

Abstract
Purpose
T cells can be genetically modified to express an anti-CD19 chimeric antigen receptor (CAR). We assessed the safety and efficacy of administering autologous anti-CD19 CAR T cells to patients with advanced CD19+ B-cell malignancies.
Patients and Methods
We treated 15 patients with advanced B-cell malignancies. Nine patients had diffuse large B-cell lymphoma (DLBCL), two had indolent lymphomas, and four had chronic lymphocytic leukemia. Patients received a conditioning chemotherapy regimen of cyclophosphamide and fludarabine followed by a single infusion of anti-CD19 CAR T cells.
Results
Of 15 patients, eight achieved complete remissions (CRs), four achieved partial remissions, one had stable lymphoma, and two were not evaluable for response. CRs were obtained by four of seven evaluable patients with chemotherapy-refractory DLBCL; three of these four CRs are ongoing, with durations ranging from 9 to 22 months. Acute toxicities including fever, hypotension, delirium, and other neurologic toxicities occurred in some patients after infusion of anti-CD19 CAR T cells; these toxicities resolved within 3 weeks after cell infusion. One patient died suddenly as a result of an unknown cause 16 days after cell infusion. CAR T cells were detected in the blood of patients at peak levels, ranging from nine to 777 CAR-positive T cells/μL.
Conclusion
This is the first report to our knowledge of successful treatment of DLBCL with anti-CD19 CAR T cells. These results demonstrate the feasibility and effectiveness of treating chemotherapy-refractory B-cell malignancies with anti-CD19 CAR T cells. The numerous remissions obtained provide strong support for further development of this approach.
INTRODUCTION
Recent advances have improved the treatment of B-cell malignancies, but many patients still succumb to these diseases.1–7 Among patients with diffuse large B-cell lymphoma (DLBCL) refractory to second-line chemotherapy, < 50% of patients respond to third-line chemotherapy, and few experience long-term survival.1–3 In patients with DLBCL that has progressed after autologous stem-cell transplantation, median overall survival is < 10 months.4,8 Improved treatments for chemotherapy-refractory B-cell malignancies are clearly needed.
CD19 is an antigen expressed on malignant and normal B cells but not on other normal cells.9 Chimeric antigen receptors (CARs) are fusion proteins incorporating antigen-recognition domains and T-cell activation domains.10–14 T cells expressing anti-CD19 CARs recognize and kill CD19+ target cells.15–21 In our previous studies of anti-CD19 CAR T cells, multiple patients with indolent B-cell malignancies had specific depletion of normal B cells and lengthy remissions.22,23 Other groups have also reported regressions of B-cell malignancies in patients receiving infusions of anti-CD19 CAR T cells.24–31
We now report the first patients to our knowledge to obtain complete remissions (CRs) in chemotherapy-refractory DLBCL after receiving anti-CD19 CAR T cells. We have significantly changed our anti-CD19 CAR T-cell production process and clinical treatment protocol since our last report.23 After treatment with our modified anti-CD19 CAR protocol, 12 of 13 evaluable patients with a variety of B-cell malignancies obtained partial (PRs) or CRs.
PATIENTS AND METHODS
Clinical Trial and Patient Information
All enrolled patients provided informed consent. The protocol was approved by the institutional review board of the National Cancer Institute. CD19 expression by malignancies was confirmed by either flow cytometry or immunohistochemistry (IHC).
Preparation of Anti-CD19 CAR T Cells and Ex Vivo Assays
The gammaretroviral vector encoding the CAR (Fig 1A) has been described.21 Anti-CD19 CAR T cells were produced by adding the anti-CD3 monoclonal antibody OKT3 directly to whole peripheral-blood mononuclear cells (PBMCs) suspended in culture medium containing interleukin-2 (IL-2), as described in the Data Supplement.23,24 CAR T cells were dosed as a number of CD3+ CAR-positive cells/kg bodyweight (Table 1). The percentage of CAR-positive T cells was determined by flow cytometry and used to calculate the total number of cells to infuse to achieve the target dose. Flow cytometry, IHC, and quantitative polymerase chain reaction (qPCR) are described in the Data Supplement.21,23,32 L. Cooper and B. Jena provided a CAR-specific antibody used in certain experiments.33
Fig 1.

Anti-CD19 chimeric antigen receptor (CAR) design and function. (A) Schematic of anti-CD19 CAR. Single-chain (sc) Fv region that recognizes CD19 was derived from FMC63 monoclonal antibody. CAR contained CD28 costimulatory domain and T-cell receptor (TCR) –ζ T-cell activation domain. (B) Anti-CD19 CAR T cells were produced by activating peripheral-blood mononuclear cells (PBMCs) with anti-CD3 antibody OKT3 on day 0 and transducing T cells on day 2. Cells were ready for infusion on day 10. (C) CAR expression on T-cell surface of infused cells of patient No. 1 was detected with anti-Fab antibodies. Isotype control staining of same T cells is also shown. Plots are gated on live CD3+ lymphocytes. (D) Plots show isotype control staining and CD45RA versus CCR7 staining of CD3+ CAR positive–infused cells of patient No. 1. (E) Anti-CD19 CAR-transduced T cells of patient No. 1 were cultured for 4 hours with either CD19-K562 cells expressing CD19 or nerve growth factor receptor (NGFR) –K562 cells not expressing CD19. CAR T cells upregulated CD107a, indicating degranulation, in CD19-specific manner. Plots gated on live CD3+ lymphocytes. Anti-CD19 CAR T cells of patient No. 1 were cultured for 6 hours with CD19-K562 or NGFR-K562 cells, and intracellular cytokine staining for (F) interferon gamma (IFNγ), (G) tumor necrosis factor (TNF), and (H) interleukin-2 (IL-2) was performed. CAR T cells produced cytokines in CD19-specific manner. Plots gated on CD3+ lymphocytes. For (E) to (H), experiments were performed on T cells at time of infusion into patient No. 1. LTR, long terminal repeat.
