

Abstract
Excessive loss of functional pancreatic β-cell mass, mainly due to apoptosis, is a major factor in the development of hyperglycemia in both type 1 and type 2 diabetes (T1D and T2D). In T1D, β-cells are destroyed by immunological mechanisms. In T2D, while metabolic factors are known to contribute to β-cell failure and subsequent apoptosis, mounting evidence suggests that islet inflammation also plays an important role in the loss of β-cell mass. Therefore, it is of great importance for clinical intervention to develop new therapies. γ-Aminobutyric acid (GABA), a major neurotransmitter, is also produced by islet β-cells, where it functions as an important intraislet transmitter in regulating islet-cell secretion and function. Importantly, recent studies performed in rodents, including in vivo studies of xenotransplanted human islets, reveal that GABA exerts β-cell regenerative effects. Moreover, it protects β-cells against apoptosis induced by cytokines, drugs, and other stresses, and has anti-inflammatory and immunoregulatory activities. It ameliorates the manifestations of diabetes in preclinical models, suggesting potential applications for the treatment of diabetic patients. This review outlines the actions of GABA relevant to β-cell regeneration, including its signaling mechanisms and potential interactions with other mediators. These studies increase our understanding of the regenerative processes of pancreatic β-cells, and help pave the way for the development of regenerative medicine for diabetes.
Keywords: β-cell, proliferation, apoptosis, GABA, ion channel, insulin, glucagon, inflammation, diabetes
Introduction
γ-Aminobutyric acid (GABA) was initially identified as a major inhibitory neurotransmitter in the brain by Roberts and Frankel in 1950.1 The biological functions of GABA are mediated by activation of the GABA receptors. There are two basic types, ie, type A and type B receptors (GABAAR and GABABR, respectively).2 In the adult brain GABAAR is the most prevalent receptor, and upon binding GABA, it exerts an inhibitory effect manifested by hyperpolarization of the cell membrane.2 In sharp contrast, in the developing brain GABA has a depolarizing effect, acting as the principal excitatory transmitter and exerting trophic effects, including cell proliferation and dendritic maturation.2–4
In addition to neurons, some nonneuronal cells, such as pancreatic islet cells, are found to produce GABA in large quantities.5–10 In α-cells, GABA induces membrane hyperpolarization and suppresses glucagon secretion,11–12 whereas in islet β-cells it induces membrane depolarization and increases insulin secretion.13–15 Moreover, GABA has multiple beneficial effects on β-cells, which include the stimulation of cell proliferation and antiapoptotic activities,13–16 making it an attractive agent for diabetes treatment. Importantly, it prevents insulitis to a remarkable degree in preclinical models,13–16 suggesting applications in the prevention of this disease, as well as treatment particularly in the context of clinical islet transplantation.
GABA
GABA biology
GABA is the predominant inhibitory neurotransmitter in the central nervous system (CNS). It is located in about 30% of cerebral neurons, and affects almost all neuronal activities.17 In the neuron, GABA is synthesized in the cytosol from its precursor glutamate by glutamate decarboxylase (GAD), and then transported into the synaptic vesicles against a proton electrochemical gradient.18 Exocytosis of synaptic vesicles is triggered when the voltage-gated calcium channels (VGCCs) open, resulting in a transient rise in cytosolic calcium. GABA is released into the synaptic space, and exerts its effect through binding to corresponding receptors.18 The effect of GABA is terminated rapidly after it is removed from the synaptic space by the actions of four types of plasma membrane GABA cotransporters (GAT1–4) located in the presynaptic terminal and in the glial cells.19
GABA is also present in peripheral organs, such as the testes, gastrointestinal tract, ovaries, placenta, uterus, and adrenal medulla, as well as the pancreas, where its concentration is the highest and comparable to that in the CNS.8,17 In accord with this, high levels of GAD have been detected in the islets of Langerhans.20 Moreover, it has been reported that both pancreatic α- and β-cells express a vesicular GABA transporter, which transports GABA into the intracellular vesicles for packaging before it is released, and GAT3, which mediates cellular uptake of GABA.21 The abundance of GABA and the presence of molecular machinery for GABA synthesis and release suggest an important role in pancreatic physiology.
GABA receptors
GABAARs are heteropentamers of different subunits that form fast-acting chloride channels.22 There are 19 known GABAAR subunits: α1–6, β1–3, γ1–3, δ, ε, θ, π, and ρ1–3.22–24 GABAARs with different subunit combinations possess different pharmacological properties. In the CNS, a functional GABAAR is mostly found in a configuration containing two α-subunits, two β-subunits, and one γ-subunit.25,26 Many of these subunits are found in islets or β-cell lines, and it appears that the αβγ configuration also represents a functional GABAAR in the islet cells.
GABABRs are composed of two invariable subunits: B1 and B2.27 This slow-acting receptor is linked to K+ channels. Activation of GABABR stimulates the opening of K+ channels, leading to hyperpolarization of the membrane potential and a reduction in the activity of adenylyl cyclase.28 This prevents the opening of sodium channels and the VGCC to convey inhibitory effects.29,30
In the adult brain, GABA exerts inhibitory effects that are primarily due to activation of the GABAAR Cl− ion channel, which leads to Cl− influx and membrane hyperpolarization.2,31 However, during brain development, GABA induces depolarizing effects, acting as the principal excitatory transmitter.3,4,32 This is because immature neurons have a higher intracellular Cl− concentration, and activation of GABAAR initiates an efflux of Cl−, leading to membrane depolarization and excitatory actions. GABA-induced depolarizing effects results in Ca2+ influx via VGCCs, and modulates a variety of Ca2+-dependent cellular processes, such as proliferation and differentiation, which are important in the formation of synapses and activity in the neuronal networks.
The shift from depolarizing to hyperpolarizing effects of GABA is associated with the onset of K+–Cl− cotransporter -2 (KCC2) expression.32 The neuron-specific KCC2 is responsible for establishing the Cl− gradient in neurons through the maintenance of low intracellular Cl− concentrations.32 In the islets, a functional KCC2 is found in the α-cells, but not in the β-cells,33 providing a molecular mechanism underlying the opposite actions of GABA in the islet β- and α-cells.
