

Abstract
There are severe neurological complications that arise from HIV infection, ranging from peripheral sensory neuropathy to cognitive decline and dementia for which no specific treatments are available. The HIV proteins secreted from infected macrophages, gp120 and Tat, are neurotoxic. The goal of this study was to screen, identify and develop neuroprotective compounds relevant to HIV-associated neurocognitive disorders (HAND). We screened more than 2000 compounds that included FDA approved drugs for protective efficacy against oxidative stress-mediated neurodegeneration and identified selective serotonin reuptake inhibitors (SSRIs) as potential neuroprotectants. Numerous SSRIs were then extensively evaluated as protectants against neurotoxicity as measured by changes in neuronal cell death, mitochondrial potential, and axodendritic degeneration elicited by HIV Tat and gp120 and other mitochondrial toxins. While many SSRIs demonstrated neuroprotective actions, paroxetine was potently neuroprotective (100 nM potency) against these toxins in vitro and in vivo following systemic administration in a gp120 neurotoxicity model. Interestingly, the inhibition of serotonin reuptake by paroxetine was not required for neuroprotection, since depletion of the serotonin transporter had no effect on its neuroprotective properties. We determined that paroxetine interacts selectively and preferentially with brain mitochondrial proteins and blocks calcium-dependent swelling but had less effect on liver mitochondria. Additionally, paroxetine induced proliferation of neural progenitor cells in vitro and in vivo in gp120 transgenic animals. Therefore, SSRIs such as paroxetine may provide a novel adjunctive neuroprotective and neuroregenerative therapy to treat HIV-infected individuals.
Electronic supplementary material
The online version of this article (doi:10.1007/s13311-014-0315-9) contains supplementary material, which is available to authorized users.
Keywords: Paroxetine, SSRI, HAND, Neuroprotective, Mitochondria
Introduction
HIV infection can result in severe complications involving the nervous system. Even with the advent and implementation of combination antiretroviral therapy (cART) to diminish HIV viral load, the occurrence of HIV-associated neurocognitive disorders (HAND) persists. As cART allows HIV patients to live longer, 36-45 % individuals experience some form of neurocognitive deficits [1]. These complications are a result of immune activation, oxidative stress and neurotoxicity that results from either persistent HIV replication in cellular reservoirs in the brain or the release of viral products such as Tat and gp120 which may be directly toxic to neurons or act on glial cells to block glutamate uptake, release cytokines and free radicals which may also lead to oxidative neuronal injury [2]. HIV-infected patients also report high rates of depression [3, 4]. Major depressive disorder is twice as likely to occur in patients infected with HIV than those who don’t have the disease [5]. Often neurocognitive impairment and depression may co-exist making the management challenging [6]. Neuroinflammatory pathways and oxidative stress have also been implicated in the pathophysiology of depression [7]. Thus neuroprotective compounds, if identified and developed preclinically, may provide a treatment for HIV-related complications such as HAND and depression.
Identification and development of therapeutic strategies to protect against this neuronal damage and the ensuing clinical deficits is a major unmet medical need. To date, however, despite several clinical trials targeting multiple neuroprotective pathways, there is no effective treatment for HAND. All studies thus far have shown little or no clinical benefit [8, 9]. Although multiple factors may contribute to failure of clinical trials, we reasoned that neuroprotection alone may not be sufficient. Brain atrophy that accompanies HAND may in part reflect a failure of neurogenesis as well [10]. Clearly, neural progenitor cells (NPCs) may get infected with HIV [11] and HIV proteins have been shown to impair adult neurogenesis [12–14]. We thus initially screened more than 2000 compounds that included FDA approved drugs for protective efficacy against oxidative stress-mediated neuronal injury. Several classes of compounds were found to be neuroprotective. This included the selective serotonin reuptake inhibitors (SSRIs). While SSRIs are routinely used for treatment of depression, recent studies also show that these drugs can promote adult neurogenesis in healthy and in transgenic animals expressing HIV proteins in the brain [12]. Using paroxetine as a prototype of this class of compounds, we determined if the mechanism of neuroprotection involves the serotonergic pathways, and if the phamacokinetics of neuroprotection was different from its effects on the serotonergic transporters. We discovered that the neuroprotective mechanisms of paroxetine are distinct from its antidepressive effects on the serotonergic transporters.
Materials and Methods
Compound Screening
The Spectrum Collection of compounds by Microsource Discovery contains 2,000 compounds, of which about half are FDA-approved drugs and the remaining compounds are natural products or other compounds with some prior human exposure and safety testing data. The compound collection is dispensed and maintained in 96-well plates at a concentration of 10 mM in 100 % DMSO and stored at −80 °C. The compound mother plates were thawed one time in order to make four sets of daughter plates, and one set of daughter plates was used for these screening assays.
HIV-1 Proteins, Reagents and Engineering RNA
The preparation of recombinant Tat1–72 protein has been described previously [15]. Recombinant Tat displayed as a single band and a single peak by SDS-PAGE and HPLC, respectively. gp120 derived from HIV-IIIB and de-glycosylated gp120 was acquired from the NIH-AIDS repository (Rockville, MD, USA). 3-nitropropionic acid (3-NP), N-met7hyl-D-aspartic acid (NMDA) and H2O2 were obtained from Sigma Chemical Co. (St. Louis, MO, USA). All siRNA pools were purchased from Invitrogen (Grand Island, NY, USA).
Cell Culture
Primary neuronal cultures were prepared from Sprague–Dawley (SD) rat fetal brains (Charles River Laboratories, Wilmington, MA, USA) and SERT knock-out (SERT−/−) mice (Charles River Laboratories) at 18 days of gestation. Brains were harvested from 10 to 15 pups, all meninges were removed and the cortex and hippocampus was dissected. Tissues were dissociated by gentle trituration with a firepolished glass pipette in calcium-free Hank’s balanced salt solution. The single cell suspension was centrifuged at 200 × g and re-suspended in minimal essential medium containing 10 % (v/v) heat-inactivated fetal bovine serum and 1 % (v/v) antibiotic and antimycotic solution (Sigma, St Louis, MO, USA). Cells were allowed to attach for 3 h before the media was replaced with serum-free neurobasal medium containing 2 % (v/v) B-27 supplement (Gibco, Rockville, MD, USA) and 1 % antibiotic and antimycotic solution. Rat neuronal cultures were used between 10 and 14 days in vitro which consisted of >98 % neurons that expressed microtubule-associated protein 2 (MAP2) and the remainder of the cells were predominantly astrocytes which expressed glial fibrillary acidic protein (GFAP). Alternatively, rat mixed hippocamal neurons were generated from freshly cultured rat hippocampi in media containing 5 % fetal bovine serum and 2 % B-27 supplement. Hippocampal neurons were plated in 96-well plates at a density of 4× 105 cells per ml on 35-mm dishes for neurotoxicity studies. Mixed rat hippocampal cultures were plated into 96-well plates at a density of 4 × 105 cells per ml. These mixed rat hippocampal cultures consisted of 40-45 % βIII tubulin expressing neurons, 50-55 % GFAP expressing astrocytes and about 1 % microglia.
Human NPC cultures were prepared as described previously [16]. Briefly, the tissues were triturated after removing meninges and blood vessels. After centrifugation at 200 × g, cells were resuspended in DMEM/F12 media [containing 8 mm glucose, 1× N2 supplement, 1 % antibiotics, 0.1 % (w/v) albumin (Sigma-Aldrich), human fibroblast growth factor-β (hFGFb) (20 ng/ml), and human epidermal growth factor (hEGF) (20 ng/ml)] and plated inpoly-d-lysine (Sigma-Aldrich)-coated T 25 cm2 tissue culture flasks. When cell cultures reached 60 % confluence, they were subcultured by treatment with 0.0125 % trypsin (Sigma-Aldrich) and plated at a density of 2 × 104 cells/ml in poly-d-lysine-coated 96-well plates or on coverslips in 24-well plates. Medium was replaced every other day. NPC cultures were ready for experiments 4–5 d after replating and >98 % of the cells expressed the neural stem cell marker nestin, whereas <1 % of the cells expressed GFAP (a marker for astrocyte) or β-III-tubulin (a neuronal cell marker) as determined by immunocytochemistry. To label proliferative cells, cells were incubated with bromodeoxyuridine (BrdU) labeling reagent (1:100; Zymed Laboratories, San Francisco, CA, USA) for 6 h followed by immunostaining for BrdU.
All primary cell culture procedures used in this study were performed in accordance with National Institutes of Health (NIH) guidelines and following approval by the Institutional Review Board at The Johns Hopkins University.
Cell Viability Assay
Cell viability was evaluated using two methods; the 3-(4,5-dimethylthiazol-2-yl)- 2,5-diphenyltetrazolium bromide (MTT) assay [17] and CellQuanti-Blue™ cell viability assay kit (BioAssay Systems, Hayward, CA). For MTT assay, 10 μl of 5 mg/ml MTT tetrazolium salt (Invitrogen) was added to the media of treated cells followed by incubation for 2 hr at 37 °C in 5 % CO2 incubator. The medium was then aspirated and was replaced with 100 μl DMSO to dissolve the Formosan crystals in the cells. The amount of Formosan crystals was measured by absorbance using a microplate reader (Bio-Rad, Hercules, CA, USA) at test and reference wavelengths of 570 and 630 nm respectively.
In assay using CellQuanti-Blue™ cell viability assay kit, cells were cultured at 1 × 105/ml in 90 μl serum-free opti-MEM medium in 96-well plates. The cells were transfected with a pooled siRNA to SERT, Non-specific control siRNA (NsiRNA) (Invitrogen) or vehicle only using lipofectamine 2000. The cultures were exposed to Tat (50 nM) and morphine (1 μM) with or without paroxetine (1 μM) for 24 hrs. CellQuanti-blue solution (10 μl/well) was then added for 30 min and fluorescence intensity was detected at 590 nm (530 nm excitation, 590 nm emission filters).