Table 1.
Patient Clinical Characteristics
| Patient No. | Age (years) | Sex | Malignancy | No. of Prior Therapiesa | sAAIPI Risk Group | Total Cyclophosphamide Dose (mg/kg)b | No. of CAR-Positive T Cells Infused (× 106/kg) | Responsec |
Grade ≥ 3 Toxicitiesd | |
|---|---|---|---|---|---|---|---|---|---|---|
| Type | Duration (months) | |||||||||
| 1e | 56 | Male | SMZL | 4 | NA | 120 | 5 | PR | 23+f | Hypotension, confusion, acute renal failure, fever |
| 2 | 43 | Female | PMBCLg | 4 | Low | 60 | 5 | CR | 22+f | Fever, confusion, aphasia, facial nerve palsy, headache, urinary tract infection |
| 3 | 61 | Male | CLL (FR) | 2 | NA | 60 | 4 | CR | 23+f | Headache, fever, confusion, hypotension |
| 4 | 30 | Female | PMBCLg | 3 | High | 120 | 2.5 | NE | Nausea, hypoxia, dyspnea, tachycardia, fever, bacteremia, malaise, vascular leak syndrome, death | |
| 5e | 63 | Male | CLL | 4 | NA | 120 | 2.5 | CR | 15+f | None |
| 6 | 48 | Male | CLL (FR) | 1 | NA | 60 | 2.5 | CR | 14+f | None |
| 7 | 42 | Male | DLBCL NOSg | 5 | High | 60 | 2.5 | CR | 9+f | Influenza, fever, headache, bacteremia |
| 8 | 44 | Female | PMBCLg | 10 | High | 60 | 2.5 | CR | 12+f | Fever, pneumonitis, hypotension, hypoxia, bacteremia, obtundation, elevated creatinine |
| 9 | 38 | Male | PMBCLg | 3 | High | 120 | 2.5 | SD | 1 | Fever, aphasia, myoclonus |
| 10 | 57 | Female | Low-grade NHLh | 4 | NA | 60 | 1 | CR | 11+f | Bacteremia, fever, fatigue |
| 11 | 58 | Female | DLBCLg from CLL | 12 | High | 60 | 1 | PR | 1 | Bacteremia, urinary tract infection, fever |
| 12 | 60 | Female | DLBCL NOSg | 3 | High | 60 | 1 | NEi | Fever, urinary tract infection, bacteremia, upper extremity thrombosis | |
| 13 | 68 | Male | CLL | 4 | NA | 60 | 1 | PR | 4 | Dyspnea, upper extremity thrombosis, urinary tract infection, creatinine increase, hypotension |
| 14 | 43 | Male | DLBCL NOSg | 2 | High | 60 | 1 | CR | 6 | Fever |
| 15 | 64 | Female | DLBCL NOSh | 3 | Intermediate | 60 | 1 | PR | 6+f | Fever, aphasia, encephalopathy, neuropathy, gait disturbance |
Abbreviations: CAR, chimeric antigen receptor; CLL, chronic lymphocytic leukemia; CR, complete response; CT, computed tomography; DLBCL, diffuse large B-cell lymphoma; FR, fludarabine refractory; NA, not applicable; NE, not evaluable; NHL, non-Hodgkin lymphoma; NOS, not otherwise specified; PET, positron emission tomography; PMBCL, primary mediastinal B-cell lymphoma; PR, partial response; sAAIPI, second-line age-adjusted international prognostic index; SD, stable disease; SMZL, splenic marginal zone lymphoma.
All prior therapies for each patient are listed in Data Supplement.
This chemotherapy uniformly caused profound lymphocyte depletion on day of CAR T-cell infusion (Data Supplement).
Response duration is time from first documentation of response, which was 1 month after cell infusion in all patients, until progression. Some patients with CRs had initial response of PR that evolved into CR over time as PET scans and CT scans normalized. FR defined as progression ≤ 6 months after fludarabine administration.
All patients had cytopenias, including neutropenia, thrombocytopenia, and anemia, resulting from chemotherapy; these are not listed.
Patients No. 1 and 5 were previously treated on our anti-CD19 CAR T-cell protocol; they are patients No. 4 and 3, respectively, in study by Kochenderfer et al.23
Indicates ongoing response.
Chemotherapy refractory, defined as no achievement of PR or CR after most recent chemotherapy.
Relapse after autologous transplantation.
Lost to follow-up because patient refused to come to appointments.
