

Summary
Mutations of androgen receptor (AR) are the most frequent cause of 46, XY disorders of sex development and associated with a variety of phenotypes, ranging from phenotypic women (complete androgen insensitivity syndrome (CAIS)) to milder degrees of undervirilization (partial form or PAIS) or men with only infertility (mild form or MAIS). From 2009 to 2012, two young Chinese female individuals with CAIS from two families were referred to our hospital due to primary amenorrhea. Defects in testosterone (T) and dihydrotestosterone (DHT) synthesis were excluded. Physical examination revealed that the patients have normal female external genitalia, normal breast development, vellus hair in the axilla and on the arms and legs, but absence of pubic hair, and a blind-ending vagina. Two different types of AR mutations have been detected by sequencing of genomic DNA: Family A showed deletion of exon 2 in AR gene; Family B showed a single nucleotide C-to-T transition in exon 8 of AR gene resulting in a proline 893-to-leucine substitution (Pro893Leu). Testicular histology showed developmental immaturity of seminiferous tubules with the absence of spermatogenic cells or spermatozoa. No AR immunoreactivity was observed in either case. Three adult patients recovered well from bilateral orchiectomy. The juvenile patient of family B was followed up. Our present study on these two families revealed two different types of AR mutation. The definitive diagnosis of AIS was based on clinical examination and genetic investigations. Our findings verified the mechanism of CAIS and also enriched AR Gene Mutation Database.
Keywords: Complete androgen insensitivity syndrome, androgen receptor, disorder of sex development, AR domains, deletion and transition
1. Introduction
Androgen plays a key role in the control of male sexual differentiation and the maintenance of normal male reproductive function. Androgen actions are mediated by ligand-dependent transcription factors, and the androgen receptor, which is translocated into the nucleus and binds to the regulatory regions of specific chromosomal DNA sequences to activate androgen dependent genes. The androgen-AR complex functions in conjunction with co-regulatory proteins (1–3).
Inability to respond to circulating androgens named as androgen insensitivity syndrome (AIS), formerly known as “testicular feminization syndrome", was first described by Morris in 1953 (4). Androgen receptor (AR) gene mutations are the most frequent cause of 46, XY disorders of sex development and associated with a variety of phenotypes, ranging from phenotypic women (complete androgen insensitivity syndrome (CAIS)) to milder degrees of undervirilization (partial form or PAIS) or men with only infertility (mild form or MAIS) (5). The mutated AR gene products which lost androgen-binding ability and transcriptional activity abolished the target cells' response to testosterone and dihydrotestosterone (6,7).
The estimated prevalence of this disorder was 1:20,000 to 1:64,000 live male births (5,8–10). To date, over 500 unique mutations in the AR gene causing androgen insensitivity syndrome had been reported from more than 850 patients (http://androgendb.mcgill.ca) (11). Most cases of AR mutation were inherited and transmitted from parents to offspring generation (9).
We reported our experience in the diagnosis and treatment of four patients with CAIS from two unrelated Chinese families at Huashan Hospital of Fudan University. Diagnosis was made by physical examination, imaging examination (B-ultrasound, CT scan) and laboratory tests (including measurement of blood sexual hormones and karyotype analysis). Polymerase chain reaction (PCR) and DNA sequencing of AR gene were also carried out using peripheral blood leukocytes of the probands and siblings.
2. Materials and Methods
This study was approved by the institutional review board of Huashan Hospital, Fudan University. From 2009 to 2012, two young Chinese female individuals and their siblings with CAIS from two families were referred to our hospital.