Immunocytochemistry
To characterize cell types in primary cultures, cells were fixed in 4 % (w/v) paraformaldehyde (Sigma) and rinsed with TBS with 0.5 % (v/v) Triton- × 100. Primary antibodies were diluted in blocking solution as follows: mouse anti-MAP2 (1: 1000, Sigma-Aldrich), mouse anti-nestin (1:1000, Millipore), mouse anti-beta-III tubulin (1:1000, Promega) and rabbit anti-GFAP (1:1000, Sigma) and incubated with cells for 1 hr at room temperature. For detection of BrdU, the fixed cells were denatured in a 2 N HCl bath for 30 min at 37 °C followed by neutralization of the acid in 0.5 M sodium borate, pH 8.5, for 10 min at room temperature. After washing with TBS, cells were incubated with rat anti-bromodeoxyuridine (BrdU) (1:1000; Accurate, Westbury, NY, USA) for 1 hr at room temperature. After washing with TBS, cells were incubated with the secondary antibodies (anti-mouse Alexa Flour 597, 1:400 anti-rabbit Alexa Flour 488, 1:400, Invitrogen) followed by washing and counterstaining with 4′,6-diamidino-2-phenylindole (DAPI, 1:5000) to label all nuclei. Stained cells were mounted on slides and imaged by a fluorescence microscope (model AMF-4302, Advanced Microscopy Group, Bothell, WA, USA).
Multiplex Assay
Rat mixed hippocampal cultures were plated onto 6 well plates at 7.5 × 105 cells per well and pretreated with 1 μM paroxetine for 1 h, prior to exposure to 500 nM HIV Tat for 24 h at 37 C. After 24 h, the culture supernatants were removed and simultaneous quantitation of 23 cytokines and chemokines in the culture supernatants was performed on a Luminex 100 imager (Luminex Corp., Austin, TX) with a rat cytokine assay system (Milliplex; EMD Millipore, Billerica, MA). Simultaneous quantification of 23 cytokines and chemokines from culture supernatants of rat mixed hippocampal cultures was achieved with a rat cytokine assay system (Milliplex; EMD-Millipore, Billerica, MA). The analytes included IL-1α, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-9, IL-10, IP-10, IL-12 (p70), IL-13, IL-17, IL-18, eotaxin, G-CSF, GM-CSF, GRO/KC, IFN-γ, MCP-1, MIP-1α, RANTES, TNF-α and VEGF. We determined that a number of these were not consistently secreted from our cultures at levels that could be accurately quantitated, so we present only those cytokines and chemokines that were within the dynamic range of the standard curves. Standard calibration curves were prepared using sequential 4 fold dilutions or recombinant cytokine standards, according to the manufacturer’s instructions. All assays were performed in 96 well microplate format Milliplex assays at room temperature and protected from light as per the manufacturer’s instructions. 3–5 independent values for each cytokine were determined for each cytokine and expressed as pg/ml. Data were analyzed by ANOVA, and post hoc comparison of groups were made with Dunnett’s multiple comparison test. An alpha level of p ≤ 0.05 was considered significant for all statistical tests used. Data are expressed in the table as mean (±SEM). Statistical significance is depicted as *p < 0.05, **p < 0.01, ***p < 0.001. The significant Tat dependent cytokine levels were compared to basal cytokine levels, while significance of Tat + paroxetine cytokine levels were compared to those of Tat treatment alone.
Production and Stereotaxic Injection of SV(gp120), TUNEL Assay and NT Staining
An SV40-derived gene delivery vector, SV(gp120), were used to express HIV-1 envelope protein gp120 and induce neuronal cell death in the brain of rats [18]. The vector was generated according to previously described methodology [18]. Briefly, a 1.6 kb DNA fragment encoding gp120 from HIV-1NL4-3 was made by PCR using primers with engineered restriction sites. This PCR product was cloned into pT7A5 (a plasmid containing an SV40 genome, in which large T antigen gene was replaced by cytomegalovirus (CMV) immediate early promoter and downstream polylinker), giving pT7A5-gp120. To make SV(gp120), the SV(gp120) genome was released from the carrier plasmid by restriction digestion, and used to make recombinant virus in COS-7 cells. Virus stocks were purified and titered. For injection of SV(gp120), 12 female SD rats (275–300 g, Charles River Laboratories) housed under standard conditions were anaesthetized ketamine (100 mg/kg) and xyline (50 mg/kg) cocktail and placed in a stereotaxic apparatus (Stoelting Corp, Wood Dale, IL) for cranial surgery. 5 μL of SV(gp120) viral vector, which contains approximately 107 infectious units, was sterotaxically injected into the caudate-putamen (CPu) using coordinates obtained from the rat brain atlas of Paxinos and Watson (Paxinos and Watson, 2006). Target coordinates are anterior-posterior (AP) = −0.26 mm, medial-lateral (ML) = ±4.2 mm, and dorsal-ventral (DV) = −4.0 mm with respect to bregma. All animal procedures were in accordance with institutional guidelines. Controls received saline in the CPu. After surgery, animals were individually housed with free access to water and food. All animal procedures used in this study were performed in accordance with the protocol approved by the Institutional Animal Care and Use Committee at Thomas Jefferson University.
SV(gp120)-induced apoptosis was measured by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay. The TUNEL assay was performed according to the manufacturer's instructions (In Situ Cell Death Detection kit; Roche Diagnostics, Indianapolis, IN, USA). Apoptosis in the CPu was quantitated as number of TUNEL-positive cells in microscopic fields. NeuroTrace (NT) staining was used as a neuronal marker and was performed as according to the manufacturer's instructions (NeuroTraceTM Fluorescent Nissl Stains; Molecular Probes, Inc, Eugene, OR, USA). All quantitative assessments were done by investigators blinded to the treatment.
Quantitative Western Blot Analysis
For Western blot analysis, rat hippocampal neurons were homogenized and lysed directly in the sodium dodecyl sulphate (SDS) sample buffer (62.5 mM Tris pH 6.8, 10 % (v/v) glycerol, 2 % (w/v) SDS, 0.01 % (w/v) bromphenol blue). Protein homogenates were boiled for 5 minutes and loaded onto 10-15 % SDS PAGE gel for electrophoresis (Bio-Rad, Philadelphia, PA, USA). Nitrocellulose membranes with transferred proteins were blocked with 4 % (w/v) BSA in TBS (10 mM Tris–HCl, pH 7.5, 150 mM NaCl), incubated in TBST (TBS + 0.05 % Tween-20) with mouse anti-inducible nitric oxide synthase (iNOS) / nitric oxide synthase (NOS) (1:1000, BD Biosciences, Bedford, MA, USA), washed and reacted with horse radish peroxidase-conjugated secondary antibodies (Roche Molecular Systems, Inc., Branchburg, NJ, USA). Membranes were stripped and re-blotted with mouse anti-Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) (1:5000, Santa Cruz Biotechnology, Santa Cruz, CA, USA) for loading control. The relative intensities of the blots were measured by densitometry.
Animals, Paroxetine Administration and BrdU Labeling
All animal procedures used in this study were performed in accordance with the protocol approved by the Institutional Animal Care and Use Committee at Johns Hopkins University School of Medicine. Adult (8–9 weeks old) male gp120 transgenic mice in which the expression of gp120 was driven by a GFAP promoter [19] and littermate wild type controls were used. A minimum of five animals per group were used for each experimental condition. Chronic administration of paroxetine (Tocris Cookson, Ellisville, MO, USA) was accomplished by subcutaneous implantation of an osmotic mini-pump (model 2004, Alzet, Palo Alto, CA, USA) according to manufacturer's instructions. Pumps containing either paroxetine or saline were activated prior to implantation by overnight incubation at 37 °C as per manufacturer's instructions. Pumps were placed subdermally in the interscapular region of mice under deep anaesthesia, and a pump flow rate of 0.25 μl/h allowed for delivery of paroxetine at a dose of 10 mg/kg per day for 20 or 28 days. Upon completion of the experiments, pumps were removed and residual volume was determined, to ensure that the appropriate amount of drug had indeed been delivered to each mouse.
A group of the animals were injected with a single dose of BrdU labeling reagent (200 mg/kg, Sigma) after paroxetine treatment and were euthanized 2 hours later for cell proliferative analysis. To assess BrdU + NeuN + newly generated neurons, an additional group of mice were injected with BrdU (50 mg/kg, Sigma) for seven consecutive days during the second week of the experiment and were euthanized 3 weeks after the first injection of BrdU.
Immunohistochemistry
Animals were transcardially perfused with saline followed by 4 % paraformaldehyde (PFA). After postfixing in PFA overnight, brains were immersed in a 30 % (v/v) sucrose solution. On the following day, brains were cut in 40 μm thick coronal sections on a sliding microtome. Every sixth brain sections spanning the entire hippocampal dentate gyrus were incubated with rat anti-BrdU (1:000, Accurate) and mouse anti-NeuN (1:250, Millipore) overnight at 4 °C. For detection of BrdU, slices were treated with 2 N HCl at 37 °C for 30 min prior to application of the primary antibody. After washing with TBS, sections were incubated with the appropriate fluorescent conjugated secondary antibodies (1:250, Jackson ImmunoResearch, West Grove, PA, USA). The sections were counterstained with DAPI (1:5000, Sigma) for 10 min before washing and mounting. Images were acquired on a Zeiss LSM 510 Meta multiphoton confocal system (Carl Zeiss Microimaging Inc., Thornwood, NY, USA) using a multitrack configuration. Z-stacks (2 μm thick, spanning the entire 40 mm thickness of each slice) were constructed for each image. Stereological quantification of immunopositive cells within the subgranular zone and granule cell layer were carried out as previously described [20]. Double-immunolabeling was confirmed for each cell by orthogonal analysis using LSM 5 Image Browser. Positively labeled cells were expressed as cells per volume of the granule cell layer, as assessed by staining of the nuclei with DAPI. All quantitative assessments were done by investigators blinded to the treatment.
