

Abstract
Many neurodegenerative disorders are characterized by the aberrant accumulation of aggregate-prone proteins. Alzheimer’s disease (AD) is associated with the buildup of β-amyloid peptides and tau, which aggregate into extracellular plaques and neurofibrillary tangles, respectively. Multiple studies have linked dysfunctional intracellular degradation mechanisms with AD pathogenesis. One such pathway is the autophagy–lysosomal system, which involves the delivery of large protein aggregates/inclusions and organelles to lysosomes through the formation, trafficking, and degradation of double-membrane structures known as autophagosomes. Converging data suggest that promoting autophagic degradation, either by inducing autophagosome formation or enhancing lysosomal digestion, provides viable therapeutic strategies. In this review, we discuss compounds that can augment autophagic clearance and may ameliorate disease-related pathology in cell and mouse models of AD. Canonical autophagy induction is associated with multiple signaling cascades; on the one hand, the best characterized is mammalian target of rapamycin (mTOR). Accordingly, multiple mTOR-dependent and mTOR-independent drugs that stimulate autophagy have been tested in preclinical models. On the other hand, there is a growing list of drugs that can enhance the later stages of autophagic flux by stabilizing microtubule-mediated trafficking, promoting lysosomal fusion, or bolstering lysosomal enzyme function. Although altering the different stages of autophagy provides many potential targets for AD therapeutic interventions, it is important to consider how autophagy drugs might also disturb the delicate balance between autophagosome formation and lysosomal degradation.
Electronic supplementary material
The online version of this article (doi:10.1007/s13311-014-0320-z) contains supplementary material, which is available to authorized users.
Keywords: Autophagy, Alzheimer’s disease, Neurodegeneration, Therapeutics, Tau, Amyloid (Aβ), Flux, Lysosomes
Introduction
Alzheimer’s disease (AD), like many neurodegenerative diseases, is largely characterized by the aberrant accumulation of endogenous proteins, resulting in the formation of cytotoxic aggregates and inclusions. Multiple lines of evidence have shown that intracellular degradation mechanisms can be activated to remove pathological forms of these proteins, and thus serve as viable druggable targets. One such pathway is the ubiquitin-proteasome system, which breaks down short-lived and soluble proteins. While the ubiquitin-proteasome system degrades disease-linked proteins associated with neurodegenerative disease [1, 2], the narrow pore of the proteasomal barrel may impede clearance of larger protein oligomers and aggregates [3]. In contrast, the autophagy–lysosome system degrades long-lived and large protein complexes and organelles through a multistep process that requires a fine balance between initial induction and end-stage degradation. Dysfunction at either end of the pathway has been linked to AD pathogenesis [4, 5], thus providing multiple targets for pharmacological intervention. This review focuses on the different stages of autophagic clearance as targets for AD therapeutics, which are also relevant to other proteinopathies associated with neurodegenerative disorders.
Autophagic Process and Machinery
Autophagy is a highly conserved pathway that delivers intracellular cytoplasmic components to lysosomes. Three distinct forms have been identified based on their mode of delivery to lysosomes: macroautophagy, chaperone-mediated autophagy, and microautophagy [6–9]. Macroautophagy (herein referred to as autophagy) involves the formation, trafficking, maturation, and subsequent degradation of double-membrane structures known as autophagosomes. The cumulative process is referred to as autophagic flux and is essential for neuronal and brain health. Disruption of any of these highly regulated steps will result in incomplete autophagic digestion and impaired autophagic flux, and represents distinct targets for drug development. Following induction, an isolation membrane (also known as a phagophore) elongates to engulf damaged organelles and proteins, and encloses to form an autophagosome. Autophagosomes can fuse directly with lysosomes to form autolysosomes, or, alternatively, with late endosomes/multivesicular bodies to form amphisomes. These acidified structures fuse with lysosomes, where their cargo and inner membrane are digested by lysosomal hydrolases [10] (Fig. 1). To date, 36 autophagy-related genes that are essential for autophagosome biogenesis have been identified in yeast [11], and many have known mammalian orthologs [12, 13].
Fig. 1.
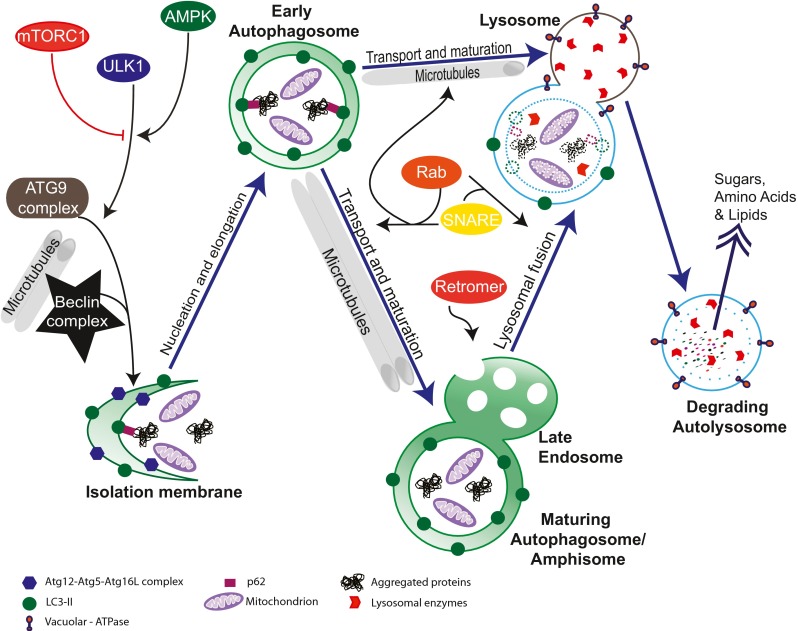
Key molecular players in autophagic flux. Autophagy induction is mediated by mammalian target of rapamycin (mTOR) complex (mTORC)1, adenosine monophosphate-activated protein kinase (AMPK), and Unc-51 like kinase 1 (ULK1), which interact with autophagy-related protein (ATG)9 and Beclin 1 complexes during nucleation of the isolation membrane. Isolation membrane elongation is dependent upon the Atg12–Atg5•Atg16L complex and microtubule-associated protein 1A/1B-light chain 3 (LC3)-II. The scaffolding protein p62 recruits autophagic protein substrates to LC3-bound autophagosomal membranes. The structure then encloses to form an autophagosome. During maturation, autophagosomes can directly fuse with lysosomes or with endosomes and multivesicular bodies to form amphisomes, which is regulated by Rab, solube N-ethylmaleamide-sensitive factor attachment protein receptor (SNARE), and retromer proteins. Autophagic cargo (including organelles and protein aggregates) and the inner membranes of autophagosomes are digested by lysosomal enzymes during lysosomal fusion, which is mediated by the SNARE and Rab proteins. The acidic environment of lysosomes is maintained by vacuolar-ATPase
Regulation of Autophagy
Mammalian target of rapamycin (mTOR) is a ubiquitously expressed protein kinase at the center of a complex signaling network that regulates mRNA translation [14], ribosome biogenesis [15], metabolism [16], and autophagy [17]. mTOR is part of 2 core complexes: the mTOR complex 1 (mTORC1), which includes regulatory-associated protein of mTOR, and the mTOR complex 2 (mTORC2) [18]. mTORC1 plays a major role in autophagy regulation through interactions with serine/threonine kinase autophagy-related protein (Atg)1 or its mammalian ortholog, Unc-51 like kinase 1 (ULK1). ULK1 forms a protein complex with Atg13 and FAK-family interacting protein of 200 kDa (FIP200). Under basal conditions, mTORC1 binds directly to the ULK1 complex to suppress its activity [19–21]. However, amino acid deprivation or pharmacological inhibition of mTOR initiates ULK1–FIP200–Atg13 dissociation [19, 22], and enhances its interaction with the energy sensing kinase adenosine monophosphate-activated protein kinase (AMPK) [23, 24]. AMPK, in turn, activates ULK1 [24], and also directly inhibits mTOR through phosphorylation of tuberous sclerosis complex 2 and regulatory-associated protein of mTOR [25–27]. Therefore, both mTOR inhibitors and AMPK activators can be used to regulate positively ULK1 [28–31].
Nucleation
Until recently, little was known about how the ULK1–FIP200–Atg13 complex was involved in the initiation of autophagosome formation. Beclin 1, the mammalian ortholog of Atg6, is essential for isolation membrane nucleation. It forms a core complex with Atg14L, (activating molecule in Beclin 1-regulated autophagy (AMBRA1), p150, and hVps34/class III phosphatidylinositol 3-kinase (PI3K), which is tethered to the cytoskeleton through the association between AMBRA1 and dynein motor complex. ULK1 phosphorylation of AMBRA1, releases the Beclin 1/PI3K complex so that it can be translocated to the endoplasmic reticulum (ER), which is thought to be the site of autophagosome formation [32–34] (Fig. 1). Atg14L directs the Beclin 1/PI3K complex to autophagosomes by identifying curved membrane structures with a high content of phosphatidylinositol 3-phosphate and is also involved in the maintenance of membrane curvature [35]. While Beclin 1 interaction with UVRAG induces autophagy [36], RUN domain and cysteine-rich domain containing Beclin 1-interacting protein binds with Beclin 1 to inhibit autophagy [37].
The ULK1–FIP200–Atg13 complex plays an essential role in recruiting membranes to phagophore assembly sites in yeast or preautophagosomal structures in mammals. In yeast, Atg1 mediates the trafficking of Atg9 [38], which is an integral membrane protein critical for autophagosome formation that cycles between the phagophore assembly sites and cytosol. In mammals, Atg9 cycles between the trans-Golgi network and late endosomes [38], and requires Atg2 and tryptophan–aspartic acid (WD)-repeat protein interacting with phosphoinositides, as well as the Beclin 1/PI3K complex, for its normal function [39]. Tryptophan–aspartic acid (WD)-repeat protein interacting with phosphoinositides and Atg2 bind to phosphatidylinositol 3-phosphate on phagophores and autophagosomal membranes [40], and are thought to mediate the conversion of omegasomes (phosphatidylinositol 3-phosphate-enriched ER membrane structures) to autophagosomes [41].
Elongation, Closure, and Cargo Recruitment
Autophagosomal membrane elongation involves two ubiquitin-like conjugation reactions. Atg12 and Atg8 are both conjugated by E1-like activating enzyme, Atg7, but are processed by 2 different E2-like conjugating enzymes (Atg10 and Atg3, respectively). Atg12 is conjugated with Atg5 [42], and noncovalently binds with Atg16L to form a complex that is localized to elongating isolation membranes [43, 44]. Small GTPase protein, Rab5 facilitates Atg12–Atg5 conjugation [45, 46].