2.1. Clinical features
Family A: A 24-year-old female (III-B in the pedigrees of family A, Figure 1) was referred to our hospital due to primary amenorrhea. Physical examination revealed a 171 cm height, 55 kg weight patient with normal female external genitalia, normal breast development, but absence of pubic hair, and 4 cm deep blind-ending vagina. B-ultrasound showed one testicle located at right inguinal area accompanying right inguinal hernia, another testicle located in the left side of pelvic cavity, and without internal female genital organs (uterus and ovaries). A peripheral leukocyte chromosome analysis gave a 46, XY karyotype. The patient expressed satisfaction with her sexual life and accepted female gender very well. These data supported the diagnosis of CAIS, and the patient underwent bilateral orchiectomy without vaginal lengthening. Her 22-year-old cousin (III-H in the pedigrees of family A, Figure 1) with 46, XY karyotype was also diagnosed with CAIS. The presentation, diagnosis, and treatment were the same as that of the proband, except that testes were found in bilateral inguinal regions.
Figure 1.
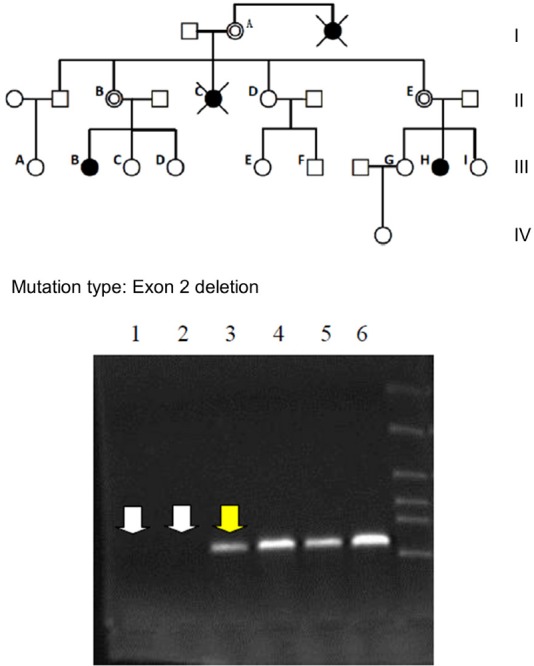
Amplification of the AR gene exon 2 in family members. Lanes 1 and 2 = No amplification in the CAIS patients (III-B and III-H). Lane 3 = Intermediate amplification in an obligate carrier mother of a CAIS patient (II-B).Lanes 4 to 6 = Strong amplification in three normal family members. Lane 5 is a normal male family member. PCR amplification and sequencing of AR gene exons in the probands showed a deletion of exon 2. DNA sequencing confirmed that there was no point mutation, except the deletion of exon 2, and showed that the remaining exons and introns were intact in the AR genes of the probands. (Note: The pedigrees with complete androgen insensitivity syndrome (CAIS); Black circles = CAIS 46, XY individuals; open circles = normal females; double open circles = obligate carrier women; open squares = normal males; crossed black circles = deceased individuals suspected of having CAIS.)
Family B: A 20-year-old female (III-B in the pedigrees of family B, Figure 2) was referred to our hospital due to primary amenorrhea with bilateral solid inguinal mass. Physical examination revealed 170 cm height, 50 kg weight young girl with normal female external genitalia, normal breast development, and an 8 cm deep blind-ending vagina (Figure 3A). Imaging examinations (computer tomography and B-ultrasound) showed testis-like gonad located at each side of inguinal canal (Figure 3B). Sex chromosome analysis reported 46, XY karyotype. This patient was also diagnosed with CAIS and underwent bilateral orchiectomy. Her 13-year-old half-sister (III-C in the pedigrees of family B, Figure 2) was also diagnosed with CAIS after clinical examination. The juvenile patient was followed up.
Figure 2.