GABA and diabetes
GABA release in the endocrine system
A screen of selected peripheral organs of rats revealed that GABA was present at the highest concentration in the pancreas.8 Indeed, it has been reported that islet GABA content and release are increased in response to increasing extracellular L-glutamine concentration; when L-glutamine is at a physiological concentration (0.5 mM), the content of GABA reaches a maximum that remains stable at 5 and 10 mM. This suggests that GABA synthesis is always saturated under normal circumstances.34 GABA is further metabolized in the mitochondria through the GABA-shunt pathway, which may play a role in insulin secretion.20,35
The basal release of GABA from the β-cells is relatively constant,6,12,36 but it is modulated depending on the metabolic state of these cells.37 GABA is localized in synaptic-like microvesicles,6 and partially localized to large dense-core vesicles containing insulin.21 Therefore, β-cells secrete GABA by both a glucose-dependent (exocytosis of insulin-containing granules) and a glucose-independent mechanism.38 It appears that GABA is part of the fine-tuning machinery that is critical in maintaining islet-cell glucose competence (Figure 1).
Figure 1.
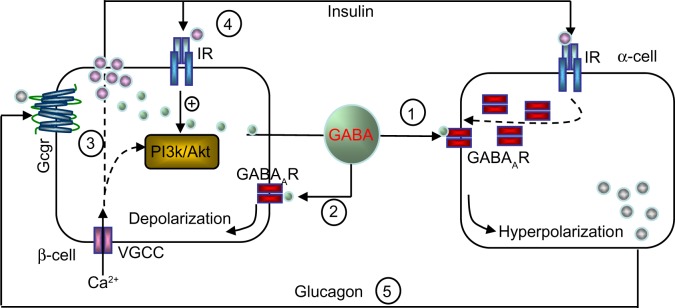
Hypothetical model of intraislet cell interaction.
Notes: ① Paracrine hyperpolarizing effects of GABA. ② Autocrine depolarizing effects of GABA. ③ GABA–GABAAR-Ca2+ -PI3K/Akt pathway. ④ Autocrine insulin action. ⑤ Intraislet glucagon action. There is an expanded discussion of these events in other reviews.91,92(\p)(p)Abbreviations: GABAAR, γ-aminobutyric acid type A receptor; IR, insulin receptor; VGCC, voltage-gated calcium channel; Gcgr, glucagon receptor; PI3K, phosphatidylinositol 3 kinase; Akt, protein kinase B.
Signaling mechanisms of GABA action
α-Cells
The signaling pathway is not fully elucidated. In neurons, GABA evokes Cl− currents that alter resting membrane potential and intracellular Ca2+ concentration.39–41 In this context, Ca2+ often acts as an important messenger initiating intracellular downstream signaling. During neuronal development, GABAAR-mediated depolarization and Ca2+ influx via VGCCs can activate the PI3K/PKC signaling pathway.42 Activation of these protein kinases can phosphorylate GABAARs and induce receptor-trafficking events.40,43–45
This is also the case with α-cells.11 In response to increasing glucose levels, insulin released from β-cells activates the insulin receptor present on α-cells. Subsequent activation of Akt leads to phosphorylation of β-subunits of GABAAR that causes rapid translocation of the receptor to the plasma membrane. Although GABA is constantly released, the efficacy of the receptor-mediated inhibitory currents (Cl−) and membrane hyperpolarization are enhanced, due to the increased GABAAR numbers at the cell surface. In turn, membrane hyperpolarization shuts down the VGCC and inhibits α-cell exocytosis and glucagon release.11 An impaired insulin/Akt/GABAAR/glucagon secretory pathway in the islet might be an underlying mechanism for unsuppressed glucagon secretion, despite hyperglycemia, in diabetic subjects.
β-Cells
Signaling differs sharply in β-cells, where GABA induces membrane depolarization.14,46,47 In isolated rodent and human islets, GABA was shown to stimulate Akt activation, promoting β-cell proliferation and survival in a GABAAR antagonist- and/or Ca2+ channel blocker-sensitive fashion.13–14 This suggests that the GABAAR-mediated Ca2+-dependent PI3K/Akt pathway is a major mediator in conveying the trophic effects of GABA on β-cells.
GABABR is a G-protein-coupled receptor that initiates cyclic adenosine monophosphate signaling and Ca2+-dependent signaling. Previous studies demonstrated that in neurons GABAAR activation induced VGCC-dependent extracellular Ca2+ influx and Ca2+ release from intracellular stores, whereas GABABR evoked intracellular Ca2+ only.48 This is consistent with our observation that GABA-mediated elevation of intracellular Ca2+ in human β-cells was blocked by the type A receptor antagonist (picrotoxin), while it was only partially attenuated by the type B receptor antagonist (saclofen).13 Notably, our studies suggested that GABA stimulated CREB activation in a cyclic adenosine monophosphate/PKA-dependent signaling pathway mediated by GABABR.13 CREB plays a key role in regulating β-cell mass homeostasis, as mice lacking CREB in their β-cells have diminished expression of IRS249 and display excessive β-cell loss.50 CREB is also a target gene of Akt signaling.51 Interestingly, our study showed that the GABA-GABABR-induced CREB activation was independent of the PI3K/Akt pathway, because upon inhibition of PI3K/Akt, activation of CREB was not suppressed, whereas blockade of PKA-dependent CREB did not affect GABA-stimulated Akt activation.13 This is of relevance, and provides a plausible mechanism by which GABA may be bypassing Akt activation in subjects with insulin resistance.
Studies examining the role of GABABR in modulating insulin secretion have shown variable results. It has been reported that in the presence of glucose at high concentration (over 10 mmol/L) GABABR displays an inhibitory effect on insulin secretion,52,53 whereas in the presence of lower glucose levels it has no effect.53 In vivo studies suggest an important role for GABABR in regulating β-cell function. For instance, the treatment of nonobese diabetic (NOD) mice with a GABABR agonist delayed onset of type 1 diabetes (T1D),54 and other studies showed GABABR-dependent improvement of β-cell survival and proliferation.13,16,55 In apparent contradiction, GABAB1R-deficient mice (global knockout of B1 subunit) displayed improved glucose tolerance, increased pancreatic insulin content, and elevated glucose-stimulated insulin secretion, associated with enlarged islets and insulin resistance.56,57 The GABABR agonist baclofen inhibited glucose-stimulated insulin secretion in wild-type but not GABABR-knockout islets.57 At this point, it appears that this receptor can have inhibitory or stimulatory effects on β-cell functions under various circumstances, and this merits further investigation.
