

Introduction
Metaphyseal dysplasia, Schmid type (MS, OMIM #156500) is a bone dysplasia that presents with autosomal dominant inheritance. MS belongs to the “Metaphyseal dysplasia” group in the “Nosology and Classification of Genetic Skeletal Disorders” (1). Its clinical symptoms include coxa vara, bowed leg, short stature, short limbs, and normal facial appearance. An orthopedic operation can be conducted for coxa vara, depending on its severity. Among MS patients, genetic mutations of the COL10A1 gene have been identified (2).
The COL10A1 gene encodes the type X collagen α1 chain. Three type X collagen α1 chains form a homotrimer. Type X collagen expresses restrictedly in hypertrophic chondrocytes during endochondral ossification, and the role of type X collagen is not entirely clear.
A large part of the pathogenic mutation in the COL10A1 gene is located in the carboxyl-terminal non-collagenous (NC1) domain and at Glycine 18 located at the boundary between the signal peptide domain and N-terminal non-collagenous (NC2) domain (3).
This report describes a familial case with MS accompanied by a previously unreported mutation, p.T555P, located on the NC1 domain of a type X collagen α1 chain.
Case Report
A 1-yr 11-mo old boy was referred to our hospital because of coxa vara at 1 yr and 6 mo of age. He was born by vaginal delivery at 39 wk. Asphyxia was not observed. His height and wt at birth were, respectively, 49.0 cm (0.00 SD) and 2506 g (–1.17 SD). His height at first arrival at our hospital was 77.5 cm (–2.40 SD) in a standing position. In the spine position, five fingers could be inserted between his knees. His facial appearance was normal. Metaphyseal flaring and fraying were evident in radiographs (Fig. 1-a and Fig. 1-b). This patient showed no abnormality in biochemical analysis, such as analysis of his serum levels of calcium, inorganic phosphate and parathyroid hormone and alkaline phosphatase activity. His father was 175 cm tall (+0.72 SD) and had normal proportions. His mother showed short stature (140.0 cm, –3.42 SD) and a normal facial appearance. She had undergone osteotomy for correction of coxa vara when she was 6 yr of age. Based on the clinical symptoms shown by the patient and his mother, we diagnosed the patient as having MS. His height decreased from –2.40 SD at first arrival to –3.51 SD at 4 yr and 5 mo old (Fig. 1-c). At 5 yr of age, abduction osteotomy was conducted to correct coxa vara.
Fig. 1.
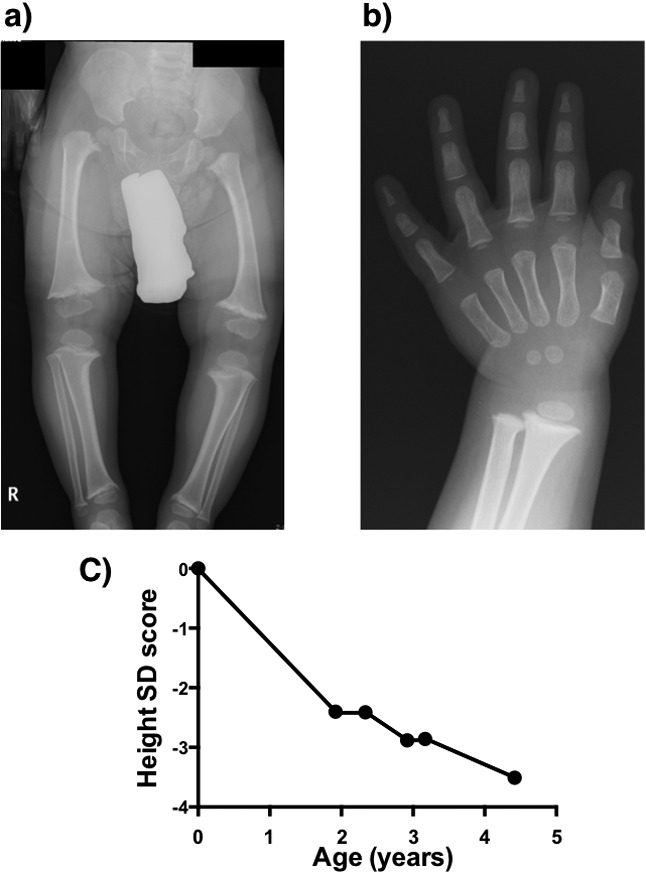
Radiographs of the patient at 1 yr and 11 mo old. a) Lower limbs: Metaphyseal flaring and fraying and coxa vara are evident. b) Left hand: Flaring is evident in the metaphysis of distal radius and ulna in contrast to short bones and metacarpals. c) Transition of the height SD score before abduction osteotomy.