Yeast Atg8 has several mammalian orthologs, including microtubule-associated protein 1A/1B-light chain 3 (LC3), which is cleaved at the C-terminal and retrieved from nonautophagosomal membranes by Atg4 to form cytosolic LC3-I [47]. LC3-I conjugation to phosphatidylethanolamine is facilitated by the unique protein-lipid E3-like activity of the Atg12–Atg5•Atg16L complex to form lipidated LC3-II [48–51]. Lipidated LC3-II association with isolation membrane is required for elongation and closure of autophagosomal membrane, and remains bound to the autophagosome until it fuses with lysosomes [52], whereas the Atg12–Atg5•Atg16L complex dissociates upon autophagosomal closure [53] (Fig. 1).
Sequestosome1/p62 is a multifunctional scaffolding protein commonly found in ubiquitinated inclusion bodies and rapidly accumulates when autophagy is suppressed [54]. p62 has multiple protein–protein interaction motifs and binds both polyubiquitinated proteins and LC3 to recruit protein cargo to autophagosomes [55, 56]. As aggregated forms are degraded by autophagy [57], p62 also serves a useful marker for autophagic flux. Additionally, p62 interacts with polyubiquitinated forms of AD-associated protein tau and accumulates in other related tauopathies [58, 59].
Maturation of Autophagic Vacuoles
Maturation of autophagosomes involves fusion with early/late endosomes or with multivesicular bodies [7, 60–65]. Amongst the key players responsible for these fusion events are microtubules [66–70], soluble N-ethylmaleamide-sensitive factor attachment protein receptor (SNARE) proteins [71, 72], and ultraviolet radiation resistance-associated gene protein [73]. Additionally, the small GTPase Rab family of proteins, especially Rab7, has been implicated in maturation and fusion of amphisomes with lysosomes [74, 75]. Rab11 has been shown to promote late endosome–autophagosome fusion by interacting with Hook protein [64], which normally anchors late endosomes tightly to microtubules [67]. In addition to these, other Rab proteins have been implicated in autophagy [76, 77]. Rab protein inactivation by specific GTPase-activating proteins may provide a potential drug target that could increase Rab function, thereby enhancing autophagic maturation.
Retromer is a conserved protein sorting and trafficking complex that contains an assembly of proteins that associate with endosomes and regulate protein transport from endosomes to the trans-Golgi network and to cell membrane [78–80]. A study in Caenorhabditis elegans indicated a role for a Beclin 1 ortholog in retromer function [81], suggesting crosstalk between the endosomal and autophagy systems. Furthermore, mutations in vacuolar protein sorting 35, part of the retromer trimer complex, disrupt Atg9 trafficking and ultimately impair autophagy [82].
Axonal Transport
Mammalian autophagosomes are formed in various regions of the cytoplasm and, upon maturation, are transported retrogradely to lysosomes, which are primarily located in the cell body or juxta-nuclear region. Microtubules and the dynein motor complex mediate retrograde transport and have been directly implicated in autophagosomal transport and fusion of autophagic/endocytic vesicles with lysosomes [83–85]. Live imaging studies in primary dorsal root ganglion neurons revealed that punctate green fluorescent protein–LC3 structures form in distal neurites, where they initially undergo bidirectional transport, but are eventually transported predominantly in the retrograde direction, which is associated with autophagosome maturation [86]. A recent study suggests that competitive binding of kinesin and dynein contributes to the initial bidirectional movement of autophagic structures; however, when scaffolding protein c-Jun NH2-terminal kinase-interacting protein-1 binds to LC3, the kinesin motor complex is inhibited, thus allowing for sustained dynein-mediated transport of autophagosomes in the retrograde direction [66]. Rab7 has also been implicated in the transport of autophagosomes along microtubules via its interaction with FYVE and coiled-coil domain containing 1 [87]. As autophagic vesicles in neurons are often transported across long distances from distal axons to the cell bodies where they fuse with lysosomes, drugs that promote transport may be effective in alleviating impaired autophagic flux in neurodegenerative disease.
Lysosomal Fusion and Degradation
Autolysosomal fusion depends on at least 3 independent factors: transport of autophagosomes/amphisomes to lysosomes, acidic lysosomal pH, and lysosomal membrane proteins (Fig. 1). Membrane protein complexes—class C core vacuole/endosome tethering factor, SNARE, and homotypic fusion and vacuole protein sorting, which is thought to be modulated by Rab7 [88]—act as tethers that facilitate fusion between autophagic vesicles and lysosomes [89, 90]. SNAREs are localized on both mature autophagosomes/amphisomes and lysosomes, and act as a bridge between the 2 structures to initiate fusion [72, 91]. While the majority of SNARE proteins are localized to endosomes and synaptic vesicles, syntaxin17 has recently been identified as an autophagosome-specific SNARE [92, 93].
The acidic pH of lysosomes is critical for their function and is maintained largely by vacuolar-type H+-ATPase, which pumps protons into the lysosomal lumen [94, 95]. Homeostatic autophagic flux requires proper lysosomal acidity and is completely halted by vacuolar-type H+-ATPase inhibitor bafilomycin A1 [96]. Similarly, a deficiency of lysosomal cathepsin enzymes disrupts autophagic flux causing the accumulation of autophagosomes and undigested autophagic cargo [97, 98]. Additionally, the loss of lysosomal associated membrane protein-2, which is involved in the selective uptake of proteins by the lysosome [99], leads to the accumulation of autophagic vacuoles [100]. As the final effectors of the autophagic machinery, lysosomal dysfunction leads to the blockage of the whole system, as evidenced in the lysosomal storage diseases [101].
AD Pathogenesis and Link to Autophagy
AD is characterized pathologically by the formation of extracellular plaques consisting of insoluble aggregated amyloid-β (Aβ) peptides and neurofibrillary tangles (NFTs) containing aggregated microtubule-associated tau. β-amyloid precursor protein (APP) is a transmembrane protein that is cleaved by α-, β-, and γ-secretases to form amyloidogenic and nonamyloidogenic peptides [102]. Familial mutations in APP and γ-secretase components presenilin-1 (PS1) or presenilin-2, cause enhanced production of Aβ peptides, which are prone to self-aggregation and the formation of oligomers, fibrils, and plaques [103]. Tau normally binds to and stabilizes microtubules; however, genetic tau mutations or hyperphosphorylation leads to detachment from microtubules and misfolding, followed by formation of pretangle aggregates and eventual deposition into filamentous inclusions (NFTs) [104]. AD pathology is also closely associated with inflammatory responses, including microglial clustering in and around dense core Aβ plaques [105], elevated levels of proinflammatory cytokines [106], and microglial activation that precedes NFT formation [107]. Additionally, genetic variants of triggering receptor expressed on myeloid cells-2 and CD33, which encode microglial receptors that regulate phagocytosis and inflammatory response, are significant risk factors in developing AD [108, 109]. As the disease progresses, there is widespread synaptic and neuritic degeneration, and subsequent neuronal cell death.
A growing body of evidence points to defective autophagic clearance as one of the disease-causing culprits of AD pathogenesis. Autophagy plays a vital neuroprotective role in central nervous system neurons, as it facilitates the removal of aggregated ubiquitinated protein inclusions and is essential for the prevention of neurodegeneration [110, 111]. Dysfunctional autophagy has been linked to many chronic proteinopathies, including frontotemporal dementia (FTD), amyotrophic lateral sclerosis, Parkinson’s disease (PD), Huntington’s disease (HD), and AD [1, 112–119].
In response to increased levels of cytosolic proteins and/or aggregates, autophagic induction or degradation may be heightened in affected neurons. In both patients with AD and the PS1/APP mouse model, there is a massive proliferation of autophagosomes and autolysosomes in dystrophic neurites [114, 120], which are enriched in Aβ and γ-secretase subunits [120, 121]. Whether the marked increase of these structures reflects elevated autophagosome formation or incomplete lysosomal/autolysosomal digestion is still under debate; however, multiple studies point to the latter [4, 122–124]. Strong induction of autophagy in primary cortical neurons leads to robust accumulation of mature autophagic vesicles, rather than early autophagosomes [122, 125]. Lysosomal protease cathepsin D is highly upregulated in affected regions of AD brains [126], suggesting a compensatory mechanism for insufficient lysosomal processing or degradation. Impaired lysosomal degradation may also be connected to early-onset familial AD mutations. PS1 is required for lysosomal acidification, and fibroblasts from mutated PS1 from patients with familial AD have defective autophagic–lysosomal proteolysis, along with impaired autolysosome acidification and cathepsin activation [4]. These studies suggest that promoting lysosomal clearance would reverse AD pathogenesis and improve cognitive function. For example, enhancing proteolysis in CRND8 transgenic mice through the deletion of cathepsin inhibitor, cystatin B, ameliorates Aβ plaque load, and learning and memory deficits [127].
While it is clear that improper lysosomal fusion and degradation are closely associated with AD neuropathology and enhancing lysosomal function reverses disease progression in mice, there is growing evidence that autophagy induction is also a viable therapeutic target that can ameliorate the pathological features associated with AD. Autophagosome formation progressively declines during normal aging [128], and may contribute to toxic protein accumulation in sporadic cases of age-related neurodegenerative diseases. Mice with central nervous system-specific deletion of essential autophagy genes, Atg5 or Atg7, display progressive neurodegeneration and pervasive polyubiquitinated protein inclusions [110, 111]. Genetic ablation of Atg7 has also been shown to exacerbate Aβ pathology. APP transgenic mice crossed with conditional knockout mice with forebrain-specific Atg7 deletion display increased intracellular Aβ accumulation in neurons [129], while mice with microglial-specific Atg7 ablation accumulate higher levels of Aβ following stereotaxic injection of fibril Aβ [130]. Similarly, the loss of Beclin 1 in the APP transgenic mouse model promotes Aβ deposition, and can be rescued by Beclin 1 viral gene transfer [5]. Postmortem examination of brains from patients with AD reveals reduced levels of Beclin 1, which is involved in regulating APP processing and turnover [5, 131], while microglia isolated from postmortem AD brains display lower levels of Beclin 1, which is associated with defective microglial phagocytosis of Aβ in APP transgenic mice [132]. These studies suggest that promoting levels of proteins associated with autophagy may have dual beneficial effects in AD models by enhancing autophagic degradation of Aβ in neurons and phagocytosis and degradation by microglia. This may also be relevant in microglial clearance of circulating Aβ peptides or tau released from neurons, as well as antibodies targeting these substrates.
Impaired autophagy increases levels of intracellular Aβ in APP mice, but also reduces extracellular deposits of Aβ, suggesting that while autophagy-stimulating compounds may reduce toxic intracellular Aβ levels, they may also exacerbate secretion of extracellular Aβ over time [129]. Together, these studies indicate an important role for autophagy in maintaining homeostatic levels of Aβ, and suggest that therapies targeted at autophagy may reduce Aβ-induced toxicity, but have detrimental effects if the level of induction exceeds lysosomal clearance of autophagic vacuoles. This is an important element in developing autophagy drugs and assays for the screening of new compounds.
