

Abstract
Specific therapies for neurologic diseases such as Alzheimer’s disease provide the potential for better clinical outcomes. Expression of caspases in the brain is developmentally regulated, and dysregulated in neurologic disease, supporting that caspases may be therapeutic targets. The activity of caspases is carefully regulated via binding partners, cleavage, or endogenous inhibitors to prevent spontaneous activation, which could lead to aberrant cell death. This review serves as a brief examination of the current understanding of the regulation and function of caspases, and approaches to specifically target aberrant caspase activity. The use of proper tools to investigate individual caspases is addressed. Moreover, it summarizes the reports of various caspases in Alzheimer’s disease studies. A better understanding of specific caspase pathways in heath and neurodegenerative disease is crucial for identifying specific targets for the development of therapeutic interventions.
Electronic supplementary material
The online version of this article (doi:10.1007/s13311-014-0307-9) contains supplementary material, which is available to authorized users.
Keywords: Caspase, Inhibitor of apoptosis protein, Alzheimer’s disease, Cerebral ischemia, Stroke
Over 100 years ago Alois Alzheimer identified the clinical and pathologic hallmarks of a dementing illness that came to be known as Alzheimer’s disease (AD) [1]. AD is characterized clinically by progressive loss of cognition and pathologically by the accumulation of amyloid plaques, neurofibrillary tangles, synaptic loss, and neuronal death [2]. It is estimated that patients have already lost as much as 50 % of their neurons at the time of their first clinical symptoms [3], supporting the relevance of neuronal death in the disease. In the century since Alzheimer’s studies, β-amyloid (Aβ) and tau have been identified as the protein components of plaques and tangles, respectively, and genetic studies of familial AD identified mutations in genes regulating the production of Aβ, supporting a critical role for Aβ in the disease [4]. Yet, despite much work by many laboratories, there is still no treatment for AD. What has been lacking is a deeper understanding of the molecular events that lead to the development of AD.
Current therapeutic options for AD are limited. A variety of approaches has been tried but have yet to yield promising results. By understanding the molecular pathways leading to neuronal dysfunction therapeutic targets can be identified for intervention. As AD is a chronic disease, it is probable that different pathways may dominate at different points of disease progression.
By focusing on a signaling pathway that is activated in the brains of patients with AD but not in brains of unaffected controls we can begin to identify therapeutically relevant targets. As noted above, the major hallmarks of AD are loss of neuronal function and loss of neurons. Over the years, the potential contribution of the caspase family of proteases to these processes has been studied. Here we review the evidence for specific caspase pathway activation in AD and discuss the therapeutic targeting of these pathways.
Caspases
Caspases are a conserved family of metazoan proteases best studied for their roles in cell death and inflammation. In mammals, there are 13 caspases, numbered 1–12 and 14 (number 13 was an erroneously identified bovine caspase that was likely an ortholog of human caspase-4) [5, 6]. In 1992 interleukin (IL)-1β cleaving enzyme (ICE), now known as caspase-1, was identified [7, 8], and within a few months ced-3 was identified in Caenorhabditis elegans as a key molecule in the execution of apoptosis, and noted to have significant homology to ICE [9]. In the ensuing years more homologs and orthologs of ICE were identified in mammals and insects [6]. As the number of mammalian homologs increased the term “caspase” for cysteine-dependent, aspartate-specific protease was agreed upon for the mammalian family, with numbering to indicate the order of identification.
The mammalian caspases can be classified structurally into 2 groups—those with long prodomains (caspases 1, 2, 4, 5, 8, 9, 10, 11, and 12) and those with short prodomains (caspases 3, 6, 7, and 14). Caspase-14, found to be critical for keratinocyte differentiation, does not appear to have a function in the nervous system [10]. The others with short prodomains (caspases 3, 6, and 7) are also termed effectors of apoptosis. The long prodomain caspases are further subdivided into initiators of apoptosis (caspases 2, 8, 9, and10) and inflammatory caspases (caspases 1, 4, 5, 11, and 12), which process cytokines. Caspase-2 may also function as both an initiator and an effector, similar to ced-3 in C. elegans; ontogenically, caspase-2 is most closely related to ced-3 [6]. The inflammatory caspases can also participate in cell death by autocrine activity where caspase-1 cleaves pro-IL-1β to its active form, IL-1β which is secreted and then binds to the IL-1 receptor to initiate death signaling in the cell [11].