It is interesting to note that in addition to GABA-receptor signaling, GABA catabolism also contributes to insulin secretion. GABA is catabolized in the GABA-shunt pathway after its transamination with 2-oxoglutarate by GABA transaminase into succinic semialdehyde and glutamate.58 It is reported that both α-ketoisocaproic acid58,59 and glucose35 promote GABA metabolism and stimulate insulin secretion in a GABA-shunt-dependent fashion. This mechanism appears to be important, especially in rendering β-cells more competent in the face of a restriction in the metabolic flow of the citric acid cycle set by low 2-oxoglutarate dehydrogenase activity in response to high glucose stimulation.35
Effects of GABA on the immune system and inflammatory pathways in diabetes
The effects of GABA on the immune system are generally inhibitory. In humans, GABAAR is expressed by T cells (CD4+ and CD8+), B cells, and some mononuclear cells, and GABAAR agonists suppress lymphocyte proliferation.60–62 In mice, Tian et al63 identified GABAAR on T cells and showed that GABA (0.1–3 mM) partially inhibited (~50%) T-cell responses. These effects were duplicated by a GABAAR agonist (muscimol), but not by a GABABR agonist (baclofen), indicating that only GABAAR is involved. In a subsequent study, Tian et al64 reported that GABA protected NOD mice against T1D, and reduced the response of their T-helper (Th)-1 cells to islet antigens.
Although T1D is a T-cell-dependent autoimmune disease, inflammatory mechanisms that are not directly T-cell-mediated also appear to play a major role in the pathogenesis.65 This is particularly apparent in multiple low-dose streptozotocin-induced diabetes (MDSD).66 Notably, anti-inflammatory treatment is beneficial in both MDSD and autoimmune NOD mice.65,67 In agreement with this, we found that GABA therapy lowered the levels of inflammatory cytokines (eg, IL-1β, TNFα, IFNγ, and IL-12) in the serum of MDSD mice.14 In vitro, GABA also reduced the secretion of these cytokines. In contrast, it increased TGFβ1 production, which is a key regulatory (suppressive) cytokine.14
It is clear that GABA acts on T cells of both effector and regulatory function. It suppresses the action of Th1 cells,14,64 as well as cytotoxic T lymphocytes, as demonstrated in a T-cell receptor (TCR) transgenic mouse model of T1D.14 Importantly, we found that GABA increases regulatory T cells (Tregs) of the Foxp3+/neuropilin-1+ phenotype,14 currently denoted thymus-derived Tregs. Tian et al68 reported similar findings in a mouse type 2 diabetes (T2D) model. We hypothesize that this contributes to the immunosuppressive effects of GABA, and the underlying molecular mechanisms warrant further investigation. Other investigators have also shown immunosuppressive effects. For instance, Bjurstöm et al69 reported that low levels of GABA (as low as 100 nM) suppressed CD4+ encephalitogenic T cells that causes experimental autoimmune encephalitis. These levels are similar to the concentration of GABA in normal plasma. Bhat et al70 also reported protective effects in autoimmune encephalitis, but they concluded that GABA acted by suppressing macrophages.
We found that the immunosuppressive effects of GABA first observed in mice are also present in humans.15 Indeed, it suppressed CD3-stimulated human T-cell proliferation in a GABAAR-dependent manner. Until recently, the immunosuppressive mechanisms were unclear, but we observed that GABA blocks calcium influx in human T cells. The calcium signal is a very early T-cell-activation signal, and its blockade is expected to lead to the inhibition of all subsequent activation-related events. Moreover, GABA suppressed NFκB activation in both human T cells and islet cells.15 This is a key observation, because NFκB is a critically important pathway involved in both innate and adaptive immunity. Indeed, its activation appears closely linked to β-cell apoptosis initiated by inflammatory cytokines and other cell injuries.
However, it is unclear how GABA mediates these effects. Interestingly, we observed that GABA-induced effects in β-cells are associated with elevated activity of SIRT1, suggesting the involvement of this enzyme.71 SIRT1 is an NAD+-dependent deacetylase that increases insulin secretion, and can block NFκB signaling. We found that the incubation of INS-1 insulinoma cells with GABA augmented SIRT1 expression, as did agonists acting on either GABAAR or GABABR. In addition, NAD+, which is essential for SIRT1 enzymatic action, was increased. GABA increased SIRT1 activity, which resulted in deacetylation of the p65 component of NFκB. This type deacetylation has previously been shown to interfere with the activation of NFκB. Of note, GABA stimulated insulin production and reduced apoptosis, and these effects were negated by SIRT1 inhibitors. We investigated whether SIRT1 and NAD+ are similarly induced by GABA in human islet cells, and found that this was indeed the case. As in INS-1 cells, the protective effect of GABA against human islet-cell apoptosis was SIRT1-dependent. Therefore, it appears that important beneficial effects of GABA on β-cells are due to increased SIRT1 and NAD+.
Of importance to islet transplantation, we found that GABA alleviates the toxic effects of immunosuppressive drugs, including rapamycin, tacrolimus (FK506), and mycophenolate mofetil.15 These drugs are used to prevent rejection of transplanted organs or islets of Langerhans, and have all been implicated in impairing β-cell function and survival.72–74 Interestingly, while GABA reduced the toxicity of rapamycin, it collaborated with rapamycin to improve T-cell suppression. This suggests that GABA could be combined with other immunosuppressive drugs in a manner that protects islet cells and improves immunotherapy.
GABA and therapy of diabetes
GABA therapy of diabetes in preclinical models
T1D is an autoimmune disease characterized by infiltration of the pancreatic islets by T lymphocytes, macrophages, and other immune cells, and consequent loss of β-cells.65,75,76 At the onset of T1D, the majority of β-cells are destroyed, resulting in a severe lack of insulin production.77 In T2D, insulin resistance and β-cell failure remain the major pathophysiological defects; however, mounting evidence suggests that islet inflammation also plays an important role in the loss of functional β-cell mass in T2D. Therefore, therapies of T1D and T2D require both suppression of the inflammatory process and restoration of islet β-cells.
GABA induces membrane depolarization and Ca2+-dependent activation of cell-growth and survival pathways involving PI3K/Akt.13–14 This is in accord with our findings that GABA therapy preserves β-cell mass and prevents diabetes in three mouse models of T1D, ie, in the MDSD model, wild-type NOD mice, and transgenic TCR-8.3 NOD mice. Remarkably, in severely diabetic mice (MDSD model), GABA therapy regenerated β-cell mass and completely reversed hyperglycemia.14 This was associated with anti-inflammatory and immunoregulatory events, which also appear to contribute to the success of therapy.