Mitochondrial Studies
Female SD rats (275–300 g, Charles River Laboratories) were decapitated and the liver and brain tissues were transferred within 1 min to ice-cold isolation buffer (0.32 M sucrose, 2 mM EGTA, 10 mM Tris, pH 7.4). Mitochondria from the liver and brain were prepared and mitochondrial swelling assays were conducted as described [21]. Briefly, mitochondria were de-energized by addition of 0.5 μM rotenone, 0.5 μM antimycin and 2 μM ionomycin. Mitochondria were then pretreated with fluoxetine or paroxetine for 10 min on ice, and mitochondrial swelling was initiated by addition of 10 μM calcium ions. Mitochondrial swelling was monitored by measuring the decrease in optical density (OD) at 520 nm for 8 min post calcium ions addition. 1 μM cyclosporin A (CsA) was run in parallel assays as a positive control for inhibition of mitochondrial swelling.
Calcium Imaging
Cytosolic calcium ([Ca2+]C) was determined using the ratiometric Ca2+-specific fluorescent probe acetoxy-methyl-ester fura-2 (fura-2 AM) as previously described [22]. Rat hippocampal neurons were incubated with 2 μM fura-2 AM for 15 min at 37 °C in neurobasal medium and washed with Locke’s buffer (154 mM NaCl, 3.6 mM NaHCO3, 5.6 mM KCl, 1 mM MgCl2, 5 mM HEPES, 2.3 mM CaCl2, 10 mM glucose) to remove extracellular fura-2. Cells loaded with fura-2 were then incubated at 37 °C for 10 min to allow complete de-esterfication of the probe. Coverslips containing the cells were maintained at 37 °C in a recording chamber and perfused with Locke’s buffer at the rate of 2 mL/min (Warner Instruments Inc., Hamden, CT, USA). Image pairs were acquired every 1.0-5.0 s and 340 : 380 ratios were converted to nM Ca2+ using a video based imaging system (Intracellular Imaging Inc., Cincinnati, OH, USA) and commercial reference standards (Molecular Probes). For studies of effect of paroxetine on calcium responses, neurons were pretreated with 10 μM paroxetine for 1 h before they were stimulated with 40 mM potassium chloride (KCl) to induce depolarization.
Identification of Paroxetine Binding Proteins
Paroxetine was coupled to cyanogen bromide (CNBr)-activated Sepharose 4B (Sigma) according to instructions supplied by the manufacturer. The amount of paroxetine bound to the beads was determined by spectrophotometric depletion of paroxetine from the coupling solution over time. Typically, 3–4 mg per milliliter of paroxetine was covalently attached to the Sepharose beads for these experiments. As a control for non-specific absorption to the Sepharose beads, a control column was generated using Tris base [tris(hydroxylmethyl)amino methane], which was utilized in parallel experiments. Rat cortical neurons (106 cells per well) were scraped from 6 well plates and collected by centrifugation. The cells were washed, then the pellets were snap frozen on dry ice. The cell pellets were thawed on ice, resuspended in 10 mM Hepes, 150 mM NaCl, 1 mM NaEGTA, 1 mM NaN3, 1 mM DTT, pH 7.4 plus 1 mM PMSF, 10 ug/mL of leupeptin and pepstatin (protease inhibitors) and homogenized in a glass/Teflon homogenizer with 4–5 up/down strokes on ice. The homogenates were centrifuged at 900 × g for 10 min to remove cell nuclei, and the supernatants were centrifuged at 100,000 × g for 60 min. The neuronal membranes were resuspended in 10 volumes of the Hepes resuspension buffer and Triton × -100 was added to a final concentration of 1 % for incubation at 4 °C for 60 min. The membrane debris was removed by centrifugation at 100,000 × g for 60 min, and the triton-extracted proteins were applied to paroxetine-Sepharose or Tris-Sepharose for 16 h overnight incubation at 4 °C. After this incubation, the column flow through was collected, and the column was washed with at least 10 column volumes of resuspension buffer plus 0.1 % Triton × -100, followed by a 5 column volumes of 1 M KCl in resuspension buffer +0.1 % Triton × -100 buffer. The target proteins were then eluted by addition of 25 mM paroxetine. Eluted proteins were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Resulting protein gels are stained by silver staining and immunoblotting. Next, specific protein bands were cut from the gel and subjected to protein identification by mass-spectrometry analysis using a LC-MS/MS at the Johns Hopkins University Mass Spectrometry/ Proteomics facility. Additionally, for identification of mitochondrial proteins, rat brain mitochondria preparations were made and extracted with 1 % Triton × -100. The extracts were loaded onto either the paroxetine- Sepharose or Tris-Sepharose columns. The flow-through, wash fractions as well as a material that bound to both columns was eluted. Eluted proteins were analyzed by SDS-PAGE and immunoblotting.
Inhibition Assay of HIV-1 Replication
TZM-bl cells expressing CD4 and CCR5 were pre-seeded in 96-well plates. Cells with 60–80 % confluence were pretreated with the serially diluted paroxetine at concentrations of 0.1, 1 or 10 μM for 1 h at 37 °C and then infected with HIV-1 JRCSF at 2000 TCID50/ml in a final 200 μl volume of culture medium. Levels of β- galactosidase were quantitated in the cell lysates 2 days later using β- galactosidase ELISA Kit (Roche Applied Science, Indianapolis, IN, USA).
Results
Neuroprotection by Paroxetine and Fluoxetine Against Neurotoxic Stress In Vitro
Because neuronal cell death by oxidative and excitotoxic stress is seen in the brains of patients with HAND [23, 24], we used 3-NP-, H2O2- and NMDA - induced neurotoxicity in an in vitro model to test neuroprotective effect of SSRIs. Initially, we screened the Microsource Spectrum collection for neuroprotectants against the oxidative stresser 3-NP (see [25]. From these studies, a number of tricyclic antidepressants as well as selective serotonin reuptake inhibitors were identified as protective against oxidative stress (Table 1). Compounds such as nortriptyline, trimipramine, fluoxetine and paroxetine displayed greater than 50 % protection at the 10 μM screening dose. The overall intra-assay variability of the screen was 8-10 %.
Table 1.
Neuroprotective antidepressants against 3-NP mediated oxidative stress
| Compound (10 μM) | 3-NP Neurotoxicity |
|---|---|
| % Protection | |
| Desipramine | 42.5 ± 8.4 |
| Imipramine | 23.5 ± 3.1 |
| Doxepin | 51.1 ± 14.3 |
| Maprotiline | 23.5 ± 4.0 |
| Amitriptyline | 44.1 ± 6.9 |
| Nortriptiyline | 143.4 ± 37.9 |
| Paroxetine | 56.3 ± 3.8 |
| Fluoxetine | 101.8 ± 45.4 |
| Clomipramine | 25.1 ± 10.2 |
| Amoxepine | 32.4 ± 11.0 |
| Quipazine | 19.1 ± 7.4 |
| AcetylTryptophan | 30.9 ± 9.3 |
| Trimipramine | 95.2 ± 27.9 |
Values presented are Mean ± SEM, n = 3.
Rat hippocampal neurons were exposed to mitochondria toxin, 3-NP at a concentration of 3 mM for 18 h, resulting in cell death of ~20 %. Paroxetine administered 1 h prior to 3-NP exposure significantly reduced the cell death in a concentration-dependent manner. Paroxetine at doses of 5 and 10 μM provided almost complete protection against neuronal death. Pretreatment of fluoxetine for 1 hour followed by 3-NP exposure also significantly reduced cell death against 3-NP at a concentration of 10 μM by 500 %. Paroxetine exerted a more pronounced cytoprotective effect compared to fluoxetine (Fig. 1a). The neurons were exposed to 100 μM NMDA for 18 h, thereby inducing a decrease in cell viability. Both paroxetine and fluoxetine at a concentration of 10 μM were able to protect cells against NMDA-induced neurotoxicity (Fig. 1b). Next, cultured neurons exposed to 100 μM H2O2 for 2 h showed significant decrease in the viability. Paroxetine treatment significantly increased the cell viability in a concentration-dependent manner. The results showed that 50 % and 30 % cells underwent cell death in 1 μM and 5 μM paroxetine, respectively, while 80 % cells underwent cell death in H2O2-treated cultures (Fig. 1c).
Fig. 1.
