

Abstract
The retromer is an evolutionary conserved multiprotein complex involved in the sorting and retrograde trafficking of cargo from endosomal compartments to the Golgi network and to the cell surface. The neuronal retromer traffics the amyloid precursor protein away from the endosomes, a site where amyloid precursor protein is enzymatically cleaved into pathogenic fragments in Alzheimer’s disease. In recent years, deficiencies in retromer-mediated transport have been implicated in several neurological and non-neurological diseases, including Parkinson’s disease, suggesting that improving the efficacy of the retromer trafficking pathway would result in decreased pathology. We recently identified a new family of small molecules that appear to stabilize the interaction between members of the retromer complex and enhance its function in neurons: the retromer pharmacological chaperones. Here we discuss the role of these molecules in the improvement of retromer trafficking and endosomal dysfunction, as well as their potential as therapeutics for neurological and non-neurological disorders.
Electronic supplementary material
The online version of this article (doi:10.1007/s13311-014-0321-y) contains supplementary material, which is available to authorized users.
Key Words: Retromer, Alzheimer’s disease, neurodegeneration, pharmacological chaperones
Endosomes, Retromer, and Alzheimer’s Disease
A common emerging theme in the research field of age-related neurodegenerative diseases is that of endosomal dysfunction [1]. Specifically in Alzheimer’s disease (AD), primary evidence pinpointing the endosome as the intracellular site of dysfunction was described in a seminal series of histological studies performed by Cataldo et al. [2]. They showed that endosomes in the entorhinal cortex and the cornu ammonis subfields of the hippocampal formation are abnormally enlarged during the earliest stages of disease. Remarkably, the distribution of abnormally enlarged endosomes was nearly diagnostic of the disease, with very little overlap in the size of endosomes observed in age-matched controls. Additionally, they showed that carrying the apolipoprotein E4 gene variant, the strongest genetic risk factor for late-onset AD, further enhanced endosomal enlargement, and that abnormal enlargement did not occur as part of normal aging. The near uniformity of the abnormality in patients with late-onset “sporadic” AD suggests a convergence of pathogenic mechanisms leading to endosomal enlargement. In the case of AD, this is particularly important as the endosomal compartment of the cell is one of the main sites where amyloid precursor protein (APP) interacts with β-site APP-cleaving enzyme 1 (BACE1), initiating a pathogenic process leading to the accumulation of a number of putatively neurotoxic proteolytic fragments, including beta-amyloid (Aβ), the main component of the amyloid plaques found in AD brains. The fact that enlarged endosomes represent a cell-biological phenotype of AD has recently been validated in neurons derived from patients via induced pluripotent stem cells technology [3]. Endosomes are a hub of membrane trafficking, and mechanistically it can be assumed that they become enlarged either because of accelerated delivery, via cell-surface endocytosis or by reducing the transport of molecules out of endosomes. Taken together, these observations establish that endosomal dysfunction is a pathogenic mechanism, and validate endosomal transport as a “cell biological” target for drug discovery.
As we will discuss below, proteins and mechanisms that modulate the endosomal residency time of APP, BACE1, and other key molecular players, and hence their physical interaction, could thus regulate the production of Aβ and have implications for AD. One such mechanism is the retromer-dependent retrieval of transmembrane proteins, such as APP, from endosomes to the trans-Golgi network and the plasma membrane. We will begin by looking in some detail at these key molecular entities, as their basic biology is important to further understand their roles in neuronal AD-related retromer-mediated trafficking.