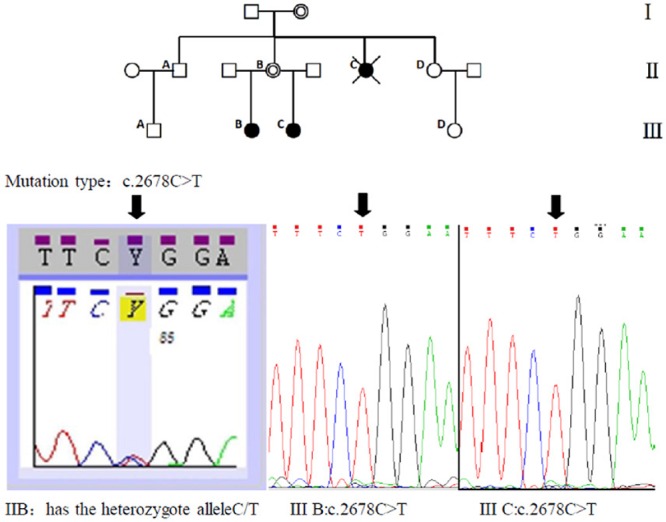
Direct sequencing analysis of PCR products revealed in the patients with the presence of a C to T transition in exon 8 resulting in the proline 893 leucine substitution (Pro893Leu) in CAIS patients of this family. Their mother was detected with the same mutation in heterozygous form. (Note: The pedigrees with complete androgen insensitivity syndrome (CAIS); Black circles = CAIS 46, XY individuals; open circles = normal females; double open circles = obligate carrier women; open squares = normal males; crossed black circles = deceased individuals suspected of having CAIS.)
Figure 3.
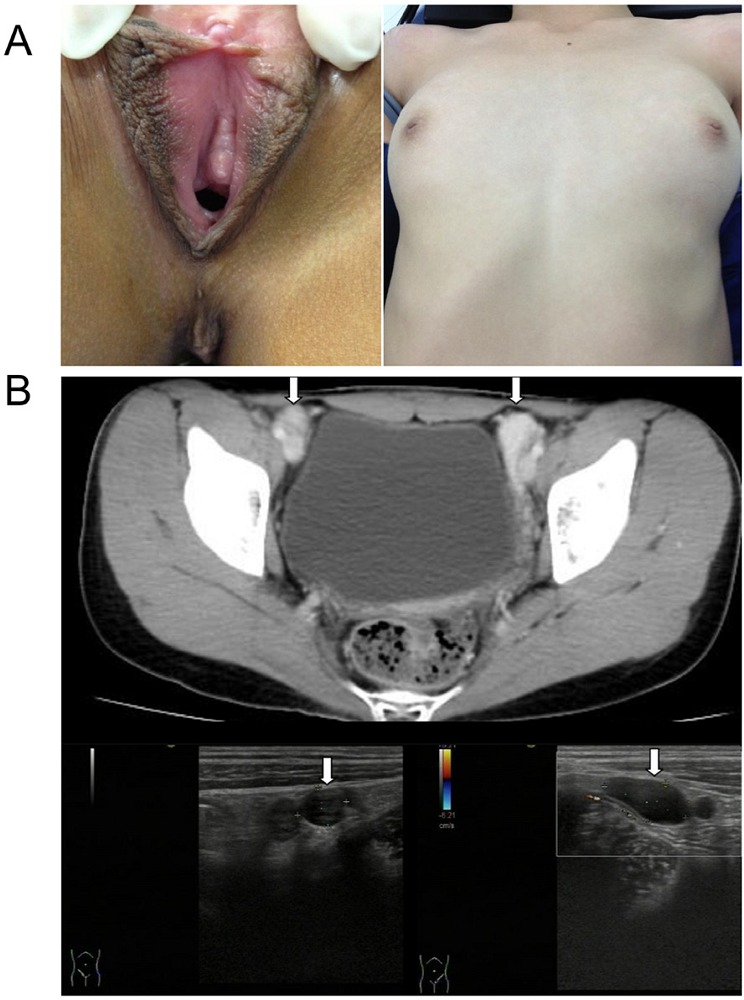
Patient III-B from Family B. (A), showed normal female external genitalia, normal breast development, absence of pubic hair and a blind-ending vagina. (B), Imaging examinations (computer tomography and B-ultrasound) showed testis-like gonads located in the inguinal canals bilaterally.