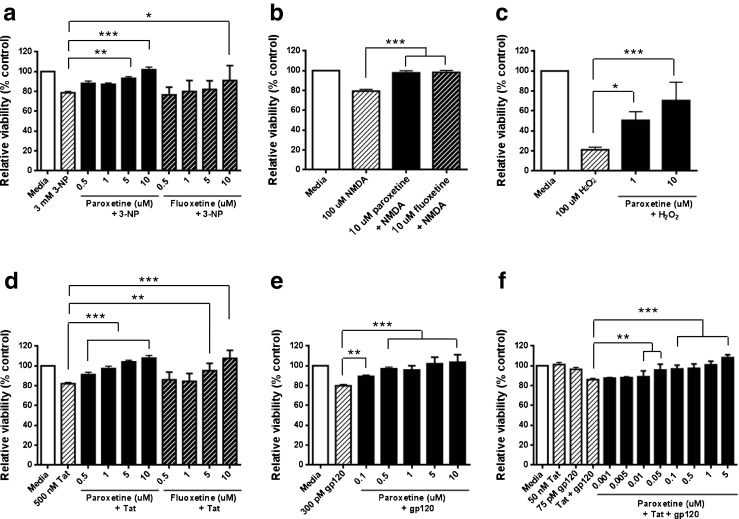
SSRIs protect neurons against diverse neurotoxic agents in vitro. (A-C) Rat mixed hippocampal cultures were incubated with increasing concentrations of paroxetine and/or fluoxetine for 1 hour prior to addition of (a) 3-NP, (b) NMDA, or (c) H2O2. Cultures were exposed to 3-NP or NMDA for 18 hours, or 2 hour exposure to H2O2 at 37 °C, and cell viability was quantitated by MTT Assay. An analysis of variance showed that the effects of paroxetine and fluoxetine were significant: Panel A F(9,82) = 14.0, p < 0.0001; Panel B F[3, 28] = 33.87, p < 0.0001; Panel C F[3, 27] = 12.42, p < 0.0001). Post hoc analyses were done using Tukey’s multiple comparison test. Values represent mean ± SEM (3 experiments per condition, *p <0.05; **p < 0.01; ***p < 0.001). (d-f) Neuroprotection effect of fluoxetine and paroxetine were tested against HIV-1 proteins, (d) Tat, (e) gp120, or (f) Tat and gp120. Rat mixed hippocampal cultures were pretreated with fluoxetine and/or paroxetine for 1 hour, and then 500 nM Tat, 300 pM gp120, or 50 nM Tat and 75 pM gp120 were added for 18 hours at 37 C. Cell survival was quantitated by MTT Assay. An analysis of variance showed that the effects of paroxetine and fluoxetine were significant: Panel D F(9,75) = 19.07, p < 0.0001; Panel E F[6, 49] = 21.98, p < 0.0001; Panel F F(11,84) = 19.55, p < 0.0001). Post hoc analyses were done using Tukey’s multiple comparison test. Values represent mean ± SEM (3 experiments per condition, **p < 0.01; ***p < 0.001)
HIV-1 proteins, Tat and gp120 trigger neuronal apoptosis and excitotoxicity as a result of oxidative stress, perturbed cellular calcium homeostasis and mitochondrial alterations [23, 26]. We next determined whether SSRIs, paroxetine and fluoxetine, provide neuroprotection against HIV proteins, Tat, gp120 or a combination of Tat and gp120. Rat hippocampal cultures were challenged with 500 nM Tat for 18 hours, inducing ~20 % cell death. Tat-induced cell death was significantly reduced by paroxetine in a concentration-dependent manner. The cell viability was increased by 50 % in the lowest dose of paroxetine tested (0.5 μM) and over 100 % by the highest dose tested (10 μM). Fluoxetine also protected against Tat-induced cell death at concentrations higher than 5 μM (Fig. 1d). 300 pM gp120 also induced cell death by ~20 %. Pretreatment of paroxetine followed by gp120 increased cell viability in a concentration-dependent manner, ranging from 0.1 (50 %) to 10 μM (100 %) (Fig. 1e). To further test if paroxetine is neuroprotective against the combination toxicity of Tat and gp120, we used subtoxic dosages of each of the viral proteins (50 nM Tat and 75 pM gp120) which caused synergistic neurotoxicity [27]. Under these conditions, the combination of Tat and gp120 induced ~15 % cell death. Concentrations of paroxetine as low as 0.01 μM showed significant neuroprotection and complete protection at 1 μM or above (Fig. 1f). Taken together, these results indicate that SSRIs, paroxetine and fluoxetine, have neuroprotective effects against neurotoxic stress in vitro. Neuroprotection of paroxetine was more effective as compared with fluoxetine treatment. Therefore, 10 μM paroxetine was used in the succeeding experiments.
Neuroprotection by Paroxetine Against gp120-induced Cell Death In Vivo
gp120 can be successfully delivered to the rodent brain by SV40 viral vector delivery [18]. To study possible neuroprotection by paroxetine against SV(gp120)-induced apoptosis in an in vivo model, rats were given either paroxetine or saline (10 mg/kg/day in each) via an osmotic pump. 7 days later, 5 μL of SV(gp120) or saline were stereotaxically injected into the caudate putamen (CPu) of rats that had been administered with paroxetine or saline. Brains were harvested 7 days after injection and studied for apoptosis by TUNEL assay (Fig. 2a). Rare apoptotic cells were seen in the control group of animals which had received saline and paroxetine. Injection of SV(gp120) increased the number of apoptotic cells in the CPu (67.00 ± 3.00 cells per area). This neuronal death by SV(gp120) was strikingly reduced by paroxetine treatment, showing a significant reduction of TUNEL- positive neurons to greater than 50 % (Fig. 2b and c). Thus, there was a significant neuroprotective effect of paroxetine treatment in vivo following systemic delivery of paroxetine.
Fig. 2.

Paroxetine protects neurons against gp120-induced cell death in vivo. (a) Experimental design; rats were given either paroxetine or saline at 10 mg/kg/day via an osmotic pump. 7 days later, rats were then challenged with intra-caudate-putamen (CPu) injection of SV(gp120). Brains were harvested 7 days after SV(gp120) challenge and studied for apoptosis by TUNEL assay. (b) Shown are sample images of neurotrace (NT, a neuronal marker)- and TUNEL- positive neurons. (c) Shown is a stereological quantification of TUNEL- positive cells in the Cpu. Values represent mean ± SEM (n =4 animals in each group, *p <0.05, one-way ANOVA followed by Tukey’s multiple comparison test)
Paroxetine Stimulated Proliferation of NPCs and Generation of Newborn Neurons
We determined whether paroxetine could impact proliferation of NPCs using an established in vitro model of human fetal cells. Dissociated human cultures were exposure to varying concentrations of paroxetine for 24 hours, and BrdU incorporation was assessed. Addition of paroxetine increased human NPC proliferation in a concentration-dependent manner. 2 μM paroxetine significantly enhanced NPC proliferation (Fig. 3a and b).
Fig. 3.

Paroxetine enhances proliferation of neural progenitor cells (NPC). (a, b) Human NPC were cultured in proliferation medium on poly-D-lysine coated cover slips and treated with paroxetine for 24 hours and with BrdU reagent overnight. (a) BrdU incorporation was determined by immunostaining (red fluorescence). DIC images were also taken to count total number of cells. (b) Quantification shows significant proliferation at 2 μM paroxetine. Data represents two independent experiments performed with duplicate wells at each dosage. An analysis of variance showed that the effect of paroxetine was significant F[3, 19] = 19.41, p < 0.0001). Post hoc analysis was done using Tukey’s multiple comparison test. Values represent mean ± SEM (3 experiments per condition, ***p < 0.001). (c, d) gp120 transgenic mice were implanted with either saline- or paroxetine-containing pumps for 28 days and were analyzed 2 hours after BrdU injection. (c) Representative images of cells labeled with BrdU to identify proliferative cells (BrdU+, in red). Scale bar = 200 μm. SGZ, subgranular zone. GCL, granule cell layer. (#) quantification of proliferating hippocampal cells show that paroxetine administration increased NPC proliferation in gp120 transgenic mice. An analysis of variance showed that the effect of paroxetine was significant: F[3, 37] = 9.92, p < 0.0001). Post hoc analyses were done using Tukey’s multiple comparison test. Values represent mean ± SEM (n =5 ~ 6 per group, ***p < 0.001). (e). Mice were administered daily injections of BrdU (50 mg/kg) for seven days, beginning 2 weeks after pump implantation, and analyzed 28 days after the first BrdU injection. Quantification of newly generated (BrdU + NeuN+) neurons by analysis of variance (F[2, 9] = 0.0088) shows the rescue of adult hippocampal neurogenesis in gp120 transgenic mice by paroxetine. Values represent mean ± SEM (n =5 ~ 6 per group, *p <0.05, one-way ANOVA followed by Tukey’s multiple comparison test)
To further test the effect of paroxetine on NPC proliferation in gp120 transgenic mice, adult mice were euthanized 2 h after BrdU injection to label proliferating cells. We found that a 40 % reduction of BrdU+cells in the dentate gyrus of gp120 transgenic mice as compared to their littermate wt mice, suggesting that expression of gp120 inhibits proliferation of adult hippocampal NPCs. Subcutaneous administration of paroxetine for 28 days was sufficient to completely rescue the deficit in NPC proliferation in the dentate gyrus of gp120 transgenic mice, increasing NPC proliferation by over 100 % to reach levels similar to wild type animals. However, a 20 day period of paroxetine treatment failed to rescue the deficit in NPC proliferation observed in gp120 transgenic mice (Fig. 3c and d).
We next asked whether rescue of the NPC population resulted in rescue of the generation of new neurons in gp120 transgenic mice. We observed that paroxetine administration for 28 days in gp120 transgenic animals led to a significant increase in BrdU+ NeuN+ cells in the dentate gyrus, to levels comparable to wild type mice (Fig. 3e). Thus, paroxetine increased both proliferating NPCs and generation of new neurons in gp120 transgenic mice.
Anti-inflammatory Effect of Paroxetine and Fluoxetine
Next, we determined the effects of the SSRIs on Tat-mediated induction of iNOS. Pretreatment with SSRIs, fluoxetine or paroxetine, followed by Tat attenuated Tat-induced iNOS expression in mixed rat hippocampal cultures. iNOS expression in the cultures was markedly reduced by treatment of fluoxetine or paroxetine in dose dependent manner. The effect of paroxetine on iNOS expression was more pronounced than that of fluoxetine, revealing complete block of iNOS expression with 10 μM fluoxetine and 5 μM paroxetine treatments (Fig. 4). Thus both SSRIs exhibited anti-nitrosative stress effects.
Fig. 4.