Anti-CD19 CAR Treatment Plan
The clinical treatment plan consisted of a course of chemotherapy, followed 1 day later by a single infusion of anti-CD19 CAR T cells. The chemotherapy was cyclophosphamide at a total dose of either 120 or 60 mg/kg (Table 1), followed by five daily doses of fludarabine 25 mg/m2. Chemotherapy was administered before anti-CD19 CAR T cells to deplete endogenous leukocytes that can inhibit the antimalignancy activity of adoptively transferred T cells.34–37 Because of toxicity, the dose of cells was reduced from 5 to 1 × 106 CAR-positive T cells/kg during the study. Exogenous IL-2 was not administered to patients in this study. Treatment responses to chronic lymphocytic leukemia (CLL) or lymphoma were defined according to standard international criteria.38,39
RESULTS
Anti-CD19 CAR T Cells Generated From PBMCs of Heavily Treated Patients
The anti-CD19 CAR used in our work contained a CD28 costimulatory moiety (Fig 1A). CAR-expressing T cells were produced from autologous PBMCs with a 10-day cell-production process (Fig 1B). CAR T cells were successfully produced for all patients on the first attempt, despite the extensive prior treatment received by the patients. This work adds to previous evidence of the feasibility of autologous T-cell therapies for advanced hematologic malignancies.22,23,25,29 Some treated patients underwent apheresis for PBMC collection only 1 to 2 months after chemotherapy. Anti-CD19 CAR T cells were produced despite low blood lymphocyte counts at the time of the apheresis; for patients in the trial, the mean lymphocyte count at the time of apheresis was 632/μL, with a range of 140 to 1,470/μL (normal blood lymphocyte range, 1,320 to 3,570/μL; Data Supplement). CAR expression was measured on cells 1 to 3 days before infusion by flow cytometry (Fig 1C). A mean of 70% (range, 54% to 84%) of the infused T cells expressed the CAR. A mean of 34% of infused CAR-positive T cells had a C-C chemokine receptor type-7 (CCR7)–positive, CD45RA-negative phenotype. This phenotype is consistent with central memory T cells (Fig 1D; Data Supplement).40 T cells expressing the CAR upregulated CD107a in a CD19-specific manner (Fig 1E). CD107a upregulation indicates degranulation and correlates with cytotoxicity.41 The anti-CD19 CAR T cells produced cytokines in a CD19-specific manner (Figs 1F to 1H).
Patient Characteristics
Nine patients with DLBCL and six with indolent B-cell malignancies were treated (Table 1). We were able to evaluate malignancy response in 13 patients. One patient died soon after treatment, and one patient was lost to follow-up because of noncompliance. The nine patients with DLBCL had lymphomas of three different subtypes. Four patients had primary mediastinal B-cell lymphoma (PMBCL). Four patients had DLBCL not otherwise specified (NOS), and one patient had DLBCL transformed from CLL. Eight of nine patients with DLBCL had chemotherapy-refractory lymphoma. We defined chemotherapy refractoriness as no achievement of at least a PR with the most recent chemotherapy-containing salvage regimen before enrolling onto the anti-CD19 CAR protocol. Seven of the nine patients wtih DLBCL met the criteria for high risk by the second-line age-adjusted international prognostic index (Table 1).
Patients With Chemotherapy-Refractory DLBCL Obtained Remissions After Infusion of Anti-CD19 CAR T Cells
Of the seven evaluable patients with DLBCL, four obtained CRs, two obtained PRs, and one had stable disease (SD) after infusion of CAR T cells. All six patients with indolent B-cell malignancies obtained either a PR or CR (Table 1). Among patients with CLL, three of four are in ongoing CRs confirmed by multicolor flow cytometry of the bone marrow. The cyclophosphamide and fludarabine conditioning chemotherapy used in this study has activity against B-cell malignancies and could have made a direct contribution to antimalignancy responses.
Patient No. 2 was diagnosed with PMBCL. She underwent treatment with six cycles of rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP); the result was progressive lymphoma. She received mediastinal radiation therapy, which resulted in a CR that lasted 5 months before relapse. Next, she received two cycles of rituximab, ifosfamide, carboplatin, and etoposide (R-ICE) chemotherapy; the result was SD. Finally, she received a regimen of rituximab, cytarabine, and methotrexate, which also led to SD. Patient No. 2 was treated on the anti-CD19 CAR protocol and entered a CR that is ongoing after 22 months (Fig 2A).
Fig 2.

Complete remissions (CRs) of chemotherapy-refractory large-cell lymphomas in patients receiving anti-CD19 chimeric antigen receptor T cells. (A) Positron emission tomography (PET)/computed tomography (CT) scans show CR of chemotherapy-refractory primary mediastinal B-cell lymphoma (PMBCL) in patient No. 2. (B) PET/CT scans demonstrate CR of lymphoma in patient No. 8 who had chemotherapy-refractory PMBCL with extensive liver involvement. (C) PET/CT images show CR of diffuse large B-cell lymphoma, not otherwise specified, in patient No. 14, who had extensive splenic lymphoma.
Patient No. 7, with DLBCL NOS, was treated with five different treatment regimens before enrolling onto the anti-CD19 CAR protocol. His lymphoma progressed after his last salvage chemotherapy regimen. After treatment on the anti-CD19 CAR protocol, he entered a CR that is ongoing after 9 months.
Patient No. 8 was diagnosed with PMBCL. She was treated with 10 prior regimens before enrollment onto the anti-CD19 CAR protocol. The resistance of this lymphoma to chemotherapy was demonstrated by the fact that it progressed < 1 month after the patient received each of four different chemoimmunotherapy regimens: R-CHOP, R-ICE, rituximab plus high-dose cytarabine, and rituximab, gemcitabine, dexamethasone, and cisplatin. At the time of enrollment onto the anti-CD19 CAR protocol, patient No. 8 had a large burden of lymphoma in her liver and other areas. After treatment on the anti-CD19 CAR protocol, patient No. 8 entered a CR that is ongoing after 12 months (Fig 2B).