Caspase Activation and Regulation
Caspases are inactive zymogens that require activation (see [5, 12, 13] for detailed discussions of activation and regulation). Most of the caspases exist as monomers in the zymogen form, caspase-2 zymogen is a dimer. The structural classification above is also implicated in the mode of activation of the 2 groups of caspases. Those with short prodomains, the effectors, require cleavage at an aspartate residue in the intersubunit linker to be activated. The cleavage site is a caspase cleavage site and both caspases 8 and 9 have been shown to cleave and activate effector caspases. The activation of initiator caspases is more complex. The initial activation step for long prodomain caspases is proximity induced dimerization. The dimerization is effected through the binding of adaptor proteins to the prodomain caspase activation and recruitment domain (CARD; caspases 1, 2, 4, 5, 9, 11 and 12) or death-effector domain (caspases 8 and 10) motifs. There are specific adaptor proteins for each long prodomain caspase. After dimerization there is a conformation change that induces activity; no cleavage is required for this step—the longer intersubunit linker allows flexibility to form an active site without cleavage. This is followed by autocleavage which can enhance activation, as shown for caspase-8, or provide a new binding site through which the caspase can be inhibited, as shown for caspase-9, discussed below. If a long prodomain caspase undergoes cleavage without the initial dimerization change the caspase will not be active.
The long prodomain caspases have unique activation platforms. Best studied are the intrinsic (caspase-9) and extrinsic (caspase-8) platforms. The intrinsic pathway centers on mitochondrial regulation of caspase-9 activation. With certain death stimuli, such as DNA damage, mitochondria are permeabilized and release cytochrome c, which leads to an adenosine triphosphate-dependent recruitment of caspase-9 by apoptotic protease activating factor 1, the caspase-9 death adaptor, in a structure termed the apoptosome. In the apoptosome caspase-9 undergoes proximity dimerization-induced activation, which is followed by limited autocleavage to p35 and p12 fragments, which can be inhibited by X-linked inhibitor of apoptosis protein (XIAP). As cleavage is not required for caspase-9 activation, and autocleavage produces a fragment of caspase-9, which is inhibited by XIAP, autocleavage results in decreased activity of caspase-9. Once caspase-9 is activated by dimerization it can activate caspase-3, which, in turn, can cleave active caspase-9 to p37 and p10 fragments, which cannot be inhibited by XIAP. So, caspase-3 can potentiate caspase-9 activation [14].
The extrinsic death pathway is initiated by ligand binding to death receptors, a multimembered family of receptors. The death inducing signaling complex is the caspase-8 activation platform. Binding of the ligand to the cell surface death receptor leads to recruitment of the adaptor protein, Fas-associated protein with death domain, which recruits and dimerizes caspase-8. Dimerized caspase-8 is active but activity is further enhanced by autocleavage. As with caspase-9, cleavage without dimerization by an effector caspase or granyme B does not produce activation of caspase-8.
The activation platform for caspase-2 has been proposed to be the PIDDosome, a complex of p53-inducible protein with a death domain (PIDD), RAIDD {RIP [receptor-interacting protein]-associated ICH-1 [ICE (IL-1β-converting enzyme)/CED-3 homologue 1] protein with a DD} and caspase-2. However, 2 different lines of PIDD null mice appear to undergo caspase-2 dependent death [15–17]. Specifically in neurons, PIDD is not required for caspase-2 activation or for caspase-2 dependent death, while RAIDD is required [15]. Caspase-2 activity is also regulated by phosphorylation of the prodomain, which prevents activation [18, 19].
The inflammosome is the activation platform for caspase-1 [20]. There are several different inflammasomes, the ICE-protease activating factor, and the NACHT-, LRR-, and pyrin-domain containing proteins (NRLP) 1, 2, and 3 inflammasomes, which are expressed differentially in various cell types in the nervous system [21–23]. The assembly involves the proteins ICE-protease activating factor or NRLP, which contain pyrin domains, caspase-1, which contains a CARD domain, and an adaptor protein with a CARD domain, such as apoptosis-associated speck-like protein containing a CARD, or CARD inhibitor of nuclear factor kappa B-activating ligands. The inflammasomes are activated by signaling through Toll-like receptors or in the cytosol by the NOD-like receptor family in response to stress signals.
The Inhibitor of Apoptosis Protein Family of Endogenous Caspase Inhibitors
The inhibitor of apoptosis protein (IAP) family is classified by the presence of baculoviral inhibitor repeat (BIR) domains [24]. There are 8 mammalian IAPs, 3 of which have been shown to bind directly caspases, cIAP1, cIAP2, and XIAP. Of these, only XIAP has been shown to inhibit caspase activity; the other IAPs can inhibit cell death but not by directly inhibiting caspase activity [25]. Several IAPs are E3 ligases and target proteins for proteosomal degradation; others inhibit binding of the IAP inhibitor second mitochondria-derived activator of caspases (SMAC) to XIAP [26]. XIAP binds to and inhibits active caspases 3 and 7 via the BIR2-linker domain; inhibition of active caspase-9 is by the BIR3 domain. cIAP1 and cIAP2 bind to active caspases 3 and 7 via the BIR2-linker domain but do not exert inhibition; the BIR3 domain of cIAP1 or cIAP2 does not bind to caspase-9 [27]. Thus, XIAP is the only IAP to provide modulation of activity of these caspases. SMAC is an endogenous antagonist of XIAP, which displaces the caspase from XIAP leading to an increase in caspase activity, providing another level of regulation, whereby the relative abundance of XIAP or SMAC determines whether apoptosis proceeds [28]. Of the other caspases, only caspase-8 has an endogenous regulator, FLICE-like inhibitory protein (FLIP), which can dimerize with caspase-8 and block activation of caspase-8. But there are multiple forms of FLIP, which increase the complexity of FLIP function with regard to caspase-8 activation [29].