SSRIs attenuate Tat-induced iNOS expression in rat hippocampal neurons. Rat mixed hippocampal neurons were pretreated with fluoxetine or paroxetine at different concentrations (1, 5 and 10 μM) for 4 hr, and then exposed to 500 nM Tat for 18 hr at 37 °C. Cell lysates were then resolved by SDS-PAGE, electroblotted and then probed with anti-NOS or anti-GAPDH. Densitometric analysis of the immunoblots was quantitated from 4 separate determinations. Analysis of variance revealed significant effects of fluoxetine and paroxetine on Tat-mediated iNOS expression (F[8, 26] = 2.797, p = 0.0223. Values represent the mean + SEM (n = 4 per group), *p < 0.05, one-way ANOVA followed by Tukey’s multiple comparison test
Tat has been shown to induce inflammatory pathways in neurons and glial cells [28–31] Consistent with these findings exposure of mixed rat hippocampal cultures to Tat for 24 h resulted in significant production of numerous cytokines (Table 2) including IL-1α, IL-1β, IL-5, IL-6, IL-17, MCP-1, MIP-1α and TNF-α proteins Pretreatment of these cultures with 1 μM paroxetine prior to Tat exposure resulted in significant attenuation of the Tat dependent IL-1α, IL-1β, IL-5, IL-6 and TNF-α proteins. Paroxetine did not modulate the Tat-dependent elevation in IL-17, and MCP-1, and produced about a 20 % decrease in Tat stimulated MIP-1α and VEGF levels. Therefore paroxetine displayed significant anti-inflammatory activity in these mixed rat hippocampal cultures.
Table 2.
Tat mediated cytokine and chemokine production in rat mixed hippocampal cultures
| Cytokine | Basal | Tat | Tat +1 uM Paroxetine |
|---|---|---|---|
| IL-1α | 11.4 (2.05) | 271.0 (11.31)*** | 193.5 (11.84)*** |
| IL-1β | 3.66 (1.93) | 813.5 (94.6)*** | 402.5 (81.3)* |
| IL-2 | 42.83 (11.17) | 34.05 (9.51) | 56.65 (28.47) |
| IL-4 | 9.56 (3.35) | 10.62 (2.79) | 8.20 (4.63) |
| IL-5 | 1.935 (0.34) | 73.4 (4.60)* | 51.75 (3.23)* |
| IL-6 | 15.73 (5.11) | 8935 (1515)** | 4611 (760.5)* |
| IL-9 | 1545 (157.4) | 1850 (131.0) | 1862.5 (317.8) |
| IL-10 | 31.8 (14.872) | 2160 (806.7)* | 1991.5 (766.5) |
| Il-17 | 3.025 (1.361) | 7.345 (1.694)* | 7.635 (0.853) |
| IL-18 | 105.6 (33.34) | 211.78 (75.74) | 214.18 (87.9) |
| G-CSF | 10.35 (0.35) | 11.75 (0.389) | 12.05 (0.15) |
| GMCSF | 28.75 (3.134) | 38.375 (6.427) | 28.65 (2.218) |
| MCP-1 | 96.9 (44.30) | 1590 (261.2)* | 1546 (392.9) |
| MIP-1α | 56.1 (6.44) | 192 (18.50)* | 160.3 (24.55) |
| TNF-α | 6.29 (0.91) | 430.7 (61.7)*** | 239.5 (34.9)* |
| VEGF | 951.8 (131.4) | 2567.5 (246.0)** | 1949.3 (472.7) |
Cytokine protein levels are expressed in pg/mL. In the Tat treatment column, asterisks represent statistical significance compared to basal treatment conditions. In the Tat + Paroxetine column, asterisks represent statistical significance versus Tat treatment. *p<0.05, **p<0.01, ***p<0.001
The Effects of Paroxetine and Fluoxetine on Ca2+-induced Mitochondrial Swelling
One of the important factors inducing mitochondrial dysfunction is Ca2+ overload, which primarily occurs during neuroexcitotoxicity [32]. Mitochondria were isolated from rat liver and brain. The addition of 10 μM Ca2+ to the mitochondria initiated a rapid decrease in absorbance, indicating mitochondrial swelling both in the liver and brain. Ca2+-induced mitochondrial swelling was inhibited by 1 μM CsA, which is a specific mitochondrial permeability transition (mPT) blocker (Fig. 5a and b). In the rat liver, treatment of 10 μM fluoxetine significantly slowed the calcium-induced mitochondrial swelling by about 50 % (Fig. 5a). This Ca2+-dependent swelling of mitochondria in the brain was blocked by paroxetine in a concentration-manner dependent, blocking a 50 % of swelling by 0.5 and 5 μM, and completely blocking by 50 μM. However the Ca2+-induced swelling in the liver mitochondria was not significantly inhibited by paroxetine (Fig. 5b).
Fig. 5.

Paroxetine inhibits Ca2+-induced swelling in the brain mitochondria. Rat liver and brain mitochondria were de-energized by addition of 0.5 mM rotenone, 0.5 mM antimycin and 2 mM ionomycin. Mitochondria were then pretreated with fluoxetine (a) or paroxetine (b) for 10 minutes on ice and mitochondrial swelling was initiated by addition of 10 μM Ca2+. Mitochondrial swelling was monitored by measuring the decrease in optical density (OD) at 520 nm for 8 min post calcium addition. An analysis of variance showed that the effect of paroxetine on brain mitochondria was significant F[5, 13] = 15.37 , p < 0.0001). Post hoc analysis was done using Tukey’s multiple comparison test. Values represent mean ± SEM (3 experiments per condition, *p <0.05; **p < 0.01; ***p < 0.001)
Paroxetine Attenuated the KCl-induced Calcium Responses
We next determined whether paroxetine modulated voltage-sensitive calcium channel activity and calcium responses. KCl-induced calcium responses were measured by a ratiometric technique using fura-2 AM in rat hippocampal neurons. The neurons were preincubated with 10 μM paroxetine for 1 hour before stimulation with 40 mM KCl and then calcium responses were recorded. Depolarization with KCl induced excess cytosolic calcium responses in neurons. Paroxetine significantly inhibited the peak amplitudes of KCl-induced calcium responses by ~70 % (Fig. 6a and b).
Fig. 6.

KCl-induced calcium responses are decreased by paroxetine. (a) Shown is cytosolic calcium imaging. Rat hippocampal neurons were pretreated with 10 μM paroxetine for 1 hour before they were stimulated with 40 mM potassium chloride (KCl) to induce depolarization. KCl-evoked calcium responses in neurons were decreased by pretreatment cultures with paroxetine. (b) The peak amplitudes of depolarization-evoked calcium responses were significantly reduced in neurons pre-incubated with 10 μM paroxetine. Values represent mean ± SD (3–5 experiments per condition, *p <0.05, Student’s t-test)
Neuroprotective Effects of Paroxetine are not Mediated by Serotonin Transporter
To determine whether paroxetine has the neuroprotective effect by acting upon SERT, cortical cultures from wild type or SERT −/− mice were used. The percentage of live cells was equivalent between wild type and SERT−/− cultures treated with Tat. 5 μM paroxetine pretreatment significantly increased cell viability from 80 to over 100 % after Tat treatment in wild-type cultures. Unexpectedly, 1 and 5 μM paroxetine pretreatment significantly increased the viability from 75 to over 100 % after Tat treatment in SERT−/− cultures, suggesting that high-affinity for SERT is not necessary for the neuroprotective effects of paroxetine (Fig. 7a).
Fig. 7.

Neuroprotective effects of paroxetine are not mediated by serotonin transporter. (a) Mouse mixed cortical cultures from both wild type and SERT KO mice were preincubated with paroxetine or 10 μM reseveratrol (R) as indicated for 1 hour prior to 18 exposure to 500 nM Tat. Cell Survival was quantitated with MTT endpoint. An analysis of variance showed that the effect of paroxetine in cultures from both wild type and SERT KO mice was significant F(14,75) = 7.80, p < 0.0001). Post hoc analyses were done using Tukey’s multiple comparison test. Values represent mean ± SD (3–5 experiments per condition, ***p < 0.001). (b) Mixed neuronal cultures were transfected with a pooled siRNA to SERT or Non-specific control siRNA (NsiRNA) using lipofectamine 2000. The next day cultures were exposed to 50 nM Tat and 1 μM Morphine with/without paroxetine (1 μM) for 24 hours. Cell viability was measured by a fluorescence-based Cellquanti-blue assay. An analysis of variance showed that the effect of paroxetine was significant in both siRNA and non-specific control RNA treated cultures F [8, 45] = 45.71, p < 0.0001). Post hoc analyses were done using Tukey’s multiple comparison test. Values represent mean ± SD (3–5 experiments per condition, *p <0.05; **p < 0.01)
To confirm the observation from SERT−/− mice, we employed siRNA-mediated knockdown of the SERT mRNA in the mixed neuronal cultures. SERT mRNA expression was determined 48 hr post transfection by real-time PCR. Nearly a 50 % decrease in SERT receptor transcripts was observed (data not shown). Mixed neuronal cultures were transfected with siRNA to SERT, non-specific siRNA or vehicle only. For these experiments we used a combination of Tat and morphine to cause neurotoxicity since the two are synergistic and induce multiple pathways to cause neuronal injury [33]. The next day cultures were exposed to 50 nM Tat and 1 μM morphine with/without paroxetine (1 μM) for 24 hours. Cell viability was measured by a fluorescence-based Cellquanti-blue assay. In non-transfected control cells and non-specific siRNA-transfected cells, the combination of Tat and morphine caused cell death. 1 μM paroxetine was sufficient to protect neurons from Tat and morphine-induced cell death. Most importantly, siRNA-induced reduction in SERT mRNA failed to protect cells against neurotoxicity by Tat and morphine, while neuroprotective actions of paroxetine were still evident (Fig. 7b). Taken together, these results indicate that the efficacy of paroxetine for neuroprotection is independent of SERT inhibition.