Patient No. 14 had DLBCL NOS that progressed after R-CHOP and also progressed after the rituximab, etoposide, methylprednisolone, high-dose cytarabine, and cisplatin regimen. He obtained a CR after treatment on the anti-CD19 CAR protocol (Fig 2C), but his lymphoma recurred after 6 months.
Infusion of Anti-CD19 CAR T Cells Was Associated With Significant but Transient Toxicity
Grade 3 and 4 toxicities experienced by patients are listed in Table 1. Toxicities mostly occurred during the first 2 weeks after infusion. Patient No. 4, who had chemotherapy-refractory PMBCL with extensive fibrotic mediastinal lymphoma involvement, died suddenly 16 days after infusion of anti-CD19 CAR T cells. The patient was not experiencing signs of cytokine-release toxicities such as fever at the time of death. She had a modestly decreased left ventricular ejection fraction and sinus tachycardia before infusion of CAR T cells. Because no cause of death was discovered at autopsy, a likely cause of death was cardiac arrhythmia. Four of the 15 patients in the trial experienced grade 3 or 4 hypotension. All patients had elevations in serum interferon gamma and/or IL-6 around the time of peak toxicity, but most patients did not develop elevations in serum tumor necrosis factor (Data Supplement).
Patients experienced a variety of neurologic toxicities that have been previously reported in those receiving infusions of CAR T cells or high-dose IL-2.23,42 These toxicities included confusion and obtundation. In addition, three of the 15 patients developed different and unexpected neurologic abnormalities. On day 5 after anti-CD19 CAR T-cell infusion, patient No. 2 developed aphasia that occurred intermittently for 7 days before resolving. She also had right-sided facial paresis that lasted approximately 20 minutes on day 8 after CAR T-cell infusion. At the time of these neurologic abnormalities, the CSF contained 14 WBC/μL; qPCR analysis showed that 1.9% of these WBCs contained the anti-CD19 CAR gene. Patient No. 9 developed aphasia 5 days after CAR T-cell infusion. Subsequently, he developed confusion and severe generalized myoclonus; all of these abnormalities resolved by 11 days after CAR T-cell infusion, except for a mild tremor that resolved over the next month. Patient No. 15 developed aphasia 5 days after CAR T-cell infusion, which was rapidly followed by onset of confusion, hemifacial spasms, apraxia, and gait disturbances; these abnormalities varied in severity until dramatically improving 20 days after CAR T-cell infusion. Eleven days after the CAR T-cell infusion, the CSF of patient No. 15 contained 3 WBC/μL, and flow cytometry showed that CSF lymphocytes were 97% T cells and that 32.9% of the T cells were CAR positive. The CNS has been shown to lack CD19 expression by other investigators.9 We assessed CD19 expression in multiple brain regions by qPCR and IHC and found no CD19 expression (Data Supplement).
Anti-CD19 CAR T Cells Infiltrated Malignant Lymph Node Mass
Patient No. 13 had CLL manifesting in part as a bulky cervical lymph node mass that dramatically regressed after treatment (Fig 3A). A fine-needle aspiration was performed on this lymph node mass 19 days after infusion of anti-CD19 CAR T cells; 70% of the aspirated lymphocytes were T cells, and 31% of the T cells expressed the anti-CD19 CAR as measured by flow cytometry with a CAR-specific antibody (Fig 3B).33
Fig 3.

Chimeric antigen receptor (CAR) –expressing T cells were detected in regressing lymph node mass of patient with chronic lymphocytic leukemia (CLL). (A) Computed tomography scans show regression of large cervical lymph node mass in patient No. 13. (B) Fine-needle aspiration of lymph node mass shown in (A) was performed 19 days after infusion of anti-CD19 CAR T cells. Aspirated cells were analyzed by flow cytometry with CAR-specific monoclonal antibody. Among lymphoid cells from mass, 70% were T cells, and 31% of T cells expressed anti-CD19 CAR. Plot gated on CD3+ lymphocytes. (C) Before treatment, flow cytometry of blood of patient No. 3 revealed large population of CLL cells as indicated by aberrant CD19+CD5+ phenotype; 91% of pretreatment blood B cells were CLL cells. (D) Ten weeks after treatment, all B cells were absent from blood of patient No. 3, as shown by complete lack of CD19+ cells. CD20+ and CD22+ cells were also absent, which confirmed lack of B cells. (E) One year after treatment, recovering B cells with normal CD19+CD5− phenotype were detected in blood of patient No. 3. (F) Polyclonality of recovering B cells was confirmed by kappa/lambda staining on CD19+ population from (E). In (C), (D), and (E), plots gated on lymphocytes.
Monoclonal CLL B Cells Were Eradicated and Replaced by Polyclonal B Cells
Most patients in the trial were not evaluable for B-cell depletion because of preexisting B-cell depletion resulting from rituximab. All three patients who entered the trial with polyclonal blood B-cell counts in the normal range had B-cell depletion for at least 4 months after CAR T-cell infusion (Data Supplement). In patient No. 3, a monoclonal population of CLL cells was present before treatment in the trial. This CLL population had an aberrant CD19+CD5+ phenotype (Fig 3C) and was monoclonal as determined by kappa/lambda ratio staining. Complete eradication of B cells occurred after infusion of anti-CD19 CAR T cells (Fig 3D). Thirteen months after the CAR T-cell infusion, recovery of polyclonal B cells and continued absence of the CLL cells were evident (Figs 3E and 3F). Patient No. 3 remains in a CR that is ongoing after 23 months.
