to a lower level in MiTF-negative melanoma cell lines c8161, c81-46A, WM3211, Lu1205, and A375, as compared to MiTF-positive normal melanocytes and melanoma SK-Mel-28, MNT1, and c83-2C, the APE-1 level did not diminish in the MiTF-negative cell lines. This suggests that the basal level expression of APE-1 does not require MiTF, although MiTF can activate Ape-1 promoter. Therefore we next examined whether MiTF was required for upregulation of APE-1 upon H2O2 treatment. The APE-1 response to H2O2 treatment in MiTF-negative cell lines A375 and c81-46A were examined (Figure 3d, top and bottom). In both cell lines, APE-1 did not exhibit a significant increase after treatment with 0.15mM H2O2. In contrast, JunB, one of the AP1 proteins that usually respond to increased cellular ROS levels, increased 3 hours after the treatment and reached its highest level at 6–9 hours (Figure 3d).
To investigate whether APE-1 promoter responded to MiTF levels after H2O2 treatment, pAPE-Luc was co-transfected with one of the following constructs: pcDNA3.1, pcDNA-MiTF, pTER-GFPi, and pTER-mi3. After 36 hours, cells were treated with 0.15mM H2O2 and collected for luciferase assay 12 hours later. As shown in Figure 3e, again overexpression of MiTF increased APE-1 promoter activity (open box, lanes 1 and 2), whereas depletion of MiTF by shRNA decreased APE-1 activity (open box, lane 3 and 4). When treated with H2O2, APE-1 promoter activity increased in cells transfected with pcDNA3.1, pcDNA-MiTF, and pTER-GFPi, where MiTF is present (lanes 1–6). APE-1 promoter activity was not stimulated by H2O2 when MiTF was depleted by pTER-mi3 (lanes 7–8), suggesting that MiTF is required for this stimulation. Comparing cells transfected with pcDNA3.1 alone or pcDNA-MiTF (lanes 2 and 4, filled box), overexpression of MiTF did not lead to more dramatic induction of APE-1 after H2O2 treatment, presumably due to saturation of MiTF on the APE-1 promoter in this cell line. These results demonstrate that upon H2O2 treatment APE-1 upregulation requires MiTF.
MiTF-positive cells are more resistant to H2O2-induced cell death
To examine the cellular effect of MiTF under the elevated ROS condition, we knocked down MiTF in both SK-Mel-28 and MNT1 human melanoma cells by different approaches. The plasmid carrying tandem repeats of MiTF shRNA pTER-mi3 was transfected into SK-Mel-28 cells, whereas the same vector carrying GFP shRNA was used as control. No obvious cell death or growth arrest was observed up to 4 days after transfection. As examined by western blot analysis, MiTF levels decreased significantly 24 hours after transfection with pTER-mi3, and maintained a low level until 96 hours post transfection (Figure 4a). Cells transfected with control GFP shRNA did not show changes in MiTF levels (Figure 4a). APE-1 protein level also decreased when MiTF was knocked down, but not depleted (Figure 4a). These transfected cells were treated with H2O2 and collected for western blot analysis. APE-1 protein level remained constant after H2O2 treatment (Figure 4b), whereas JunB induction was obvious 3–12 hours after treatment. However, an induction of APE-1 protein was observed in SK-Mel-28 cells with no shRNA transfection 6–12 hours after H2O2 treatment (Figure 4c). These results indicated that MiTF not only activated the basal level of APE-1, it also regulated the induction of APE-1 by ROS stress.
Figure 4. Knockdown of MiTF led to poor survival under ROS stress in melanoma cells.