Autophagy Therapeutics in Disease-related Protein Clearance
The goal of most preclinical AD research programs is to focus on discovering compounds that reduce tauopathy and Aβ accumulation, and reverse cognitive impairment. To this end, numerous transgenic animal models of AD have been generated to express different familial mutations of APP, PS1, tau, and combinations of these mutations. These models recapitulate aspects of AD neuropathology, behavioral deficits, and disease time course to varying degrees; therefore, selecting the appropriate mouse model to test potential therapeutics warrants careful consideration. Additionally, conflicting results within the same mouse models may be due to the route of administration, duration of treatment, and disease stage at onset of treatment. These factors should be considered in evaluating the data in the studies presented below. To date, multiple compounds have been identified in the context of AD and other neurodegenerative diseases that stimulate autophagic clearance by promoting induction, trafficking/maturation, or lysosomal fusion (Fig. 2).
Fig. 2.
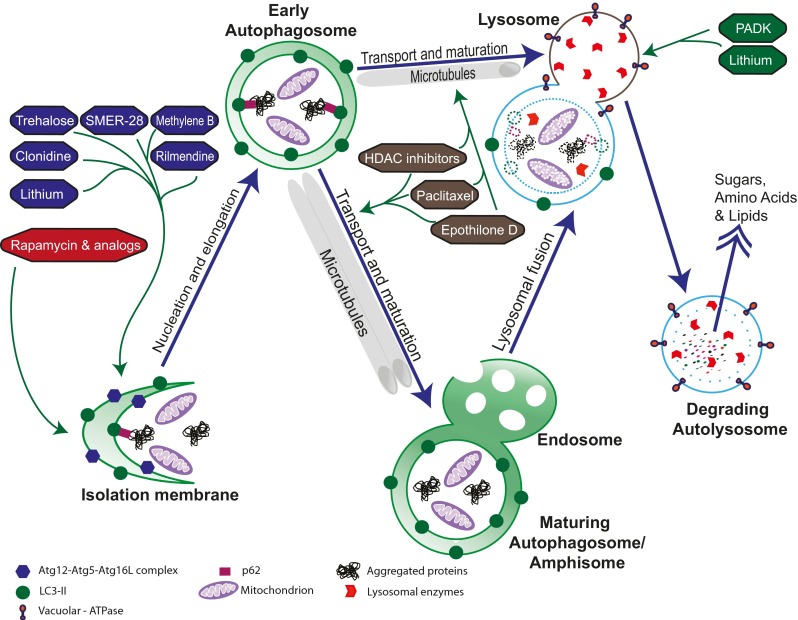
Autophagy-enhancing drugs and their targets. Compounds that stimulate autophagosome biogenesis include canonical mammalian target of rapamycin (mTOR)-dependent inhibitors (red) and multiple mTOR-independent autophagy inducers (blue), which likely act through different mechanisms. Compounds that promote maturation, trafficking, and degradation include microtubule stabilizers that aid in transport (gray) and enhancers of lysosomal fusion (green). SMER-28 = small molecule enhancer of autophagy; PADK = Z-Phe-Ala-diazomethylketone; HDAC = histone deacetylase; Atg = autophagy-related protein; LC3 = microtubule-associated protein 1A/1B-light chain 3
Targeting Canonical Autophagy Induction Through mTOR Signaling
Rapamycin
Multiple studies indicate that inhibition of mTOR increases lifespan in yeast, C. elegans, and Drosophila [133–135], and evidence suggests this effect is conserved in mammals [136–138]. Rapamycin, a US Food and Drug Administration (FDA)-approved antifungal antibiotic [139], anticancer cytostatic agent [140], and immunosuppressant [141, 142], inhibits mTOR signaling by forming a drug–receptor complex with FKBP12, which binds with the mTORC1 complex [143]. In addition to enhancing longevity [144, 145], rapamycin treatment has been shown, through autophagic induction, to clear aggregate-prone forms of disease-related proteins in cell and animal models of PD [1], HD [2, 119], and AD [146, 147].
Rapamycin was initially identified as an autophagy inducer capable of clearing aggregate-prone forms of huntingtin and α-synuclein [1, 148]. Rapamycin treatment induces clearance of wild-type and mutant R406W tau, reduces eye toxicity, and increases lifespan in a Drosophila model of AD, and promotes clearance of nonmicrotubule-bound aggregate-prone tau in COS-7 cells [119]. In 7PA2 cells (Chinese hamster ovary cells that stably express mutant APP), Aβ levels are reduced after rapamycin treatment [149]. These findings were extended to the 3 × TgAD mouse model of AD (a triple transgenic mouse that displays both Aβ plaques and tau tangles) [150]. Rapamycin leads to a reduction in both phospho-tau (Thr181) and Aβ levels in the CA1 region of hippocampus and reversal of early learning and memory deficits in 3 × TgAD mice [149]. Interestingly, mTOR kinase activity increases in the hippocampus and cortex of these mice with age, suggesting a reduction in autophagic activity over time [149]. These studies further support the notion that induction of autophagy may be beneficial in clearance of aggregate-prone proteins/peptides. In another study, 3 × TgAD mice treated with rapamycin early in life (starting at 2 months) exhibited reduced phospho-tau (at the Ser212, Thr214, and Thr231 residues) and Aβ pathology, and showed preserved learning and memory function without causing overt side effects [147]. Studies in hAPP-J20 mice, which have high hippocampal levels of Aβ1-42 peptide and synaptic degeneration [151], confirmed that rapamycin boosted autophagy, which improved behavioral deficits and also decreased Aβ42 levels, but not Aβ40 [146].
Rapamycin Analogs
Although rapamycin crosses the blood–brain barrier (BBB), it has poor water solubility and stability in solution [152]. Rapamycin analog, cell cycle inhibitor 779 (also known as temsirolimus), is an FDA-approved cancer drug, with improved pharmacological properties comparable to rapamycin. Like rapamycin, temsirolimus upregulates autophagy through inhibition of the mTOR pathway, and has been shown to ameliorate motor dysfunction and neuropathology in mouse models of 2 polyglutamine disorders [148, 153]. Two recent studies suggest a neuroprotective role for temisirolimus in AD models. Temsirolimus administration in APP/PS1 mice reduces Aβ plaque load and alleviates spatial learning and memory impairments through its activation of autophagy [154]. Likewise, the same group showed the beneficial effects of temsirolimus on tauopathy. Temsirolimus treatment in okadaic acid-treated SH-SY5Y cells (which induces tau hyperphosphorylation) and P301S tau transgenic mice reduces levels of hyperphosphorylated tau, while rescuing spatial learning deficits in mice through autophagy induction [155].
The Benefits and Risks of mTOR-dependent Autophagy Induction
Despite the promising effects of drugs that enhance autophagy through inhibition of mTOR, long-term administration of compounds that impede mTOR activity may have detrimental effects. The mTOR signaling cascade is required for protein synthesis associated with synaptic plasticity and memory formation in hippocampus (reviewed in [156]); therefore, the reversal of cognitive dysfunction observed in mice treated with mTOR inhibitors may be transitory, while long-term administration required in chronic disorders like AD may eventually exacerbate memory loss over time owing to untoward effects from inhibiting protein synthesis. Additionally, long-term use of rapamycin and its analogs may cause a broad array of side effects, including, but not limited to, increased infections, skin disorders, swelling of the lower extremities, reduced male fertility, hyperlipidemia, insulin resistance, and diabetes mellitus [157–161].
mTOR-independent Autophagy Inducers
Small-molecule Screening
Initial screens for small-molecule autophagy enhancers that activate autophagy in an mTOR-independent manner identified several compounds that reduced mutant forms of huntingtin and A53T α-synuclein aggregates in cell models, and were neuroprotective in Drosophila models of HD [162]. Small-molecule enhancer of rapamycin-28, an autophagy inducer with minimal cytotoxic effects, reduces Aβ and APP/C-terminal fragment accumulation in N2a-APP neuroblastoma cells [163]. FDA-approved hypertensive drugs, clonidine and the related drug rilmendine, were identified as autophagy enhancers in a screen of compounds that cleared aggregate-prone forms of α-synuclein and huntingin in PC12 cells [164]. While both drugs bind α2-adrenoceptors and I1 imidazoline receptors (I1R), rilmendine has a much greater specificity for I1R, and therefore fewer potential side effects. Rilmendine rescues motor dysfunction and decreases soluble mutant huntingtin levels, but not aggregates, in a transgenic mouse model of HD; however, owing to high dosing, treated mice experienced adverse effects that would limit its therapeutic potential. Nevertheless, converging data support the view that compounds that induce autophagic clearance can ameliorate disease pathogenesis and restore physiological function (Fig. 2).
Methylene Blue
In a screen of compounds that inhibit heparin-induced tau aggregation into filaments without disrupting microtubule interactions, several phenothiazines were identified with IC50 (half maximal inhibitory concentration) values in the low micromolar range [165]. Phenothiazines are clinically used as neuropsychotic agents. They readily cross the BBB and are generally well tolerated without major side effects. Methylthioninium chloride, also known as methylene blue (MB), is a non-neuroleptic phenothiazine that has been shown to reduce aggregation of truncated tau and Aβ oligomerization in vitro [166, 167], and acts as a memory-enhancing drug [168]. As MB has pleiotropic effects, including modulation of cyclic guanosine monophosphate signaling and synaptic neurotransmission, as well as inhibition of chaperone protein heat shock protein 70 (which promotes degradation) [169], it is unclear exactly how MB mediates its aggregate-inhibiting properties. However, a study in a mouse hippocampal cell line demonstrated that MB induces autophagy [170].
Studies from our laboratory have shown that MB reduces aggregated and phosphorylated tau in the JNPL3 mouse model (which expresses the P301L tau mutation) following treatment in ex vivo organotypic brain slice culture or administration in vivo by oral gavage [171]. MB increased levels of autophagy markers LC3, Beclin 1, and cathepsin D, suggesting that MB-induced clearance of tau is associated with autophagy activation [171]. While focal infusion of MB, by osmotic minipump, improved cognitive function and reduced tau levels in rTg4510 mice (another transgenic line expressing the P301L tau mutation), only long-term, high-dose administration of MB (ad libitum) beginning before the formation of NFTs led to the reduction of soluble tau and reversal of spatial learning impairments [172]. These effects were not observed following long-term, low-dose treatments at the same age, and tau NFT pathology was not reversed in either dosing regimen. In another study, MB treatment in 17-month-old rTg4510 mice (when mice already display substantial hippocampal and cortical neurofibrillary tangles) did not reduce existing NFT pathology [173]. In the triple transgenic 3 × TgAD model, where mice develop both plaques and tangles, MB significantly decreases soluble Aβ, but does not reduce tau phosphorylation or mislocalization [174]. The conflicting results of these studies may be attributed to the use of various mouse models (under the control of different promoters), time points of intervention (where late stage intervention is less promising than earlier time points), brain bioavailability [172], as well as dosing regimen (routes of administration and dosing schedules).