Measurement of Caspase Activation/Activity
In order to ascribe a function for a specific caspase in a death paradigm, evidence of caspase activation and activity must be obtained. A casual perusal of the literature would suggest that these measures would be easy to obtain. Multiple companies market caspase substrates that are identified as specific for individual caspases. Unfortunately, owing to the high homology of caspases, particularly at the active sites, the substrates, which are small peptides, do not have enough structure to distinguish between different caspases. An elegant study showed that these substrates are not specific (nor are the caspase inhibitors) and cannot be used as an indicator of activation of a specific caspase [30].
Another tool frequently used to assess caspase activation is the generation of caspase cleavage products and neoepitopes, using either Western blotting or immunoctyochemistry. While this can be used as indicative of effector caspase activation, as cleavage is a required step, it cannot be used to indicate initiator caspase activation as cleavage is not required for activation. And for the effector caspases 3 and 7, while cleavage indicates activation, it does not indicate activity as XIAP could be bound to the active caspase preventing activity [13].
There are methods that can be used to detect an active caspase. For identifying the initial caspase active in a death pathway the broad spectrum caspase affinity ligand biotin-Val-Ala-Asp(OMe)-fluoromethylketone (VADfmk) is very useful. To detect initiator caspases the ligand is delivered prior to the death insult; preloading the cells allows the ligand to interact with the first caspase activated. The ligand binds irreversibly to the active site of the first active caspase it encounters, and also inhibits any downstream events. The ligand–caspase complex is isolated with streptavidin, which binds to the biotin and then analyzed by Western blotting with specific antibodies for the different caspases. In support of proximity induced dimerization being the first step in activation of initiator caspases the isolated active initiator caspases are full length [31]. With this technique our group has identified active caspases in primary neurons [15], in mouse and rat brains in vivo and in rat retina in vivo [32, 33]. Other methods are specific fluorescent read-outs of caspase activity. For caspase-2 such a system has been used in cultured cell lines to show dimerization and activation of caspase-2 after heat shock [34].
After identifying the active caspases it is critical to determine functional relevance. As with substrates, there is a series of caspase inhibitors available commercially and sold as specific for individual caspases. As with the substrates, the inhibitors are not specific and cannot provide information about individual caspases. Currently, molecular approaches, such as knockdown or knockout, are the best indication of functional relevance. In neurons, small interfering RNA and antisense approaches have been used to knockdown successfully individual caspases. Caspase-null mice have proved informative for the function of some caspases in development, such as the critical role for caspase-9 in development of the forebrain [35]. Use of the caspase-null animals has elucidated functions of caspase-6 [32]. Other caspase-null animals have exhibited compensatory changes in other caspases in the central nervous system, obscuring the functional role of the targeted caspase, as found in the caspase-2-null mice [36]. Inducible knockouts are beginning to be developed and will provide more specific data.
Targets of Caspases
Caspases can potentially cleave many different proteins. Cleavage of an individual protein is limited, usually to no more than a few cleavage sites. This is perhaps best illustrated by the cleavage of effector caspases by initiator caspases, which leads to activation of the effectors. Cleavage can also change the function of the substrate, such as converting a proapoptotic protein into an antiapoptotic protein, or vice versa. These cleavage events can lead to amplification of a signaling pathways leading to death or survival. But despite much effort the target substrates of each caspase have not been determined. Predictions of substrates have been made, but proof will require analysis of the time course of substrate cleavage, combined with specific block of the cleavage events and further determination of the relevance of such events for death signaling.
Nonapoptotic Functions of Caspases in the Brain
Evidence continues to accumulate that caspases may have nonapoptotic functions in the brain [37]. Compelling data support a role for caspase-3 in neurogenesis and in synaptic activity. Caspase-3 activity has been shown to promote neurogenesis in neuronal progenitors. Inhibition of caspase activity also blocked neurite extension, suggesting a function in of caspases in morphologic changes associated with neurogenesis.