(a) MiTF is knocked down by pTER-mi3 in SK-Mel-28 cells. SK-Mel-28 cells were transiently transfected by vector pTER-GFPi or pTER-mi3, cells were collected at the indicated time point for western blot analysis. p84 serves as loading control. Ctrl, cells without transfection. (b) Western blot analysis of APE-1 protein level in cells with depleted MiTF after 0.1mM H2O2 treatment. The pTER-mi3-transfected cells were treated with 0.1mM H2O2 24 hours after transfection, and collected for western blot analysis. Again p84 serves as loading control and JunB serves as a positive control. (c) Western blot analysis of APE-1 protein induction in SK-Mel-28 cells treated with 0.1mM H2O2. (d) Cell viability and cell-death analysis after MiTF knockdown. SK-Mel-28 cells were transfected with pTER-GFPi or pTER-mi3, 12 hours later cells were trypsinized and plated in 24-well plates. At 24 hours after transfection, 0 (lane 1), 0.1mM (lane 2), or 0.15mM (lane 3) of H2O2 was added into the media and continued to incubate for another 16 hours, when 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay (left) or Trypan blue staining (right) was performed. (e, f) Cell death after H2O2 (e) or GOX (f) treatment in MNT1 infected with either adenovirus Ad-shLuc or Ad-shMiTF. Time 0 is 48 hours after infection, when treatment started. Cell death was analyzed 24 hours after treatment.
APE-1 is a critical protein for redox sensing and DNA damage repair (Evans and Smith, 1997), therefore it is important for cell survival. To examine the consequence of MiTF regulation on APE-1, SK-Mel-28 cells was first transfected with pTER-GFPi or pTER-mi3, and then treated with 0.1 or 0.15mM H2O2. Cell proliferation was assayed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide method and cell death was assayed by Trypan blue staining. As shown in Figure 4d (left panel), cell viability decreased more dramatically when MiTF was knocked down. For example, after treatment with 0.15mM H2O2, cell viability decreased to 45% for those transfected with GFP shRNA as compared to the untreated cells, whereas viability decreased to about 10% for those with MiTF knocked down (Figure 4d, left panel, lane 3). Meanwhile, the percentages of cell death in GFP shRNA- and pTER-mi3-transfected cells were both about 8.5% without treatment; this rate increased to 15% in vector-transfected cells, and to 38% in MiTF-knockdown cells after 0.15mM H2O2 treatment (Figure 4d, right panel, lane 3). This observation was repeated in MNT1 cells when MiTF was knocked down by adenovirus carrying an shRNA targeting to a different area in MiTF mRNA. This method depleted MiTF 72 hours after infection (Figure 2a). As shown in Figures 4e and f, after various dosage of H2O2 or GOX treatment, MNT1 cells exhibited more cell death when cells were depleted of MiTF and under ROS stress.
Overexpression of APE-1 partially rescued the MiTF-mediated H2O2-induced cell death
Previously our laboratory demonstrated that overexpression of APE-1 protected melanoma cells from H2O2-induced cell death (Yang et al., 2005). To further demonstrate that the enhanced H2O2-induced cell death in MiTF-depleted cells is mediated by APE-1, we overexpressed flag-tagged APE-1 in SK-MEL-28 cells, whereas MiTF was knocked down, and then examined the cell proliferation and cell-death rate after H2O2 treatment. As shown in Figure 5a, 60 hours after transfection of pFlag-APE-1 and 48 hours after infection with Ad-shMiTF, MiTF protein level decreased dramatically, as did the endogenous APE-1 protein level (Figure 5a, lane 3). Overexpression of flag-APE-1 slightly rescued viability after GOX or H2O2 treatment (Figure 5b, rows 1 and 3). Knocking down MiTF without APE-1 overexpression led to a poorer viability (Figure 5b, comparing rows 1 and 2), and expression of flag-APE-1 increase viability in these cells (rows 2 and 4).
Figure 5. Over-expression of APE-1 rescued ROS-induced cell viability in MiTF-depleted cells.
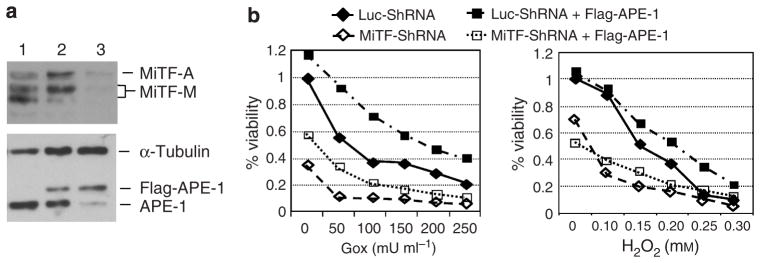
(a) Concomitant knock-down of MiTF and over-expression of APE-1 in SK-MEL-28 cells. Cells were transiently transfected with pFlag-APE-1, and then infected with Ad-shLuc or Ad-shMiTF 24 hours later. (1) Control SKMel- 28 cells; (2) Cells transfected with pFlag-APE-1 and infected with Ad-shLuc; (3) Cells transfected with pFlag-APE-1 and infected with Ad-shMiTF. α-Tubulin serves as a loading control. (b) Cells with or without MiTF knock-down or APE-1 overexpression were treated with various concentration of glucose oxidase (Gox, left) or H2O2 (right) 48 hours post-infection, and MTT assay was performed to measure cell viability. All data is normalized to cells with Luc-ShRNA transfection.