2.2. Pedigree analysis
Two pedigree analyses of these two unrelated families were performed. The confirmed CAIS patients were subjects III-B and III-H of family A; subjects III-B and III-C of family B. Subject I-B, subject II-C of family A and subject II-C of family B were reported infertile with female phenotype and were also suspected to have CAIS.
2.3. Hormone assays
Serum levels of testosterone (T), estradiol (E2), luteinizing hormone (LH), follicle-stimulating hormone (FSH), and dihydrotestosterone (DHT) were measured by radioimmunoassay for the probands and their siblings.
2.4. DNA extraction and sequencing
Genomic DNA was extracted from peripheral blood samples of family members using a Qiagen Pure Gene Blood Core Kit C according to the manufacturer's instructions (QIAGEN, Shanghai, China). The coding region of the AR gene was screened by polymerase chain reaction (PCR) amplification and direct sequencing was screened using the ABI 3730 XL DNA analyzer (Applied Biosystems). PCR primers were designed as reported before (12). The AR gene variations were identified between the patient with CAIS and the reference genome using the BLAT tool of the UCSC Genome Browser (available from: http://genome.ucsc.edu).
2.5. Testicular histology
Gonadal tissue of the adult patients (III-B and III-H of family A; subjects III-B of family B.) was fixed with 10% buffered neutral formalin solution. Histopathological change was observed by hematoxylin-eosin stain microscopically. Immunohistochemistry was conducted on paraffin-embedded tissue sections of the viable testicle using AR antibody (DAKO, Glostrup, Denmark).
3. Results
3.1. Probands' clinical characteristics
The results of hormone levels and physical examination are shown in Table 1. Elevated blood LH level was found in the probands and their siblings. Three adult CAIS patients revealed slightly increased E2 level, but T and DHT were within normal range. Detailed physical examination performed on our three adult CAIS patients didn't show any prominent difference compared to a normal woman. The external genitalia and blind-ending vagina didn't affect their sexual life.
Table 1. Summary of clinical characteristics of CAIS patents.
| Items | Family A |
Family B |
|||
|---|---|---|---|---|---|
| Patient III-B | Patient III-H | Patient III-B | Patient III-C | ||
| Age | 24 | 22 | 20 | 13 | |
| Height (cm) | 171 | 172 | 170 | 155 | |
| Weight (kg) | 55 | 52 | 50 | 40 | |
| Clitoral length (cm) | 1.4 | 1.2 | 1.5 | 1.0 | |
| Clitoral to urethral length (cm) | 2.0 | 2.2 | 2.0 | 1.5 | |
| vaginal depth (cm) | 4 | 5.5 | 8 | (not measured) | |
| (Hormone analysis results) | (Normal male range) | ||||
| Testosterone | 9.90–27.80 nM | 23.5 nM | 31.1 nM | 40.57 nM | 20.1 nM |
| Estradiol | 28.00–156 pM | 171.8 pM | 143.3 pM | 187.5 pM | 121.3 pM |
| Luteinizing hormone | 1.70–8.60 IU/L | 45.81 IU/L | 33.70 IU/L | 47.04 IU/L | 22.9 IU/L |
| Follicle-stimulating hormone | 1.50–12.40 IU/L | 2.35 IU/L | 13.80 IU/L | 25.04 IU/L | 7.30 IU/L |
| Dihydrotestosterone | 55.10–386.5 ng/dL | 51.0 ng/dL | 49.1 ng/dL | 60.4 ng/dL | 55.4 ng/dL |
3.2. Identification of the genetic mutation
Different types of AR mutations have been detected on genomic DNA. Family A: PCR amplification and sequencing of AR gene exons in the probands showed the deletion of exon 2. DNA sequencing confirmed that there was no point mutation, except for the deletion of exon 2, and showed that the remaining exons and introns were intact in the AR genes of the probands (Figure 1). Family B: Direct sequencing analysis of PCR products revealed the presence of a single nucleotide C-to-T transition in exon 8 resulting in a 893 proline-to-leucine substitution (Pro893Leu) (Figure 2) in CAIS patients of this family. Their mother has the same mutation in heterozygous form.