Our studies showed that activation of both GABAAR and GABABR are important in mediating GABA trophic effects to promote β-cell replication and survival, in both rodents and humans,13 which is consistent with a study by Tian et al.16 To analyze whether GABA could exert therapeutic effects on human islet cells, we used a suboptimal (marginal mass) islet-xenotransplantation model. This suboptimal mass of human islets was transplanted into immunodeficient NOD-severe combined immunodeficiency-γ mice after induction of diabetes with streptozotocin.13 This in vivo approach revealed that oral GABA treatment increased graft-cell proliferation and decreased apoptosis, leading to a significantly enhanced β-cell mass. Furthermore, GABA lowered blood glucose levels and ameliorated glucose tolerance. Our results suggest that the Ca2+-dependent PI3K/Akt and CREB/IRS2 are two synergistic and independent signaling pathways that mediate the trophic effect of GABA in human islet cells.13
In addition, GABA appears to be beneficial to T2D. Tian et al68 demonstrated that oral treatment with GABA improves glucose tolerance and insulin sensitivity in high-fat diet-fed mice. They concluded that this was due to the inhibition of obesity-related inflammation and upregulation of Treg responses.
GAD as a target antigen in T1D
GAD is colocalized with GABA6 in islets of rodents and humans, and the predominant isoform (GAD65 or GAD67) varies between species.78 GAD65 has long been identified as one of the major target antigens recognized by self-reactive T cells in T1D.79 We hypothesize that autoimmunity against GAD in the islet depletes this enzyme and hence reduces GABA levels, and this promotes the progression of this disease. Indeed, immunomodulation with GAD65 vaccination has been extensively investigated for the prevention or treatment of T1D.79 In 1994, the Swedish pharmaceutical company Diamyd Medical licensed the rights to GAD65 as the active substance in the antigen-based diabetes therapy Diamyd®. Unfortunately, despite promising results from Phase I and II trials,80–82 the Phase III study failed to produce a therapeutic effect.83 These results are disappointing, but a recent publication suggests that the antigen therapy in combination with other agents, such as GABA, may hold promise for intervention in T1D.84
Combined islet-antigen and GABA therapy
Antigen therapy of T1D by administration of insulin, GAD, peptides, or other islet antigens with various adjuvant formulations or delivery methods has proven effective in mice, but thus far clinical trials have shown only limited or no benefits.79,85 However, preclinical studies of combined antigen vaccination and GABA therapy suggest a possible avenue for improvement. Recently, Tian et al84 reported that combined therapy with proinsulin/alum and GABA synergizes to restore normoglycemia in newly diabetic NOD mice, sometimes with permanent remission of the disease. In this study, proinsulin/alum alone failed to correct hyperglycemia, whereas GABA monotherapy restored normoglycemia for a limited period of time. Therefore, combined therapy proved necessary to induce remission, and was found to inhibit pathogenic T-cell responses and to promote β-cell proliferation.
In another study,86 the combination of oral GABA treatment and immunization with a GAD/alum formulation was also found to be effective in a transplantation model. In diabetic NOD mice receiving syngeneic pancreas grafts, combined therapy was much more effective than monotherapy in prolonging the survival of β-cells.
GABA administration to humans
Studies published as far back as 1960 showed that GABA can be administered orally in large amounts to humans (several grams/day) without serious adverse effects.87 To date, there are at least three reports studying the effects of GABA on endocrine pancreatic function in humans. In one study, a single oral dose of GABA (5 or 10 g) significantly increased plasma insulin and C-peptide levels in 12 healthy subjects.88 In other study, 20 mg GABABR agonist baclofen was administered orally to ten healthy subjects 1 hour prior to glucose challenge and posttreatment test, which resulted in significantly increased insulin responses to glucose challenge and increased basal glucagon levels. However, glucose tolerance was not found to be changed after baclofen treatment.89 In another study, it was shown that intravenous administration of GABA (2–4 mg) significantly reduced blood glucose levels in the majority of diabetic individuals, but not in the nondiabetic subjects.90 These human studies were quite limited in scope, but nevertheless suggest that GABA plays a role in regulating endocrine pancreatic function. We are conducting clinical trials to study the pharmacokinetics and pharmacodynamics of GABA in healthy subjects (http://clinicaltrials.gov/show/NCT01917760), as well as its efficacy in patients with newly diagnosed T1D (http://clinicaltrials.gov/show/NCT01781884). Our Phase I GABA study in health volunteers showed that GABA orally administered was rapidly absorbed in the gastrointestinal tract and had a favorable safety profile.
Conclusion
In the past five decades, the function of GABA in the CNS has been well documented. However, the presence of a GABAergic system within the pancreas as a potential target for treating diabetes mellitus emerged only recently. In α-cells, GABA induces membrane hyperpolarization and inhibits glucagon secretion, and this involves an insulin-mediated GABAAR-trafficking mechanism. In β-cells, GABA induces membrane depolarization and enhances insulin secretion. GABA also has beneficial effects on β-cell survival and regeneration, which results in enlarged β-cell mass. Furthermore, GABA suppresses insulitis and systemic inflammatory cytokine production. All these data hold promise for GABA therapy in regulating islet cell function, glucose homeostasis, and autoimmunity. Of note, similarly to rodent studies, GABA shows trophic effects on human islets. This is important, because while a number of agents exert protective and proliferative effects on rodent islet cells, very few show similar activities on human islet cells. Orally administered GABA is safe for humans, and acts on peripheral GABA receptors but does not affect CNS functions, since it does not cross the blood–brain barrier, and thus it represents a promising new therapeutic agent for diabetes.