APP is a type I transmembrane protein that can undergo proteolytic processing in different ways by different sets of enzymes—in normal healthy conditions, a minor pathway leads to amyloid plaque formation (the amyloidogenic pathway), while the other major pathway does not (the nonamyloidogenic pathway). In the nonamyloidogenic pathway, APP is cleaved first by α-secretase to yield a soluble N-terminal fragment (sAPPα) and a C-terminal fragment (αCTF). The sAPPα fragment may be involved in the enhancement of synaptogenesis, neurite outgrowth, and neuronal survival, and is considered to be neuroprotective [4]. In the amyloidogenic pathway, APP is cleaved first by BACE1 (a transmembrane aspartic protease), yielding a soluble N-terminal fragment (sAPPβ) and a membrane-bound C-terminal fragment (βCTF). βCTF is then acted upon by γ-secretase yielding a membrane-bound C-terminal fragment and a soluble N-terminal fragment, Aβ. Aβ can then accumulate in the extracellular space of the brain, where it can form oligomers, insoluble fibrils, and amyloid plaques if they are insufficiently cleared, as observed in advanced AD. The progression to AD is thus determined by the proportion of APP that is processed by the pathways initiated by BACE1 (i.e., amyloidogenic) and α-secretases (i.e., nonamyloidogenic). APP is constitutively actively sorted and trafficked through membrane compartments of the cell. After internalization from the cell surface and while it temporarily resides in endosomes, APP is most likely to interact and be cleaved by BACE1 [5]. Previous studies have shown that retromer-related defects reduced trafficking of APP out of endosomes and caused an elevation in Aβ levels in vivo [6, 7]. That said, a recent study performed on HEK293 cells using an RNA interference approach suggests that Aβ40 is generated predominantly in the TGN [8]; however, in this case no other APP proteolytic fragments were studied. Yet another study suggests that APP and BACE1 can physically interact at the plasma membrane level and along the late secretory pathway and result in APP proteolysis [9]. So there is still conflicting evidence in the literature regarding the intracellular compartment(s) where the amyloidogenic processing of APP takes place; however, many of the studies suggesting a nonendosomal locus of amyloidogenic APP processing have been performed in experimental conditions that do not resemble the normal physiological neuronal state. Nevertheless, one aspect of Aß production is clear: the more time APP and BACE1 reside and interact with each other in any given cellular compartment under the right conditions (e.g., an acidic pH for optimal BACE1 enzymatic activity), the more Aβ is likely to be produced by the cell. This interplay will be determined by the dynamic trafficking routes of each of these molecular components separately. While we know that retromer regulates APP trafficking, less is known about the role of retromer affecting BACE1 transport. BACE1 also shuttles between different membranous subcellular compartments [9], hence raising the critical, and still unanswered, question of how both these important AD molecular players come in closer contact and trigger amyloid-related AD pathophysiology.
Another key player in the regulation of APP processing at the endosomal level is a sorting protein-related receptor containing low-density lipoprotein receptor class A repeats (SorLA). Like APP, SorLA is a type I transmembrane protein that is expressed in neurons and is a critical participant in the intracellular transport and processing of APP. Through its Vps10-like structural similarity, SorLA binds to the retromer complex and serves as an adaptor protein for the trafficking of APP from endosomes to the TGN [10]. It has been shown that SorLA levels are reduced in affected brain areas in patients with AD [10], and that overexpression of SorLA redistributes APP to the Golgi apparatus thereby reducing the residency and interaction time of APP and BACE1 in the early endosomes. Consequently, Aβ levels are decreased in SorLA overexpression conditions. These findings became more relevant when variants in SORL1 (coding for SorLA) associated with late-onset AD were identified a few years ago [11].