3.3. Histology report
The specimen from the gonadectomy was identified as testis with epididymis and vas deferens attached. Testicular histology showed developmental immaturity of seminiferous tubules containing monolayers of Sertoli cells without spermatogenic cells or spermatozoa together with hyperplasia of mesenchymal cells and fibrous tissue (Figure 4A). No AR immunoreactivity was observed in all cases (Figure 4B). Negative AR immunostaining was attributed to the absence of AR production at the protein level. The absence of AR immunostaining in our cases could reflect that either Sertoli cell immaturity or AR gene mutation could result in no expression of AR protein at all.
Figure 4.
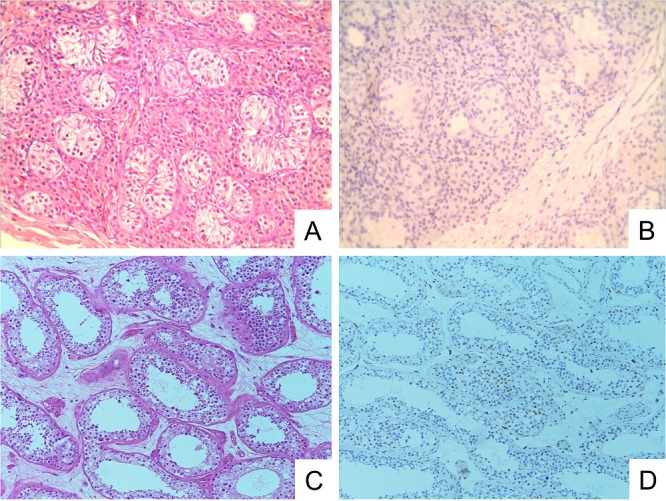
Patient III-B from family B: (A), showed developmental immaturity of seminiferous tubules containing monolayers of Sertoli cells without spermatogenic cells or spermatozoa. (B), showed no immunoreactivity for androgen receptors. (C), showed normal seminiferous tubules with spermatogenic cells and spermatozoa (positive control). (D), the seminiferous tubule normal testicular tissue demonstrates strong nuclear immunoreactivity for androgen receptors.
4. Discussion
The androgen receptor gene is more than 90 kb long with 8 exons and located at Xq11–12 (13) The subsections, or domains, consist of the N-terminal domain (NTD, residues 1–534) harboring AR transcriptional activation function encoded by exon 1, a central DNA-binding domain (DBD, residues 559–624) encoded by exons 2 and 3, the “hinge” region which binds the NTD and DBD, and ligand binding domain (LBD, residues 664–919) encoded by exons 4–8 (14,15).
The DNA-binding domain (DBD) is the region of the protein that interacts with DNA. The androgen receptor DBD determines androgen selectivity of transcriptional response (16). The ligand binding domain (LBD) is the site of interaction of the ligand (the androgen hormone), binding of which will turn on the androgen function that will lead to a migration of the receptor to the cellular nucleus and the activation of the receptor's target genes (17).
According to the AR mutation database (ARDB http://androgendb.mcgill.ca), out of 314 unique AR mutations causing CAIS, 89 mutations were located at the NTD, 49 mutations located at the DBD, 158 mutations located in the LBD, and 18 mutations located in the intron and splice site. Not surprisingly, most mutations (207/314) are found in the DBD and the LBD. Most of mutations of these two domains would make the crystal structure change and cause the mutated AR to be completely inactive (18).
Our present study of these two families revealed two different types of AR mutation in CAIS patients: deletion of exon 2 and a single nucleotide mutation transition in exon 8. Although there were a few similar reports about these mutations, it was the first found in Chinese people.
In our study, the probands' mothers were carriers of the mutant allele and the patients' fathers exhibited the normal allele. AR mutation was inherited and transmitted from mother to the offspring generation. Our study provided useful information in prenatal diagnosis and recommendation of appropriate counseling for these two families.