Acknowledgments
Our studies were supported by the Juvenile Diabetes Research Foundation (JDRF), the Canadian Institute for Health Research (CIHR), the Canadian Diabetes Association (CDA), and the National Science Foundation China (NSFC).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Roberts E, Frankel S. γ-Aminobutyric acid in brain: its formation from glutamic acid. J Biol Chem. 1950;187(1):55–63. [PubMed] [Google Scholar]
- 2.Owens DF, Kriegstein AR. Is there more to GABA than synaptic inhibition? Nat Rev Neurosci. 2002;3(9):715–727. doi: 10.1038/nrn919. [DOI] [PubMed] [Google Scholar]
- 3.Represa A, Ben-Ari Y. Trophic actions of GABA on neuronal development. Trends Neurosci. 2005;28(6):278–283. doi: 10.1016/j.tins.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 4.Fiszman ML, Schousboe A. Role of calcium and kinases on the neurotrophic effect induced by gamma-aminobutyric acid. J Neurosci Res. 2004;76(4):435–441. doi: 10.1002/jnr.20062. [DOI] [PubMed] [Google Scholar]
- 5.Adeghate E, Ponery AS. GABA in the endocrine pancreas: cellular localization and function in normal and diabetic rats. Tissue Cell. 2002;34(1):1–6. doi: 10.1054/tice.2002.0217. [DOI] [PubMed] [Google Scholar]
- 6.Reetz A, Solimena M, Matteoli M, Folli F, Takei K, De Camilli P. GABA and pancreatic beta-cells: colocalization of glutamic acid decarboxylase (GAD) and GABA with synaptic-like microvesicles suggests their role in GABA storage and secretion. EMBO J. 1991;10(5):1275–1284. doi: 10.1002/j.1460-2075.1991.tb08069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taniguchi H, Okada Y, Seguchi H, et al. High concentration of gamma-aminobutyric acid in pancreatic beta cells. Diabetes. 1979;28(7):629–633. doi: 10.2337/diab.28.7.629. [DOI] [PubMed] [Google Scholar]
- 8.Gerber JC, 3rd, Hare TA. Gamma-aminobutyric acid in peripheral tissue, with emphasis on the endocrine pancreas: presence in two species and reduction by streptozotocin. Diabetes. 1979;28(12):1073–1076. doi: 10.2337/diab.28.12.1073. [DOI] [PubMed] [Google Scholar]
- 9.Vincent SR, Hökfelt T, Wu JY, Elde RP, Morgan LM, Kimmel JR. Immunohistochemical studies of the GABA system in the pancreas. Neuroendocrinology. 1983;36(3):197–204. doi: 10.1159/000123456. [DOI] [PubMed] [Google Scholar]
- 10.Okada Y, Taniguchi H, Schimada C. High concentration of GABA and high glutamate decarboxylase activity in rat pancreatic islets and human insulinoma. Science. 1976;194(4265):620–622. doi: 10.1126/science.185693. [DOI] [PubMed] [Google Scholar]
- 11.Xu E, Kumar M, Zhang Y, et al. Intra-islet insulin suppresses glucagon release via GABA-GABAA receptor system. Cell Metab. 2006;3(1):47–58. doi: 10.1016/j.cmet.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 12.Rorsman P, Berggren PO, Bokvist K, et al. Glucose-inhibition of glucagon secretion involves activation of GABAA-receptor chloride channels. Nature. 1989;341(6239):233–236. doi: 10.1038/341233a0. [DOI] [PubMed] [Google Scholar]
- 13.Purwana I, Zheng J, Li X, et al. GABA promotes human β-cell proliferation and modulates glucose homeostasis. Diabetes. 2014;63(12):4197–4205. doi: 10.2337/db14-0153. [DOI] [PubMed] [Google Scholar]
- 14.Soltani N, Qiu H, Aleksic M, et al. GABA exerts protective and regenerative effects on islet beta cells and reverses diabetes. Proc Natl Acad Sci U S A. 2011;108(28):11692–11697. doi: 10.1073/pnas.1102715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prud’Homme GJ, Glinka Y, Hasilo C, Paraskevas S, Li X, Wang Q. GABA protects human islet cells against the deleterious effects of immunosuppressive drugs and exerts immunoinhibitory effects alone. Transplantation. 2013;96(7):616–623. doi: 10.1097/TP.0b013e31829c24be. [DOI] [PubMed] [Google Scholar]
- 16.Tian J, Dang H, Chen Z, et al. γ-Aminobutyric acid regulates both the survival and replication of human β-cells. Diabetes. 2013;62(11):3760–3765. doi: 10.2337/db13-0931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gladkevich A, Korf J, Hakobyan VP, Melkonyan KV. The peripheral GABAergic system as a target in endocrine disorders. Auton Neurosci. 2006;124(1–2):1–8. doi: 10.1016/j.autneu.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Thomas-Reetz AC, De Camilli P. A role for synaptic vesicles in non-neuronal cells: clues from pancreatic beta cells and from chromaffin cells. FASEB J. 1994;8(2):209–216. doi: 10.1096/fasebj.8.2.7907072. [DOI] [PubMed] [Google Scholar]
- 19.Glykys J, Mody I. Activation of GABAA receptors: views from outside the synaptic cleft. Neuron. 2007;56(5):763–770. doi: 10.1016/j.neuron.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 20.Sorenson RL, Garry DG, Brelje TC. Structural and functional considerations of GABA in islets of Langerhans. Beta-cells and nerves. Diabetes. 1991;40(11):1365–1374. doi: 10.2337/diab.40.11.1365. [DOI] [PubMed] [Google Scholar]
- 21.Gammelsaeter R, Frøyland M, Aragón C, et al. Glycine, GABA and their transporters in pancreatic islets of Langerhans: evidence for a paracrine transmitter interplay. J Cell Sci. 2004;117(Pt 17):3749–3758. doi: 10.1242/jcs.01209. [DOI] [PubMed] [Google Scholar]
- 22.Rudolph U, Knoflach F. Beyond classical benzodiazepines: novel therapeutic potential of GABAA receptor subtypes. Nat Rev Drug Discov. 2011;10(9):685–697. doi: 10.1038/nrd3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olsen RW, Tobin AJ. Molecular biology of GABAA receptors. FASEB J. 1990;4(5):1469–1480. doi: 10.1096/fasebj.4.5.2155149. [DOI] [PubMed] [Google Scholar]
- 24.Lüddens H, Korpi ER, Seeburg PH. GABAA/benzodiazepine receptor heterogeneity: neurophysiological implications. Neuropharmacology. 1995;34(3):245–254. doi: 10.1016/0028-3908(94)00158-o. [DOI] [PubMed] [Google Scholar]