The retromer complex was first identified in yeast, mediating the retrieval of the acid hydrolase receptors from endosomes (vacuoles in yeast) to the Golgi apparatus [12]. Further studies showed that the function and components of the retromer complex is evolutionary conserved across species. Retromers are heteropentameric protein complexes composed mainly of 2 components: 1) a structural component made of a dimer of sorting nexins (SNX), which mediates the binding to lipids in the endosomal membrane; and 2) a core cargo recognition element, a heterotrimer comprised of ‘vacuolar protein sorting’ (VPS) proteins Vps35–Vps29–Vps26. Vps35 is the core component of the complex, which then binds Vps26 (there are 2 isoforms: Vps26a and Vps26b), Vps29, and the SNX proteins [13, 14]. This core trimeric complex binds to be-transported transmembrane proteins while they reside in endosomal membranes. Most relevant to AD, these transmembrane proteins are typically receptors, such as VPS10 domain-containing receptors [e.g., SorLA, Sortilin, and sortilin-related VPS10 domain containing receptor (SORCS)1, SORCS2, and SORCS3], and the mannose-6-phosphate receptor. These receptors often bind other proteins, which are trafficked together with their receptors. To date, the most disease-relevant among these trafficked proteins is APP. A few years ago, by using model-guided microarray techniques, our laboratory first identified deficiencies in the levels of the core retromer component Vps35 in the hippocampus of AD brains, suggesting a possible involvement of retromer function and trafficking in the pathogenic processing of APP [15]. In addition, protein levels of Vps26 were also reduced in the entorhinal cortex of patients with AD. In parallel in vitro cell culture studies, reductions of Vps35 levels by using siRNA in HeLa cells increased the production of Aβ40 levels, while overexpression of Vps35 led to decreased production. Further in vivo studies showed that Vps26 knockdown mice exhibit altered hippocampal-dependent memory function and long-term potentiation, and retromer-deficient flies expressing human APP and BACE1 showed an increase in neuronal loss [7]. This neuronal loss was accompanied by accumulation of Aβ, suggesting a functional correlation between retromer and APP processing and trafficking.
These data, together with that of other laboratories, led to the hypothesis that increasing the levels and stability of the retromer complex would improve its function and rescue deficiencies in the trafficking and transport cellular machinery. From a more molecular and structural point of view, it has been observed that knocking down just 1 protein of the Vps35–Vps29–Vps26 heterotrimer core complex leads to secondary reductions in the other retromer members, suggesting that the interactions among the individual proteins are not only important for complex function, but also for its stability and half-life. We hypothesized that increasing the interaction between the retromer proteins would increase complex stability and enhance retromer-mediated trafficking and transport. Moreover, we hypothesized that small molecules could achieve these goals.
Small Molecules as Retromer Chaperones
Most proteins in all cell types in the human body turn over regularly. If such proteins have a mutation, or become targets of a pathological mechanism, they may be degraded more rapidly, thereby lowering the steady-state levels below those needed to maintain a healthy living cell. In the context of pharmacology, the term “chaperone” refers to small molecules that stabilize an already native folded macromolecule (usually a protein) by binding to it and stabilizing it against thermal denaturation and proteolytic degradation [16]. Pharmacological chaperones are designed specifically to bind to their target proteins, acting as a sort of molecular ‘glue’, holding various parts of the protein structure together. The greatest challenge in isolating effective pharmacological chaperones is that most pathogenic proteins do not possess natural binding sites. Prior knowledge of a protein’s 3-dimensional (3D) structure provides a tractable solution to this problem, as this information can be used to identify a protein’s potential “docking sites”, to which chaperones can bind [17]. Noteworthy, a recent study introduced a novel high-throughput method for the identification of pharmacological chaperones, particularly those related to protein–protein interactions. The investigators were able to generate a novel compound library enriched with pharmacological chaperones, which could potentially obviate the need to predetermine the 3D structure of a targeted protein, relying on the observation that short α-helical peptide segments, spanning 2–3 helical turns, often play key roles in protein–protein interactions [18]. Most of the work up to now has been focused on chaperoning unstable mutant proteins, but there is no reason, in principle, to use chaperones to stabilize the normal form of a protein and thereby increasing its steady-state cellular levels, as we have done with the mammalian retromer complex.