In the female infant or toddler, no immediate therapy was needed for CAIS patients. These patients who had normal female hormonal levels would develop into phenotypically normal females. Once final height and breast development had been obtained, the gonads should be removed because of risk of testicular tumors (19). Our three adult CAIS patients accepted orchiectomy surgery and estrogen replacement therapy was applied afterwards.
It was reported that 90% of women with CAIS had sexual difficulties when compared to the general female population, including most commonly sexual infrequency and vaginal penetration difficulty (20). Women with CAIS may have vaginal hypoplasia, clitoral hypoplasia, and psychological problems that might contribute to sexual dysfunction. Detailed physical examination performed on our three adult CAIS patients didn't show a remarkable difference compared to a normal woman. We assumed that sexual difficulties were related to the degree of feminization.
After orchiectomy surgery, the regular follow-up included three aspects: sexual hormone levels, sexual function and psychological state. Our adult CAIS patients accepted their female gender with psychological gratification pre- and post-operatively. We were more willing to encourage our patients to accept their female gender because of the concern that the female-to-male sexual transition could be more challenging (21,22).
In conclusion, the definitive diagnosis of CAIS was based both on clinical examination and the results of appropriate investigations. Our clinical experience revealed that the mutated AR gene resulted in primary amenorrhea and absence of internal genitalia. Our findings of AR mutations verified the mechanism of CAIS and also enriched the AR Gene Mutations Database.
Acknowledgements
This work was sponsored by National Natural Science Foundation of China (81170697) and National Basic Research Program (973 plan) of China (2011CB944503)
References
- 1. La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991; 352:77-79. [DOI] [PubMed] [Google Scholar]
- 2. Eder IE, Culig Z, Putz T, Nessler-Menardi C, Bartsch G, Klocker H. Molecular biology of the androgen receptor: From molecular understanding to the clinic. Eur Urol. 2001; 40:241-251. [DOI] [PubMed] [Google Scholar]
- 3. Gelmann EP. Molecular biology of the androgen receptor. J Clin Oncol. 2002; 20:3001-3015. [DOI] [PubMed] [Google Scholar]
- 4. Morris JM. The syndrome of testicular feminization in male pseudohermaphrodites. Am J Obstet Gynecol. 1953; 65:1192-1211. [DOI] [PubMed] [Google Scholar]
- 5. Audi L, Fernandez-Cancio M, Carrascosa A, et al. Novel (60%) and recurrent (40%) androgen receptor gene mutations in a series of 59 patients with a 46, XY disorder of sex development. J Clin Endo Metab. 2010; 95:1876-1888. [DOI] [PubMed] [Google Scholar]
- 6. Heemers HV, Tindall DJ. Androgen receptor (AR) coregulators: A diversity of functions converging on and regulating the AR transcriptional complex. Endocr Rev. 2007; 28:778-808. [DOI] [PubMed] [Google Scholar]
- 7. Heinlein CA, Chang CS. Androgen receptor (ar) coregulators: An overview. Endocr Rev. 2002; 23:175-200. [DOI] [PubMed] [Google Scholar]
- 8. Ahmed SF, Cheng A, Dovey L, Hawkins JR, Martin H, Rowland J, Shimura N, Tait AD, Hughes IA. Phenotypic features, androgen receptor binding, and mutational analysis in 278 clinical cases reported as androgen insensitivity syndrome. J Clin Endo Metab. 2000; 85:658-665. [DOI] [PubMed] [Google Scholar]