MB may have multiple targets, as a recent study suggests that AMPK mediates MB-induced neuroprotection, independent of mTOR signaling [170]. MB (also known as Rember) may act as a potent tau aggregation inhibitor and has been shown to slow cognitive decline in patients with mild-to-moderate AD in a phase 2 trial [175]. Rember (TauRx, Singapore) is currently in phase 3 clinical trials in patients with behavioral-variant FTD [176].
Inhibitors of Myo-inositol-1,4,5,-Triphosphate and Glycogen Synthase Kinase-3β Signaling
Autophagy can be activated through several known mTOR-independent mechanisms. One such pathway involves the reduction of myo-inositol-1,4,5,-triphosphate (IP3) levels to induce autophagy. IP3 is a second messenger that mediates the release of calcium from the ER through its receptor, IP3R. A recent study suggests that the IP3R negatively regulates autophagy via interactions with the Beclin 1 complex [177]. The IP3R antagonist xestospongin B disrupts the IP3R–Beclin 1 interaction, thereby inducing autophagy [177]. Lithium, a major treatment for bipolar affective disorder, inhibits inositol monophosphatase, which reduces IP3 signaling and thus upregulates autophagy. Lithium treatment in cell models of HD and PD reduced the accumulation of aggregate-prone forms of huntingtin and α-synuclein, respectively [178]. Lithium also inhibits glycogen synthase kinase (GSK)-3β (a kinase that hyperphosphorylates tau leading to NFT formation), which indirectly suppresses autophagy through its activation of mTOR [179]. Owing to its opposing effects on autophagy, Sarkar et al. [179] have proposed that lithium, in combination with rapamycin, is an effective brain-penetrant therapy to induce autophagy, and suggest that lower doses for each drug can be used in combination to induce autophagy while potentially reducing harmful side effects. Although this combinatorial therapy has only been tested in Drosophila models of HD, lithium may also provide beneficial effects in ameliorating tauopathy. In addition, GSK-3β inhibition directly improves lysosomal function by increasing biogenesis and acidification, and increasing APP and C-terminal fragment degradation [180, 181]. NP12, a specific GSK-3β inhibitor, reduces neuronal loss, astrocyte activity, and Aβ/tau pathology, and improves cognitive function in mice carrying APPSwedish mutations K670N and M671L and tau mutations G272V, P301L, and R406W [182].
Trehalose
Trehalose is a naturally occurring nonreducing disaccharide, consisting of 2 glucose molecules, that is produced endogenously in bacteria, fungi, plants, and invertebrates as a response to environmental stressors [183]. Trehalose treatment in vertebrates and mammalian-derived cell lines confers protection against oxidative damage, prevents protein aggregation, and promotes protein interactions by acting as a molecular chaperone [184]. Trehalose was initially identified as being neuroprotective through chemical screening in mutant huntingtin models and its inhibition of oligomeric Aβ40 aggregation [185, 186], and has since been shown to reduce disease-related protein aggregates associated with several neurodegenerative diseases in vitro through its induction of autophagy [162, 187].
In animal AD models, trehalose protects against Aβ and tau accumulation. Direct injection of trehalose into the lateral ventricles of APP/PS1 mice rescues deficits in spatial memory and learning, and reduces Aβ plaque load [188]. In transgenic mutant P301S tau models, ad libitum administration of trehalose reduces sarkosyl-insoluble tau aggregates and rescues cell death in the cortex and brainstem, but does not prevent motor deficits [189]. p62 levels are also reduced following trehalose treatment, indicating that autophagic induction is associated with tau removal. Data from our laboratory reveal similar effects in rTg4510 and JNPL3 transgenic tau mice treated with trehalose. Treated mice exhibit enhanced levels of autophagy markers, decreased tau aggregation, and improved performance in motor and cognitive behavioral tests [190].
Trehalose decreases the levels of endogenous tau in primary cortical neurons and reduces aggregates of tau with an FTD-17 mutation in N2a neuroblastoma cells, presumably by autophagy [191]. In addition to promoting autophagy, trehalose directly prevents tau aggregation in vitro [191], likely through its ability to stabilize proteins in their native form, thereby suppressing aggregation [192]. Therefore, trehalose may be a viable preventative therapy owing to its dual neuroprotective mechanisms.
Although multiple studies have demonstrated that trehalose confers beneficial effects on protein aggregation, behavior, and cell survival in an mTOR-independent manner [193], and leads to a dose- and time-dependent increase in the number of autophagosomes and autophagic markers [194], the specific autophagy-related molecular mechanisms have yet to be identified. A recent study in the mutant superoxide dismutase (SOD)1(G93A) mouse model of ALS suggests that trehalose treatment enhances autophagic flux. Ad libitum administration of trehalose reduced p62, ubiquitin, and SOD1 accumulation, while alleviating impaired autophagic flux [195]. While SOD1(G93A) mice exhibited elevated levels of Atg5, LC3-II, and increased numbers of autophagosomes compared with wild-type mice, transgenic mice treated with trehalose showed no significant change in autophagic induction markers compared with sucrose-treated control mice, suggesting that trehalose may not induce autophagy, but possibly enhances clearance (lower autophagosomes to autolysosomes ratio). Alternatively, these results may indicate that trehalose treatment combined with neuropathological conditions leads to more efficient lysosomal clearance. More studies in AD models will be necessary to pinpoint the mechanisms underlying the beneficial effects of trehalose in alleviating tauopathies and Aβ accumulation.
Trehalose is found in plants such as kelp and aloe, and is currently used as a dietary supplement and sugar substitute in food. Its druggable properties (e.g., trehalose is nontoxic at high concentrations, tolerated by humans, and readily bioavailable in the periphery and the brain) make it a suitable candidate for chronic treatment in neurodegenerative diseases. However, as trehalose is a sugar, chronic administration would require monitoring for diabetic-like complications. Furthermore, the chemical structure of trehalose does not lend itself to modification as a lead compound. Therefore, uncovering the mechanism of action for trehalose-mediated clearance of tau through autophagy is of particular interest. Identifying the signaling pathways that govern these effects also holds therapeutic potential as drug targets.
Compounds Promoting Autophagosome Maturation, Transport, and Fusion
Microtubule stabilizers have great potential as an adjuvant therapy to autophagy inducers. Histone deacetylase inhibitors, such as sodium valproate, sodium butyrate, and suberoylanilide hydroxamic acid, have been shown to reduce memory deficits in the APPSwedish/PS1ΔE9 mouse model of AD [196, 197], and have recently been shown to activate autophagy in rabbit cardiomyocytes [198]. Autophagy induction by histone deacetylase inhibitors has been attributed to microtubule stabilization due to increased tubulin acetylation [199–201]. Paclitaxel, another microtubule stabilizer, ameliorates motor impairment while improving fast axonal transport and axonal morphology in tau transgenic mice [202]. Conflicting reports, however, have emerged regarding the role of paclitaxel in autophagy induction [203, 204]. Through its stabilization of microtubules, paclitaxel may be a viable therapy for enhancing autophagic transport and fusion. This drug, however, requires further investigation as the bioavailability of paclitaxel in brain is suboptimal [205, 206]. Microtubule stabilizer, Epothilone D readily crosses the BBB [206], and has recently been shown to increase axonal stability, function, and transport, reduce tau pathology, preserve hippocampal morphology, and prevent cognitive deficits in PS19, 3 × Tg, and rTg4510 animal models of AD [207–209]. Epothilone D (also known as BMS-241027) was tested in phase 1 clinical trials for safety and tolerability; however, further studies were discontinued based on unfavorable study outcomes (ClinicalTrials.gov identifier NCT01492374). Dictyostatin, triazolopyrimidines, and phenylpyrimidines have recently been identified as brain-penetrant, microtubule-stabilizing agents [210, 211], though their effects on autophagic flux have not yet been validated.
Compounds that modulate lysosomal fusion include potential cystatin B and C antagonists [127], lysosomal cathepsin B modulators [212], and GSK-3β inhibitors, such as lithium, valproate, and NP12 [180, 181]. Genetic deletion of cystatin B and C has been shown to improve neurological deficits and restore autophagic dysfunction in the TgCRND8 and hAPP-J20 mouse models of AD [127, 213]. However, conflicting reports indicate that cystatin C is neuroprotective in several neurodegenerative diseases, and these effects may be mediated by inducing autophagy [214–217]. Future studies should examine the effects of cystatins in AD [218], and whether neuroprotection is attenuated in the absence of autophagy. Cathepsin B is a lysosomal/endosomal enzyme with neuroprotective properties based on its protease activity, and in particular, its Aβ1-42 cleavage function [219]. Z-Phe-Ala-diazomethylketone, a positive modulator of cathepsin B activity, reduces Aβ levels in 10–11-month-old APP mice with Swedish and Indiana mutations (APPSwe/Ind) and in 20–22-month-old APPswedish/PS1ΔE9 mice, protects against synaptic degeneration, and mitigates behavioral deficits [212]. Similarly, cathepsin D, another lysosomal/endosomal enzyme, has been implicated in clearance of Lewy bodies in PD [220]. One way to pharmacologically enhance cathepsin B and D activity is by administering pharmacological chaperones similar to the ones used in lysosomal storage diseases [221]. To date, no known molecule has been tested for this property.
Conclusions
Enhancing autophagic clearance of toxic protein aggregates through mTOR-dependent or mTOR-independent compounds ameliorates protein aggregation, neuron survival, and behavior. However, while converging evidence suggests that augmenting autophagy is a promising therapeutic intervention [162, 222], it may also overwhelm the lysosomal machinery, leading to increased numbers of autophagosomes and undigested autolysosomes that can impair axonal trafficking and lead to dystrophic axons. In a mouse model of excitotoxicity, constitutive activation of a glutamate receptor induced the buildup of green fluorescent protein–LC3-labelled autophagosomes in dystrophic axons [223]. These data suggest that excessive autophagosome formation may eventually form roadblocks to normal retrograde transport, causing axonal swellings. However, in diseases like AD, inducing autophagy may exacerbate already impaired lysosomal clearance [125]. Therefore, it is important to consider the downstream effects of drug therapies targeted at each stage of autophagy in different diseases, and how they might disturb the delicate balance between autophagosome formation and lysosomal degradation. We propose that a more viable therapeutic strategy for stimulating autophagic clearance of disease-related proteins in AD and other neurodegenerative diseases incorporates not only drugs that increase autophagic induction, but also those that target later steps in the autophagy–lysosome system, thus improving autophagic flux (Fig. 2). Although the right combination of drugs may differ based on the disease type, stage of progression, underlying cause (i.e., genetic vs idiopathic), and a variety of other factors, it is clear that modulation of neuronal autophagy may be a promising therapeutic intervention to attenuate AD pathogenesis and rescue cognitive impairment.