DISCUSSION
In this study we investigated the role of MiTF in response to a moderate increase of intracellular ROS. We have demonstrated that APE-1 is a transcriptional target for MiTF, and that MiTF regulates cellular ROS response by upregulating APE-1. Interestingly, the MiTF-positive melanoma cells accumulated relatively higher levels of APE-1 than MiTF-negative cells. This increased APE-1 protein level may reflect an increased APE-1 activity, therefore it is not surprised that the MiTF-positive melanoma cells were more resistant to H2O2-induced cell death and exhibited better survival as compared to MiTF-negative cells. Currently there is a substantial effort directed toward developing chemoprevention agents against cancers, including melanoma, based on their elevated ROS levels (Meyskens et al., 2001; Cen et al., 2002). For example, cells treated with buthionine sulfoximine, an inhibitor of γ-glutamyl-cysteine synthetase, exhibited lower cellular glutathione level and therefore higher level of ROS (Fruehauf et al., 1997). In melanoma cells, the endogenous cellular ROS level is relatively high; when these cells are treated with drugs such as buthionine sulfoximine, they have an even higher level of ROS, which leads to apoptosis (Fruehauf et al., 1997). Our finding that MiTF-positive melanoma cells are more resistant to H2O2-induced cell death suggests that the therapeutic approach by increasing cellular ROS may be more effective in MiTF-negative melanoma than in MiTF-positive ones, and therefore it may be necessary to characterize each melanoma based on its MiTF levels to maximize the therapeutic approach. We examined 11 primary melanoma tissues by immunohistochemistry, our preliminary data indicated that 2 out of 11 samples are MiTF-positive and 9 are MiTF-negative (F Liu and FL Meyskens, unpublished data), giving more weights on seeking an MiTF-based differential treatment approach.
Several lines of evidence support our conclusion that APE-1 is a downstream transcriptional target for MiTF: (1) three E boxes were found on the APE-1 promoter, (2) the MiTF protein directly binds to APE-1 promoter through the E-boxes, (3) MiTF-positive cell lines accumulate higher level of APE-1 protein, and (4) induction of APE-1 by ROS requires MiTF. Factors required for APE-1 upregulation upon H2O2 treatment are not fully understood. It has been reported that the cAMP response element on APE-1 promoter (Figure 1a) is required for APE-1 upregulation upon H2O2 treatment, inferring that the cAMP response element-binding protein was also required (Grosch and Kaina, 1999). Interestingly, APE-1 has been shown to interact with cAMP response element-binding protein and activate Hif1α and its own expression. All of these proteins form a transcription activation complex to activate VEGF under hypoxia conditions (Ramphal et al., 2006). cAMP response element-binding protein binds p300 or CBP to modify chromatin structure for transcription activation (Rouaux et al., 2004). MiTF binds DNA through its basic helix-loop-helix domain and activates transcription in both p300-dependent and -independent manners (Sato et al., 1997; Vachtenheim et al., 2007). In the current study MiTF was phosphorylated in both melanoma cells and normal melanocytes after elevated ROS treatment (Figures 3a–c), suggesting that MiTF itself is responsive to cellular ROS change. Hif1α, another key transcription factor involved in hypoxia-induced VEGF activation, is also a transcription target for MiTF (Busca et al., 2005). Both APE-1 and Hif1α have key roles in response to cellular ROS stress. The fact that both genes are MiTF transcriptional targets suggests that MiTF regulates a broad range of gene expressions in response to ROS stimuli.
In response to ROS stimulation, MiTF was phosphorylated and then degraded (Figures 3a–c). This observation is consistent with previous reports that dual phosphorylation of MiTF leads to transient activation and subsequent degradation of MiTF (Wu et al., 2000). The phosphorylation of MiTF upon H2O2 treatment can be inhibited by Mek1 inhibitor U0126 (Figure S1), suggesting that the phosphorylation of MiTF is mediated by MAPK-Erk1/2 pathway, likely on serine 73 (Wu et al., 2000; Molina et al., 2005). This phosphorylation is probably important for activating Ape-1 transcription after ROS stress.
