

Abstract
Recent reports have indicated the role of the prokineticin receptor 2 gene (PROKR2) in the etiology of congenital hypopituitarism, including septo-optic dysplasia and Kallmann syndrome. In the present study, using next-generation targeted sequencing, we identified a novel heterozygous PROKR2 variant (c.742C>T; p.R248W) in a female patient who had combined pituitary hormone deficiency (CPHD), morning glory syndrome and a severely malformed pituitary gland. No other mutation was present in 27 genes related to hypogonadotropic hypogonadism, pituitary hormone deficiency and optic nerve malformation. The substituted amino acid was located on the third intracellular loop of the PROKR2 protein, which is a G protein-coupled receptor. Computational analyses with two programs (SIFT and PolyPhen-2) showed that the substitution was deleterious to PROKR2 function. The p.R248W mutation was transmitted from the patient’s mother, who had a slightly delayed menarche. Collectively, we provide further genetic evidence linking heterozygous PROKR2 mutations and the development of CPHD.
Keywords: combined pituitary hormone deficiency (CPHD), morning glory syndrome, pituitary dysplasia, PROK2, PROKR2
Introduction
Loss-of-function mutations in PROK2 and PROKR2 (encoding prokineticin-2 and prokineticin receptor-2, respectively) have been implicated in Kallmann syndrome (KS), which is characterized by hypogonadotropic hypogonadism (HH) and anosmia (1,2,3). In general, subjects with biallelic PROKR2 mutations exhibit a fairly severe reproductive phenotype in both mice and humans (4, 5); however, the majority of PROKR2 mutation-carrying patients harbor one heterozygous mutation and have variable expressivity of both the reproductive and olfactory phenotypes (1,2,3,4,5). Recently, PROKR2 mutations were reported in patients with combined pituitary hormone deficiency (CPHD), including septo-optic dysplasia (SOD) (6,7,8), suggesting a potential role for the PROK2 pathway in pituitary development, in addition to its role in GnRH neuron development.
In the present study, we used next-generation targeted sequencing to screen for the genetic cause(s) of CPHD and identified a novel heterozygous PROKR2 substitution (c.742C>T; p.R248W) in a patient with CPHD accompanied by marked pituitary dysplasia and morning glory syndrome.
Case Report
Clinical history
We report the case of a female Japanese patient who was 21 yr old at the commencement of our study. She was the second child born to unrelated healthy parents after a full-term pregnancy. Her mother attained menarche at the age of 14 yr, had regular menstrual cycles until 48 yr of age, and had not undergone treatment for infertility. At birth, the weight and height of the patient were 3110 g and 48 cm, respectively. She required hospitalization for 2 weeks after delivery for respiratory distress caused by nasopharyngeal stenosis. At the age of 2 mo, she was diagnosed with morning glory syndrome in both eyes, resulting in amblyopia. Her early developmental milestones were reported to be within the normal range, but she did not walk until 18 mo of age. A dentist who treated the patient for misaligned teeth referred her to our Department of Endocrinology for an evaluation of short stature at the age of 11 yr. At this time, she was 122.4 cm tall (–3.3 SD) and weighed 24.6 kg, and she did not present with any other apparent anomalies.
Medical evaluation of the blood chemistry of the patient showed normal results. The results of endocrine provocation tests are shown in Table 1. The GH responses to insulin-induced hypoglycemia and arginine were low. The serum IGF-I level was low (129 ng/mL; –3.24SD; age-specific reference) (9). The responses of gonadotropins to GnRH stimulation were prolonged, although the peak gonadotropin levels were appropriate for her pubertal stage (Table 1). The patient’s thyroid axis and adrenal axis were intact. Recombinant GH at a physiological dose induced a remarkable change in her growth rate (Fig. 1). Breast development occurred spontaneously at the age of 11 yr and 11 mo, and menarche occurred at the age of 13 yr and 6 mo, but she had never had regular menstrual cycles. By the age of 15 yr, she had developed amenorrhea, and hormone replacement therapy was started. Her adult height was improved by supplementation with recombinant GH, and she reached a height of 148.8 cm (–1.8 SD).