- 9. Brown TR, Lubahn DB, Wilson EM, Joseph DR, French FS, Migeon CJ. Deletion of the steroid-binding domain of the human androgen receptor gene in one family with complete androgen insensitivity syndrome: Evidence for further genetic heterogeneity in this syndrome. Proc Natl Acad Sci U S A. 1988; 85:8151-8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Raicu F, Giuliani R, Gatta V, Palka C, Franchi PG, Lelli-Chiesa P, Tumini S, Stuppia L. Novel mutation in the ligand-binding domain of the androgen receptor gene (1790p) associated with complete androgen insensitivity syndrome. Asian J Androl. 2008; 10:687-691. [DOI] [PubMed] [Google Scholar]
- 11. Gottlieb B, Beitel LK, Nadarajah A, Paliouras M, Trifiro M. The androgen receptor gene mutations database: 2012 update. Hum Mutat. 2012; 33:887-894. [DOI] [PubMed] [Google Scholar]
- 12. Li BK, Ding Q, Wan XD, Wang X. Clinical and genetic characterization of complete androgen insensitivity syndrome in a Chinese family. Gen Molec Res. 2011; 10:1022-1031. [DOI] [PubMed] [Google Scholar]
- 13. Brown CJ, Goss SJ, Lubahn DB, Joseph DR, Wilson EM, French FS, Willard HF. Androgen receptor locus on the human X chromosome: Regional localization to Xq11–12 and description of a DNA polymorphism. Am J Hum Genet. 1989; 44:264-269. [PMC free article] [PubMed] [Google Scholar]
- 14. Liao G, Chen LY, Zhang A, Godavarthy A, Xia F, Ghosh JC, Li H, Chen JD. Regulation of androgen receptor activity by the nuclear receptor corepressor SMRT. J Biol Chem. 2003; 278:5052-5061. [DOI] [PubMed] [Google Scholar]
- 15. Bain DL, Heneghan AF, Connaghan-Jones KD, Miura MT. Nuclear receptor structure: Implications for function. Annu Rev Physiol. 2007; 69:201-220. [DOI] [PubMed] [Google Scholar]
- 16. Verrijdt G, Tanner T, Moehren U, Callewaert L, Haelens A, Claessens F. The androgen receptor DNA-binding domain determines androgen selectivity of transcriptional response. Biochem Soc Trans. 2006; 34:1089-1094. [DOI] [PubMed] [Google Scholar]
- 17. Sack JS, Kish KF, Wang C, Attar RM, Kiefer SE, An Y, Wu GY, Scheffler JE, Salvati ME, Krystek SR, Jr, Weinmann R, Einspahr HM. Crystallographic structures of the ligand-binding domains of the androgen receptor and its t877A mutant complexed with the natural agonist dihydrotestosterone. Proc Natl Acad Sci U S A. 2001; 98:4904-4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Matias PM, Donner P, Coelho R, Thomaz M, Peixoto C, Macedo S, Otto N, Joschko S, Scholz P, Wegg A, Bäsler S, Schäfer M, Egner U, Carrondo MA. Structural evidence for ligand specificity in the binding domain of the human androgen receptor. Implications for pathogenic gene mutations. J Biol Chem. 2000; 275:26164-26171. [DOI] [PubMed] [Google Scholar]
- 19. Deans R, Creighton SM, Liao LM, Conway GS. Timing of gonadectomy in adult women with complete androgen insensitivity syndrome (CAIS): Patient preferences and clinical evidence. Clin Endocrinol (Oxf). 2012; 76:894-898. [DOI] [PubMed] [Google Scholar]
- 20. Minto CL, Liao KL, Conway GS, Creighton SM. Sexual function in women with complete androgen insensitivity syndrome. Fertil Steril. 2003; 80:157-164. [DOI] [PubMed] [Google Scholar]
- 21. Meyer-Bahlburg HF. Concerns regarding gender change to male in a 46,XY child with complete androgen insensitivity syndrome: Comment of Kulshreshtha et al. (2009). Arch Sex Behav. 2009; 38:876-877. [DOI] [PubMed] [Google Scholar]
- 22. T'Sjoen G, De Cuypere G, Monstrey S, Hoebeke P, Freedman FK, Appari M, Holterhus PM, Van Borsel J, Cools M. Male gender identity in complete androgen insensitivity syndrome. Arch Sex Behav. 2011; 40:635-638. [DOI] [PubMed] [Google Scholar]