Electronic supplementary material
(PDF 499 kb)
Acknowledgments
During the writing of this manuscript we were supported by grants from the National Institutes of Health (NS074593; W.H,Y.), BrightFocus (A2012057; W.H.Y.), Alzheimer’s Association IIRG (11-205828; W.H.Y.), Alzheimer’s Drug Discovery Foundation (W.H.Y.), Merck Initiatives for Neuroscience Therapeutics (W.H.Y.), and Weston Brain Institute (L.G.F.).
Footnotes
Lauren G. Friedman and Yasir H. Qureshi contributed equally.
References
- 1.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 2.Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107–1117. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 3.Verhoef LG, Lindsten K, Masucci MG, Dantuma NP. Aggregate formation inhibits proteasomal degradation of polyglutamine proteins. Hum Mol Genet. 2002;11:2689–2700. doi: 10.1093/hmg/11.22.2689. [DOI] [PubMed] [Google Scholar]
- 4.Lee JH, Yu WH, Kumar A, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pickford F, Masliah E, Britschgi M, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118:2190–2199. doi: 10.1172/JCI33585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu XC, Yu JT, Jiang T, Tan L. Autophagy modulation for Alzheimer's disease therapy. Mol Neurobiol. 2013;48:702–714. doi: 10.1007/s12035-013-8457-z. [DOI] [PubMed] [Google Scholar]
- 7.Eskelinen EL. Maturation of autophagic vacuoles in Mammalian cells. Autophagy. 2005;1:1–10. doi: 10.4161/auto.1.1.1270. [DOI] [PubMed] [Google Scholar]
- 8.Cuervo AM. Autophagy: many paths to the same end. Mol Cell Biochem. 2004;263:55–72. doi: 10.1023/B:MCBI.0000041848.57020.57. [DOI] [PubMed] [Google Scholar]
- 9.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Araki Y, Ku WC, Akioka M, et al. Atg38 is required for autophagy-specific phosphatidylinositol 3-kinase complex integrity. J Cell Biol. 2013;203:299–313. doi: 10.1083/jcb.201304123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol. 2009;335:1–32. doi: 10.1007/978-3-642-00302-8_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang J, Klionsky DJ. Autophagy and human disease. Cell Cycle. 2007;6:1837–1849. doi: 10.4161/cc.6.15.4511. [DOI] [PubMed] [Google Scholar]
- 14.Richter JD, Sonenberg N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature. 2005;433:477–480. doi: 10.1038/nature03205. [DOI] [PubMed] [Google Scholar]
- 15.Hannan KM, Brandenburger Y, Jenkins A, et al. mTOR-dependent regulation of ribosomal gene transcription requires S6K1 and is mediated by phosphorylation of the carboxy-terminal activation domain of the nucleolar transcription factor UBF. Mol Cell Biol. 2003;23:8862–8877. doi: 10.1128/MCB.23.23.8862-8877.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng T, Golub TR, Sabatini DM. The immunosuppressant rapamycin mimics a starvation-like signal distinct from amino acid and glucose deprivation. Mol Cell Biol. 2002;22:5575–5584. doi: 10.1128/MCB.22.15.5575-5584.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998;273:3963–3966. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- 18.Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol. 2014;15:155–162. doi: 10.1038/nrm3757. [DOI] [PubMed] [Google Scholar]
- 19.Hosokawa N, Hara T, Kaizuka T, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284:12297–12305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jung CH, Jun CB, Ro SH, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee JW, Park S, Takahashi Y, Wang HG. The association of AMPK with ULK1 regulates autophagy. PLoS One. 2010;5:e15394. doi: 10.1371/journal.pone.0015394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Egan D, Kim J, Shaw RJ, Guan KL. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy. 2011;7:643–644. doi: 10.4161/auto.7.6.15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334:678–683. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 27.Gwinn DM, Shackelford DB, Egan DF, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67:10804–10812. doi: 10.1158/0008-5472.CAN-07-2310. [DOI] [PubMed] [Google Scholar]
- 30.Kalender A, Selvaraj A, Kim SY, et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010;11:390–401. doi: 10.1016/j.cmet.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ben Sahra I, Regazzetti C, Robert G, et al. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011;71:4366–4372. doi: 10.1158/0008-5472.CAN-10-1769. [DOI] [PubMed] [Google Scholar]
- 32.Axe EL, Walker SA, Manifava M, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11:1433–1437. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- 34.Di Bartolomeo S, Corazzari M, Nazio F, et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol. 2010;191:155–168. doi: 10.1083/jcb.201002100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fan W, Nassiri A, Zhong Q. Autophagosome targeting and membrane curvature sensing by Barkor/Atg14(L) Proc Natl Acad Sci U S A. 2011;108:7769–7774. doi: 10.1073/pnas.1016472108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex—at the crossroads of autophagy and beyond. Trends Cell Biol. 2010;20:355–362. doi: 10.1016/j.tcb.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhong Y, Wang QJ, Li X, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11:468–476. doi: 10.1038/ncb1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Young AR, Chan EY, Hu XW, et al. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci. 2006;119:3888–3900. doi: 10.1242/jcs.03172. [DOI] [PubMed] [Google Scholar]
- 39.Reggiori F, Tucker KA, Stromhaug PE, Klionsky DJ. The Atg1–Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Develop Cell. 2004;6:79–90. doi: 10.1016/s1534-5807(03)00402-7. [DOI] [PubMed] [Google Scholar]
- 40.Proikas-Cezanne T, Ruckerbauer S, Stierhof YD, Berg C, Nordheim A. Human WIPI-1 puncta-formation: a novel assay to assess mammalian autophagy. FEBS Lett. 2007;581:3396–3404. doi: 10.1016/j.febslet.2007.06.040. [DOI] [PubMed] [Google Scholar]
- 41.Polson HE, de Lartigue J, Rigden DJ, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy. 2010;6:506–522. doi: 10.4161/auto.6.4.11863. [DOI] [PubMed] [Google Scholar]
- 42.Mizushima N, Sugita H, Yoshimori T, Ohsumi Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J Biol Chem. 1998;273:33889–33892. doi: 10.1074/jbc.273.51.33889. [DOI] [PubMed] [Google Scholar]
- 43.Mizushima N, Kuma A, Kobayashi Y, et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci. 2003;116:1679–1688. doi: 10.1242/jcs.00381. [DOI] [PubMed] [Google Scholar]
- 44.Mizushima N, Noda T, Ohsumi Y. Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J. 1999;18:3888–3896. doi: 10.1093/emboj/18.14.3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Christoforidis S, Miaczynska M, Ashman K, et al. Phosphatidylinositol-3-OH kinases are Rab5 effectors. Nat Cell Biol. 1999;1:249–252. doi: 10.1038/12075. [DOI] [PubMed] [Google Scholar]
- 46.Ravikumar B, Imarisio S, Sarkar S, O'Kane CJ, Rubinsztein DC. Rab5 modulates aggregation and toxicity of mutant huntingtin through macroautophagy in cell and fly models of Huntington disease. J Cell Sci. 2008;121:1649–1660. doi: 10.1242/jcs.025726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu ZQ, Ni T, Hong B, et al. Dual roles of Atg8-PE deconjugation by Atg4 in autophagy. Autophagy. 2012;8:883–892. doi: 10.4161/auto.19652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walczak M, Martens S. Dissecting the role of the Atg12-Atg5-Atg16 complex during autophagosome formation. Autophagy. 2013;9:424–425. doi: 10.4161/auto.22931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tanida I, Tanida-Miyake E, Komatsu M, Ueno T, Kominami E. Human Apg3p/Aut1p homologue is an authentic E2 enzyme for multiple substrates, GATE-16, GABARAP, and MAP-LC3, and facilitates the conjugation of hApg12p to hApg5p. J Biol Chem. 2002;277:13739–13744. doi: 10.1074/jbc.M200385200. [DOI] [PubMed] [Google Scholar]
- 50.Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117:2805–2812. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- 51.Hanada T, Noda NN, Satomi Y, et al. The Atg12–Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282:37298–37302. doi: 10.1074/jbc.C700195200. [DOI] [PubMed] [Google Scholar]
- 52.Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010;29:1792–1802. doi: 10.1038/emboj.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mizushima N, Yamamoto A, Hatano M, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152:657–668. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Komatsu M, Waguri S, Koike M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 55.Shvets E, Fass E, Scherz-Shouval R, Elazar Z. The N-terminus and Phe52 residue of LC3 recruit p62/SQSTM1 into autophagosomes. J Cell Sci. 2008;121:2685–2695. doi: 10.1242/jcs.026005. [DOI] [PubMed] [Google Scholar]
- 56.Ichimura Y, Kumanomidou T, Sou YS, et al. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem. 2008;283:22847–22857. doi: 10.1074/jbc.M802182200. [DOI] [PubMed] [Google Scholar]
- 57.Bjorkoy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nogalska A, Terracciano C, D'Agostino C, King Engel W, Askanas V. p62/SQSTM1 is overexpressed and prominently accumulated in inclusions of sporadic inclusion-body myositis muscle fibers, and can help differentiating it from polymyositis and dermatomyositis. Acta Neuropathol. 2009;118:407–413. doi: 10.1007/s00401-009-0564-6. [DOI] [PubMed] [Google Scholar]
- 59.Babu JR, Geetha T, Wooten MW. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J Neurochem. 2005;94:192–203. doi: 10.1111/j.1471-4159.2005.03181.x. [DOI] [PubMed] [Google Scholar]
- 60.Gordon PB, Seglen PO. Prelysosomal convergence of autophagic and endocytic pathways. Biochem Biophys Res Commun. 1988;151:40–47. doi: 10.1016/0006-291x(88)90556-6. [DOI] [PubMed] [Google Scholar]
- 61.Dunn WA., Jr Studies on the mechanisms of autophagy: maturation of the autophagic vacuole. J Cell Biol. 1990;110:1935–1945. doi: 10.1083/jcb.110.6.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lucocq J, Walker D. Evidence for fusion between multilamellar endosomes and autophagosomes in HeLa cells. Eur J Cell Biol. 1997;72:307–313. [PubMed] [Google Scholar]
- 63.Morvan J, Kochl R, Watson R, Collinson LM, Jefferies HB, Tooze SA. In vitro reconstitution of fusion between immature autophagosomes and endosomes. Autophagy. 2009;5:676–689. doi: 10.4161/auto.5.5.8378. [DOI] [PubMed] [Google Scholar]
- 64.Fader CM, Sanchez D, Furlan M, Colombo MI. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic. 2008;9:230–250. doi: 10.1111/j.1600-0854.2007.00677.x. [DOI] [PubMed] [Google Scholar]
- 65.Fader CM, Colombo MI. Autophagy and multivesicular bodies: two closely related partners. Cell Death Differ. 2009;16:70–78. doi: 10.1038/cdd.2008.168. [DOI] [PubMed] [Google Scholar]