In summary, here we show evidence that MiTF is involved in regulating the cellular stress response. This study also provides an alternative explanation for the role of MiTF in melanoma carcinogenesis by regulating cellular ROS response: loss of MiTF at an early stage may lead to reduced APE-1 and therefore persistent high level of ROS. However, at later stages of transformation, MiTF accumulation may lead to increased APE-1 and other survival factors such as Bcl2, which in turn leads to better survival outcomes for some cells. We note that all three MiTF-positive melanoma cell lines were derived from malignant metastatic melanomas, whereas the others were obtained from various earlier stages of melanoma. This is consistent with clinical reports that high level of MiTF in blood samples was correlated with poor outcome of patients with melanoma (Koyanagi et al., 2006), as well as the fact that low levels of MiTF promotes invasiveness, whereas high level promotes proliferation (Carreira et al., 2006).
MATERIALS AND METHODS
Cell lines and cell culture
Normal human melanocytes isolated from newborn foreskin from circumcision surgery followed the procedure by Eisinger and Marko (1982), and cultured in MCDB153 medium supplied with 2% fetal bovine serum, 10 ng ml−1 of 12-O-tetradecanoylphorbol-13-acetate and 0.15% of bovine pituitary extract unless otherwise stated (Yang et al., 2005). The medical ethnical committee of University of California Irvine approved all described studies. The study was conducted according to the Declaration of Helsinki Principles. Patient consent for experiments was not required because US laws consider human tissue left over from surgery as discarded material. Melanoma cell lines c8161, c81-46A, and c83-2C were cultured in F10 media; Lu1205 cells were cultured in L15/MCDB153 (1:1) media; WM3211 were cultured in RPMI; A375 and MNT1 cells were cultured in Dulbecco’s modified Eagle’s medium; SK-Mel-28 cells were cultured in Eagle’s minimal essential medium. All these media were supplied with 5% fetal bovine serum, 5% newborn bovine sera, and 2% penicillin and streptomycin, except that MNT1 media contained doubled concentration of fetal bovine serum and newborn bovine serum (10% each). All cells were kept at 37 °C in 5% CO2 incubator.
Cell treatment
H2O2 is first diluted in H2O, and then added to media directly at the desired concentration. GOX was purchased from Sigma (G2133; St Louis, MO) and prepared according to the manufacturer’s instruction. GOX was added directly into cell media and incubated for 1 hour. Cells were then washed with 1 × phosphate-buffered saline three times, and returned to incubator with fresh media.
DNA and shRNA constructs
MiTF shRNA expression cassette (mi3) was constructed by inserting a DNA fragment derived from two annealed synthetic oligos (Table S1) into pTER+ vector (van de Wetering et al., 2003), with the targeting sequence on MiTF: 5′-GTACCACATACAGCAAGCCCA-3′. The SalI–XhoI fragment containing the H1-shRNA construct was engineered to repeated four times (tandem repeat) in the plasmid pTER-mi3. The pTER-mi5 construct was made the same way except that different oligos targeting a different sequence were used (Table S1). The targeting sequence on MiTF is: 5′-GGACAATCACAACCTGATT GA-3′. The shRNA expression cassette from pTER-mi5 was cloned into pAdTrack (He et al., 1998) for generating adenovirus Ad-shMiTF. The pTER-GFPi was made the same way using annealed oligos targeting GFP (Table S1). The Luc-ShRNA adenovirus Ad-shLuc was reported before (Liu and Lee, 2006). The pcDNA-MiTF is a gift from Dr Lee Bardwell (University of California, Irvine) (Molina et al., 2005). The p3xFLAG-APE1 is a gift from Dr Kaikobad Irani (University of Pittsburgh Medical Center). Adenovirus carrying nonsense polypeptide (control virus, Adv-polyP), wild-type MiTF (Adv-MiTF-WT), or dominant-negative MiTF (Adv-MiTF-DN) were gifts from Dr David Fisher (Dana-Farber Cancer Institute, Children’s Hospital, Harvard Medical School) (Miller et al., 2004).