- 25.Mody I, Pearce RA. Diversity of inhibitory neurotransmission through GABA(A) receptors. Trends Neurosci. 2004;27(9):569–575. doi: 10.1016/j.tins.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 26.Lüscher B, Keller CA. Regulation of GABAA receptor trafficking, channel activity, and functional plasticity of inhibitory synapses. Pharmacol Ther. 2004;102(3):195–221. doi: 10.1016/j.pharmthera.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 27.White JH, Wise A, Main MJ, et al. Heterodimerization is required for the formation of a functional GABA(B) receptor. Nature. 1998;396(6712):679–682. doi: 10.1038/25354. [DOI] [PubMed] [Google Scholar]
- 28.Hashimoto T, Kuriyama K. In vivo evidence that GABA(B) receptors are negatively coupled to adenylate cyclase in rat striatum. J Neurochem. 1997;69(1):365–370. doi: 10.1046/j.1471-4159.1997.69010365.x. [DOI] [PubMed] [Google Scholar]
- 29.Bettler B, Kaupmann K, Mosbacher J, Gassmann M. Molecular structure and physiological functions of GABA(B) receptors. Physiol Rev. 2004;84(3):835–867. doi: 10.1152/physrev.00036.2003. [DOI] [PubMed] [Google Scholar]
- 30.Bowery NG. GABAB receptor pharmacology. Annu Rev Pharmacol Toxicol. 1993;33:109–147. doi: 10.1146/annurev.pa.33.040193.000545. [DOI] [PubMed] [Google Scholar]
- 31.Kittler JT, Moss SJ. Modulation of GABAA receptor activity by phosphorylation and receptor trafficking: implications for the efficacy of synaptic inhibition. Curr Opin Neurobiology. 2003;13(3):341–347. doi: 10.1016/s0959-4388(03)00064-3. [DOI] [PubMed] [Google Scholar]
- 32.Ludwig A, Li H, Saarma M, Kaila K, Rivera C. Developmental up-regulation of KCC2 in the absence of GABAergic and glutamatergic transmission. Eur J Neurosci. 2003;18(12):3199–3206. doi: 10.1111/j.1460-9568.2003.03069.x. [DOI] [PubMed] [Google Scholar]
- 33.Davies SL, Roussa E, Le Rouzic P, et al. Expression of K+-Cl- cotransporters in the alpha-cells of rat endocrine pancreas. Biochim Biophys Acta. 2004;1667(1):7–14. doi: 10.1016/j.bbamem.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 34.Fernández-Pascual S, Mukala-Nsengu-Tshibangu A, Martín Del Río R, Tamarit-Rodríguez J. Conversion into GABA (gamma-aminobutyric acid) may reduce the capacity of L-glutamine as an insulin secretagogue. Biochem J. 2004;379(Pt 3):721–729. doi: 10.1042/BJ20031826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pizarro-Delgado J, Braun M, Hernández-Fisac I, Martín-Del-Río R, Tamarit-Rodriguez J. Glucose promotion of GABA metabolism contributes to the stimulation of insulin secretion in beta-cells. Biochem J. 2010;431(3):381–389. doi: 10.1042/BJ20100714. [DOI] [PubMed] [Google Scholar]
- 36.Smismans A, Schuit F, Pipeleers D. Nutrient regulation of gamma-aminobutyric acid release from islet beta cells. Diabetologia. 1997;40(12):1411–1415. doi: 10.1007/s001250050843. [DOI] [PubMed] [Google Scholar]
- 37.Winnock F, Ling Z, De Proft R, et al. Correlation between GABA release from rat islet beta-cells and their metabolic state. Am J Physiol Endocrinol Metab. 2002;282(4):E937–E942. doi: 10.1152/ajpendo.00071.2001. [DOI] [PubMed] [Google Scholar]
- 38.Braun M, Wendt A, Karanauskaite J, et al. Corelease and differential exit via the fusion pore of GABA, serotonin, and ATP from LDCV in rat pancreatic beta cells. J Gen Physiol. 2007;129(3):221–231. doi: 10.1085/jgp.200609658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moulder KL, Cormier RJ, Shute AA, Zorumski CF, Mennerick S. Homeostatic effects of depolarization on Ca2+ influx, synaptic signaling, and survival. J Neurosci. 2003;23(5):1825–1831. doi: 10.1523/JNEUROSCI.23-05-01825.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vithlani M, Moss SJ. The role of GABAAR phosphorylation in the construction of inhibitory synapses and the efficacy of neuronal inhibition. Biochem Soc Trans. 2009;37(Pt 6):1355–1358. doi: 10.1042/BST0371355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fukura H, Komiya Y, Igarashi M. Signaling pathway downstream of GABAA receptor in the growth cone. J Neurochem. 1996;67(4):1426–1434. doi: 10.1046/j.1471-4159.1996.67041426.x. [DOI] [PubMed] [Google Scholar]
- 42.Porcher C, Hatchett C, Longbottom RE, et al. Positive feedback regulation between gamma-aminobutyric acid type A (GABA(A)) receptor signaling and brain-derived neurotrophic factor (BDNF) release in developing neurons. J Biol Chem. 2011;286(24):21667–21677. doi: 10.1074/jbc.M110.201582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Terunuma M, Pangalos MN, Moss SJ. Functional modulation of GABAB receptors by protein kinases and receptor trafficking. Adv Pharmacol. 2010;58:113–122. doi: 10.1016/S1054-3589(10)58005-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chou WH, Wang D, McMahon T, et al. GABAA receptor trafficking is regulated by protein kinase Cε and the N-ethylmaleimide-sensitive factor. J Neurosci. 2010;30(42):13955–13965. doi: 10.1523/JNEUROSCI.0270-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Birnir B, Korpi ER. The impact of sub-cellular location and intracellular neuronal proteins on properties of GABA(A) receptors. Curr Pharm Des. 2007;13(31):3169–3177. doi: 10.2174/138161207782341330. [DOI] [PubMed] [Google Scholar]
- 46.Braun M, Ramracheya R, Bengtsson M, et al. Gamma-aminobutyric acid (GABA) is an autocrine excitatory transmitter in human pancreatic beta-cells. Diabetes. 2010;59(7):1694–1701. doi: 10.2337/db09-0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dong H, Kumar M, Zhang Y, et al. Gamma-aminobutyric acid up- and downregulates insulin secretion from beta cells in concert with changes in glucose concentration. Diabetologia. 2006;49(4):697–705. doi: 10.1007/s00125-005-0123-1. [DOI] [PubMed] [Google Scholar]