Identification of Paroxetine Interacting Proteins
We next identified proteins which interact with paroxetine to determine cellular targets of paroxetine in rat cortical neurons. Using a paroxetine-Sepharose affinity column, we identified SLC6A4, which is the integral membrane serotonin transporter protein as paroxetine targets in rat cortical neurons . Two mitochondrial proteins, adenine nucleotide translocase (ANT) and the voltage dependent anion channel (VDAC) were identified by LC-MS/MS analysis (Table 3). These proteins were not found in similar apparent size ranges of the SDS-polyacrylamide gels containing the Tris-sepharose fractions. To confirm interaction of ANT and VDAC with paroxetine that was observed by LC-MS/MS analysis, rat brain mitochondria were extracted and loaded onto either the paroxetine-sepharose or Tris-sepharose columns. The anti-ANT immunoreactive protein bands showed a depletion of ANT protein in the paroxetine-sepharose flow through lane and concentration of ANT protein in the paroxetine-sepharose bound fraction. No ANT protein was bound to the Tris-Sepharose control bead column (Fig. 8a). Similarly, there was a partial depletion of VDAC protein from the paroxetine-sepharose column and elution from the paroxetine-sepharose beads. VDAC did not interact with the Tris-sepharose column in these studies (Fig. 8b).
Table 3.
Identified peptide sequences of paroxetine binding proteins
| Adenine nucleotide translocase (ANT) protein |
| AAYFGVYDTAK |
| DFLAGGVAAAISK |
| YFPTQALNFAFK |
| Voltage dependent anion channel (VDAC) |
| TKSENGLEFTSSGSANTETTK |
| LETAVNLAWTAGNSNTRFGIAAK |
Fig. 8.

Paroxetine interacts with ANT and VDAC of mitochondria. Rat brain mitochondria were extracted and loaded onto either the paroxetine-sepharose or Tris-sepharose columns. (a) The anti-ANT immunoreactive protein bands showed a depletion of ANT protein in the paroxetine-sepharose flow through lane and concentration of ANT protein in the paroxetine-sepharose bound fraction. (b) Similarly, there was a partial depletion of VDAC protein from the paroxetine-sepharose column and elution from the paroxetine-sepharose beads
No Effect of Paroxetine on HIV Replication
To function as an adjunctive neuroprotective therapy for HIV associated neurocognitive disorders, the effects of paroxetine on HIV replication need to be understood. TZM-bl cells expressing CD4 and CCR5 were treated with paroxetine prior to infection with HIV and paroxetine was maintained in the culture supernatant throughout the experiment. Untreated cells were readily infected by HIV, and exposure to paroxetine up to 10 μM concentrations had no effect on HIV LTR transactivation (Fig. 9).
Fig. 9.

Paroxetine doesn’t have effect on HIV replication in TZM-bl cells. TZM-bl cells expressing CD4 and CCR5 were preincubated with 0.1, 1 or 10 μM paroxetine for 1 hour prior to infection with HIV-1 JRCSF. Infection continued for 48 hours, and the levels of HIV β-galactosidase were quantified by β-galactosidase ELISA. Values represent mean ± SEM (3 experiments per condition)
Discussion
In this study, we provide evidence that paroxetine and fluoxetine reduced neuronal cell death induced by 3NP, NMDA, H2O2 and HIV-1 proteins in vitro and in vivo. We also showed that paroxetine and fluoxetine prevented Tat-induced iNOS and inflammatory cytokine expression, and Ca2+-induced mitochondrial swelling, and attenuated the KCl-induced calcium responses. In addition, paroxetine and fluoxetine had NPC proliferative and neuroregenerative effects. The neuroprotective effect was independent of its effects on the serotonergic transporter. The effects were mediated by direct binding to the mPT pore. Activation of the ANT and VDAC in the form of the mPT pore can be triggered by oxidative and inflammatory mediators, so it may be that these mechanisms may be linked to these proteins. It is possible that aberent calcium signaling may also be linked to the mitochondrial MPT. The proproliferative effects of paroxetine on neural progenitor cells seem less likely to be driven by paroxetine–mitochondrial protein interactions, but may involve serotonin or other pathways.
SSRIs, were developed to selectively target the serotonin reuptake pump, a property shared with the tricyclic antidepressants (TCAs), but without affecting the other neurotransmitter systems such as norepinephrine, acetylcholine, histamine or dopamine, or fast acting sodium channels. SSRIs have a broader safety profile and larger therapeutic index than TCAs. The four SSRIs, fluoxetine, paroxetine, sertraline and citalopram, inhibit serotonin reuptake with IC50 of 7 nM, 0.3 nM, 0.2 nM and 1.8 nM respectively [34]. Three of these drugs possess active metabolites with significant inhibitory effects on the serotonin reuptake pump, with paroxetine as the SSRI which is apparently active as the parent compound only [34, 35].
SSRIs have been used to reduce depression symptom severity in HIV-infected populations. A number of clinical studies demonstrated efficacy and safety of SSRIs as an antidepressant in adult HIV-infected patients [36–38]. We demonstrated that paroxetine could impact proliferation of NPCs in an in vitro model of human fetal cells. Further, paroxetine increased both proliferating NPCs and generation of new neurons in a transgenic mouse model of HIV neurologic disease. These studies confirm our previous observations of paroxetine on NPC [12]. Several studies have shown that antidepressants increase hippocampal neurogenesis in both animals and humans [39, 40] and hippocampal neurogenesis has been suggested to be a key factor in the action of antidepressant drugs [41, 42]. Suppression of hippocampal neurogenesis by irradiation prevents the effects of fluoxetine and imipramine [41].
We provide evidence that paroxetine and fluoxetine reduced neuronal cell death induced by 3NP, NMDA, H2O2 and HIV-1 proteins in vitro and in vivo. Neuronal cell death is seen in the brains of patients with HAND. HIV-1 proteins, Tat and gp120 are known to trigger neuronal apoptosis and excitotoxicity as a result of oxidative stress, perturbed cellular calcium homeostasis and mitochondrial alterations [23, 26]. Our data suggests that the neuroprotective properties of paroxetine and fluoxetine are seen at concentrations of 0.5 to10 μM. Similar concentrations are also needed to enhance proliferation of neural progenitor cells. These concentrations can be easily achieved in the brain at therapeutic dosages since these compounds are preferentially taken up and enriched in brain tissue with 10–20 fold higher concentrations in the human brain compared to plasma [43]. Steady state levels are achieved over several weeks to months [44]. These studies also show that the SSRI are evenly distributed throughout the brain. This is also supported by our in vivo studies in which paroxetine showed neuroprotection and proliferation of neural progenitor cells following systemic administration. Therefore, treatment with SSRIs can be an effective neurorestorative approach for ameliorating neuronal death as well as defective neurogenesis in HIV-infected individuals with neurologic dysfunction.
Overproduction of nitric oxide through increased iNOS production has been implicated in the pathogenesis of many disorders including HAND. iNOS is increased in the brain of individuals with HIV encephalitis [45, 46] and in the cerebrospinal fluid of individuals with progressive neurocognitive dysfunction [47]. HIV-infected macrophages release proinflammatory factors that induce iNOS in astrocytes (Hori et al., 1999; Hu et al., 1999). Tat increased iNOS induction to produce NO via activation of NF-κB and C/EBPβ in human astroglial cells [48]. The NO produced by this mechanism may initiate apoptotic events in neighboring neurons. HIV-1 Tat inducing iNOS may participate in the pathogenesis of HAND. We found that both paroxetine and fluoxetine inhibited Tat-induced iNOS, and the effect was more pronounced in paroxetine than fluoxetine.
We could envision the paroxetine treatment to be given concurrently with the combination antiretroviral therapy as an adjunctive neuroprotective therapy for neural cells at risk to exposure to HIV in the central nervous system. Even in the era of effective combined antiretroviral therapy, HIV patients continue to display signs of chronic immune activation and persistent chronic inflammation [49] In our experiments in cultures containing neurons, astrocytes and microlia, we found that paroxetine treatment attenuated the production of many of the inflammatory cytokines induced by Tat. However these effects were seen at higher concentrations compared to the neuroprotective effects .
NO plays an important role in regulating several aspects of mitochondrial function including control of respiration and mitochondrial biogenesis [50]. Many studies have suggested a direct interaction of the NO derived from iNOS in contributing to mitochondrial dysfunction [51, 52]. We determined if SSRIs had a direct effect on mitochondrial function. We found that paroxetine was able to potently inhibit Ca2+-induced mitochondrial swelling. These effects were specific for brain mitochondria as no significant effect was seen in the liver mitochondria.
The mechanisms of the neural protective effects of paroxetine have not been well-characterized. Because the most likely target may be the SERT itself, which is the key high affinity receptor protein for the SSRIs, we determined whether paroxetine has the neuroprotective effect by acting upon SERT. We found that neuroprotective effects of paroxetine were independent of SERT inhibition. The neuroprotective efficacy is provided by both SSRIs paroxetine and fluoxetine, with paroxetine demonstrating better potency. In neurons lacking the SERT, paroxetine maintains its neuroprotective efficacy, perhaps acting via the mitochondria proteins ANT and/or VDAC. While it is true that both compounds block the serotonin transporter, they both also have some effects on blocking calcium dependent swelling of the mitochondria. At concentrations that elicit the neuroprotective actions of paroxetine (100 nM), the SERT will also be bound to and occupied by this SSRI.
SSRIs also have significant interactions with other target proteins. Fluoxetine is known to interact with the volume regulated anion channel (VRAC) [53, 54], voltage dependent potassium channels (Kv1.1, 1.3, 1.4 and 1.5) [55–58], inward rectifying potassium channels [59] and HERG channels [60]. In particular, it has been shown that fluoxetine interacted with the VDAC of mitochondria and prevented mitochondria-mediated cell death [61, 62]. Under conditions of oxidative stress and elevated cytoplasmic calcium, the VDAC from the outer mitochondrial membrane can interact selectively with the inner mitochondrial membrane protein ANT and forms the mitochondrial mPT pore [63]. Activation of this pore is an early step in mitochondrial dysfunction, in which nonselective transport of small solutes (<2000 Daltons) into the mitochondria, abolishes the mitochondria membrane potential, inhibits the electron transport system and depletes ATP, eventually resulting in cell death. Mitochondrial swelling occurs when the inner mitochondrial membrane swells in response to ion and small molecule flux across the inner mitochondrial membrane as the mitochondrial membrane potential is abolished and mPT pore is activated [64]. In the study, we showed paroxetine bound to mitochondrial membrane proteins, ANT and VDAC. This can elucidate the mechanism of neuroprotective effect of paroxetine, with blocking calcium-dependent mitochondrial swelling.