- 66.Fu MM, Nirschl JJ, Holzbaur EL. LC3 Binding to the scaffolding protein JIP1 regulates processive dynein-driven transport of autophagosomes. Develop Cell. 2014;29:577–590. doi: 10.1016/j.devcel.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Szatmari Z, Kis V, Lippai M, et al. Rab11 facilitates cross-talk between autophagy and endosomal pathway through regulation of Hook localization. Mol Biol Cell. 2014;25:522–531. doi: 10.1091/mbc.E13-10-0574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ejlerskov P, Rasmussen I, Nielsen TT, et al. Tubulin polymerization-promoting protein (TPPP/p25alpha) promotes unconventional secretion of alpha-synuclein through exophagy by impairing autophagosome-lysosome fusion. J Biol Chem. 2013;288:17313–17335. doi: 10.1074/jbc.M112.401174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xie R, Nguyen S, McKeehan K, Wang F, McKeehan WL, Liu L. Microtubule-associated protein 1S (MAP1S) bridges autophagic components with microtubules and mitochondria to affect autophagosomal biogenesis and degradation. J Biol Chem. 2011;286:10367–10377. doi: 10.1074/jbc.M110.206532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aplin A, Jasionowski T, Tuttle DL, Lenk SE, Dunn WA., Jr Cytoskeletal elements are required for the formation and maturation of autophagic vacuoles. J Cell Physiol. 1992;152:458–466. doi: 10.1002/jcp.1041520304. [DOI] [PubMed] [Google Scholar]
- 71.Ishihara N, Hamasaki M, Yokota S, et al. Autophagosome requires specific early Sec proteins for its formation and NSF/SNARE for vacuolar fusion. Mol Biol Cell. 2001;12:3690–3702. doi: 10.1091/mbc.12.11.3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Furuta N, Fujita N, Noda T, Yoshimori T, Amano A. Combinational soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins VAMP8 and Vti1b mediate fusion of antimicrobial and canonical autophagosomes with lysosomes. Mol Biol Cell. 2010;21:1001–1010. doi: 10.1091/mbc.E09-08-0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liang C, Lee JS, Inn KS, et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol. 2008;10:776–787. doi: 10.1038/ncb1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gutierrez MG, Munafo DB, Beron W, Colombo MI. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci. 2004;117:2687–2697. doi: 10.1242/jcs.01114. [DOI] [PubMed] [Google Scholar]
- 75.Jager S, Bucci C, Tanida I, et al. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci. 2004;117:4837–4848. doi: 10.1242/jcs.01370. [DOI] [PubMed] [Google Scholar]
- 76.Chua CE, Gan BQ, Tang BL. Involvement of members of the Rab family and related small GTPases in autophagosome formation and maturation. Cell Mol Life Sci. 2011;68:3349–3358. doi: 10.1007/s00018-011-0748-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Munafo DB, Colombo MI. Induction of autophagy causes dramatic changes in the subcellular distribution of GFP-Rab24. Traffic. 2002;3:472–482. doi: 10.1034/j.1600-0854.2002.30704.x. [DOI] [PubMed] [Google Scholar]
- 78.Seaman MN. Recycle your receptors with retromer. Trends Cell Biol. 2005;15:68–75. doi: 10.1016/j.tcb.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 79.Feinstein TN, Wehbi VL, Ardura JA, et al. Retromer terminates the generation of cAMP by internalized PTH receptors. Nat Chem Biol. 2011;7:278–284. doi: 10.1038/nchembio.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Temkin P, Lauffer B, Jager S, Cimermancic P, Krogan NJ, von Zastrow M. SNX27 mediates retromer tubule entry and endosome-to-plasma membrane trafficking of signalling receptors. Nat Cell Biol. 2011;13:715–721. doi: 10.1038/ncb2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ruck A, Attonito J, Garces KT, et al. The Atg6/Vps30/Beclin 1 ortholog BEC-1 mediates endocytic retrograde transport in addition to autophagy in C. elegans. Autophagy. 2011;7:386–400. doi: 10.4161/auto.7.4.14391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zavodszky E, Seaman MN, Moreau K, et al. Mutation in VPS35 associated with Parkinson's disease impairs WASH complex association and inhibits autophagy. Nat Commun. 2014;5:3828. doi: 10.1038/ncomms4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kochl R, Hu XW, Chan EY, Tooze SA. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic. 2006;7:129–145. doi: 10.1111/j.1600-0854.2005.00368.x. [DOI] [PubMed] [Google Scholar]
- 84.Kimura S, Noda T, Yoshimori T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct Funct. 2008;33:109–122. doi: 10.1247/csf.08005. [DOI] [PubMed] [Google Scholar]
- 85.Jahreiss L, Menzies FM, Rubinsztein DC. The itinerary of autophagosomes: from peripheral formation to kiss-and-run fusion with lysosomes. Traffic. 2008;9:574–587. doi: 10.1111/j.1600-0854.2008.00701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Maday S, Wallace KE, Holzbaur EL. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol. 2012;196:407–417. doi: 10.1083/jcb.201106120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pankiv S, Alemu EA, Brech A, et al. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol. 2010;188:253–269. doi: 10.1083/jcb.200907015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Huotari J, Helenius A. Endosome maturation. EMBO J. 2011;30:3481–3500. doi: 10.1038/emboj.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brocker C, Engelbrecht-Vandre S, Ungermann C. Multisubunit tethering complexes and their role in membrane fusion. Curr Biol. 2010;20:R943–R952. doi: 10.1016/j.cub.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 90.Starai VJ, Hickey CM, Wickner W. HOPS proofreads the trans-SNARE complex for yeast vacuole fusion. Mol Biol Cell. 2008;19:2500–2508. doi: 10.1091/mbc.E08-01-0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fader CM, Sanchez DG, Mestre MB, Colombo MI. TI-VAMP/VAMP7 and VAMP3/cellubrevin: two v-SNARE proteins involved in specific steps of the autophagy/multivesicular body pathways. Biochim Biophys Acta. 2009;1793:1901–1916. doi: 10.1016/j.bbamcr.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 92.Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012;151:1256–1269. doi: 10.1016/j.cell.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 93.Takats S, Nagy P, Varga A, et al. Autophagosomal Syntaxin17-dependent lysosomal degradation maintains neuronal function in Drosophila. J Cell Biol. 2013;201:531–539. doi: 10.1083/jcb.201211160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lukacs GL, Rotstein OD, Grinstein S. Phagosomal acidification is mediated by a vacuolar-type H(+)-ATPase in murine macrophages. J Biol Chem. 1990;265:21099–21107. [PubMed] [Google Scholar]
- 95.Lukacs GL, Rotstein OD, Grinstein S. Determinants of the phagosomal pH in macrophages. In situ assessment of vacuolar H(+)-ATPase activity, counterion conductance, and H+ "leak". J Biol Chem. 1991;266:24540–24548. [PubMed] [Google Scholar]
- 96.Liu B, Fang M, Hu Y, et al. Hepatitis B virus X protein inhibits autophagic degradation by impairing lysosomal maturation. Autophagy. 2014;10:416–430. doi: 10.4161/auto.27286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Koike M, Nakanishi H, Saftig P, et al. Cathepsin D deficiency induces lysosomal storage with ceroid lipofuscin in mouse CNS neurons. J Neurosci. 2000;20:6898–6906. doi: 10.1523/JNEUROSCI.20-18-06898.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shacka JJ, Klocke BJ, Young C, et al. Cathepsin D deficiency induces persistent neurodegeneration in the absence of Bax-dependent apoptosis. J Neurosci. 2007;27:2081–2090. doi: 10.1523/JNEUROSCI.5577-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273:501–503. doi: 10.1126/science.273.5274.501. [DOI] [PubMed] [Google Scholar]
- 100.Tanaka Y, Guhde G, Suter A, et al. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature. 2000;406:902–906. doi: 10.1038/35022595. [DOI] [PubMed] [Google Scholar]
- 101.Lieberman AP, Puertollano R, Raben N, Slaugenhaupt S, Walkley SU, Ballabio A. Autophagy in lysosomal storage disorders. Autophagy. 2012;8:719–730. doi: 10.4161/auto.19469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 103.Zhang YW, Thompson R, Zhang H, Xu H. APP processing in Alzheimer's disease. Mol Brain. 2011;4:3. doi: 10.1186/1756-6606-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 105.Itagaki S, McGeer PL, Akiyama H, Zhu S, Selkoe D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol. 1989;24:173–182. doi: 10.1016/0165-5728(89)90115-x. [DOI] [PubMed] [Google Scholar]
- 106.Tan ZS, Beiser AS, Vasan RS, et al. Inflammatory markers and the risk of Alzheimer disease: the Framingham Study. Neurology. 2007;68:1902–1908. doi: 10.1212/01.wnl.0000263217.36439.da. [DOI] [PubMed] [Google Scholar]
- 107.Yoshiyama Y, Higuchi M, Zhang B, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 108.Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. 2013;368:107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013;368:117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 111.Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:88588–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 112.Sapp E, Schwarz C, Chase K, et al. Huntingtin localization in brains of normal and Huntington's disease patients. Ann Neurol. 1997;42:604–612. doi: 10.1002/ana.410420411. [DOI] [PubMed] [Google Scholar]
- 113.Anglade P, Vyas S, Javoy-Agid F, et al. Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol Jistopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- 114.Nixon RA, Wegiel J, Kumar A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 115.Lee JA, Gao FB. Inhibition of autophagy induction delays neuronal cell loss caused by dysfunctional ESCRT-III in frontotemporal dementia. J Neurosci. 2009;29:8506–8511. doi: 10.1523/JNEUROSCI.0924-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Morimoto N, Nagai M, Ohta Y, et al. Increased autophagy in transgenic mice with a G93A mutant SOD1 gene. Brain Res. 2007;1167:112–117. doi: 10.1016/j.brainres.2007.06.045. [DOI] [PubMed] [Google Scholar]
- 117.Rideout HJ, Lang-Rollin I, Stefanis L. Involvement of macroautophagy in the dissolution of neuronal inclusions. Int J Biochem Cell Biol. 2004;36:2551–2562. doi: 10.1016/j.biocel.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 118.Martinez-Vicente M, Talloczy Z, Wong E, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat Neurosci. 2010;13:567–576. doi: 10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Berger Z, Ravikumar B, Menzies FM, et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet. 2006;15:433–442. doi: 10.1093/hmg/ddi458. [DOI] [PubMed] [Google Scholar]