CAR T Cells Had Variable Peak Blood Levels and Persistence
We measured blood cells containing the anti-CD19 CAR gene by performing qPCR on DNA from total PBMCs collected before treatment and at multiple time points after CAR T-cell infusion. The peak level of CAR-positive blood cells varied considerably among patients. CAR-positive cells were detected by qPCR at peak blood levels, ranging from nine to 777 CAR-positive cells/μL (Fig 4). The number of CAR-positive blood cells peaked between 7 and 17 days after infusion.
Fig 4.
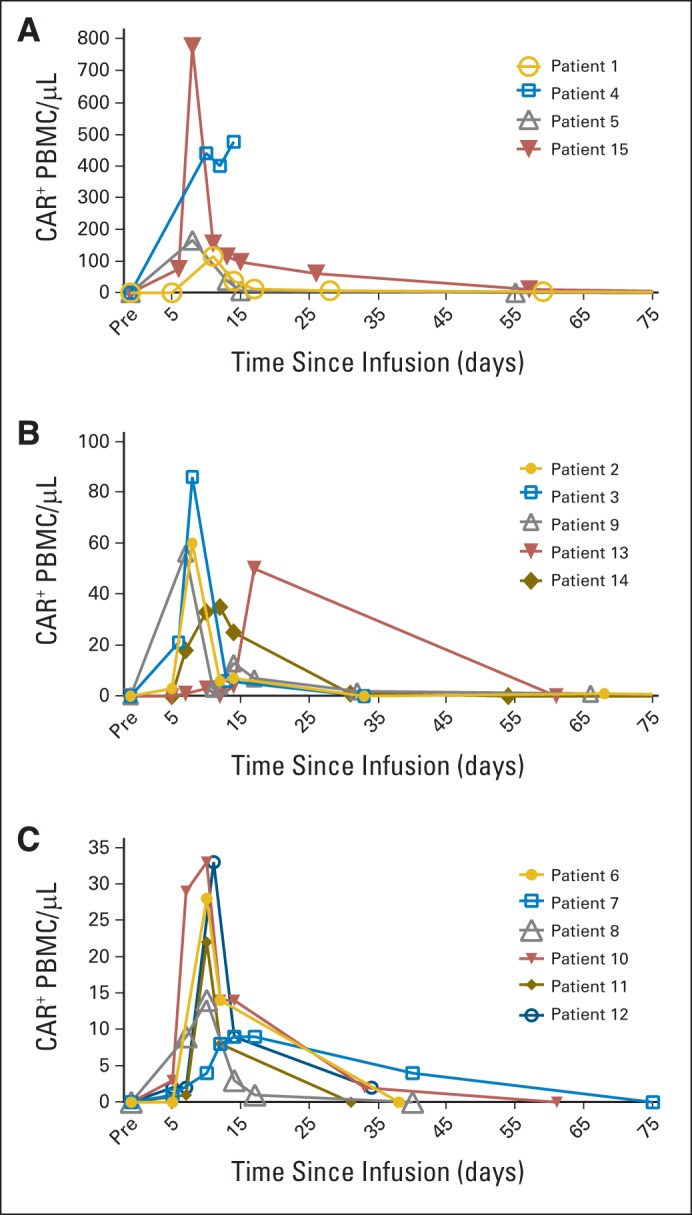
Variable levels of anti-CD19 chimeric antigen receptor (CAR) T cells were detected in blood of patients. CAR-positive cells were detected in blood of all patients after infusion by quantitative polymerase chain reaction. Graphs show absolute number of total peripheral-blood mononuclear cells (PBMC) containing CAR gene. Peak levels of CAR-positive cells varied considerably from patient to patient, so patients were divided into three groups: patients with (A) peak blood CAR-positive cell levels of 115 to 777/μL, (B) blood CAR-positive cell levels of 35 to 86/μL, and (C) blood CAR-positive cell levels of 9 to 33/μL. No matter what peak CAR-positive cell level was, number of CAR-positive cells in blood followed similar pattern, with blood CAR-positive cell numbers increasing to peak between day 7 and 17 after infusion, followed by rapid decrease in CAR-positive cells. Low numbers of CAR-positive cells persisted for variable lengths of time. Note that CAR-positive cell curve for patient No. 4 ends abruptly, because she died 16 days after infusion of CAR T cells.
Anti-CD19 CAR T Cells Acquired a More Differentiated Phenotype From Time of Infusion to Time of Peak Blood Levels
We assessed the number and phenotype of blood CAR-positive cells at the time of the peak number of blood CAR-positive cells by using a CAR-specific monoclonal antibody (Figs 5A and 5B; Data Supplement).33 The time of peak CAR-positive T cells was determined by qPCR (Fig 4). The absolute number of CAR-positive cells at the time of the peak number of blood CAR-positive cells was determined by both qPCR and flow cytometery, and the results of the different methods were closely correlated (Pearson correlation coefficient, r2 = 0.95; P < .001; Data Supplement provides all absolute numbers determined by both methods.). At the time of peak blood CAR-positive cells, a majority of CD3+ CAR-positive T cells were CD8+ in 12 of 15 patients, and for all 15 patients, at the time of the peak of blood CAR-positive cells, the mean ratio of CD3+CD8+ CAR-positive cells to CD3+CD4+ CAR-positive cells was 9.4.
Fig 5.