Cell viability assay and Trypan blue staining
Cell viability was determined by standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Cell death was analyzed by Trypan blue staining. Both attached and floating cells were collected and incubated with 50% Trypan blue solutions for 5 minutes before blue cells and dye-repelling viable cells were counted under a microscope.
Cell lysate and western blot analysis
Cell pellet was lyzed in a lysis 250 buffer (Liu and Lee, 2006) and quantified by the Bradford protein assay method (Bio-Rad, Richmond, CA). Western blot was performed using antibodies against MiTF C5 plus D5 (MS-773-P; Lab Vision, Fremont, CA), APE-1 (Novus Biologicals, Littleton, CO), p84 (Abcam, Cambridge, MA), and α-tubulin (T9026; Sigma).
Reporter constructs and luciferase reporter assay
The APE-1 promotor reporter plasmid was constructed by ligating the pGL3-basic vector (digested with NheI and HindIII) and an AvrII–HindIII fragment from Ape-1 promoter plasmid pCB2 (Harrison et al., 1995). The resulting plasmid is termed pAPE1-Luc. Luciferase activities were measured according to the manufacturer’s (Promega, Madison, WI) instructions in an ML 3000 Microtiter Plate Luminometer from Dynatech Laboratories Inc. (Chantilly, VA). The co-transfected β-galactosidase activities (pCH110) were measured by a Beckman DU-64 spectrophotometer at 410 nm. The primers used for making the deletion mutant Δ1E and Δ3E, point mutants E1, E2, and E3 are listed in Table S1. The resulting point mutations for E1, E2, and E3 are from original E box sequence CACGTG to GATATC (E1), GTCGAC (E2), and GAATTC (E3).
Chromatin immunoprecipitation assays
Chromatin immunoprecipitation assays were based on the previously published protocol, with minor modifications (Chakrabarti et al., 2002; Liu and Lee, 2006). Three 10-cm dishes of cells were crosslinked with 1% formaldehyde in phosphate-buffered saline for 15 minutes at room temperature, and washed in phosphate-buffered saline three times. The fixed cells were resuspended in 0.6 ml of sonication buffer (Liu and Lee, 2006) and sonicated with an ultrasonic processor (Cole-Parmer 4710 series; Cole-Parmer Instruments Co., Chicago, IL) at 60% amplitude. After 15 cycles of 12-second sonication pulses, the sheared chromatin was clarified by centrifugation and incubated with 5 μg of primary antibodies at 4 °C overnight. Protein A/G-Sepharose slurry (25 μg) was added and incubated for 1 hour. The Sepharose beads were spun down, washed, eluted, and precipitated as described before (Liu and Lee, 2006). In total, 1-μl aliquots out of 30-μl DNA were used for PCR. AT1 and AT2 primers (Table S1) were used for α-tubulin PCR; ref1p4 and ref1p6 (Table S1) were used for amplifying APE-1 promoter. The PCR was performed for 35 cycles for all samples and primers.
Supplementary Material
Mek1/2–Erk1/2 MAP kinase pathway-mediated ROS-induced MiTF phosphorylation.
Oligos for shRNA construction and primers for PCR and site mutagenesis.
Acknowledgments
We thank Dr Lee Bardwell (University of California at Irvine) for his gift of pcDNA-MiTF, Dr Kaikobad Irani (University of Pittsburg) for his gift of pFlag-Ref-1, Dr Bruce Demple (Harvard School of Public Health) for his gift of human APE-1 promoter construct pCB2, Dr David Fisher (Dana-Farber Cancer Institute) for his MiTF-WT, MiTF-DN and control adenovirus, and Dr Sun Yang (University of California at Irvine) for her stimulating discussion and suggestions. This research is supported in part by CA62230 to FLM and the Waltmar and Oxnard Foundations, as well as donations from grateful patients.
Abbreviations
- APE
apurinic/apyrimidinic endonuclease
- GOX
glucose oxidase
- ROS
reactive oxygen species
- shRNA
short hairpin RNA
Footnotes
CONFLICT OF INTEREST
The authors state no conflict of interest.