Table 1. Hormonal evaluations of the proband at 11 yr of age.
Fig. 1.

Growth chart of the patient. Recombinant GH at a physiological dose induced a remarkable change in her growth rate. Breast development occurred spontaneously at the age of 11 yr and 11 mo, and menarche occurred at the age 13 yr and 6 mo, but she had never had regular menstrual cycles; hormone replacement therapy was started due to amenorrhea.
Computed tomography and magnetic resonance imaging
Computed tomography revealed a bony defect in the floor of the sella turcica and a tumorous soft tissue structure that extended downward through the defect (Fig. 2A). Magnetic resonance imaging of the hypothalamo-pituitary region with a 1.5-T scanner showed marked dysmorphic structures. A duplicated pituitary stalk with the right stalk continuing to the pituitary gland in the deformed sella was found (Fig. 2, B and C). A tumorous structure extending to the nasopharynx was also detected (Fig. 2D); a dermoid tumor or a teratoma was suspected based on its fat signal intensity. The olfactory sulci and bulbs were normal (data not shown).
Fig. 2.
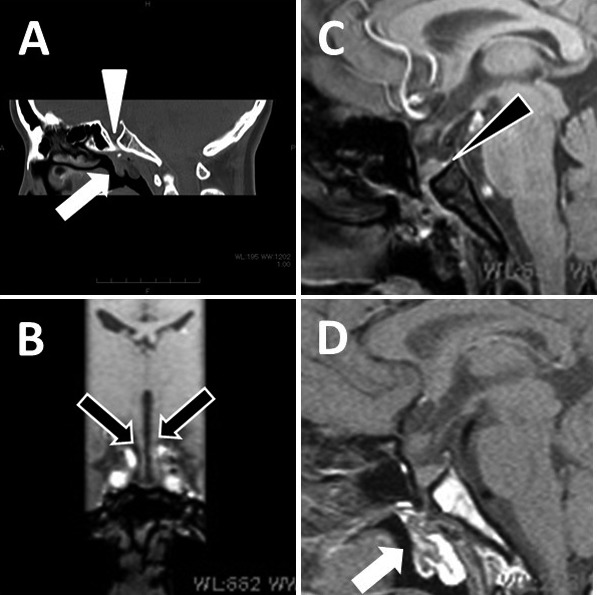
A, Midsagittal reconstruction in plane computed tomography. The bony floor of the sella was defective (white arrowhead), and a downward extension of the tumorous structure through the defect (white arrow) was identified. B and C, T1-weighted three-dimensional images (1 mm thickness) B, reconstructed coronal image showing duplicated pituitary stalks (black arrows) C, pituitary gland identified in the deformed sella. The posterior lobe showed a region of high intensity (black arrowhead). D, Midsagittal spin echo image (2 mm thickness) revealed a tumorous structure below the sella turcica that extended to the nasopharynx through the defective sella floor; the fat signal intensity indicates a dermoid tumor or teratoma.