- 48.Schwirtlich M, Emri Z, Antal K, Máté Z, Katarova Z, Szabó G. GABA(A) and GABA(B) receptors of distinct properties affect oppositely the proliferation of mouse embryonic stem cells through synergistic elevation of intracellular Ca(2+) FASEB J. 2010;24(4):1218–1228. doi: 10.1096/fj.09-143586. [DOI] [PubMed] [Google Scholar]
- 49.Jhala US, Canettieri G, Screaton RA, et al. cAMP promotes pancreatic beta-cell survival via CREB-mediated induction of IRS2. Genes Dev. 2003;17(13):1575–1580. doi: 10.1101/gad.1097103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Withers DJ, Gutierrez JS, Towery H, et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998;391(6670):900–904. doi: 10.1038/36116. [DOI] [PubMed] [Google Scholar]
- 51.Du K, Montminy M. CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem. 1998;273(49):32377–32379. doi: 10.1074/jbc.273.49.32377. [DOI] [PubMed] [Google Scholar]
- 52.Gu XH, Kurose T, Kato S, et al. Suppressive effect of GABA on insulin secretion from the pancreatic beta-cells in the rat. Life Sci. 1993;52(8):687–694. doi: 10.1016/0024-3205(93)90229-v. [DOI] [PubMed] [Google Scholar]
- 53.Brice NL, Varadi A, Ashcroft SJ, Molnar E. Metabotropic glutamate and GABA(B) receptors contribute to the modulation of glucose-stimulated insulin secretion in pancreatic beta cells. Diabetologia. 2002;45(2):242–252. doi: 10.1007/s00125-001-0750-0. [DOI] [PubMed] [Google Scholar]
- 54.Beales PE, Hawa M, Williams AJ, Albertini MC, Giorgini A, Pozzilli P. Baclofen, a gamma-aminobutyric acid-b receptor agonist, delays diabetes onset in the non-obese diabetic mouse. Acta Diabetol. 1995;32(1):53–56. doi: 10.1007/BF00581047. [DOI] [PubMed] [Google Scholar]
- 55.Ligon B, Yang J, Morin SB, Ruberti MF, Steer ML. Regulation of pancreatic islet cell survival and replication by gamma-aminobutyric acid. Diabetologia. 2007;50(4):764–773. doi: 10.1007/s00125-007-0601-8. [DOI] [PubMed] [Google Scholar]
- 56.Crivello M, Bonaventura MM, Chamson-Reig A, et al. Postnatal development of the endocrine pancreas in mice lacking functional GABAB receptors. Am J Physiol Endocrinol Metab. 2013;304(10):E1064–E1076. doi: 10.1152/ajpendo.00569.2012. [DOI] [PubMed] [Google Scholar]
- 57.Bonaventura MM, Catalano PN, Chamson-Reig A, et al. GABAB receptors and glucose homeostasis: evaluation in GABAB receptor knockout mice. Am J Physiol Endocrinol Metab. 2008;294(1):E157–E167. doi: 10.1152/ajpendo.00615.2006. [DOI] [PubMed] [Google Scholar]
- 58.Pizarro-Delgado J, Hernández-Fisac I, Martín-Del-Río R, Tamarit-Rodriguez J. Branched-chain 2-oxoacid transamination increases GABA-shunt metabolism and insulin secretion in isolated islets. Biochem J. 2009;419(2):359–368. doi: 10.1042/BJ20081731. [DOI] [PubMed] [Google Scholar]
- 59.Hernández-Fisac I, Fernández-Pascual S, Ortsäter H, et al. Oxo-4-methylpentanoic acid directs the metabolism of GABA into the Krebs cycle in rat pancreatic islets. Biochem J. 2006;400(1):81–89. doi: 10.1042/BJ20060173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alam S, Laughton DL, Walding A, Wolstenholme AJ. Human peripheral blood mononuclear cells express GABAA receptor subunits. Mol Immunol. 2006;43(9):1432–1442. doi: 10.1016/j.molimm.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 61.Dionisio L, Jose De Rosa M, Bouzat C, Esandi Mdel C. An intrinsic GABAergic system in human lymphocytes. Neuropharmacology. 2011;60(2–3):513–519. doi: 10.1016/j.neuropharm.2010.11.007. [DOI] [PubMed] [Google Scholar]
- 62.Mendu SK, Bhandage A, Jin Z, Birnir B. Different subtypes of GABA-A receptors are expressed in human, mouse and rat T lymphocytes. PloS One. 2012;7(8):e42959. doi: 10.1371/journal.pone.0042959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tian J, Chau C, Hales TG, Kaufman DL. GABA(A) receptors mediate inhibition of T cell responses. J Neuroimmunol. 1999;96(1):21–28. doi: 10.1016/s0165-5728(98)00264-1. [DOI] [PubMed] [Google Scholar]
- 64.Tian J, Lu Y, Zhang H, Chau CH, Dang HN, Kaufman DL. Gamma-aminobutyric acid inhibits T cell autoimmunity and the development of inflammatory responses in a mouse type 1 diabetes model. J Immunol. 2004;173(8):5298–5304. doi: 10.4049/jimmunol.173.8.5298. [DOI] [PubMed] [Google Scholar]
- 65.Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol. 2009;5(4):219–226. doi: 10.1038/nrendo.2009.21. [DOI] [PubMed] [Google Scholar]
- 66.Kolb-Bachofen V, Epstein S, Kiesel U, Kolb H. Low-dose streptozocin-induced diabetes in mice. Electron microscopy reveals single-cell insulitis before diabetes onset. Diabetes. 1988;37(1):21–27. doi: 10.2337/diab.37.1.21. [DOI] [PubMed] [Google Scholar]
- 67.Cockfield SM, Ramassar V, Urmson J, Halloran PF. Multiple low dose streptozotocin induces systemic MHC expression in mice by triggering T cells to release IFN-gamma. J Immunol. 1989;142(4):1120–1128. [PubMed] [Google Scholar]
- 68.Tian J, Dang HN, Yong J, et al. Oral treatment with gamma-aminobutyric acid improves glucose tolerance and insulin sensitivity by inhibiting inflammation in high fat diet-fed mice. PloS One. 2011;6(9):e25338. doi: 10.1371/journal.pone.0025338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bjurstöm H, Wang J, Ericsson I, et al. GABA, a natural immunomodulator of T lymphocytes. J Neuroimmunol. 2008;205(1–2):44–50. doi: 10.1016/j.jneuroim.2008.08.017. [DOI] [PubMed] [Google Scholar]
- 70.Bhat R, Axtell R, Mitra A, et al. Inhibitory role for GABA in autoimmune inflammation. Proc Natl Acad Sci U S A. 2010;107(6):2580–2585. doi: 10.1073/pnas.0915139107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Prud’homme GJ, Glinka Y, Udovyk O, Hasilo C, Paraskevas S, Wang Q. GABA protects pancreatic beta cells against apoptosis by increasing SIRT1 expression and activity. Biochem Biophys Res Commun. 2014;452(3):649–654. doi: 10.1016/j.bbrc.2014.08.135. [DOI] [PubMed] [Google Scholar]