Relying on the already known 3D structure of the complex formed between the retromer core proteins Vps35 and Vps29, our team performed a computerized virtual in silico screen that identified several small molecules that were predicted to bind to specific pockets created at the interface of the Vps35–Vps29 interaction. We decided to focus on the Vps35–Vps29 complex interaction based on the results of our initial thermal stability assays, which showed that the interface between these 2 members within the retromer core complex was the weakest link within the complex as a whole [19]. Of the 10 docking sites we found on the Vps35–Vps29 protein complex, 6 were located at the interface between the 2 proteins, suggesting that any ligand binding at these sites would most likely interact with residues from both, possibly providing stabilization to the entire complex. Nonetheless, we screened all 10 of the sites against the entire library of compound structures. The docking procedure allowed full flexibility in the conformation of the ligand and limited flexibility in the conformations of the amino acid side chains in the binding sites. Only 4 of the 10 predicted docking sites on the Vps35–Vps29 complex generated computed interaction scores with test molecules worth testing in the secondary in vitro screen. Each of these four sites was at the interface of the 2 proteins. Further in vitro thermal stabilization screenings followed by in vivo studies in primary neuronal cultures led to the identification of 2 chaperones, named R55 and R33 (both thiophene thiourea derivatives; Fig. 1), which were able to stabilize the retromer complex against thermal denaturation and were also able to increase the levels of the retromer complex in hippocampal neurons [19]. We specifically focused on hippocampal neurons because the hippocampal formation is a brain structure enriched in retromer proteins and hippocampal cultured neurons have been previously used to investigate retromer-mediated trafficking of APP. These small molecules were found to enhance the function of retromer in neurons, as indicated by the shifting of the localization of APP and SorLA out of endosomes. We were also able to show that the retromer pharmacological chaperones resulted in an APP fragment profile indicative of a reduced BACE1 cleavage, characterized by a reduction in the BACE1-dependent fragments (βCTF, sAPPβ, Aβ40, and Aβ42) and a concomitant increase in sAPPα [19]. Documenting a reduction in BACE1-dependent processing biochemically confirmed the microscopic observation that the pharmacological chaperones reduce endosomal APP levels, indicating that the chaperones enhance retromer’s function, at least for APP. These results confirmed our hypothesis, namely that increasing the interaction between retromer proteins would increase the stability of the complex as a whole and enhance retromer-mediated trafficking function. To our knowledge, this is the first class of small molecules that can apparently act as pharmacological retromer chaperones by stabilizing a multiprotein complex and improving its physiological function.
Fig. 1.
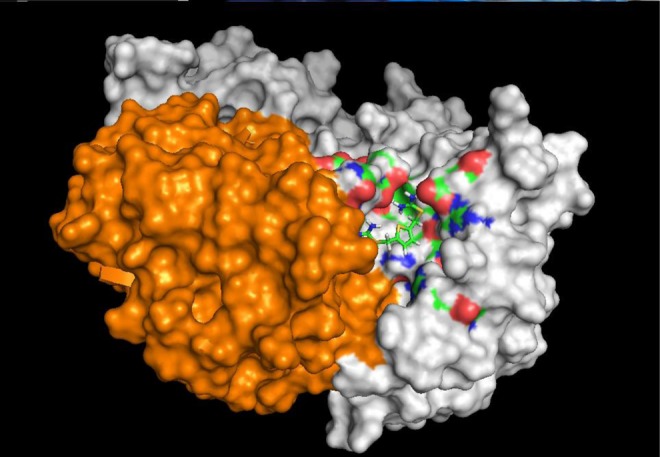
Space-filling diagram depicting the retromer chaperone R55 docked at its interface pocket between vacuolar protein sorting (Vps)29 (in orange) and Vps35 (in gray). R55 is predicted to make interactions with side chains from both proteins (multicolored surfaces). R55 has the systematic chemical name thiophene-2,5-diylbis(methylene) dicarbamimidothioate dihydrochloride. Binding studies using microscale thermophoresis established its Kd for the retromer core complex to be about 5 μM [19]
The retromer chaperones show decent in vitro water solubility and cell penetrability. Many of the in vivo properties of the retromer chaperones such as blood–brain barrier permeability, bioavailability, and effective doses and administration regimes/routes are yet unknown. Unpublished data from the National Cancer Institute and preliminary data from our laboratory show that these molecules are relatively nontoxic to mice. Further studies aimed at characterizing the effects of the retromer chaperones on retromer complex stability and on APP processing in vivo are actively under way.