Selective pruning of synapses, axons, and dendrites regulates neuronal circuits. The Drosphila initiator caspase Dronc is required for pruning [38]. Caspases have also been linked to neuronal plasticity. In zebra finch, caspase-3 activity is required for the birds to learn a new song [39]. In the zebra finch, a complex of XIAP and caspase-3 was detected in the synapse, suggesting that XIAP modulates caspase-3 activity, allowing synaptic remodeling but not death. In rats, Asp(OMe)-Glu(OMe)-Val-Asp(OMe)-fluoromethylketone (DEVDfmk) inhibition of caspase activity support a function of caspases in long-term potentiation and in active avoidance learning [40]. Caspase-3 activity is also required for long-term depression [41]. These nonapoptotic functions of caspase-3 indicate why it is not appropriate to equate the presence of cleaved caspase-3 with apoptosis. It has been suggested that caspase-3 acts as a bifurcation point between plasticity and death [42].
Caspases in AD
What is the evidence for caspases playing a critical role in AD? In postmortem AD brains there is an increase in mRNA expression of multiple caspases, including caspases 1, 2, 3, 5, 6, 7, 8, and 9, compared with control brains [43]. At the protein level, there is increased expression of caspase-2 [44], cleaved caspase-3 [45], cleaved caspase-6 [46], and caspase-9; caspase-9 has been isolated in synaptosomal fractions from AD brains [47]. Negative correlations have been found for caspase-8 levels and the age of disease onset and age of patient death, suggesting a role for caspase-8 in disease regulation [48]. Recent studies have also found association of NRLP3, an activator of caspase-1, with AD [49, 50]. Overall, there are much correlative data supporting changes in caspases in AD but sparse specific data for individual caspases in the disease. Data are summarized in Table 1. One issue has been the lack of adequate animal models for the study of AD. Animal models currently available do not replicate the human disease fully but can be useful for studies of preclinical AD [13]. However, combining human, animal model, and primary neuronal studies it may be possible to begin to assess a function of specific caspase pathways. Here we will focus on the data for caspase-2 in AD, as an illustration of how this can be approached.
Table 1.
Caspases implicated in Alzheimer’s Disease
| Caspase | Paradigm | Measure | Citation |
|---|---|---|---|
| 1 | AD brain Aβ treated microglia |
mRNA Casp1 cleavage In vitro Tau cleavage |
[43, 51–53] |
| 2 | AD brain Aβ treated hippocampal neurons Aβ production from H4-C99 cells |
mRNA , protein Casp2 null neurons siRNA depletion Casp2 cleavage z-VDVAD-fmk |
[43, 44, 51, 54, 55] |
| 3 | AD brain Aβ production from H4-C99 cells Tg4519 mice |
mRNA z-DEVD-fmk In vitro Tau cleavage cleaved Casp3 IHC Casp3 cleavage |
[43, 45, 51, 53, 54, 56–58] |
| 4 | Aβ treatment of SK-N-SH cells | Casp4 cleavage siRNA depletion |
[59, 60] |
| 5 | AD brain | mRNA | [43, 51] |
| 6 | AD brain Tg4510 mice |
mRNA In vitro Tau cleavage Casp6 cleavage |
[43, 46, 51, 53, 55] |
| 7 | AD brain Tg4510 mice |
mRNA In vitro Tau cleavage |
[43, 51, 53, 56] |
| 8 | AD brain Aβ production from H4-C99 cells |
mRNA siRNA depletion z-IETD-fmk In vitro Tau cleavage cleaved Casp8 IHC |
[43, 48, 51, 53, 54, 61, 62] |
| 9 | AD brain | mRNA cleaved Casp9 IHC |
[51, 62, 63] |
| 12 | Aβ treated cortical neurons Hippocampal organotypic slices treated with Aβ |
Antisense to Casp12 Casp12 null neurons Casp12 cleavage |
[63, 64] |
Ab = antibody; siRNA = small interfering RNA; IHC = immunohistochemistry
Caspase-2 and AD
The function of caspase-2 has been somewhat elusive. It is the caspase most related to ced-3, the C. elegans caspase, and like ced-3 can execute cell death without effector caspases [6]. In primary neuronal cultures treated with oligomeric Aβ1-42 caspase-2 is activated within 30 min and regulates transcriptional events leading to death, including induction of phospho-c-Jun and Bim [44]. This, surprisingly, places caspase-2 at a proximal place in the neurodegeneration pathway. In vivo, injection of oligomeric Aβ1-42 into the hippocampus induces an increase in caspase-2 and Bim in the same neurons, co-expression of caspase-2 and Bim is also found in neurons in AD brains [44]. Additional work has shown that caspase-2 regulates the decrease in spine density found in primary neuronal cultures exposed to oligomeric Aβ1-42 and in the J20 mouse model of AD; and that crossing J20 mice with caspase-2 null mice protects from the behavioral changes found in the J20 mice [65]. Taken together these data suggest that caspase-2 is an excellent target for AD. It is active proximally in the pathway and regulates synapse loss, as well as neuron death. In control brain caspase-2 is undetectable, supporting a degenerative function for caspase-2. The issue is specific targeting of caspase-2.
