

Abstract
We have generated a photoactivatable form of sonic hedgehog protein by modifying the N-terminal cysteine with the heterobifunctional photocrosslinker 4-maleimidobenzophenone (Bzm). The Bzm modification on ShhN imparted a significant increase in activity as assessed in the C3H10T1/2 functional assay with potency comparable to that of the endogenous dual-lipidated form of ShhN (ShhNp). Reversed-phase HPLC analysis indicated that the increase in activity compared to unmodified ShhN may be due in part to the hydrophobic nature of the benzophenone group. In contrast to the fully processed ShhNp, Bzm-ShhN is monomeric as assessed by analytical SEC and does not require detergent to be soluble. Further, we demonstrated that the Bzm-ShhN was able to crosslink in vitro in the presence of a known binding partner, heparin. We suggest that Bzm-ShhN can serve as a relatively facile and preferred source of ShhNp for in vitro assays and as a probe to identify novel Hh protein interactions.
Keywords: Sonic hedgehog, benzophenone, crosslinking, photoactivatable, heparin, palmitoyl
Introduction
The Hedgehog (Hh) family of secreted proteins plays fundamental roles in vertebrate and invertebrate embryonic development and continues to control the development of many tissues in adults [1], as well as being a major force in driving Hh-related cancers [2]. Three Hh homologs, Indian, Desert and Sonic have been described in mammals and their activity characterized [3]. Sonic hedgehog (Shh) is the most well characterized of the three, having a central role in limb [4], craniofacial [5, 6], and central nervous system [7, 8] development. Shh is expressed in the notochord and zone of polarizing activity in the limb bud, and its release as a morphogen gradient specifies patterning of the ventral neural tube [9] and digit formation [10] respectively.
Correct post-translational processing, particularly lipid modifications, are important for proper Hh signaling. The ~45 kDa translated product of shh is autocatalytically cleaved to generate a ~25 kDa C-terminal fragment involved in the autoprocessing reaction, and a 20 kDa N-terminal fragment (ShhN) [7] responsible for all known Shh activity [11]. ShhN is doubly lipidated with cholesterol [12] and palmitoyl [13] adducts on the C- and N- termini, respectively. This doubly-lipidated form of ShhN is the fully active form [14]. These lipid modifications are involved in ShhN secretion, its migration to receiving cells, and modulation of ShhN signal intensity. Palmitoylation of Hh is required for processing [15], activity [16] and association with receiving cells [17]. Hh protein lacking palmitoylation is unable to signal or diffuse normally (reviewed in [18]), with the absence of cholesterol and palmitoylation substantially reducing signaling activity [19, 20]. Fetal exposure to alcohol disrupts cholesterol modification of Hh during post-translational processing [21] and its trafficking to membranes [22].
Although Patched (Ptc) on responding cells is the primary receptor for Shh [23, 24], a number of other proteins, receptors and factors have been shown to participate in modulating its activity either positively or negatively (reviewed in [25]). Membrane proteins Cdo, Boc and Gas1 [26] bind Hh and positively regulate signaling [27–29]. Cdo and Boc are localized to microdomains and actively distribute Shh in filopodia [30]. In contrast, the cell surface Hh-interacting protein (Hhip) acts as a sink to sequester Hh from Ptc and restrict Hh activity (reviewed in [25, 31]). Likewise heparan sulfate proteoglycans (HSPG) have been implicated in modulating Hh diffusion and signaling [32, 33] either positively or negatively [34, 35], and via either their protein [35] or sugar [36] regions with the latter implied from previous observations that Hh can bind heparin directly [37, 38].
How all these components co-operate to fine-tune Hh secretion and signaling is a challenge to model. Purified proteins and HSPGs can be used, although it can be a problem to demonstrate that binding is physiologically relevant. A number of biochemical approaches can be taken to assess interactions including pull-downs, co-crystallization and cross-linking studies. Crystallographic studies of Hh complexed with individual components have provided some clues [38–41], including that the binding sites on Hh for Ptc and Hhip overlap suggesting they compete for binding [42]. Demonstrating direct binding of Hh to the HSPG glypican-3 was not possible with purified components [43] but has been demonstrated for Shh binding to heparin and chondroitin sulfate [38]. Interestingly, recent attempts to recapitulate Shh binding to detergent-solubilized Ptc has proved difficult suggesting that additional factors may be involved for high affinity binding of Hh to Ptc [44].
Chemical cross-linking can be employed to map protein-protein interactions and to identify specific binding sites. Binding interactions are often transient and short-lived, and as a result difficult to detect. By cross-linking however, the interactions in close proximity to a protein can be studied. Our approach to identify interactions between Shh and potential binding partners and to circumvent the challenges of reconstituting what might be low affinity binding interactions, was to generate a photo-activatable version of Shh using benzophenone [45, 46] to target and cross-link these interactions. Benzophenone photophores are used extensively for photoaffinity-labeling studies as they are one of the most stable photoreactive groups and the wavelength for UV crosslinking (~350 nm) does not typically affect proteins [47]. Benzophenone-containing molecules undergo photo-activatable cross-linking to adjacent molecules with high specificity by efficient covalent modification to C–H bonds, even in aqueous buffers. Herein we describe the characterization of benzophenone modified Shh and show that this modified form not only retains activity but has potency comparable to the lipid-modified Shh making it an effective mimetic probe for the natural fully active form of ShhN.