This novel class of retromer chaperones can, in principle, traffic APP away from a pathogenic towards a nonpathogenic processing pathway, generating a profile of proteolytic fragments that mimics a fragment profile generated by a mutation in APP shown to be protective against AD [20]. This type of pharmacological intervention would differ substantially from the current approaches aimed at targeting solely the production of Aβ for example by inhibiting BACE1, or by blocking Aβ toxicity as in the case of antiamyloid immunotherapies and anti-Aβ aggregation agents. The retromer pharmacological chaperones are designed to treat defects in the cellular biology rather than a single molecular deficiency. By acting upstream of endosomal APP processing, the retromer chaperones are able to improve APP trafficking by normalizing cargo flow towards the Golgi and also perhaps towards the plasma membrane. This “whole-cell” therapeutic approach may prove to be safer and more effective than those that only aim at targeting an individual molecule or step in the disease process.
Somehow unexpectedly, retromer has been recently linked to another pathological feature of AD, namely microglia abnormalities [21]. Glia abnormalities were actually first described by Alois Alzheimer, and together with amyloid plaques and neurofibrillary tangles, is a third histological feature of the disease that bears his name. A recent study found that Vps35 is deficient in microglia harvested from AD brains and that retromer in microglia is required for the normal cell surface delivery of 2 microglia-enriched receptors—Trem2 and CD36 [21]. AD-linked variants have been found in the gene encoding Trem2 [22], and the normal cell surface delivery of CD36 is required for the microglia-meditated Aβ-clearing effects. We are currently exploring if the retromer chaperones would be effective at improving the glia-related trafficking phenotype, as we have shown in neuronal cells.
Retromer in Other Diseases
In principle, improving endosomal sorting and increasing the trafficking of APP away from its pathogenic locus toward a more neuroprotective route is worth exploring as a potential therapeutic strategy. If viable, this therapeutic approach would not only turn out to be beneficial for AD, but perhaps also be useful for other disorders where endosomal dysfunction has been described, for example PD [23]. Several studies have recently implicated the retromer complex itself in PD [24–26]. To date, a dozen genes have been implicated in PD, including a point mutation (D620N) in Vps35, which results in autosomal dominant familial PD [27, 28]. This PD-linked Vps35 mutation does not seem to affect the stability of the core Vps35–Vps26–Vps29 retromer complex per se, nor appears to affect the function of the complex in endosome-to-Golgi cargo retrieval. However, the mutation seems to impair the ability of retromer to bind and recruit the Wiskott–Aldrich Syndrome Protein and SCAR Homolog (WASH) complex to endosomes, thereby affecting processes that rely in WASH-mediated trafficking to the plasma membrane. Interestingly, this particular PD mutation has been found to impair the formation of autophagosomes and the clearance of autophagy substrates, resulting in a clear neurodegenerative phenotype [27].
In recent years, the list of diseases involving retromer and/or retromer-related proteins/pathways has grown substantially, and this trend seems to continue. Retromer-related dysfunctions have been reported not only in neurological disorders such as Down syndrome [29, 30], hereditary spastic paraplegia [31], cerebral lipofuscinosis [32], prionopathy [33], and Nieman Pick type C1 disease [34], but have also been linked to non-neurological disorders such as type II diabetes [35, 36], osteoporosis [37], retinal degeneration [38], and Legionella disease [39] (see Table 1). Several of these diseases have a clear pathological molecular link to retromer; in other cases, the connection is less clear and more tangential. Fortunately, the existence of several animal models for most of these retromer trafficking-related disorders would allow us to test if improving the retromer pathway either by genetic overexpression of one, or several, of the retromer components, or by the use of the pharmacological chaperones would result in an amelioration of the different pathologies in vivo.
Table 1.