Specific Targeting of Caspases in Neurologic Disease
Is targeting a specific caspase possible and, if so, effective as a potential therapy for central nervous system disease? Multiple attempts have been made, without success, to develop specific small molecule inhibitors of individual caspases. Here, as an illustration of how a caspase can be targeted specifically, we discuss the development of a protein biologic as a specific inhibitor of caspase-9. For these studies we turned to the study of the cerebral ischemia. Cerebral ischemia is an acute neurologic disease characterized by loss of neuronal processes, neuronal death, and functional disability; the injury develops rapidly, over hours to days, allowing study of the mechanisms leading to injury. Cerebral ischemia is also a risk factor for the development of AD. We utilized rodent models of stroke, transient middle cerebral artery occlusion in both rats and mice, which model the injury in more than 85 % of human strokes [66]. As the literature was diffuse as to which caspases were critical for the pathology in stroke, we employed a nonbiased approach to identify the proximal caspase(s) activated [32]. As noted above, the caspase affinity ligand bVADfmk can be used to capture the first caspase activated. bVADfmk was delivered to the brain prior to stroke, the middle cerebral artery was occluded, followed by reperfusion and harvest at 1-h postreperfusion. Brains were analyzed by streptavidin pullout of bVAD-caspase complexes and Western blotting to identify the proximal caspase. Somewhat surprisingly, active caspase-9 was isolated from the striatum at 1 h postreperfusion; by 24 h postreperfusion the striatum will be the necrotic core. These data suggest that early on in ischemia there is activation of apoptotic pathways in the striatum. When bVADfmk was delivered during reperfusion active caspase-9 was isolated from the dorsal cortex, in line with the spatiotemporal progression of ischemia. Taken together, these data indicated that caspase-9 was a target. In order to target caspase-9 specifically we could either decrease expression with a siRNA approach or target caspase-9 activity. We chose to target activity as that would be the most rapid and there would not be possible confounding effects of decreasing basal expression of caspase-9. Several caspases, including caspase-9, are expressed at detectable levels in the adult brain and their function is unclear. To inhibit caspase-9 activity we utilized the endogenous inhibitor XIAP. As noted above, the Bir3 domain of XIAP is a specific inhibitor of caspase-9 (shown in Fig. 1). But there were still several hurdles to traverse. In order to deliver XBir3 to the site of active caspase-9 we needed to cross the blood–brain barrier and then deliver the compound into the cytosol where active caspase-9 is found. Linkage of XBir3 to a cell-penetrating peptide, Penetratin1, by a disulfide bond, resulted in a compound with the desired properties. Intranasal delivery of Pen1-XBir3 provided significant morphologic and functional neuroprotection to 3 weeks postinfarction [32]. This supports that specific inhibition of the critical caspase can provide therapeutic intervention.
Fig. 1.
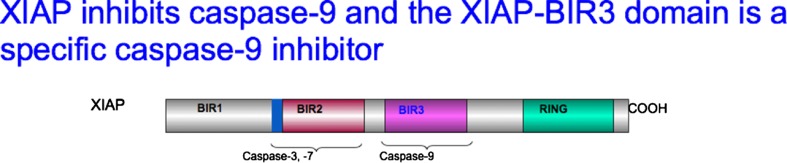
X-linked inhibitor of apoptosis protein (XIAP) inhibits caspase-9 and the XIAP-baculoviral inhibitor repeat (BIR) 3 domain is a specific caspase-9 inhibitor
The issue going forward is to identify clearly the specific caspases in a particular disease. As noted above, identification of caspase targets will require complementary studies of human tissue, animal models, and primary cultures. After the caspase pathway is identified, the next step is to develop specific inhibitors for that caspase pathway. For the intrinsic pathway, biology has provided a specific inhibitor, XIAP, the individual BIR domains of which can be used as inhibitors. For other caspases, more creativity must be employed to design specific inhibitors that are biologically active.