- 120.Yu WH, Cuervo AM, Kumar A, et al. Macroautophagy--a novel Beta-amyloid peptide-generating pathway activated in Alzheimer's disease. J Cell Biol. 2005;171:87–98. doi: 10.1083/jcb.200505082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yu WH, Kumar A, Peterhoff C, et al. Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: implications for beta-amyloid peptide over-production and localization in Alzheimer's disease. Int J Biochem Cell Biol. 2004;36:2531–2540. doi: 10.1016/j.biocel.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 122.Wolfe DM, Lee JH, Kumar A, Lee S, Orenstein SJ, Nixon RA. Autophagy failure in Alzheimer's disease and the role of defective lysosomal acidification. Eur J Neurosci. 2013;37:1949–1961. doi: 10.1111/ejn.12169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yang DS, Stavrides P, Mohan PS, et al. Therapeutic effects of remediating autophagy failure in a mouse model of Alzheimer disease by enhancing lysosomal proteolysis. Autophagy. 2011;7:788–789. doi: 10.4161/auto.7.7.15596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Coffey EE, Beckel JM, Laties AM, Mitchell CH. Lysosomal alkalization and dysfunction in human fibroblasts with the Alzheimer's disease-linked presenilin 1 A246E mutation can be reversed with cAMP. Neuroscience. 2014;263:111–124. doi: 10.1016/j.neuroscience.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Boland B, Kumar A, Lee S, et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci. 2008;28:6926–6937. doi: 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cataldo AM, Barnett JL, Berman SA, et al. Gene expression and cellular content of cathepsin D in Alzheimer's disease brain: evidence for early up-regulation of the endosomal-lysosomal system. Neuron. 1995;14:671–680. doi: 10.1016/0896-6273(95)90324-0. [DOI] [PubMed] [Google Scholar]
- 127.Yang DS, Stavrides P, Mohan PS, et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer's disease ameliorates amyloid pathologies and memory deficits. Brain. 2011;134:258–277. doi: 10.1093/brain/awq341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.McCray BA, Taylor JP. The role of autophagy in age-related neurodegeneration. Neurosignals. 2008;16:75–84. doi: 10.1159/000109761. [DOI] [PubMed] [Google Scholar]
- 129.Nilsson P, Loganathan K, Sekiguchi M, et al. Abeta secretion and plaque formation depend on autophagy. Cell Rep. 2013;5:61–69. doi: 10.1016/j.celrep.2013.08.042. [DOI] [PubMed] [Google Scholar]
- 130.Cho MH, Cho K, Kang HJ, et al. Autophagy in microglia degrades extracellular beta-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy. 2014;10:1761–1775. doi: 10.4161/auto.29647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jaeger PA, Pickford F, Sun CH, Lucin KM, Masliah E, Wyss-Coray T. Regulation of amyloid precursor protein processing by the Beclin 1 complex. PLoS One. 2010;5:e11102. doi: 10.1371/journal.pone.0011102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lucin KM, O'Brien CE, Bieri G, et al. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer's disease. Neuron. 2013;79:873–886. doi: 10.1016/j.neuron.2013.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Muller F. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature. 2003;426:620. doi: 10.1038/426620a. [DOI] [PubMed] [Google Scholar]
- 134.Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14:885–890. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kaeberlein M, Powers RW, 3rd, Steffen KK, et al. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310:1193–1196. doi: 10.1126/science.1115535. [DOI] [PubMed] [Google Scholar]
- 136.Selman C, Tullet JM, Wieser D, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;326:140–144. doi: 10.1126/science.1177221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Anisimov VN, Zabezhinski MA, Popovich IG, et al. Rapamycin increases lifespan and inhibits spontaneous tumorigenesis in inbred female mice. Cell Cycle. 2011;10:4230–4236. doi: 10.4161/cc.10.24.18486. [DOI] [PubMed] [Google Scholar]
- 138.Harrison DE, Strong R, Sharp ZD, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Vezina C, Kudelski A, Sehgal SN. Rapamycin (AY 22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J Antibiot (Tokyo) 1975;28:721–726. doi: 10.7164/antibiotics.28.721. [DOI] [PubMed] [Google Scholar]
- 140.Eng CP, Sehgal SN, Vezina C. Activity of rapamycin (AY-22,989) against transplanted tumors. J Antiobiot (Tokyo) 1984;37:1231–1237. doi: 10.7164/antibiotics.37.1231. [DOI] [PubMed] [Google Scholar]
- 141.Martel RR, Klicius J, Galet S. Inhibition of the immune response by rapamycin, a new antifungal antibiotic. Can J Physiol Pharmacol. 1977;55:48–51. doi: 10.1139/y77-007. [DOI] [PubMed] [Google Scholar]
- 142.Ingle GR, Sievers TM, Holt CD. Sirolimus: continuing the evolution of transplant immunosuppression. Ann Pharmacother. 2000;34:1044–1055. doi: 10.1345/aph.19380. [DOI] [PubMed] [Google Scholar]
- 143.Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 144.Alvers AL, Wood MS, Hu D, Kaywell AC, Dunn WA, Jr, Aris JP. Autophagy is required for extension of yeast chronological life span by rapamycin. Autophagy. 2009;5:847–849. doi: 10.4161/auto.8824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hars ES, Qi H, Ryazanov AG, et al. Autophagy regulates ageing in C. elegans. Autophagy. 2007;3:93–95. doi: 10.4161/auto.3636. [DOI] [PubMed] [Google Scholar]
- 146.Spilman P, Podlutskaya N, Hart MJ, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLoS One. 2010;5:e9979. doi: 10.1371/journal.pone.0009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Majumder S, Richardson A, Strong R, Oddo S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One. 2011;6:e25416. doi: 10.1371/journal.pone.0025416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 149.Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285:13107–13120. doi: 10.1074/jbc.M110.100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Oddo S, Caccamo A, Shepherd JD, et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 151.Mucke L, Masliah E, Yu GQ, et al. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Huang S, Houghton PJ. Mechanisms of resistance to rapamycins. Drug Resist Updat. 2001;4:378–391. doi: 10.1054/drup.2002.0227. [DOI] [PubMed] [Google Scholar]
- 153.Menzies FM, Huebener J, Renna M, Bonin M, Riess O, Rubinsztein DC. Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain. 2010;133:93–104. doi: 10.1093/brain/awp292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Jiang T, Yu JT, Zhu XC, et al. Temsirolimus promotes autophagic clearance of amyloid-beta and provides protective effects in cellular and animal models of Alzheimer's disease. Pharmacol Res. 2014;81:54–63. doi: 10.1016/j.phrs.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 155.Jiang T, Yu JT, Zhu XC, et al. Temsirolimus attenuates tauopathy in vitro and in vivo by targeting tau hyperphosphorylation and autophagic clearance. Neuropharmacology. 2014;85C:121–130. doi: 10.1016/j.neuropharm.2014.05.032. [DOI] [PubMed] [Google Scholar]
- 156.Graber TE, McCamphill PK, Sossin WS. A recollection of mTOR signaling in learning and memory. Learn Mem. 2013;20:518–530. doi: 10.1101/lm.027664.112. [DOI] [PubMed] [Google Scholar]
- 157.Mahe E, Morelon E, Lechaton S, et al. Cutaneous adverse events in renal transplant recipients receiving sirolimus-based therapy. Transplantation. 2005;79:476–482. doi: 10.1097/01.tp.0000151630.25127.3a. [DOI] [PubMed] [Google Scholar]
- 158.Zuber J, Anglicheau D, Elie C, et al. Sirolimus may reduce fertility in male renal transplant recipients. Am J Transplant. 2008;8:1471–1479. doi: 10.1111/j.1600-6143.2008.02267.x. [DOI] [PubMed] [Google Scholar]
- 159.Lamming DW, Ye L, Katajisto P, et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. 2012;335:1638–1643. doi: 10.1126/science.1215135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Gyurus E, Kaposztas Z, Kahan BD. Sirolimus therapy predisposes to new-onset diabetes mellitus after renal transplantation: a long-term analysis of various treatment regimens. Transplant Proc. 2011;43:1583–1592. doi: 10.1016/j.transproceed.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 161.McCormack FX, Inoue Y, Moss J, et al. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med. 2011;364:1595–1606. doi: 10.1056/NEJMoa1100391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sarkar S, Perlstein EO, Imarisio S, et al. Small molecules enhance autophagy and reduce toxicity in Huntington's disease models. Nat Chem Biol. 2007;3:331–338. doi: 10.1038/nchembio883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Tian Y, Bustos V, Flajolet M, Greengard P. A small-molecule enhancer of autophagy decreases levels of Abeta and APP-CTF via Atg5-dependent autophagy pathway. FASEB J. 2011;25:1934–1942. doi: 10.1096/fj.10-175158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Williams A, Sarkar S, Cuddon P, et al. Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4:295–305. doi: 10.1038/nchembio.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Taniguchi S, Suzuki N, Masuda M, et al. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J Biol Chem. 2005;28:7614–7623. doi: 10.1074/jbc.M408714200. [DOI] [PubMed] [Google Scholar]
- 166.Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci U S A. 1996;93:11213–11218. doi: 10.1073/pnas.93.20.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Necula M, Breydo L, Milton S, et al. Methylene blue inhibits amyloid Abeta oligomerization by promoting fibrillization. Biochemistry. 2007;46:8850–8860. doi: 10.1021/bi700411k. [DOI] [PubMed] [Google Scholar]
- 168.Gonzalez-Lima F, Bruchey AK. Extinction memory improvement by the metabolic enhancer methylene blue. Learn Mem. 2004;11:633–640. doi: 10.1101/lm.82404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Oz M, Lorke DE, Hasan M, Petroianu GA. Cellular and molecular actions of Methylene Blue in the nervous system. Med Res Rev. 2011;31:93–117. doi: 10.1002/med.20177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Xie L, Li W, Winters A, Yuan F, Jin K, Yang S. Methylene blue induces macroautophagy through 5' adenosine monophosphate-activated protein kinase pathway to protect neurons from serum deprivation. Front Cell Neurosci. 2013;7:56. doi: 10.3389/fncel.2013.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Congdon EE, Wu JW, Myeku N, et al. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy. 2012;8:609–622. doi: 10.4161/auto.19048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.O'Leary JC, 3rd, Li Q, Marinec P, et al. Phenothiazine-mediated rescue of cognition in tau transgenic mice requires neuroprotection and reduced soluble tau burden. Mol Neurodegener. 2010;5:45. doi: 10.1186/1750-1326-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Spires-Jones TL, Friedman T, Pitstick R, et al. Methylene blue does not reverse existing neurofibrillary tangle pathology in the rTg4510 mouse model of tauopathy. Neurosci Lett. 2014;562:63–68. doi: 10.1016/j.neulet.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Medina DX, Caccamo A, Oddo S. Methylene blue reduces abeta levels and rescues early cognitive deficit by increasing proteasome activity. Brain Pathol. 2011;21:140–149. doi: 10.1111/j.1750-3639.2010.00430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wischik C, Staff R. Challenges in the conduct of disease-modifying trials in AD: practical experience from a phase 2 trial of Tau-aggregation inhibitor therapy. J Nutr Health Aging. 2009;13:367–369. doi: 10.1007/s12603-009-0046-5. [DOI] [PubMed] [Google Scholar]