Most blood chimeric antigen receptor (CAR) –positive T cells expressed CD8, and CAR-positive T cells acquired more differentiated phenotype after infusion. (A) Example of anti-CAR antibody staining is shown. Plot gated on live lymphocytes. Lymphocytes were from blood of patient No. 14 and were collected 7 days after CAR T-cell infusion at time of peak number of blood CAR-positive cells. (B) At time of peak blood CAR-positive T cell levels, majority of CAR-positive T cells were CD8+ in 12 of 15 patients. Example of CD4 and CD8 staining at time of peak blood CAR-positive cells is shown. Plot gated on live CD3+ CAR-positive lymphocytes from patient No. 14. (C) Mean percentage of CD3+CD4+ CAR-positive lymphocytes expressing CCR7+CD45RA− central memory phenotype dropped from time of infusion to time of peak CAR-positive cell blood levels. (D) Mean percentage of CD3+CD8+ CAR-positive lymphocytes expressing CCR7+CD45RA− central memory phenotype dropped from time of infusion to time of peak CAR-positive cell blood levels. (E) Mean percentage of CD3+CD8+ CAR-positive lymphocytes expressing CCR7−CD45RA+ effector memory RA phenotype increased from time of infusion to time of peak CAR-positive cell blood levels. (F) Mean percentage of CD3+CD8+ CAR-positive lymphocytes expressing CD57 increased from time of infusion to time of peak CAR-positive cell blood levels. In (C), (D), (E), and (F), results from all 15 patients studied are included in all groups. All P values from two-tailed paired t tests comparing two groups; error bars represent SEMs.
Central memory T cells express CCR7 and lack expression of CD45RA; in contrast, effector memory T cells lack expression of both CCR7 and CD45RA.40 We found a decrease in the percentage of CAR-positive T cells with a central memory phenotype and an increase in the percentage of CAR-positive T cells with either an effector memory phenotype or a CD8+ effector memory RA phenotype when the infused CAR-positive cells were compared with CAR-positive blood cells at the time of peak CAR-positive cell numbers (Figs 5C to 5E; data not shown). An increase in the percentage of CD3+CD8+ CAR-positive T cells expressing CD57 occurred between the time of infusion and the time of peak CAR-positive blood cells (Fig 5F). We previously reported an increase in programmed death-1 (PD1) expression on CD4+ CAR-positive T cells after infusion.24 We were able to assess PD1 expression on CD4+ CAR-positive cells of 11 patients. In eight of these patients, PD1 expression increased by at least three-fold from the time of infusion to the time of the peak blood levels of CAR-positive cells. Taken together, these phenotypic changes indicate a shift toward a more differentiated T-cell phenotype between the time of infusion and the time of peak blood CAR-positive T-cell levels.40,43–45
DISCUSSION
Our results are the first to our knowledge to show CRs of DLBCL after infusions of anti-CD19 CAR T cells. Our protocol was effective against lymphomas refractory to salvage chemotherapy. These results are a significant advance from previous reports, which showed the effectiveness of anti-CD19 CAR T cells against leukemia and indolent lymphomas.22–27,29,31 Because the longest duration of CR that we have observed is ongoing at 23 months, a critical unanswered question is whether any of the CRs achieved in this trial will lead to permanent malignancy-free survival. A prerequisite for effective treatment of lymphoma with T cells is infiltration of malignant lymph node masses by the T cells. We demonstrated that anti-CD19 CAR T cells can infiltrate malignant lymph node masses by detecting a large number of CAR-positive T cells in a regressing lymph node mass of a patient with CLL (Figs 3A and 3B).
The most troublesome toxicities experienced by patients on this protocol were hypotension and neurologic toxicities (Table 1). We previously reported neurologic toxicities including confusion and obtundation occurring after infusions of anti-CD19 CAR T cells plus high-dose IL-2.23 These toxicities still occurred in some patients when CAR T cells were administered without IL-2, but we also observed other unexpected neurologic toxicities including aphasia and myoclonus (Table 1). The mechanism of these neurologic toxicities is not known and is still under investigation. We speculate that the toxicity was caused by some substance secreted from CAR T cells. Importantly, all patients recovered completely from their neurologic toxicities. Some of the neurologic toxicities that we observed were similar to toxicities observed in other trials of anti-CD19 CAR T cells31 and clinical trials of anti-CD19– and anti-CD3–bispecific antibodies.46 We treated two patients with severe toxicities by infusing the IL-6 receptor–blocking antibody tocilizumab. One of the patients had hypotension, and the other had predominantly neurologic toxicity; the toxicity did not substantially improve in either patient.
The number of CAR-positive T cells in the blood of our patients rose to a peak between 7 and 17 days after infusion and then decreased rapidly. The relative importance of peak blood CAR-positive T-cell levels versus sustained persistence of blood CAR-positive T cells is unknown; moreover, the importance of the levels of blood CAR-positive T cells in general is unknown. It is possible that a more important indicator of effectiveness in treating lymphoma could be the number of CAR-positive T cells infiltrating lymphoma masses, so every opportunity to study CAR-positive T cells within lymphoma masses should be taken.
From the time of infusion to the time of peak blood levels, anti-CD19 CAR T cells acquired a more differentiated phenotype, manifested by a decrease in cells with a central memory phenotype and increases in effector memory and CD57+ cells (Fig 5). The CD8+ effector memory RA and CD57+CD8+ phenotypes are associated with reduced proliferative capacity; acquisition of these more differentiated phenotypes might partially explain the rapid decreases in blood CAR-positive T cells in our patients.40,43–45 Generating CAR T cells that preferentially maintain a less differentiated phenotype might be one way to improve persistence of the T cells and possibly the clinical effectiveness of CAR T-cell therapies.