Molecular studies
We obtained written informed consent from the patient and her parents for molecular studies. This study complied with the Helsinki Declaration of 1975, revised in 1983, and was approved by the institutional review board of Kanagawa Children’s Medical Center. Leukocytic DNA was isolated from the patient and her parents with standard techniques. Array comparative genomic hybridization analysis showed no significant genomic rearrangement. Direct sequencing of 10 genes implicated in congenital hypopituitarism (POU1F1, PROP1, LHX3, LHX4, SOX2, SOX3, HESX1, PAX6, SIX6, and GLI2) revealed no mutation. We then performed next-generation targeted sequencing for HH and hypopituitarism. We tested 25 genes, including KAL1, FGFR1, FGF8, GNRH1, GNRHR, KISS1, KISS1R, LHB, FSHB, CHD7, PROK2, PROKR2, TAC3, TACR3, HESX1, GLI2, LHX4, OTX2, PAX6, POU1F1, PROP1, SOX2, GH1, GHRH and GHRHR using a MiSeq instrument (Illumina Inc., San Diego, CA, USA) according to the SureSelect protocol (Agilent Technologies, Santa Clara, CA, USA), as previously described (10) with minor modifications. As a result, we found a novel heterozygous PROKR2 substitution (c.742C>T, p.R248W) (Fig. 3). This substitution was not present in 150 control alleles and has not been reported in the Single Nucleotide Polymorphism Database (dbSNP) or the Human Genetic Variation Database (HGVD). The substituted amino acid was located on the third intracellular loop of the PROKR2 protein (Fig. 4). The functional effects of the substitution were analyzed using SIFT and PolyPhen-2. These tools indicated that the substitution was deleterious (score of 0.02) and probably damaging (score of 0.966), respectively. No other mutation was identified among the genes analyzed in this study. Family analysis revealed that the PROKR2 p.R248W mutation was transmitted from the mother.
Fig. 3.
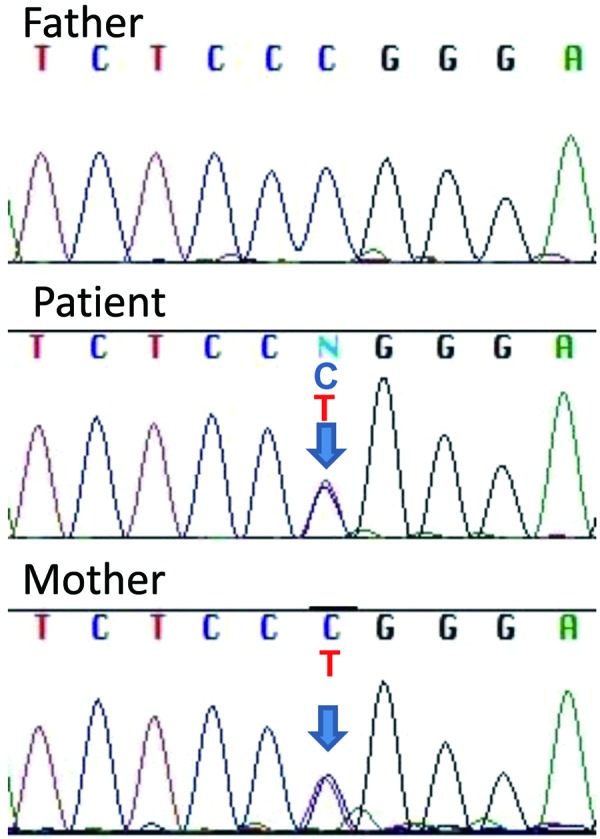
Sequence electropherograms for the PROKR2 gene. A heterozygous c.742C>T, p.R248W mutation was identified in the patient and her mother. Her father showed a wild-type sequence.
Fig. 4.

Schematic representation of the PROKR2 protein showing mutations identified in GnRH-deficient and CPHD patients. Mutations labeled in red, yellow, and green have been identified in KS patients, GnRH-deficient normosmic idiopathic hypogonadotropic hypogonadism (nIHH) probands, and SOD/CPHD/pituitary stalk interruption syndrome (PSIS) patients, respectively. An X in a circle represents homozygous mutations. Compound heterozygous mutations are labeled as c (1 or 2), and the digenic mutations are shown as d:genes (those described in red and green represent KS and SOD/CPHD/PSIS, respectively). A heterozygous PROKR2 mutation (c.742C>T, p.R248W) was identified in our patient.