- 176.Wischik CM, Bentham P, Wischik DJ, Seng KM. O3-04-07: Tau aggregation inhibitor (TAI) therapy with rember™ arrests disease progression in mild and moderate Alzheimer's disease over 50 weeks. Alzheimers Dement. 2008;4:T167. [Google Scholar]
- 177.Vicencio JM, Ortiz C, Criollo A, et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ. 2009;16:1006–1017. doi: 10.1038/cdd.2009.34. [DOI] [PubMed] [Google Scholar]
- 178.Sarkar S, Floto RA, Berger Z, et al. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol. 2005;170:1101–1111. doi: 10.1083/jcb.200504035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sarkar S, Krishna G, Imarisio S, Saiki S, O'Kane CJ, Rubinsztein DC. A rational mechanism for combination treatment of Huntington's disease using lithium and rapamycin. Hum Mol Genet. 2008;17:170–178. doi: 10.1093/hmg/ddm294. [DOI] [PubMed] [Google Scholar]
- 180.Parr C, Carzaniga R, Gentleman SM, Van Leuven F, Walter J, Sastre M. Glycogen synthase kinase 3 inhibition promotes lysosomal biogenesis and autophagic degradation of the amyloid-beta precursor protein. Mol Cell Biol. 2012;32:4410–4418. doi: 10.1128/MCB.00930-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Avrahami L, Farfara D, Shaham-Kol M, Vassar R, Frenkel D, Eldar-Finkelman H. Inhibition of glycogen synthase kinase-3 ameliorates beta-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: in vivo and in vitro studies. J Biol Chem. 2013;288:1295–1306. doi: 10.1074/jbc.M112.409250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Sereno L, Coma M, Rodriguez M, et al. A novel GSK-3beta inhibitor reduces Alzheimer's pathology and rescues neuronal loss in vivo. Neurobiol Dis. 2009;35:359–367. doi: 10.1016/j.nbd.2009.05.025. [DOI] [PubMed] [Google Scholar]
- 183.Chen Q, Haddad GG. Role of trehalose phosphate synthase and trehalose during hypoxia: from flies to mammals. J Exp Biol. 2004;207:3125–3129. doi: 10.1242/jeb.01133. [DOI] [PubMed] [Google Scholar]
- 184.Jain NK, Roy I. Effect of trehalose on protein structure. Protein Sci. 2009;18:24–36. doi: 10.1002/pro.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Tanaka M, Machida Y, Niu S, et al. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med. 2004;10:148–154. doi: 10.1038/nm985. [DOI] [PubMed] [Google Scholar]
- 186.Liu R, Barkhordarian H, Emadi S, Park CB, Sierks MR. Trehalose differentially inhibits aggregation and neurotoxicity of beta-amyloid 40 and 42. Neurobiol Dis. 2005;20:74–81. doi: 10.1016/j.nbd.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 187.Zhang L, Yu J, Pan H, et al. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc Natl Acad Sci U S A. 2007;104:19023–19028. doi: 10.1073/pnas.0709695104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Du J, Liang Y, Xu F, Sun B, Wang Z. Trehalose rescues Alzheimer's disease phenotypes in APP/PS1 transgenic mice. J Pharm Pharmacol. 2013;65:1753–1756. doi: 10.1111/jphp.12108. [DOI] [PubMed] [Google Scholar]
- 189.Schaeffer V, Lavenir I, Ozcelik S, Tolnay M, Winkler DT, Goedert M. Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain. 2012;135:2169–2177. doi: 10.1093/brain/aws143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lee LH, Shineman DW, Fillit HM. A diverse portfolio of novel drug discovery efforts for Alzheimer's disease: Meeting report from the 11th International Conference on Alzheimer's Drug Discovery, 27–28 September 2010, Jersey City, NJ, USA. Alzheimers Res Ther. 2010;2:33. doi: 10.1186/alzrt57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kruger U, Wang Y, Kumar S, Mandelkow EM. Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol Aging. 2012;33:2291–2305. doi: 10.1016/j.neurobiolaging.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 192.Singer MA, Lindquist S. Multiple effects of trehalose on protein folding in vitro and in vivo. Mol Cell. 1998;1:639–648. doi: 10.1016/s1097-2765(00)80064-7. [DOI] [PubMed] [Google Scholar]
- 193.Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009;16:46–56. doi: 10.1038/cdd.2008.110. [DOI] [PubMed] [Google Scholar]
- 194.Casarejos MJ, Solano RM, Gomez A, Perucho J, de Yebenes JG, Mena MA. The accumulation of neurotoxic proteins, induced by proteasome inhibition, is reverted by trehalose, an enhancer of autophagy, in human neuroblastoma cells. Neurochem Int. 2011;58:512–520. doi: 10.1016/j.neuint.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 195.Zhang X, Chen S, Song L, et al. MTOR-independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy. 2014;10:588–602. doi: 10.4161/auto.27710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Kilgore M, Miller CA, Fass DM, et al. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology. 2010;35:870–880. doi: 10.1038/npp.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Guan JS, Haggarty SJ, Giacometti E, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Xie M, Kong Y, Tan W, et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation. 2014;129:1139–1151. doi: 10.1161/CIRCULATIONAHA.113.002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Yoshiike Y, Yamashita S, Mizoroki T, et al. Adaptive responses to alloxan-induced mild oxidative stress ameliorate certain tauopathy phenotypes. Aging Cell. 2012;11:51–62. doi: 10.1111/j.1474-9726.2011.00756.x. [DOI] [PubMed] [Google Scholar]
- 200.Piperno G, LeDizet M, Chang XJ. Microtubules containing acetylated alpha-tubulin in mammalian cells in culture. J Cell Biol. 1987;104:289–302. doi: 10.1083/jcb.104.2.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Hubbert C, Guardiola A, Shao R, et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 202.Zhang B, Maiti A, Shively S, et al. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc Natl Acad Sci U S A. 2005;102:227–231. doi: 10.1073/pnas.0406361102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Nunes P, Ernandez T, Roth I, et al. Hypertonic stress promotes autophagy and microtubule-dependent autophagosomal clusters. Autophagy. 2013;9:550–567. doi: 10.4161/auto.23662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Xie R, Nguyen S, McKeehan WL, Liu L. Acetylated microtubules are required for fusion of autophagosomes with lysosomes. BMC Cell Biol. 2010;11:89. doi: 10.1186/1471-2121-11-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Fellner S, Bauer B, Miller DS, et al. Transport of paclitaxel (Taxol) across the blood-brain barrier in vitro and in vivo. J Clin Invest. 2002;110:1309–1318. doi: 10.1172/JCI15451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Brunden KR, Yao Y, Potuzak JS, et al. The characterization of microtubule-stabilizing drugs as possible therapeutic agents for Alzheimer's disease and related tauopathies. Pharmacol Research. 2011;63:341–351. doi: 10.1016/j.phrs.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Zhang B, Carroll J, Trojanowski JQ, et al. The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J Neurosci. 2012;32:3601–3611. doi: 10.1523/JNEUROSCI.4922-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Barten DM, Fanara P, Andorfer C, et al. Hyperdynamic microtubules, cognitive deficits, and pathology are improved in tau transgenic mice with low doses of the microtubule-stabilizing agent BMS-241027. J Neurosci. 2012;32:7137–7145. doi: 10.1523/JNEUROSCI.0188-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Brunden KR, Zhang B, Carroll J, et al. Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. J Neurosci. 2010;30:13861–13866. doi: 10.1523/JNEUROSCI.3059-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Lou K, Yao Y, Hoye AT, et al. Brain-penetrant, orally bioavailable microtubule-stabilizing small molecules are potential candidate therapeutics for Alzheimer's disease and related tauopathies. J Med Chem. 2014;57:6116–6127. doi: 10.1021/jm5005623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Brunden KR, Gardner NM, James MJ, et al. MT-stabilizer, dictyostatin, exhibits prolonged brain retention and activity: potential therapeutic implications. ACS Med Chem Lett. 2013;4:886–889. doi: 10.1021/ml400233e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Butler D, Hwang J, Estick C, et al. Protective effects of positive lysosomal modulation in Alzheimer's disease transgenic mouse models. PLoS One. 2011;6:e20501. doi: 10.1371/journal.pone.0020501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Sun B, Zhou Y, Halabisky B, et al. Cystatin C-cathepsin B axis regulates amyloid beta levels and associated neuronal deficits in an animal model of Alzheimer's disease. Neuron. 2008;60:247–257. doi: 10.1016/j.neuron.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Gauthier S, Kaur G, Mi W, Tizon B, Levy E. Protective mechanisms by cystatin C in neurodegenerative diseases. Front Biosci (Schol Ed) 2011;3:541–554. doi: 10.2741/s170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Zhong XM, Hou L, Luo XN, et al. Alterations of CSF cystatin C levels and their correlations with CSF Alphabeta40 and Alphabeta42 levels in patients with Alzheimer's disease, dementia with lewy bodies and the atrophic form of general paresis. PLoS One. 2013;8:e55328. doi: 10.1371/journal.pone.0055328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Liu Y, Cai H, Wang Z, et al. Induction of autophagy by cystatin C: a potential mechanism for prevention of cerebral vasospasm after experimental subarachnoid hemorrhage. Eur J Med Res. 2013;18:21. doi: 10.1186/2047-783X-18-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Tizon B, Sahoo S, Yu H, et al. Induction of autophagy by cystatin C: a mechanism that protects murine primary cortical neurons and neuronal cell lines. PLoS One. 2010;5:e9819. doi: 10.1371/journal.pone.0009819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Zerovnik E. The emerging role of cystatins in Alzheimer's disease. BioEssays. 2009;31:597–599. doi: 10.1002/bies.200900012. [DOI] [PubMed] [Google Scholar]
- 219.Mueller-Steiner S, Zhou Y, Arai H, et al. Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer's disease. Neuron. 2006;51:703–714. doi: 10.1016/j.neuron.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 220.Qiao L, Hamamichi S, Caldwell KA, et al. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol Brain. 2008;1:17. doi: 10.1186/1756-6606-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Valenzano KJ, Khanna R, Powe AC, et al. Identification and characterization of pharmacological chaperones to correct enzyme deficiencies in lysosomal storage disorders. Assay Drug Develop Technol. 2011;9:213–235. doi: 10.1089/adt.2011.0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Ravikumar B, Berger Z, Vacher C, O'Kane CJ, Rubinsztein DC. Rapamycin pre-treatment protects against apoptosis. Hum Mol Genet. 2006;15:1209–1216. doi: 10.1093/hmg/ddl036. [DOI] [PubMed] [Google Scholar]
- 223.Wang QJ, Ding Y, Kohtz DS, et al. Induction of autophagy in axonal dystrophy and degeneration. J Neurosci. 2006;26:8057–8068. doi: 10.1523/JNEUROSCI.2261-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 499 kb)