Electronic supplementary material
(PDF 1224 kb)
Acknowledgments
Carol M. Troy was supported in part by National Institutes of Health grant RO1NS081333.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
References
- 1.Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR. An English translation of Alzheimer's 1907 paper, "Uber eine eigenartige Erkankung der Hirnrinde". Clin Anat. 1995;8:429–431. doi: 10.1002/ca.980080612. [DOI] [PubMed] [Google Scholar]
- 2.Small GW, Rabins PV, Barry PP, et al. Diagnosis and treatment of Alzheimer disease and related disorders. Consensus statement of the American Association for Geriatric Psychiatry, the Alzheimer's Association, and the American Geriatrics Society. JAMA. 1997;278:1363–1371. doi: 10.1001/jama.1997.03550160083043. [DOI] [PubMed] [Google Scholar]
- 3.DeKosky ST, Carrillo MC, Phelps C, et al. Revision of the criteria for Alzheimer's disease: A symposium. Alzheimers Dement. 2011;7:e1–e12. doi: 10.1016/j.jalz.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 4.Karch CM, Cruchaga C, Goate AM. Alzheimer's disease genetics: from the bench to the clinic. Neuron. 2014;83:11–26. doi: 10.1016/j.neuron.2014.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mace PD, Riedl SJ, Salvesen GS. Caspase enzymology and activation mechanisms. Methods Enzymol. 2014;544:161–178. doi: 10.1016/B978-0-12-417158-9.00007-8. [DOI] [PubMed] [Google Scholar]
- 6.Lamkanfi M, Declercq W, Kalai M, Saelens X, Vandenabeele P. Alice in caspase land. A phylogenetic analysis of caspases from worm to man. Cell Death Differ. 2002;9:358–361. doi: 10.1038/sj.cdd.4400989. [DOI] [PubMed] [Google Scholar]
- 7.Miura M, Zhu H, Rotello R, Hartwieg EA, Yuan J. Induction of apoptosis in fibroblasts by IL-1 beta-converting enzyme, a mammalian homolog of the C. elegans cell death gene ced-3. Cell. 1993;75:653–660. doi: 10.1016/0092-8674(93)90486-A. [DOI] [PubMed] [Google Scholar]
- 8.Thornberry NA, Bull HG, Calaycay JR, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature. 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- 9.Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- 10.Eckhart L, Ban J, Fischer H, Tschachler E. Caspase-14: analysis of gene structure and mRNA expression during keratinocyte differentiation. Biochem Biophys Res Commun. 2000;277:655–659. doi: 10.1006/bbrc.2000.3698. [DOI] [PubMed] [Google Scholar]
- 11.Troy CM, Stefanis L, Prochiantz A, Greene LA, Shelanski ML. The contrasting roles of ICE family proteases and interleukin-1beta in apoptosis induced by trophic factor withdrawal and by copper/zinc superoxide dismutase down-regulation. Proc Natl Acad Sci U S A. 1996;93:5635–5640. doi: 10.1073/pnas.93.11.5635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pop C, Salvesen GS. Human caspases: activation, specificity, and regulation. J Biol Chem. 2009;284:21777–21781. doi: 10.1074/jbc.R800084200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Troy CM, Akpan N, Jean YY. Regulation of caspases in the nervous system implications for functions in health and disease. Prog Mol Biol Transl Sci. 2011;99:265–305. doi: 10.1016/B978-0-12-385504-6.00007-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Denault JB, Eckelman BP, Shin H, Pop C, Salvesen GS. Caspase 3 attenuates XIAP (X-linked inhibitor of apoptosis protein)-mediated inhibition of caspase 9. Biochem J. 2007;405:11–19. doi: 10.1042/BJ20070288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ribe EM, Jean YY, Goldstein RL, et al. Neuronal caspase 2 activity and function requires RAIDD, but not PIDD. Biochem J. 2012;444:591–599. doi: 10.1042/BJ20111588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim IR, Murakami K, Chen NJ, et al. DNA damage- and stress-induced apoptosis occurs independently of PIDD. Apoptosis. 2009;14:1039–1049. doi: 10.1007/s10495-009-0375-1. [DOI] [PubMed] [Google Scholar]
- 17.Manzl C, Krumschnabel G, Bock F, et al. Caspase-2 activation in the absence of PIDDosome formation. J Cell Biol. 2009;185:291–303. doi: 10.1083/jcb.200811105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nutt LK, Margolis SS, Jensen M, et al. Metabolic regulation of oocyte cell death through the CaMKII-mediated phosphorylation of caspase-2. Cell. 2005;123:89–103. doi: 10.1016/j.cell.2005.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shin S, Lee Y, Kim W, Ko H, Choi H, Kim K. Caspase-2 primes cancer cells for TRAIL-mediated apoptosis by processing procaspase-8. EMBO J. 2005;24:3532–3542. doi: 10.1038/sj.emboj.7600827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petrilli V, Dostert C, Muruve DA, Tschopp J. The inflammasome: a danger sensing complex triggering innate immunity. Curr Opin Immunol. 2007;19:615–622. doi: 10.1016/j.coi.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 21.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 22.Schroder K, Zhou R, Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger? Science. 2010;327:296–300. doi: 10.1126/science.1184003. [DOI] [PubMed] [Google Scholar]