Methods
To analyze the COL10A1 gene, genomic DNA was extracted from whole blood using a QIAamp DNA Blood Mini Kit (Qiagen Inc., Tokyo, Japan) after informed consent was obtained from the patient’s guardian. A PCR reaction was conducted using the standard PCR method. The following primer pairs were used: G18 forward, 5’- TGATCTCTCATTTATTTATGGCACA, and G18 reverse, 5’- TGGGCTAATTCAGAAGTTGGA, for analysis of glycine at 18 and NC1 forward, 5’- CAGTCATGCCTGAGGGTTTT, and NC1 reverse, 5’- GGGAAGGTTTGTTGGTCTGA, for analysis of the NC1 domain of the type X collagen α1 chain.
The PCR products were sequenced using a BigDye Terminator Cycle Sequencing FS Ready Reaction Kit and a Genetic Analyzer (ABI Prism 310; Applied Biosystems, Foster City, CA, USA).
This study was approved by the ethical committee of the Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences.
Results
A substitution from adenine at 1663 in the COL10A1 cDNA (c.1663 c>a) to cytosine that caused an amino acid change from threonine at 555 in the type X collagen α1 chain to proline (p.T555P) was identified in this patient. The c.1663 c>a substitution was also found in his mother, but it was not found in his father (Fig. 2-a); it was also not found among 120 alleles of healthy controls (data not shown). This substitution was found in neither the Japanese SNP (JSNP) nor dbSNP databases.
Fig. 2.

Results of the COL10A1 gene analysis. a) A substitution from adenine at 1663 in the COL10A1 cDNA to cytosine (c.1633 c>a) that results in an amino acid change from threonine at 555 in the type X collagen α1 chain to proline (p.T555P) was identified in this patient and his mother, but it was not identified in his father. b) Threonine at position 555 in Homo sapiens is conserved in various species. The conserved threonine is highlighted in the shaded box. c) PolyPhen-2 analysis of the p.T555P mutation.
p.T555 has been maintained in various species (Fig. 2-b). By PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) analysis (4), p.T555P was predicted to be “probably damaging” with a score of 0.991 (Fig. 2-c).
Discussion
p.T555P in the COL10A1 gene was found to be deleterious by in silico analysis using PolyPhen-2. We inferred that the p.T555P mutation was a pathogenic mutation. No previous report has previously described the p.T555P mutation in the COL10A1 gene. The p.T555P mutation is located in the NC1 domain of the type X collagen α1 chain. Among the previously reported mutations located in the NC1 domain, we found that the p.T555P mutation is located nearest to the amino-terminal of the NC1 domain.
MS patients present with various clinical severities: some patients require osteotomy for correction of coxa vara; some patients require no osteotomy (5). Although the genotype–phenotype correlation in MS has not been fully clarified, the p.T555P mutation is presumed to result in a severe phenotype compared with patients reported previously, as our patient and his mother required osteotomy.
The mutation in the NC1 domain reportedly disrupts NC1 folding and stability and subsequent molecular assembly and interaction in the cartilage matrix (3). To elucidate the pathogenicity of the p.T555P mutation, functional analysis is needed.
Acknowledgments
This work was supported by grants-in-aid from the Ministry of Health, Labour and Welfare (21210901 and 24132801).
References
- 1.Warman ML, Cormier-Daire V, Hall C, Krakow D, Lachman R, LeMerrer M, et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A 2011;155A: 943–68. doi: 10.1002/ajmg.a.33909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dharmavaram RM, Elberson MA, Peng M, Kirson LA, Kelley TE, Jimenez SA.Identification of a mutation in type X collagen in a family with Schmid metaphyseal chondrodysplasia. Hum Mol Genet 1994;3: 507–9. doi: 10.1093/hmg/3.3.507 [DOI] [PubMed] [Google Scholar]
- 3.Bateman JF, Wilson R, Freddi S, Lamandé SR, Savarirayan R.Mutations of COL10A1 in Schmid metaphyseal chondrodysplasia. Hum Mutat 2005;25: 525–34. doi: 10.1002/humu.20183 [DOI] [PubMed] [Google Scholar]
- 4.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7: 248–9. doi: 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wallis GA, Rash B, Sweetman WA, Thomas JT, Super M, Evans G, et al. Amino acid substitutions of conserved residues in the carboxyl-terminal domain of the alpha 1(X) chain of type X collagen occur in two unrelated families with metaphyseal chondrodysplasia type Schmid. Am J Hum Genet 1994;54: 169–78. [PMC free article] [PubMed] [Google Scholar]