- 72.Barlow AD, Xie J, Moore CE, et al. Rapamycin toxicity in MIN6 cells and rat and human islets is mediated by the inhibition of mTOR complex 2 (mTORC2) Diabetologia. 2012;55(5):1355–1365. doi: 10.1007/s00125-012-2475-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bell E, Cao X, Moibi JA, et al. Rapamycin has a deleterious effect on MIN-6 cells and rat and human islets. Diabetes. 2003;52(11):2731–2739. doi: 10.2337/diabetes.52.11.2731. [DOI] [PubMed] [Google Scholar]
- 74.Johnson JD, Ao Z, Ao P, et al. Different effects of FK506, rapamycin, and mycophenolate mofetil on glucose-stimulated insulin release and apoptosis in human islets. Cell Transplant. 2009;18(8):833–845. doi: 10.3727/096368909X471198. [DOI] [PubMed] [Google Scholar]
- 75.Atkinson MA, Eisenbarth GS. Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet. 2001;358(9277):221–229. doi: 10.1016/S0140-6736(01)05415-0. [DOI] [PubMed] [Google Scholar]
- 76.Lehuen A, Diana J, Zaccone P, Cooke A. Immune cell crosstalk in type 1 diabetes. Nat Rev Immunol. 2010;10(7):501–513. doi: 10.1038/nri2787. [DOI] [PubMed] [Google Scholar]
- 77.Pipeleers D, Chintinne M, Denys B, Martens G, Keymeulen B, Gorus F. Restoring a functional beta-cell mass in diabetes. Diabetes Obes Metab. 2008;10(Suppl 4):54–62. doi: 10.1111/j.1463-1326.2008.00941.x. [DOI] [PubMed] [Google Scholar]
- 78.Kim J, Richter W, Aanstoot HJ, et al. Differential expression of GAD65 and GAD67 in human, rat, and mouse pancreatic islets. Diabetes. 1993;42(12):1799–1808. doi: 10.2337/diab.42.12.1799. [DOI] [PubMed] [Google Scholar]
- 79.Ryden AK, Wesley JD, Coppieters KT, Von Herrath MG. Non-antigenic and antigenic interventions in type 1 diabetes. Hum Vaccin Immunother. 2014;10(4):838–846. doi: 10.4161/hv.26890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Agardh CD, Cilio CM, Lethagen A, et al. Clinical evidence for the safety of GAD65 immunomodulation in adult-onset autoimmune diabetes. J Diabetes Complications. 2005;19(4):238–246. doi: 10.1016/j.jdiacomp.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 81.Ludvigsson J, Faresjö M, Hjorth M, et al. GAD treatment and insulin secretion in recent-onset type 1 diabetes. N Engl J Med. 2008;359(18):1909–1920. doi: 10.1056/NEJMoa0804328. [DOI] [PubMed] [Google Scholar]
- 82.Ludvigsson J, Hjorth M, Chéramy M, et al. Extended evaluation of the safety and efficacy of GAD treatment of children and adolescents with recent-onset type 1 diabetes: a randomised controlled trial. Diabetologia. 2011;54(3):634–640. doi: 10.1007/s00125-010-1988-1. [DOI] [PubMed] [Google Scholar]
- 83.Ludvigsson J, Krisky D, Casas R, et al. GAD65 antigen therapy in recently diagnosed type 1 diabetes mellitus. N Engl J Med. 2012;366(5):433–442. doi: 10.1056/NEJMoa1107096. [DOI] [PubMed] [Google Scholar]
- 84.Tian J, Dang H, Nguyen AV, Chen Z, Kaufman DL. Combined therapy with GABA and proinsulin/alum acts synergistically to restore long-term normoglycemia by modulating T-cell autoimmunity and promoting beta-cell replication in newly diabetic NOD mice. Diabetes. 2014;63(9):3128–3134. doi: 10.2337/db13-1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Harrison LC, Wentworth JM, Zhang Y, et al. Antigen-based vaccination and prevention of type 1 diabetes. Curr Diab Rep. 2013;13(5):616–623. doi: 10.1007/s11892-013-0415-7. [DOI] [PubMed] [Google Scholar]
- 86.Tian J, Dang H, Kaufman DL. Combining antigen-based therapy with GABA treatment synergistically prolongs survival of transplanted ss-cells in diabetic NOD mice. PloS One. 2011;6(9):e25337. doi: 10.1371/journal.pone.0025337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tower DB. The administration of gamma-aminobutyric acid to man: systemic effects and anticonvulsant action. In: Tower DB, Roberts E, editors. Inhibition in the Nervous System and γ-Aminobutyric Acid. New York: Pergamon; 1960. pp. 562–578. [Google Scholar]
- 88.Cavagnini F, Pinto M, Dubini A, Invitti C, Cappelletti G, Polli EE. Effects of gamma aminobutyric acid (GABA) and muscimol on endocrine pancreatic function in man. Metabolism. 1982;31(1):73–77. [PubMed] [Google Scholar]
- 89.Passariello N, Giugliano D, Torella R, Sgambato S, Coppola L, Frascolla N. A possible role of gamma-aminobutyric acid in the control of the endocrine pancreas. J Clin Endocrinol Metab. 1982;54(6):1145–1149. doi: 10.1210/jcem-54-6-1145. [DOI] [PubMed] [Google Scholar]
- 90.Khumarian NG, Mamikonian RS. On the role of gamma aminobutyric acid in the regulation of the level of glycemia in diabetes mellitus. Zh Eksp Klin Med. 1967;7(2):3–9. Russian. [PubMed] [Google Scholar]
- 91.Bansal P, Wang Q. Insulin as a physiological modulator of glucagon secretion. Am J Physiol Endocrinol Metab. 2008;295(4):E751–E761. doi: 10.1152/ajpendo.90295.2008. [DOI] [PubMed] [Google Scholar]
- 92.Wang Q, Jin T. The role of insulin signaling in the development of beta-cell dysfunction and diabetes. Islets. 2009;1(2):95–101. doi: 10.4161/isl.1.2.9263. [DOI] [PubMed] [Google Scholar]
- 93.Egea J, Espinet C, Soler RM, et al. Neuronal survival induced by neurotrophins requires calmodulin. J Cell Biol. 2001;154(3):585–597. doi: 10.1083/jcb.200101023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: a Ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87(7):1203–1214. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- 95.Park S, Dong X, Fisher TL, et al. Exendin-4 uses Irs2 signaling to mediate pancreatic beta cell growth and function. J Biol Chem. 2006;281(2):1159–1168. doi: 10.1074/jbc.M508307200. [DOI] [PubMed] [Google Scholar]