Potential therapeutic use of the retromer chaperones in human disease. List of neurological and non-neurological diseases with established links to the retromer pathway
| Pathogenic link‡ | Established pathway§ | Animal models | |
|---|---|---|---|
| Alzheimer’s disease* | + + + | APP endosomal trafficking | + + + |
| Parkinson’s disease* | + + + | Vps35 mutations, impaired autophagy | + |
| Down’s syndrome* | + | SNX27 dysfunction | + + |
| HSP* | + | Retromer/WASH complex interaction | + |
| Cerebral lipofuscinosis* | + | Deficient retromer recruitment machinery | + |
| Nieman Pick type C1* | +/– | ESCRT-mediated endosomal cholesterol trafficking | + |
| Type II diabetes† | + + | SORCS1 (Vps10 family) dysfunction | + + |
| Osteoporosis† | + | Vps35 deficiency, deregulation of RANK signaling | + |
HSP = hereditary spastic paraplegia; APP = amyloid precursor protein; Vps = vacuolar protein sorting; WASH = Wiskott–Aldrich Syndrome Protein and SCAR Homolog; ESCRT = Endosomal Sorting Complex Required for Transport; SORCS1 = sortilin-related VPS10 domain containing receptor; RANK = Receptor activator of nuclear factor-kappa B
*Neurological disease
†Non-neurological disease
‡The number of plus symbols depicts the amount/strength of experimental evidence associated in each disease with the role of the retromer complex/pathway
§A short description of the molecular/cellular pathway affected in each disease is shown. For further details please refer to the cited bibliography in the main text
Two points that emerge by looking at the list of diseases are worth noting. First, most retromer-associated diseases are disorders of the nervous system, which suggests that neurons, and/or glia cells, are particularly dependent on retromer function and are differentially sensitive to its dysfunction. Second, in all cases, the disease is caused by a loss rather than a gain of retromer function. This agrees with observations in yeast, neuronal culture, and animal models that have shown that, whereas a slight loss of retromer function has dramatic pathological consequences, increasing retromer levels seems to be either beneficial or innocuous [40]. This speaks to the concern that increasing retromer levels in many cells, be it genetically or pharmacologically, might have negative side effects. We note that chaperones are, in principle, different from other common pharmacological agents, which typically act by inhibiting key enzymes or receptors. It is possible that a slight increase in the normal flow through the endosome might not be necessarily deleterious. Obviously, this prediction must be empirically tested in future studies.
Conclusions and Perspectives
Endosomal dysfunction seems to be a unified “cell biological” defect in AD—one that is a convergent point of multiple upstream molecular mechanisms, and also seems to occur before amyloid pathology [41]. Endosomal dysfunction, characterized by enlarged endosomes, is likely mediated by endosomal transport and trafficking defects. Retromer has emerged as a master regulator of endosomal transport [42]. And in this way, by targeting the retromer, we are targeting the unified cell biology of AD.
Our newly described retromer pharmacological chaperones provide, at the very least, proof-of-principle that the multiprotein retromer complex can be targeted pharmacologically. There is, of course, a long road ahead to determine whether this first crop of compounds, or analogues designed around them, will successfully make it through the stringent efficacy and safety testing of preclinical drug discovery. However, even if they fail, the successes to date should justify a continued focus on endosomal transport as a cell biological target for drug discovery in AD.
Electronic supplementary material
(PDF 1225 kb)
Acknowledgments
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
References
- 1.Karch CM, Cruchaga C, Goate AM. Alzheimer’s disease genetics: from the bench to the clinic. Neuron. 2014;83:11–26. doi: 10.1016/j.neuron.2014.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000;157:277–286. doi: 10.1016/S0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Israel MA, Yuan SH, Bardy C, et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 2012;482:216–220. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saitoh T, Sundsmo M, Roch JM, et al. Secreted form of amyloid beta protein precursor is involved in the growth regulation of fibroblasts. Cell. 1989;58:615–622. doi: 10.1016/0092-8674(89)90096-2. [DOI] [PubMed] [Google Scholar]