Electronic Supplementary Material
(PDF 507 kb)
Acknowledgments
We thank Dr. L. Mucke for kindly providing the gp120 transgenic mice and John Pontolillo, Tanya Malpic-Llanos and Daniella Asch for technical assistance. We thank Dr. Carlos Pardo-Villamizar and Jackie Mann for their expert technical assistance with the Luminex multiplex analysis of cytokines and chemokines. We also thank Drs. Robert Cole and Raghothama Chaerkady of the Johns Hopkins Mass Spectrometry and Proteomics Facility for their identification of paroxetine binding proteins. These studies were supported by NIH grants P30MH075673, R01DA024593 and NIH intramural funds
Conflict of Interest
The authors declare no competing financial interest
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Contributor Information
Joseph P. Steiner, Phone: 301-594-2950, Email: steinerjp@ninds.nih.gov
Avindra Nath, Phone: 301-496-1561, Email: natha@ninds.nih.gov.
References
- 1.Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol. 2011;17(1):3–16. doi: 10.1007/s13365-010-0006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McArthur JC, Steiner J, Sacktor N, Nath A. Human immunodeficiency virus-associated neurocognitive disorders: Mind the gap. Annals of neurology. 2010;67(6):699–714. doi: 10.1002/ana.22053. [DOI] [PubMed] [Google Scholar]
- 3.Badiee J, Moore DJ, Atkinson JH, Vaida F, Gerard M, Duarte NA, et al. Lifetime suicidal ideation and attempt are common among HIV + individuals. J Affect Disord. 2012;136(3):993–999. doi: 10.1016/j.jad.2011.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eller LS, Bunch EH, Wantland DJ, Portillo CJ, Reynolds NR, Nokes KM, et al. Prevalence, correlates, and self-management of HIV-related depressive symptoms. AIDS Care. 2010;22(9):1159–1170. doi: 10.1080/09540121.2010.498860. [DOI] [PubMed] [Google Scholar]
- 5.Ciesla JA, Roberts JE. Meta-analysis of the relationship between HIV infection and risk for depressive disorders. Am J Psychiatry. 2001;158(5):725–730. doi: 10.1176/appi.ajp.158.5.725. [DOI] [PubMed] [Google Scholar]
- 6.Levine AJ, Singer EJ, Sinsheimer JS, Hinkin CH, Papp J, Dandekar S, et al. CCL3 genotype and current depression increase risk of HIV-associated dementia. Neurobehav HIV Med. 2009;1:1–7. doi: 10.2147/nbhiv.s6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wolkowitz OM, Mellon SH, Epel ES, Lin J, Dhabhar FS, Su Y, et al. Leukocyte telomere length in major depression: correlations with chronicity, inflammation and oxidative stress--preliminary findings. PLoS One. 2011;6(3):e17837. doi: 10.1371/journal.pone.0017837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schifitto G, Navia BA, Yiannoutsos CT, Marra CM, Chang L, Ernst T, et al. Memantine and HIV-associated cognitive impairment: a neuropsychological and proton magnetic resonance spectroscopy study. Aids. 2007;21(14):1877–1886. doi: 10.1097/QAD.0b013e32813384e8. [DOI] [PubMed] [Google Scholar]
- 9.Turchan J, Sacktor N, Wojna V, Conant K, Nath A. Neuroprotective therapy for HIV dementia. Curr HIV Res. 2003;1(4):373–383. doi: 10.2174/1570162033485113. [DOI] [PubMed] [Google Scholar]
- 10.Kaul M. HIV's double strike at the brain: neuronal toxicity and compromised neurogenesis. Front Biosci. 2008;13:2484–2494. doi: 10.2741/2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwartz L, Civitello L, Dunn-Pirio A, Ryschkewitsch S, Berry E, Cavert W, et al. Evidence of human immunodeficiency virus type 1 infection of nestin-positive neural progenitors in archival pediatric brain tissue. J Neurovirol. 2007;13(3):274–283. doi: 10.1080/13550280701344975. [DOI] [PubMed] [Google Scholar]
- 12.Lee MH, Wang T, Jang MH, Steiner J, Haughey N, Ming GL, et al. Rescue of adult hippocampal neurogenesis in a mouse model of HIV neurologic disease. Neurobiology of disease. 2011;41(3):678–687. doi: 10.1016/j.nbd.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mishra M, Taneja M, Malik S, Khalique H, Seth P. Human immunodeficiency virus type 1 Tat modulates proliferation and differentiation of human neural precursor cells: implication in NeuroAIDS. J Neurovirol. 2010;16(5):355–367. doi: 10.3109/13550284.2010.513028. [DOI] [PubMed] [Google Scholar]
- 14.Okamoto S, Kang YJ, Brechtel CW, Siviglia E, Russo R, Clemente A, et al. HIV/gp120 decreases adult neural progenitor cell proliferation via checkpoint kinase-mediated cell-cycle withdrawal and G1 arrest. Cell Stem Cell. 2007;1(2):230–236. doi: 10.1016/j.stem.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 15.Hollman AM, Christian DA, Ray PD, Galey D, Turchan J, Nath A, et al. Selective isolation and purification of tat protein via affinity membrane separation. Biotechnology progress. 2005;21(2):451–459. doi: 10.1021/bp049804z. [DOI] [PubMed] [Google Scholar]
- 16.Wang T, Lee MH, Choi E, Pardo-Villamizar CA, Lee SB, Yang IH, et al. Granzyme B-induced neurotoxicity is mediated via activation of PAR-1 receptor and Kv1.3 channel. PLoS One. 2012;7(8):e43950. doi: 10.1371/journal.pone.0043950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. Journal of immunological methods. 1983;65(1–2):55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 18.Louboutin JP, Agrawal L, Reyes BA, Van Bockstaele EJ, Strayer DS. A rat model of human immunodeficiency virus 1 encephalopathy using envelope glycoprotein gp120 expression delivered by SV40 vectors. J Neuropathol Exp Neurol. 2009;68(5):456–473. doi: 10.1097/NEN.0b013e3181a10f83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toggas SM, Masliah E, Rockenstein EM, Rall GF, Abraham CR, Mucke L. Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature. 1994;367(6459):188–193. doi: 10.1038/367188a0. [DOI] [PubMed] [Google Scholar]
- 20.Lie DC, Colamarino SA, Song HJ, Desire L, Mira H, Consiglio A, et al. Wnt signalling regulates adult hippocampal neurogenesis. Nature. 2005;437(7063):1370–1375. doi: 10.1038/nature04108. [DOI] [PubMed] [Google Scholar]
- 21.Friberg H, Connern C, Halestrap AP, Wieloch T. Differences in the activation of the mitochondrial permeability transition among brain regions in the rat correlate with selective vulnerability. J Neurochem. 1999;72(6):2488–2497. doi: 10.1046/j.1471-4159.1999.0722488.x. [DOI] [PubMed] [Google Scholar]
- 22.Haughey NJ, Mattson MP. Calcium dysregulation and neuronal apoptosis by the HIV-1 proteins Tat and gp120. J Acquir Immune Defic Syndr. 2002;31(Suppl 2):S55–S61. doi: 10.1097/00126334-200210012-00005. [DOI] [PubMed] [Google Scholar]
- 23.Mattson MP, Haughey NJ, Nath A. Cell death in HIV dementia. Cell Death Differ. 2005;12(Suppl 1):893–904. doi: 10.1038/sj.cdd.4401577. [DOI] [PubMed] [Google Scholar]
- 24.Sacktor N, Haughey N, Cutler R, Tamara A, Turchan J, Pardo C, et al. Novel markers of oxidative stress in actively progressive HIV dementia. J Neuroimmunol. 2004;157(1–2):176–184. doi: 10.1016/j.jneuroim.2004.08.037. [DOI] [PubMed] [Google Scholar]
- 25.Nath S, Bachani M, Harshavardhana D, Steiner JP. Catechins protect neurons against mitochondrial toxins and HIV proteins via activation of the BDNF pathway. Journal of neurovirology. 2012;18(6):445–455. doi: 10.1007/s13365-012-0122-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li W, Galey D, Mattson MP, Nath A. Molecular and cellular mechanisms of neuronal cell death in HIV dementia. Neurotox Res. 2005;8(1–2):119–134. doi: 10.1007/BF03033824. [DOI] [PubMed] [Google Scholar]
- 27.Nath A, Haughey NJ, Jones M, Anderson C, Bell JE, Geiger JD. Synergistic neurotoxicity by human immunodeficiency virus proteins Tat and gp120: protection by memantine. Ann Neurol. 2000;47(2):186–194. doi: 10.1002/1531-8249(200002)47:2<186::AID-ANA8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 28.Conant K, Garzino-Demo A, Nath A, McArthur JC, Halliday W, Power C, et al. Induction of monocyte chemoattractant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(6):3117–3121. doi: 10.1073/pnas.95.6.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sawaya BE, Thatikunta P, Denisova L, Brady J, Khalili K, Amini S. Regulation of TNFalpha and TGFbeta-1 gene transcription by HIV-1 Tat in CNS cells. Journal of neuroimmunology. 1998;87(1–2):33–42. doi: 10.1016/S0165-5728(98)00044-7. [DOI] [PubMed] [Google Scholar]
- 30.McManus CM, Weidenheim K, Woodman SE, Nunez J, Hesselgesser J, Nath A, et al. Chemokine and chemokine-receptor expression in human glial elements: induction by the HIV protein, Tat, and chemokine autoregulation. The American journal of pathology. 2000;156(4):1441–1453. doi: 10.1016/S0002-9440(10)65013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pu H, Tian J, Flora G, Lee YW, Nath A, Hennig B, et al. HIV-1 Tat protein upregulates inflammatory mediators and induces monocyte invasion into the brain. Molecular and cellular neurosciences. 2003;24(1):224–237. doi: 10.1016/S1044-7431(03)00171-4. [DOI] [PubMed] [Google Scholar]