- 23.Tschopp J, Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10:210–215. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 24.Srinivasula SM, Ashwell JD. IAPs: what's in a name? Mol Cell. 2008;30:123–135. doi: 10.1016/j.molcel.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eckelman BP, Salvesen GS, Scott FL. Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep. 2006;7:988–994. doi: 10.1038/sj.embor.7400795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fulda S. Inhibitor of Apoptosis (IAP) proteins in hematological malignancies: molecular mechanisms and therapeutic opportunities. Leukemia. 2014;28:1414–1422. doi: 10.1038/leu.2014.56. [DOI] [PubMed] [Google Scholar]
- 27.Eckelman BP, Salvesen GS. The human anti-apoptotic proteins cIAP1 and cIAP2 bind but do not inhibit caspases. J Biol Chem. 2006;281:3254–3260. doi: 10.1074/jbc.M510863200. [DOI] [PubMed] [Google Scholar]
- 28.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/S0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 29.Subramaniam K, Hirpara JL, Tucker-Kellogg L, Tucker-Kellogg G, Pervaiz S. FLIP: a flop for execution signals. Cancer Lett. 2013;332:151–155. doi: 10.1016/j.canlet.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 30.McStay GP, Salvesen GS, Green DR. Overlapping cleavage motif selectivity of caspases: implications for analysis of apoptotic pathways. Cell Death Differ. 2008;15:322–331. doi: 10.1038/sj.cdd.4402260. [DOI] [PubMed] [Google Scholar]
- 31.Tu S, McStay GP, Boucher LM, Mak T, Beere HM, Green DR. In situ trapping of activated initiator caspases reveals a role for caspase-2 in heat shock-induced apoptosis. Nat Cell Biol. 2006;8:72–77. doi: 10.1038/ncb1340. [DOI] [PubMed] [Google Scholar]
- 32.Akpan N, Serrano-Saiz E, Zacharia BE, et al. Intranasal delivery of caspase-9 inhibitor reduces caspase-6-dependent axon/neuron loss and improves neurological function after stroke. J Neuroscience. 2011;31:8894–8904. doi: 10.1523/JNEUROSCI.0698-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vigneswara V, Akpan N, Berry M, Logan A, Troy CM, Ahmed Z. Combined suppression of CASP2 and CASP6 protects retinal ganglion cells from apoptosis and promotes axon regeneration through CNTF-mediated JAK/STAT signalling. Brain. 2014;137:1656–1675. doi: 10.1093/brain/awu037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bouchier-Hayes L, Oberst A, McStay GP, et al. Characterization of cytoplasmic caspase-2 activation by induced proximity. Mol Cell. 2009;35:830–840. doi: 10.1016/j.molcel.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuida K, Haydar TF, Kuan CY, et al. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/S0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- 36.Troy CM, Rabacchi SA, Hohl JB, Angelastro JM, Greene LA, Shelanski ML. Death in the balance: alternative participation of the caspase-2 and -9 pathways in neuronal death induced by nerve growth factor deprivation. J Neurosci. 2001;21:5007–5016. doi: 10.1523/JNEUROSCI.21-14-05007.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.D'Amelio M, Sheng M, Cecconi F. Caspase-3 in the central nervous system: beyond apoptosis. Trends Neurosci. 2012;35:700–709. doi: 10.1016/j.tins.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 38.Williams DW, Kondo S, Krzyzanowska A, Hiromi Y, Truman JW. Local caspase activity directs engulfment of dendrites during pruning. Nat Neurosci. 2006;9:1234–1236. doi: 10.1038/nn1774. [DOI] [PubMed] [Google Scholar]
- 39.Huesmann GR, Clayton DF. Dynamic role of postsynaptic caspase-3 and BIRC4 in zebra finch song-response habituation. Neuron. 2006;52:1061–1072. doi: 10.1016/j.neuron.2006.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gulyaeva NV, Kudryashov IE, Kudryashova IV. Caspase activity is essential for long-term potentiation. J Neurosci Res. 2003;73:853–864. doi: 10.1002/jnr.10730. [DOI] [PubMed] [Google Scholar]
- 41.Li Z, Jo J, Jia JM, et al. Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell. 2010;141:859–871. doi: 10.1016/j.cell.2010.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Snigdha S, Smith ED, Prieto GA, Cotman CW. Caspase-3 activation as a bifurcation point between plasticity and cell death. Neurosci Bull. 2012;28:14–24. doi: 10.1007/s12264-012-1057-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pompl PN, Yemul S, Xiang Z, et al. Caspase gene expression in the brain as a function of the clinical progression of Alzheimer disease. Arch Neurol. 2003;60:369–376. doi: 10.1001/archneur.60.3.369. [DOI] [PubMed] [Google Scholar]