- 5.Small SA, Gandy S. Sorting through the cell biology of Alzheimer’s disease: intracellular pathways to neurogenesis. Neuron. 2006;52:15–31. doi: 10.1016/j.neuron.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhalla A, Vetanovetz CP, Morel E, Chamoun Z, Di Paolo G, Small SA. The location and trafficking routes of the neuronal retromer and its role in amyloid precursor protein transport. Neurobiol Dis. 2012;47:126–134. doi: 10.1016/j.nbd.2012.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muhammad A, Flores I, Zhang H, et al. Retromer deficiency observed in Alzheimer’s disease causes hippocampal dysfunction, neurodegeneration, and Abeta accumulation. Proc Natl Acad Sci U S A. 2008;105:7327–7332. doi: 10.1073/pnas.0802545105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choy RW, Cheng Z, Schekman R. Amyloid precursor protein (APP) traffics from the cell surface via endosomes for amyloid β (Aβ) production in the trans-Golgi network. Proc Natl Acad Sci U S A. 2012;109:2077–2082. doi: 10.1073/pnas.1208635109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prabhu Y, Burgos PV, Schindler C, Farías GG, Magadán JG, Bonifacino JS. Adaptor protein 2-mediated endocytosis of the β-secretase BACE1 is dispensable for amyloid precursor protein processing. Mol Biol Cell. 2012;23:2339–2351. doi: 10.1091/mbc.E11-11-0944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andersen OM, Reiche J, Schmidt V, et al. Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci U S A. 2005;102:13461–13466. doi: 10.1073/pnas.0503689102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rogaeva E, Meng Y, Lee JH, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39:168–177. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seaman MN, Marcusson EG, Cereghino JL, Emr SD. Endosome to golgi retrieval of the vacuolar protein sorting receptor, Vps10p, requires the function of the VPS29, VPS30, and VPS35 gene products. J Cell Biol. 1997;137:79–92. doi: 10.1083/jcb.137.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonifacino JS, Hurley JH. Retromer. Curr Opin Cell Biol. 2008;20:427–436. doi: 10.1016/j.ceb.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Attar N, Cullen PJ. The retromer complex. Adv Enzym Regul. 2010;50:216–236. doi: 10.1016/j.advenzreg.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 15.Small SA, Kent K, Pierce A, et al. Model-guided microarray implicates the retromer complex in Alzheimer's disease. Ann Neurol. 2005;58:909–919. doi: 10.1002/ana.20667. [DOI] [PubMed] [Google Scholar]
- 16.Ringe D, Petsko GA. What are pharmacological chaperones and why are they interesting? J Biol. 2009;8:80. doi: 10.1186/jbiol186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Small SA. Pharmacological chaperones in the age of proteomic pathology. Proc Natl Acad Sci U S A. 2014;111:12274–12275. doi: 10.1073/pnas.1413111111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oh M, Lee JH, Wang W, et al. Potential pharmacological chaperones targeting cancer-associated MCL-1 and Parkinson disease-associated α-synuclein. Proc Natl Acad Sci U S A. 2014;111:11007–11012. doi: 10.1073/pnas.1320556111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mecozzi VJ, Berman DE, Simoes S, et al. Pharmacological chaperones stabilize retromer to limit APP processing. Nat Chem Biol. 2014;10:443–449. doi: 10.1038/nchembio.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jonsson T, Atwal JK, Steinberg S, et al. A mutation in APP protects against Alzheimer's disease and age-related cognitive decline. Nature. 2012;488:96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- 21.Lucin KM, O'Brien CE, Bieri G, et al. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron. 2013;79:873–886. doi: 10.1016/j.neuron.2013.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guerreiro R, Wojtas A, Bras J, Alzheimer Genetic Analysis Group et al. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013;368:117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shannon B, Soto-Ortolaza A, Rayaprolu S, et al. Genetic variation of the retromer subunits VPS26A/B-VPS29 in Parkinson’s disease. Neurobiol Aging. 2014;35:1958. doi: 10.1016/j.neurobiolaging.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacLeod DA, Rhinn H, Kuwahara T, et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron. 2013;77:425–439. doi: 10.1016/j.neuron.2012.