References
- Aksan I, Goding CR. Targeting the microphthalmia basic helix-loop-helix-leucine zipper transcription factor to a subset of E-box elements in vitro and in vivo. Mol Cell Biol. 1998;18:6930–8. doi: 10.1128/mcb.18.12.6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busca R, Berra E, Gaggioli C, Khaled M, Bille K, Marchetti B, et al. Hypoxia-inducible factor 1{alpha} is a new target of microphthalmia-associated transcription factor (MITF) in melanoma cells. J Cell Biol. 2005;170:49–59. doi: 10.1083/jcb.200501067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira S, Goodall J, Aksan I, La Rocca SA, Galibert MD, Denat L, et al. Mitf cooperates with Rb1 and activates p21Cip1 expression to regulate cell cycle progression. Nature. 2005;433:764–9. doi: 10.1038/nature03269. [DOI] [PubMed] [Google Scholar]
- Carreira S, Goodall J, Denat L, Rodriguez M, Nuciforo P, Hoek KS, et al. Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev. 2006;20:3426–39. doi: 10.1101/gad.406406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cen D, Gonzalez RI, Buckmeier JA, Kahlon RS, Tohidian NB, Meyskens FL., Jr Disulfiram induces apoptosis in human melanoma cells: a redox-related process. Mol Cancer Ther. 2002;1:197–204. [PubMed] [Google Scholar]
- Chakrabarti SK, James JC, Mirmira RG. Quantitative assessment of gene targeting in vitro and in vivo by the pancreatic transcription factor, Pdx1. Importance of chromatin structure in directing promoter binding. J Biol Chem. 2002;277:13286–93. doi: 10.1074/jbc.M111857200. [DOI] [PubMed] [Google Scholar]
- Demple B, Sung JS. Molecular and biological roles of Ape1 protein in mammalian base excision repair. DNA Repair (Amst) 2005;4:1442–9. doi: 10.1016/j.dnarep.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Eisinger M, Marko O. Selective proliferation of normal human melanocytes in vitro in the presence of phorbol ester and cholera toxin. Proc Natl Acad Sci USA. 1982;79:2018–22. doi: 10.1073/pnas.79.6.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans BL, Smith SB. Analysis of esterification of retinoids in the retinal pigmented epithelium of the Mitf-vit (vitiligo) mutant mouse. Mol Vis. 1997;3:11. [PubMed] [Google Scholar]
- Farmer PJ, Gidanian S, Shahandeh B, Di Bilio AJ, Tohidian N, Meyskens FL., Jr Melanin as a target for melanoma chemotherapy: pro-oxidant effect of oxygen and metals on melanoma viability. Pigment Cell Res. 2003;16:273–9. doi: 10.1034/j.1600-0749.2003.00046.x. [DOI] [PubMed] [Google Scholar]
- Fruehauf JP, Zonis S, al-Bassam M, Kyshtoobayeva A, Dasgupta C, Milovanovic T, et al. Selective and synergistic activity of L-S, R-buthionine sulfoximine on malignant melanoma is accompanied by decreased expression of glutathione-S-transferase. Pigment Cell Res. 1997;10:236–49. doi: 10.1111/j.1600-0749.1997.tb00490.x. [DOI] [PubMed] [Google Scholar]
- Fung H, Demple B. A vital role for Ape1/Ref1 protein in repairing spontaneous DNA damage in human cells. Mol Cell. 2005;17:463–70. doi: 10.1016/j.molcel.2004.12.029. [DOI] [PubMed] [Google Scholar]
- Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436:117–22. doi: 10.1038/nature03664. [DOI] [PubMed] [Google Scholar]
- Gidanian S, Mentelle M, Meyskens FL, Jr, Farmer PJ. Melanosomal damage in normal human melanocytes induced by UVB and metal uptake—a basis for the pro-oxidant state of melanoma. Photochem Photobiol. 2008;84:556–64. doi: 10.1111/j.1751-1097.2008.00309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445:851–7. doi: 10.1038/nature05661. [DOI] [PubMed] [Google Scholar]
- Grosch S, Kaina B. Transcriptional activation of apurinic/apyrimidinic endonuclease (Ape, Ref-1) by oxidative stress requires CREB. Biochem Biophys Res Commun. 1999;261:859–63. doi: 10.1006/bbrc.1999.1125. [DOI] [PubMed] [Google Scholar]