- 44.Jean YY, Ribe EM, Pero ME, et al. Caspase-2 is essential for c-Jun transcriptional activation and Bim induction in neuron death. Biochem J. 2013;455:15–25. doi: 10.1042/BJ20130556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stadelmann C, Deckwerth TL, Srinivasan A, et al. Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer's disease. Evidence for apoptotic cell death. Am J Pathol. 1999;155:1459–1466. doi: 10.1016/S0002-9440(10)65460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.LeBlanc A, Liu H, Goodyer C, Bergeron C, Hammond J. Caspase-6 role in apoptosis of human neurons, amyloidogenesis, and Alzheimer's disease. J Biol Chem. 1999;274:23426–23436. doi: 10.1074/jbc.274.33.23426. [DOI] [PubMed] [Google Scholar]
- 47.Lu DC, Rabizadeh S, Chandra S, et al. A second cytotoxic proteolytic peptide derived from amyloid beta- protein precursor [see comments] Nat Med. 2000;6:397–404. doi: 10.1038/74656. [DOI] [PubMed] [Google Scholar]
- 48.Matsui T, Ramasamy K, Ingelsson M, et al. Coordinated expression of caspase 8, 3 and 7 mRNA in temporal cortex of Alzheimer disease: relationship to formic acid extractable abeta42 levels. J Neuropathol Exp Neurol. 2006;65:508–515. doi: 10.1097/01.jnen.0000229238.05748.12. [DOI] [PubMed] [Google Scholar]
- 49.Tan MS, Yu JT, Jiang T, Zhu XC, Tan L. The NLRP3 inflammasome in Alzheimer's disease. Mol Neurobiol. 2013;48:875–882. doi: 10.1007/s12035-013-8475-x. [DOI] [PubMed] [Google Scholar]
- 50.Tan MS, Yu JT, Jiang T, et al. NLRP3 polymorphisms are associated with late-onset Alzheimer's disease in Han Chinese. J Neuroimmunol. 2013;265:91–95. doi: 10.1016/j.jneuroim.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 51.Cotman CW, Anderson AJ. A potential role for apoptosis in neurodegeneration and Alzheimer's disease. Mol Neurobiol. 1995;10:19–45. doi: 10.1007/BF02740836. [DOI] [PubMed] [Google Scholar]
- 52.Halle A, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gamblin TC, et al. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc Natl Acad Sci U S A. 2003;100:10032–10037. doi: 10.1073/pnas.1630428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chae SS, et al. Caspases-2 and −8 are involved in the presenilin1/gamma-secretase-dependent cleavage of amyloid precursor protein after the induction of apoptosis. J Neurosci Res 2010;88:1926-1933. [DOI] [PubMed]
- 55.Troy CM, et al. Caspase-2 mediates neuronal cell death induced by beta-amyloid. J Neurosci. 2000;20:1386–1392. doi: 10.1523/JNEUROSCI.20-04-01386.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de Calignon A, et al. Caspase activation precedes and leads to tangles. Nature 2010;464:1201-1204. [DOI] [PMC free article] [PubMed]
- 57.Su JH, Zhao M, Anderson AJ, Srinivasan A, Cotman CW. Activated caspase-3 expression in Alzheimer's and aged control brain: correlation with Alzheimer pathology. Brain Res. 2001;898:350–357. doi: 10.1016/S0006-8993(01)02018-2. [DOI] [PubMed] [Google Scholar]
- 58.Louneva N, et al. Caspase-3 is enriched in postsynaptic densities and increased in Alzheimer's disease. Am J Pathol. 2008;173:1488–1495. doi: 10.2353/ajpath.2008.080434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matsuzaki S, Hiratsuka T, Kuwahara R, Katayama T, Tohyama M. Caspase-4 is partially cleaved by calpain via the impairment of Ca2+ homeostasis under the ER stress. Neurochem Int 2010;56;352-356. [DOI] [PubMed]
- 60.Hitomi J, et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J Cell Biol. 2004;165:347–356. doi: 10.1083/jcb.200310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rohn TT, Head E, Nesse WH, Cotman CW, Cribbs DH. Activation of caspase-8 in the Alzheimer's disease brain. Neurobiol Dis. 2001;8:1006–1016. doi: 10.1006/nbdi.2001.0449. [DOI] [PubMed] [Google Scholar]
- 62.Rohn TT, et al. Caspase-9 activation and caspase cleavage of tau in the Alzheimer's disease brain. Neurobiol Dis. 2002;11:341–354. doi: 10.1006/nbdi.2002.0549. [DOI] [PubMed] [Google Scholar]
- 63.Nakagawa T, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 64.Ishige K, et al. Role of caspase-12 in amyloid beta-peptide-induced toxicity in organotypic hippocampal slices cultured for long periods. J Pharmacol Sci. 2007;104:46–55. doi: 10.1254/jphs.FP0061533. [DOI] [PubMed] [Google Scholar]
- 65.Pozueta J, Lefort R, Ribe EM, Troy CM, Arancio O, Shelanski M. Caspase-2 is required for dendritic spine and behavioural alterations in J20 APP transgenic mice. Nat Commun 2013;4:1939. [DOI] [PMC free article] [PubMed]
- 66.Liu F, McCullough LD. Middle cerebral artery occlusion model in rodents: methods and potential pitfalls. J Biomed Biotechnol 2011;2011:464701. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 1224 kb)