Discussion
The p.R248W mutation of the PROKR2 gene identified in our patient was not observed in ethnicity-matched controls or any variation/mutation database, indicating that this substitution is extremely rare. The affected residue (Arg248) is evolutionarily conserved, and p.R248W was judged to be a deleterious variant by the two most popular programs for functional effect prediction (SIFT and PolyPhen-2). A different substitution of the same amino acid, R248Q (c.743G>A), has been reported in a patient with a mild form of HH (Fig. 4), and functional analysis of the p.R248Q mutant revealed a mild decrease in calcium influx (3). This case report suggests that R248 is important for the function of PROKR2, and supports the hypothesis that the heterozygous p.R248W substitution identified in the present study is deleterious to protein function, though expression experiments will be required for a more rigorous evaluation of its pathogenicity.
The PROK2 pathway was initially studied for its involvement in gastrointestinal smooth muscle contraction, angiogenesis, hematopoiesis and circadian rhythms. The knockout models for both Prok2 and Prokr2 revealed unexpected roles of the pathway in olfactory bulb morphogenesis and sexual maturation (11, 12), In fact, sequencing of PROK2 and PROKR2 among patients with HH showed that a subset of patients had mutations in both genes (1,2,3,4,5, 13). More recently, an association between PROK2 pathway defects and the development of CPHD with or without SOD has been discussed (6,7,8). Reynaud et al. (7) screened for the PROK2 and PROKR2 genes among 72 index cases with pituitary stalk interruption syndrome from the GENHYPOPIT database and found three heterozygous PROKR2 variant carriers. These reports suggest a potential role of the PROK2 pathway in early pituitary development, as well as in the development of GnRH neurons. Morning glory syndrome is a congenital anomaly of the optic disc in which there is a funnel-shaped excavation of the posterior fundus incorporating the optic nerve, with the funnel-shaped excavation surrounded by an elevated annulus of chorioretinal pigment. The associated features of CPHD and the anomaly of the optic nerve, similar to SOD, suggest an embryonic linkage during forebrain development. In a genetic investigation of 103 patients with CPHD/SOD, four patients were found to harbor heterozygous loss-of-function PROKR2 mutations, suggesting a possible role for the PROK2/PROKR2 variants as modifier genes (6). Our case suggests that morning glory syndrome might be one of the diverse phenotypes of SOD.
The phenotypes resulting from heterozygous PROKR2 mutations are remarkably variable, ranging from isolated GnRH deficiency to CPHD with or without abnormalities of the olfactory and optic nerves (Fig. 4). Interactions between multiple genes causing GnRH deficiency have been demonstrated (1, 3, 4, 6, 7). Pitteloud et al. (14) reported evidence of IHH caused by the interaction of two gene defects (FGF receptor 1 (FGFR1) with nasal embryonic LHRH factor (NFLF) in one family and GnRH receptor (GNRHR) with FGFR1 in another) and showed that two different gene defects can synergize to increase phenotype severity in IHH. Oligogenic inheritance is currently considered to be the most plausible explanation for the phenotypes observed in patients with heterozygous mutations. We also observed a considerable phenotypic difference between the two mutation carriers in the present study (the patient and her mother), but no mutation to synergize was found other than PROKR2. Further investigations will be required to clarify the phenotypic variability among patients with only one heterozygous PROKR2 mutation. Nevertheless, our findings suggest that PROKR2 should be evaluated as a candidate responsible gene in patients with CPHD resulting from pituitary dysplasia in addition to KS patients.