- Harrison L, Ascione AG, Wilson DM, III, Demple B. Characterization of the promoter region of the human apurinic endonuclease gene (APE) J Biol Chem. 1995;270:5556–64. doi: 10.1074/jbc.270.10.5556. [DOI] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA. 1998;95:2509–14. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Cervantes C, Martinez-Esparza M, Perez C, Daum N, Solano F, Garcia-Borron JC. Inhibition of melanogenesis in response to oxidative stress: transient downregulation of melanocyte differentiation markers and possible involvement of microphthalmia transcription factor. J Cell Sci. 2001;114:2335–44. doi: 10.1242/jcs.114.12.2335. [DOI] [PubMed] [Google Scholar]
- Koyanagi K, O’Day SJ, Gonzalez R, Lewis K, Robinson WA, Amatruda TT, et al. Microphthalmia transcription factor as a molecular marker for circulating tumor cell detection in blood of melanoma patients. Clin Cancer Res. 2006;12:1137–43. doi: 10.1158/1078-0432.CCR-05-1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy C, Khaled M, Fisher DE. MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol Med. 2006;12:406–14. doi: 10.1016/j.molmed.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Liu F, Lee WH. CtIP activates its own and cyclin D1 promoters via the E2F/RB pathway during G1/S progression. Mol Cell Biol. 2006;26:3124–34. doi: 10.1128/MCB.26.8.3124-3134.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loercher AE, Tank EM, Delston RB, Harbour JW. MITF links differentiation with cell cycle arrest in melanocytes by transcriptional activation of INK4A. J Cell Biol. 2005;168:35–40. doi: 10.1083/jcb.200410115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEligot AJ, Yang S, Meyskens FL., Jr Redox regulation by intrinsic species and extrinsic nutrients in normal and cancer cells. Annu Rev Nutr. 2005;25:261–95. doi: 10.1146/annurev.nutr.25.050304.092633. [DOI] [PubMed] [Google Scholar]
- Meyskens FL, Jr, Chau HV, Tohidian N, Buckmeier J. Luminol-enhanced chemiluminescent response of human melanocytes and melanoma cells to hydrogen peroxide stress. Pigment Cell Res. 1997;10:184–9. doi: 10.1111/j.1600-0749.1997.tb00482.x. [DOI] [PubMed] [Google Scholar]
- Meyskens FL, Jr, Farmer P, Fruehauf JP. Redox regulation in human melanocytes and melanoma. Pigment Cell Res. 2001;14:148–54. doi: 10.1034/j.1600-0749.2001.140303.x. [DOI] [PubMed] [Google Scholar]
- Meyskens FL, Jr, Farmer PJ, Yang S, Anton-Culver H. New perspectives on melanoma pathogenesis and chemoprevention. Recent Results Cancer Res. 2007;174:191–5. doi: 10.1007/978-3-540-37696-5_16. [DOI] [PubMed] [Google Scholar]
- Miller AJ, Du J, Rowan S, Hershey CL, Widlund HR, Fisher DE. Transcriptional regulation of the melanoma prognostic marker melastatin (TRPM1) by MITF in melanocytes and melanoma. Cancer Res. 2004;64:509–16. doi: 10.1158/0008-5472.can-03-2440. [DOI] [PubMed] [Google Scholar]
- Molina DM, Grewal S, Bardwell L. Characterization of an ERK-binding domain in microphthalmia-associated transcription factor and differential inhibition of ERK2-mediated substrate phosphorylation. J Biol Chem. 2005;280:42051–60. doi: 10.1074/jbc.M510590200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nappi AJ, Vass E. Hydrogen peroxide generation associated with the oxidations of the eumelanin precursors 5, 6-dihydroxyindole and 5, 6-dihydroxyindole-2-carboxylic acid. Melanoma Res. 1996;6:341–9. doi: 10.1097/00008390-199610000-00001. [DOI] [PubMed] [Google Scholar]
- Ramana CV, Boldogh I, Izumi T, Mitra S. Activation of apurinic/apyrimidinic endonuclease in human cells by reactive oxygen species and its correlation with their adaptive response to genotoxicity of free radicals. Proc Natl Acad Sci USA. 1998;95:5061–6. doi: 10.1073/pnas.95.9.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramphal R, Pappo A, Zielenska M, Grant R, Ngan BY. Pediatric renal cell carcinoma: clinical, pathologic, and molecular abnormalities associated with the members of the mit transcription factor family. Am J Clin Pathol. 2006;126:349–64. doi: 10.1309/98YE9E442AR7LX2X. [DOI] [PubMed] [Google Scholar]