Infusion of anti-CD19 CAR T cells is a potentially powerful new treatment for chemotherapy-refractory B-cell malignancies. Improvements in gene therapy vectors, CAR design, and T-cell culture methods will probably improve CAR T cells in the near future. New clinical trials of CAR T cells will be needed to optimize antimalignancy efficacy and to elucidate methods of reducing toxicity. Our results should strongly encourage continued development of anti-CD19 CAR T-cell therapies for advanced B-cell malignancies.
Supplementary Material
Acknowledgment
We thank Laurence Cooper and Bipulendu Jena for kindly providing the chimeric antigen receptor–specific monoclonal antibody used in this work. We thank the staff of the National Cancer Institute (NCI) Surgery Branch cell production facility, the NCI Surgery Branch immunotherapy fellows, the staff of the three northwest nursing units of the National Institutes of Health (NIH) Clinical Center, and the staff of the intensive care unit of the NIH Clinical Center.
Footnotes
See accompanying article on page 651
Processed as a Rapid Communication manuscript.
Supported in part by intramural funding for the Center for Cancer Research, National Cancer Institute (NCI), National Institutes of Health (NIH); by federal funds from the NCI, NIH, under Contract No. HHSN261200800001E; and by Kite Pharma through a cooperative research and development agreement with the Surgery Branch, NCI.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
Clinical trial information: NCT00924326.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) and/or an author's immediate family member(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: None Consultant or Advisory Role: Steven A. Rosenberg, Kite Pharma (U) Stock Ownership: None Honoraria: None Research Funding: Steven A. Rosenberg, Kite Pharma Expert Testimony: None Patents, Royalties, and Licenses: None Other Remuneration: None
AUTHOR CONTRIBUTIONS
Conception and design: James N. Kochenderfer, Mark E. Dudley, Steven A. Rosenberg
Provision of study materials or patients: Steven Feldman, Andre Goy, Tatyana Feldman, David E. Spaner, Michael L. Wang
Collection and assembly of data: James N. Kochenderfer, Sadik H. Kassim, Robert P.T. Somerville, Robert O. Carpenter, Maryalice Stetler-Stevenson, Giao Q. Phan, Marybeth S. Hughes, Richard M. Sherry, Mark Raffeld, Steven Feldman, Lily Lu, Yong F. Li, Lien T. Ngo, Tatyana Feldman, David E. Spaner, Michael L. Wang, Clara C. Chen, Sarah M. Kranick, Debbie-Ann N. Nathan, Kathleen E. Morton, Mary Ann Toomey
Data analysis and interpretation: James N. Kochenderfer, Mark E. Dudley, Sadik H. Kassim, Robert O. Carpenter, Maryalice Stetler-Stevenson, James C. Yang, Giao Q. Phan, Mark Raffeld, Yong F. Li, Andre Goy, Clara C. Chen, Avindra Nath, Steven A. Rosenberg
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Elstrom RL, Martin P, Ostrow K, et al. Response to second-line therapy defines the potential for cure in patients with recurrent diffuse large B-cell lymphoma: Implications for the development of novel therapeutic strategies. Clin Lymphoma Myeloma Leuk. 2010;10:192–196. doi: 10.3816/CLML.2010.n.030. [DOI] [PubMed] [Google Scholar]
- 2.Friedberg JW. Relapsed/refractory diffuse large B-cell lymphoma. Hematology Am Soc Hematol Educ Program. 2011;2011:498–505. doi: 10.1182/asheducation-2011.1.498. [DOI] [PubMed] [Google Scholar]
- 3.Moore S, Kayani I, Peggs K, et al. Mini-BEAM is effective as a bridge to transplantation in patients with refractory or relapsed Hodgkin lymphoma who have failed to respond to previous lines of salvage chemotherapy but not in patients with salvage-refractory DLBCL. Br J Haematol. 2012;157:543–552. doi: 10.1111/j.1365-2141.2012.09096.x. [DOI] [PubMed] [Google Scholar]
- 4.Nagle SJ, Woo K, Schuster SJ, et al. Outcomes of patients with relapsed/refractory diffuse large B-cell lymphoma with progression of lymphoma after autologous stem cell transplantation in the rituximab era. Am J Hematol. 2013;88:890–894. doi: 10.1002/ajh.23524. [DOI] [PubMed] [Google Scholar]
- 5.Rivera-Rodriguez N, Cabanillas F. Recent advances in the management of mantle cell lymphoma. Curr Opin Oncol. 2013;25:716–721. doi: 10.1097/CCO.0000000000000010. [DOI] [PubMed] [Google Scholar]
- 6.Martelli M, Ferreri AJM, Agostinelli C, et al. Diffuse large B-cell lymphoma. Crit Rev Oncol Hematol. 2013;87:146–171. doi: 10.1016/j.critrevonc.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 7.Laport GG. Changing role of stem cell transplantation in follicular lymphoma. Hematology Am Soc Hematol Educ Program. 2012;2012:417–425. doi: 10.1182/asheducation-2012.1.417. [DOI] [PubMed] [Google Scholar]
- 8.Vose JM, Bierman PJ, Anderson JR, et al. Progressive disease after high-dose therapy and autologous transplantation for lymphoid malignancy: Clinical course and patient follow-up. Blood. 1992;80:2142–2148. [PubMed] [Google Scholar]
- 9.Uckun FM, Jaszcz W, Ambrus JL, et al. Detailed studies on expression and function of CD19 surface determinant by using B43 monoclonal antibody and the clinical potential of anti-CD19 immunotoxins. Blood. 1988;71:13–29. [PubMed] [Google Scholar]
- 10.Eshhar Z, Waks T, Gross G, et al. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90:720–724. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kochenderfer JN, Rosenberg SA. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat Rev Clin Oncol. 2013;10:267–276. doi: 10.1038/nrclinonc.2013.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dotti G, Gottschalk S, Savoldo B, et al. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev. 2014;257:107–126. doi: 10.1111/imr.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kershaw MH, Westwood JA, Darcy PK. Gene-engineered T cells for cancer therapy. Nat Rev Cancer. 2013;13:525–541. doi: 10.1038/nrc3565. [DOI] [PubMed] [Google Scholar]