References
- 1.Dodé C, Teixeira L, Levilliers J, Fouveaut C, Bouchard P, Kottler ML, et al. Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet 2006;2: e175. doi: 10.1371/journal.pgen.0020175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pitteloud N, Zhang C, Pignatelli D, Li JD, Raivio T, Cole LW, et al. Loss-of-function mutation in the prokineticin 2 gene causes Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci USA 2007;104: 17447–52. doi: 10.1073/pnas.0707173104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cole LW, Sidis Y, Zhang C, Quinton R, Plummer L, Pignatelli D, et al. Mutations in prokineticin 2 and prokineticin receptor 2 genes in human gonadotrophin-releasing hormone deficiency: molecular genetics and clinical spectrum. J Clin Endocrinol Metab 2008;93: 3551–9. doi: 10.1210/jc.2007-2654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sarfati J, Guiochon-Mantel A, Rondard P, Arnulf I, Garcia-Piñero A, Wolczynski S, et al. A comparative phenotypic study of kallmann syndrome patients carrying monoallelic and biallelic mutations in the prokineticin 2 or prokineticin receptor 2 genes. J Clin Endocrinol Metab 2010;95: 659–69. doi: 10.1210/jc.2009-0843 [DOI] [PubMed] [Google Scholar]
- 5.Martin C, Balasubramanian R, Dwyer AA, Au MG, Sidis Y, Kaiser UB, et al. The role of the prokineticin 2 pathway in human reproduction: evidence from the study of human and murine gene mutations. Endocr Rev 2011;32: 225–46. doi: 10.1210/er.2010-0007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raivio T, Avbelj M, McCabe MJ, Romero CJ, Dwyer AA, Tommiska J, et al. Genetic overlap in Kallmann syndrome, combined pituitary hormone deficiency, and septo-optic dysplasia. J Clin Endocrinol Metab 2012;97: E694–9. doi: 10.1210/jc.2011-2938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reynaud R, Jayakody SA, Monnier C, Saveanu A, Bouligand J, Guedj AM, et al. PROKR2 variants in multiple hypopituitarism with pituitary stalk interruption. J Clin Endocrinol Metab 2012;97: E1068–73. doi: 10.1210/jc.2011-3056 [DOI] [PubMed] [Google Scholar]
- 8.McCabe MJ, Gaston-Massuet C, Gregory LC, Alatzoglou KS, Tziaferi V, Sbai O, et al. Variations in PROKR2, but not PROK2, are associated with hypopituitarism and septo-optic dysplasia. J Clin Endocrinol Metab 2013;98: E547–57. doi: 10.1210/jc.2012-3067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Isojima T, Shimatsu A, Yokoya S, Chihara K, Tanaka T, Hizuka N, et al. Standardized centile curves and reference intervals of serum insulin-like growth factor-I (IGF-I) levels in a normal Japanese population using the LMS method. Endocr J 2012;59: 771–80. doi: 10.1507/endocrj.EJ12-0110 [DOI] [PubMed] [Google Scholar]
- 10.Takagi M, Narumi S, Asakura Y, Muroya K, Hasegawa Y, Adachi M, et al. A novel mutation in SOX2 causes hypogonadotropic hypogonadism with mild ocular malformation. Horm Res Paediatr 2014;81: 133–8. doi: 10.1159/000355279 [DOI] [PubMed] [Google Scholar]
- 11.Ng KL, Li JD, Cheng MY, Leslie FM, Lee AG, Zhou QY.Dependence of olfactory bulb neurogenesis on prokineticin 2 signaling. Science 2005;308: 1923–7. doi: 10.1126/science.1112103 [DOI] [PubMed] [Google Scholar]
- 12.Matsumoto S, Yamazaki C, Masumoto KH, Nagano M, Naito M, Soga T, et al. Abnormal development of the olfactory bulb and reproductive system in mice lacking prokineticin receptor PKR2. Proc Natl Acad Sci USA 2006;103: 4140–5. doi: 10.1073/pnas.0508881103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dodé C, Rondard P.PROK2/PROKR2 signaling and Kallmann syndrome. Front Endocrinol (Lausanne) 2013;4: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pitteloud N, Quinton R, Pearce S, Raivio T, Acierno J, Dwyer A, et al. Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J Clin Invest 2007;117: 457–63. doi: 10.1172/JCI29884 [DOI] [PMC free article] [PubMed] [Google Scholar]