- Rouaux C, Loeffler JP, Boutillier AL. Targeting CREB-binding protein (CBP) loss of function as a therapeutic strategy in neurological disorders. Biochem Pharmacol. 2004;68:1157–64. doi: 10.1016/j.bcp.2004.05.035. [DOI] [PubMed] [Google Scholar]
- Sander CS, Chang H, Hamm F, Elsner P, Thiele JJ. Role of oxidative stress and the antioxidant network in cutaneous carcinogenesis. Int J Dermatol. 2004;43:326–35. doi: 10.1111/j.1365-4632.2004.02222.x. [DOI] [PubMed] [Google Scholar]
- Sato S, Roberts K, Gambino G, Cook A, Kouzarides T, Goding CR. CBP/p300 as a co-factor for the microphthalmia transcription factor. Oncogene. 1997;14:3083–92. doi: 10.1038/sj.onc.1201298. [DOI] [PubMed] [Google Scholar]
- Steingrimsson E, Copeland NG, Jenkins NA. Melanocytes and the microphthalmia transcription factor network. Annu Rev Genet. 2004;38:365–411. doi: 10.1146/annurev.genet.38.072902.092717. [DOI] [PubMed] [Google Scholar]
- Tassabehji M, Newton VE, Read AP. Waardenburg syndrome type 2 caused by mutations in the human microphthalmia (MITF) gene. Nat Genet. 1994;8:251–5. doi: 10.1038/ng1194-251. [DOI] [PubMed] [Google Scholar]
- Vachtenheim J, Borovansky J. Microphthalmia transcription factor: a specific marker for malignant melanoma. Prague Med Rep. 2004;105:318–24. [PubMed] [Google Scholar]
- Vachtenheim J, Sestakova B, Tuhackova Z. Inhibition of MITF transcriptional activity independent of targeting p300/CBP coactivators. Pigment Cell Res. 2007;20:41–51. doi: 10.1111/j.1600-0749.2006.00354.x. [DOI] [PubMed] [Google Scholar]
- van de Wetering M, Oving I, Muncan V, Pon Fong MT, Brantjes H, van Leenen D, et al. Specific inhibition of gene expression using a stably integrated, inducible small-interfering-RNA vector. EMBO Rep. 2003;4:609–15. doi: 10.1038/sj.embor.embor865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance KW, Goding CR. The transcription network regulating melanocyte development and melanoma. Pigment Cell Res. 2004;17:318–25. doi: 10.1111/j.1600-0749.2004.00164.x. [DOI] [PubMed] [Google Scholar]
- Wellbrock C, Marais R. Elevated expression of MITF counteracts B-RAF-stimulated melanocyte and melanoma cell proliferation. J Cell Biol. 2005;170:703–8. doi: 10.1083/jcb.200505059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Hemesath TJ, Takemoto CM, Horstmann MA, Wells AG, Price ER, et al. c-Kit triggers dual phosphorylations, which couple activation and degradation of the essential melanocyte factor Mi. Genes Dev. 2000;14:301–12. [PMC free article] [PubMed] [Google Scholar]
- Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh k, et al. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature. 2005;434:338–45. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Irani K, Heffron SE, Jurnak F, Meyskens FL., Jr Alterations in the expression of the apurinic/apyrimidinic endonuclease-1/redox factor-1 (APE/Ref-1) in human melanoma and identification of the therapeutic potential of resveratrol as an APE/Ref-1 inhibitor. Mol Cancer Ther. 2005;4:1923–35. doi: 10.1158/1535-7163.MCT-05-0229. [DOI] [PubMed] [Google Scholar]
- Yang S, Misner B, Chiu R, Meyskens FL. Redox effector factor-1, combined with reactive oxygen species, plays an important role in the transformation of JB6 cells. Carcinogenesis. 2007;28:2382–90. doi: 10.1093/carcin/bgm128. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mek1/2–Erk1/2 MAP kinase pathway-mediated ROS-induced MiTF phosphorylation.
Oligos for shRNA construction and primers for PCR and site mutagenesis.