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zavodszky E, Seaman MN, Moreau K, et al. Mutation in VPS35 associated with Parkinson's disease impairs WASH complex association and inhibits autophagy. Nat Commun. 2014;13:3828. doi: 10.1038/ncomms4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang HS, Toh J, Ho P, Tio M, Zhao Y, Tan EK. In vivo evidence of pathogenicity of VPS35 mutations in the Drosophila. Mol Brain. 2014;7:73. doi: 10.1186/s13041-014-0073-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zavodszky E, Seaman MN, Rubinsztein DC. VPS35 Parkinson mutation impairs autophagy via WASH. Cell Cycle. 2014;13:2155–2156. doi: 10.4161/cc.29734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vilariño-Güell C, Wider C, Ross OA, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet. 2011;89:162–167. doi: 10.1016/j.ajhg.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Zhao Y, Zhang X, et al. Loss of sorting nexin 27 contributes to excitatory synaptic dysfunction by modulating glutamate receptor recycling in Down's syndrome. Nat Med. 2013;19:473–480. doi: 10.1038/nm.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones MW. Sorting receptors at Down’s syndrome synapses. Nat Med. 2013;1:404–406. doi: 10.1038/nm.3152. [DOI] [PubMed] [Google Scholar]
- 31.Freeman C, Seaman MN, Reid E. The hereditary spastic paraplegia protein strumpellin: characterisation in neurons and of the effect of disease mutations on WASH complex assembly and function. Biochim Biophys Acta. 1832;2013:160–173. doi: 10.1016/j.bbadis.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mamo A, Jules F, Dumaresq-Doiron K, Costantino S, Lefrancois S. The role of ceroid lipofuscinosis neuronal protein 5 (CLN5) in endosomal sorting. Mol Cell Biol. 2012;32:1855–1866. doi: 10.1128/MCB.06726-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goold R, McKinnon C, Rabbanian S, Collinge J, Schiavo G, Tabrizi SJ. Alternative fates of newly formed PrPSc upon prion conversion on the plasma membrane. J Cell Sci. 2013;126:3552–3562. doi: 10.1242/jcs.120477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Du X, Kazim AS, Brown AJ, Yang H. An essential role of Hrs/Vps27 in endosomal cholesterol trafficking. Cell Rep. 2012;1:29–35. doi: 10.1016/j.celrep.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 35.Lane RF, St George-Hyslop P, Hempstead BL, Small SA, Strittmatter SM, Gandy S. Vps10 family proteins and the retromer complex in aging-related neurodegeneration and diabetes. J Neurosci. 2012;32:14080–14086. doi: 10.1523/JNEUROSCI.3359-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moriboto MV, Berman DE, Schneider RT, Zhang Y, Leibel R, Small SA. Hyperleucinemia causes hippocampal retromer deficiency linking diabetes to Alzheimer's disease. Neurobiol Dis. 2014;65:188–192. doi: 10.1016/j.nbd.2013.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xia WF, Tang FL, Xiong L, et al. Vps35 loss promotes hyperresorptive osteoclastogenesis and osteoporosis via sustained RANKL signaling. J Cell Biol. 2013;200:821–837. doi: 10.1083/jcb.201207154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu W, Tang FL, Erion J, Xiao H, Ye J, Xiong WC. Vps35 haploinsufficiency results in degenerative-like deficit in mouse retinal ganglion neurons and impairment of optic nerve injury-induced gliosis. Mol Brain. 2014;7:10. doi: 10.1186/1756-6606-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Finsel I, Ragaz C, Hoffmann C, et al. The Legionella effector RidL inhibits retrograde trafficking to promote intracellular replication. Cell Host Microbe. 2013;14:38–50. doi: 10.1016/j.chom.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 40.Linhart R, Wong SA, Cao J, et al. Vacuolar protein sorting 35 (Vps35) rescues locomotor deficits and shortened lifespan in Drosophila expressing a Parkinson’s disease mutant of Leucine-rich repeat kinase 2 (LRRK2) Mol Neurodegener. 2014;9:23. doi: 10.1186/1750-1326-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nixon RA. Endosome function and dysfunction in Alzheimer’s disease and other neurodegenerative diseases. Neurobiol Aging. 2005;3:373–382. doi: 10.1016/j.neurobiolaging.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 42.Burd C, Cullen PJ. Retromer: a master conductor of endosome sorting. Cold Spring Harb Perspect Biol 2014;6. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 1225 kb)