- 32.Nicholls DG. Mitochondrial calcium function and dysfunction in the central nervous system. Biochim Biophys Acta. 2009;1787(11):1416–1424. doi: 10.1016/j.bbabio.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fitting S, Xu R, Bull C, Buch SK, El-Hage N, Nath A, et al. Interactive comorbidity between opioid drug abuse and HIV-1 Tat: chronic exposure augments spine loss and sublethal dendritic pathology in striatal neurons. The American journal of pathology. 2010;177(3):1397–1410. doi: 10.2353/ajpath.2010.090945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hyttel J. Pharmacological characterization of selective serotonin reuptake inhibitors (SSRIs) Int Clin Psychopharmacol. 1994;9(Suppl 1):19–26. doi: 10.1097/00004850-199403001-00004. [DOI] [PubMed] [Google Scholar]
- 35.Preskorn SH. Dose-effect and concentration-effect relationships with new antidepressants. Psychopharmacol Ser. 1993;10:174–189. doi: 10.1007/978-3-642-78010-3_17. [DOI] [PubMed] [Google Scholar]
- 36.Zisook S, Peterkin J, Goggin KJ, Sledge P, Atkinson JH, Grant I. Treatment of major depression in HIV-seropositive men. HIV Neurobehavioral Research Center Group. J Clin Psychiatry. 1998;59(5):217–224. doi: 10.4088/JCP.v59n0502. [DOI] [PubMed] [Google Scholar]
- 37.Rabkin JG, Wagner GJ, Rabkin R. Fluoxetine treatment for depression in patients with HIV and AIDS: a randomized, placebo-controlled trial. Am J Psychiatry. 1999;156(1):101–107. doi: 10.1176/ajp.156.1.101. [DOI] [PubMed] [Google Scholar]
- 38.Schwartz JA, McDaniel JS. Double-blind comparison of fluoxetine and desipramine in the treatment of depressed women with advanced HIV disease: a pilot study. Depress Anxiety. 1999;9(2):70–74. doi: 10.1002/(SICI)1520-6394(1999)9:2<70::AID-DA4>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 39.Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci. 2000;20(24):9104–9110. doi: 10.1523/JNEUROSCI.20-24-09104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boldrini M, Underwood MD, Hen R, Rosoklija GB, Dwork AJ, John Mann J, et al. Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology. 2009;34(11):2376–2389. doi: 10.1038/npp.2009.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 2003;301(5634):805–809. doi: 10.1126/science.1083328. [DOI] [PubMed] [Google Scholar]
- 42.Surget A, Saxe M, Leman S, Ibarguen-Vargas Y, Chalon S, Griebel G, et al. Drug-dependent requirement of hippocampal neurogenesis in a model of depression and of antidepressant reversal. Biol Psychiatry. 2008;64(4):293–301. doi: 10.1016/j.biopsych.2008.02.022. [DOI] [PubMed] [Google Scholar]
- 43.Bolo NR, Hode Y, Nedelec JF, Laine E, Wagner G, Macher JP. Brain pharmacokinetics and tissue distribution in vivo of fluvoxamine and fluoxetine by fluorine magnetic resonance spectroscopy. Neuropsychopharmacology. 2000;23(4):428–438. doi: 10.1016/S0893-133X(00)00116-0. [DOI] [PubMed] [Google Scholar]
- 44.Henry ME, Schmidt ME, Hennen J, Villafuerte RA, Butman ML, Tran P, et al. A comparison of brain and serum pharmacokinetics of R-fluoxetine and racemic fluoxetine: A 19-F MRS study. Neuropsychopharmacology. 2005;30(8):1576–1583. doi: 10.1038/sj.npp.1300749. [DOI] [PubMed] [Google Scholar]
- 45.Adamson DC, Wildemann B, Sasaki M, Glass JD, McArthur JC, Christov VI, et al. Immunologic NO synthase: elevation in severe AIDS dementia and induction by HIV-1 gp41. Science. 1996;274(5294):1917–1921. doi: 10.1126/science.274.5294.1917. [DOI] [PubMed] [Google Scholar]
- 46.Zhao ML, Kim MO, Morgello S, Lee SC. Expression of inducible nitric oxide synthase, interleukin-1 and caspase-1 in HIV-1 encephalitis. J Neuroimmunol. 2001;115(1–2):182–191. doi: 10.1016/S0165-5728(00)00463-X. [DOI] [PubMed] [Google Scholar]
- 47.Li W, Malpica-Llanos TM, Gundry R, Cotter RJ, Sacktor N, McArthur J, et al. Nitrosative stress with HIV dementia causes decreased L-prostaglandin D synthase activity. Neurology. 2008;70(19 Pt 2):1753–1762. doi: 10.1212/01.wnl.0000282761.19578.35. [DOI] [PubMed] [Google Scholar]
- 48.Liu X, Jana M, Dasgupta S, Koka S, He J, Wood C, et al. Human immunodeficiency virus type 1 (HIV-1) tat induces nitric-oxide synthase in human astroglia. J Biol Chem. 2002;277(42):39312–39319. doi: 10.1074/jbc.M205107200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eden A, Price RW, Spudich S, Fuchs D, Hagberg L, Gisslen M. Immune activation of the central nervous system is still present after >4 years of effective highly active antiretroviral therapy. The Journal of infectious diseases. 2007;196(12):1779–1783. doi: 10.1086/523648. [DOI] [PubMed] [Google Scholar]
- 50.Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299(5608):896–899. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]
- 51.Chen K, Northington FJ, Martin LJ. Inducible nitric oxide synthase is present in motor neuron mitochondria and Schwann cells and contributes to disease mechanisms in ALS mice. Brain Struct Funct. 2010;214(2–3):219–234. doi: 10.1007/s00429-009-0226-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Venkatraman A, Shiva S, Wigley A, Ulasova E, Chhieng D, Bailey SM, et al. The role of iNOS in alcohol-dependent hepatotoxicity and mitochondrial dysfunction in mice. Hepatology. 2004;40(3):565–573. doi: 10.1002/hep.20326. [DOI] [PubMed] [Google Scholar]
- 53.Maertens C, Droogmans G, Verbesselt R, Nilius B. Block of volume-regulated anion channels by selective serotonin reuptake inhibitors. Naunyn Schmiedebergs Arch Pharmacol. 2002;366(2):158–165. doi: 10.1007/s00210-002-0567-5. [DOI] [PubMed] [Google Scholar]
- 54.Hisadome K, Koyama T, Kimura C, Droogmans G, Ito Y, Oike M. Volume-regulated anion channels serve as an auto/paracrine nucleotide release pathway in aortic endothelial cells. J Gen Physiol. 2002;119(6):511–520. doi: 10.1085/jgp.20028540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yeung SY, Millar JA, Mathie A. Inhibition of neuronal KV potassium currents by the antidepressant drug, fluoxetine. Br J Pharmacol. 1999;128(7):1609–1615. doi: 10.1038/sj.bjp.0702955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Choi JS, Hahn SJ, Rhie DJ, Yoon SH, Jo YH, Kim MS. Mechanism of fluoxetine block of cloned voltage-activated potassium channel Kv1.3. J Pharmacol Exp Ther. 1999;291(1):1–6. [PubMed] [Google Scholar]
- 57.Choi BH, Choi JS, Ahn HS, Kim MJ, Rhie DJ, Yoon SH, et al. Fluoxetine blocks cloned neuronal A-type K + channels Kv1.4. Neuroreport. 2003;14(18):2451–2455. doi: 10.1097/00001756-200312190-00032. [DOI] [PubMed] [Google Scholar]
- 58.Perchenet L, Hilfiger L, Mizrahi J, Clement-Chomienne O. Effects of anorexinogen agents on cloned voltage-gated K(+) channel hKv1.5. J Pharmacol Exp Ther. 2001;298(3):1108–1119. [PubMed] [Google Scholar]
- 59.Kobayashi T, Washiyama K, Ikeda K. Inhibition of G protein-activated inwardly rectifying K + channels by fluoxetine (Prozac) Br J Pharmacol. 2003;138(6):1119–1128. doi: 10.1038/sj.bjp.0705172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thomas D, Gut B, Wendt-Nordahl G, Kiehn J. The antidepressant drug fluoxetine is an inhibitor of human ether-a-go-go-related gene (HERG) potassium channels. J Pharmacol Exp Ther. 2002;300(2):543–548. doi: 10.1124/jpet.300.2.543. [DOI] [PubMed] [Google Scholar]
- 61.Nahon E, Israelson A, Abu-Hamad S, Varda SB. Fluoxetine (Prozac) interaction with the mitochondrial voltage-dependent anion channel and protection against apoptotic cell death. FEBS Lett. 2005;579(22):5105–5110. doi: 10.1016/j.febslet.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 62.Thinnes FP. Does fluoxetine (Prozak) block mitochondrial permeability transition by blocking VDAC as part of permeability transition pores? Mol Genet Metab. 2005;84(4):378. doi: 10.1016/j.ymgme.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 63.Halestrap AP, Brenner C. The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr Med Chem. 2003;10(16):1507–1525. doi: 10.2174/0929867033457278. [DOI] [PubMed] [Google Scholar]
- 64.Friberg H, Ferrand-Drake M, Bengtsson F, Halestrap AP, Wieloch T. Cyclosporin A, but not FK 506, protects mitochondria and neurons against hypoglycemic damage and implicates the mitochondrial permeability transition in cell death. J Neurosci. 1998;18(14):5151–9. Epub 1998/07/03. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 507 kb)