- 14.Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3:388–398. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brentjens RJ, Latouche JB, Santos E, et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med. 2003;9:279–286. doi: 10.1038/nm827. [DOI] [PubMed] [Google Scholar]
- 16.Cooper LJ, Topp MS, Serrano LM, et al. T-cell clones can be rendered specific for CD19: Toward the selective augmentation of the graft-versus-B-lineage leukemia effect. Blood. 2003;101:1637–1644. doi: 10.1182/blood-2002-07-1989. [DOI] [PubMed] [Google Scholar]
- 17.Milone MC, Fish JD, Carpenito C, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17:1453–1464. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Naranjo A, Brown CE, et al. Phenotypic and functional attributes of lentivirus-modified CD19-specific Human CD8+ central memory T cells manufactured at clinical scale. J Immunother. 2012;35:689–701. doi: 10.1097/CJI.0b013e318270dec7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Savoldo B, Ramos CA, Liu E, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kochenderfer JN, Yu Z, Frasheri D, et al. Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood. 2010;116:3875–3886. doi: 10.1182/blood-2010-01-265041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kochenderfer JN, Feldman SA, Zhao Y, et al. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J Immunother. 2009;32:689–702. doi: 10.1097/CJI.0b013e3181ac6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kochenderfer JN, Wilson WH, Janik JE, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kochenderfer JN, Dudley ME, Carpenter RO, et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood. 2013;122:4129–4139. doi: 10.1182/blood-2013-08-519413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Porter DL, Levine BL, Kalos M, et al. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brentjens RJ, Rivière I, Park JH, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brentjens RJ, Davila ML, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cruz CRY, Micklethwaite KP, Savoldo B, et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: A phase 1 study. Blood. 2013;122:2956–2973. doi: 10.1182/blood-2013-06-506741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carpenter RO, Evbuomwan MO, Pittaluga S, et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res. 2013;19:2048–2060. doi: 10.1158/1078-0432.CCR-12-2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jena B, Maiti S, Huls H, et al. Chimeric antigen receptor (CAR)-specific monoclonal antibody to detect CD19-specific T cells in clinical trials. PLoS One. 2013;8:e57838. doi: 10.1371/journal.pone.0057838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gattinoni L, Finkelstein SE, Klebanoff CA, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202:907–912. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.North RJ. Cyclophosphamide-facilitated adoptive immunotherapy of an established tumor depends on elimination of tumor-induced suppressor T cells. J Exp Med. 1982;155:1063–1074. doi: 10.1084/jem.155.4.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Montero AJ, Diaz-Montero CM, Kyriakopoulos CE, et al. Myeloid-derived suppressor cells in cancer patients: A clinical perspective. J Immunother. 2012;35:107–115. doi: 10.1097/CJI.0b013e318242169f. [DOI] [PubMed] [Google Scholar]
- 37.Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer. 2013;13:739–752. doi: 10.1038/nrc3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheson BD, Pfistner B, Juweid ME, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25:579–586. doi: 10.1200/JCO.2006.09.2403. [DOI] [PubMed] [Google Scholar]
- 39.Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: A report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute Working Group 1996 guidelines. Blood. 2008;111:5446–5456. doi: 10.1182/blood-2007-06-093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sallusto F, Lenig D, Förster R, et al. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 41.Rubio V, Stuge TB, Singh N, et al. Ex vivo identification, isolation and analysis of tumor-cytolytic T cells. Nat Med. 2003;9:1377–1382. doi: 10.1038/nm942. [DOI] [PubMed] [Google Scholar]
- 42.Rosenberg SA, Lotze MT, Yang JC, et al. Experience with the use of high-dose interleukin-2 in the treatment of 652 cancer patients. Ann Surg. 1989;210:474–485. doi: 10.1097/00000658-198910000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brenchley JM, Karandikar NJ, Betts MR, et al. Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood. 2003;101:2711–2720. doi: 10.1182/blood-2002-07-2103. [DOI] [PubMed] [Google Scholar]
- 44.Focosi D, Bestagno M, Burrone O, et al. CD57+ T lymphocytes and functional immune deficiency. J Leukoc Biol. 2010;87:107–116. doi: 10.1189/jlb.0809566. [DOI] [PubMed] [Google Scholar]
- 45.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: Function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–763. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 46.Nagorsen D, Baeuerle PA. Immunomodulatory therapy of cancer with T cell-engaging BiTE antibody blinatumomab. Exp Cell Res. 2011;317:1255–1260. doi: 10.1016/j.yexcr.2011.03.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.