Materials and methods
Materials
C3H10T1/2 murine embryonic fibroblast cells were from American Type Culture Collection (Manassas, VA). N-ethylmaleimide was purchased from Thermo Scientific (Rockford, IL) and fluorescein-5-maleimide and benzophenone-4-maleimide from Invitrogen (Carlsbad, CA). Recombinant ShhNp produced in human cells (catalog #SHH-025) was from StemRD (Burlingame, CA). KAAD-cyclopamine was from Calbiochem (La Jolla, CA). Taqman primers and probes for GLI 1 (Assay ID: Mm00494654_m1) and β-actin were from Applied Biosystems (Foster City, CA). Heparin ammonium (H6279) was purchased from Sigma-Aldrich (St. Louis, MO). Primary antibody for ShhN (N-19; sc-1194) and secondary antibodies (rabbit anti-goat IgG-HRP; sc-2768) were from Santa Cruz Biotechnology, Inc. (Dallas, TX).
Preparation of modified ShhN proteins
The N-terminal domain of human Shh (unmodified ShhN, residues 24–197) was expressed and purified from E.coli essentially as previously described [48, 49]. Maleimide chemistry was used to form stable thioether bonds between each label and the exposed N-terminal cysteine of ShhN using the following reagents: N-ethylmaleimide (NEM); Mr = 125, 25 mM stock solution in dH2O, fluorescein-5-maleimide (Flu); Mr = 427, 2.5 mM stock solution in dimethylformamide (DMF), and benzophenone-4-maleimide (Bzm); Mr = 277, 25 mM stock solution in DMF).
For modification, maleimide reagent (0.5 mM final concentration) was added to ShhN (100 μM) in 20 mM HEPES pH 6.5, 200 mM NaCl, 50 μM DTT and the reaction incubated at room temperature for 2h in the dark. Reactions were quenched by adding 10-fold molar excess of DTT for 1h at room temperature and then dialyzed into 5 mM sodium phosphate pH 5.5, 150 mM NaCl, 0.5 mM DTT buffer. Negative controls were generated by incubating ShhN with either dH2O or DMF alone.
SDS-PAGE
ShhN proteins were denatured and separated using NuPAGE Novex 4–12% Bis-Tris gels with MES running buffer (Invitrogen) and then visualized by staining with Coomassie Blue.
Mass spectrometry analysis of modified ShhN proteins
The masses of modified ShhN proteins were determined by electrospray-mass spectrometry (ESI-MS). ShhN samples were diluted in Optima Grade water (Fisher Scientific) to a concentration of 50 ng/μl and desalted using a NanoAcquity UPLC system with a 1.7 μm BEH130 C18 column (100 μm x 100 mm, Waters Corporation) connected in-line to a Micromass quadrupole time-of-flight mass spectrometer (Waters). Samples were eluted in a 30 min gradient (1–50% acetonitrile (Optima Grade, Fisher Scientific)) with 0.1% formic acid and a flow rate of 0.4 μl/min. Electrospray mass spectra were collected using MassLynx 4.1 (Waters Corp.) and mass spectra (charge state envelopes) of eluted proteins deconvoluted using the MaxEnt algorithm within Masslynx software (Waters Corporation).
Assessing ShhN activity in C3H10T1/2 cell-based assay
The concentration-dependent signaling activity of modified forms of ShhN was assessed in Hh-responsive C3H10T1/2 murine embryonic fibroblast cells that induce alkaline phosphatase (AP) expression upon ShhN signaling activation. The assay was performed essentially as described previously [48, 49] with cells cultured in high glucose Dulbecco’s modified Eagle’s medium containing 10% fetal calf serum in 96-well plates at 5000 cells/well. After 24h, cells were incubated with serial dilutions of the different forms of ShhN for 5 days and AP activity assessed after incubating lysed cells with the chromogenic substrate p-nitrophenyl phosphate and measuring absorbance at 405 nm. Half maximal effective concentration (EC50) constants were determined in prism 6.0 (GraphPad Software Inc., La Jolla, CA) with non-linear regression. Recombinantly generated lipid-modified ShhN (ShhNp) produced in human cells was used for comparison in the assay and was reconstituted as recommended by the vendor with water to give a final concentration of 100 ng/μl in PBS/1% CHAPS.
For inhibition studies, the C3H10T1/2 assay was carried out essentially as above except cells were incubated with a fixed concentration of ShhN protein equivalent to its EC50 value in the presence or absence of the Hh pathway inhibitor KAAD-cyclopamine (300 nM).
RT-PCR for GLI1 in C3H10T1/2 cells
C3H10T1/2 cells were cultured at 60,000 cells/well in 12-well plates and after 24h incubated with a single concentration of ShhN protein at its EC50 value in the presence or absence of the Hh pathway inhibitor KAAD-cyclopamine (1 μM). Total RNA was isolated using the RNeasy Mini Plus Kit (Qiagen). cDNA was synthesized from 300 ng of RNA using a high capacity cDNA reverse transcription kit (Applied Biosystems). Taqman assay primers and probe specific for GLI 1 were used. For mRNA quantification, real-time PCR was performed using an ABI 7500 Fast Real-Time PCR system. Target sequences were amplified at 95 C for 20s, followed by 40 cycles of 95 C for 3s and 60 C for 30s. β-actin was used as endogenous normalization control. All assays were performed in triplicate and the fold change in mRNA expression determined according to the method of 2−ΔΔCt.
Size exclusion chromatography
ShhN proteins were analyzed by size exclusion chromatography using an Agilent 1200 HPLC system fitted with a Bio SEC-5 column (4.6 mm x 300 mm, Agilent). Sodium phosphate buffer (5 mM, pH 6.5) containing 150 mM NaCl and 0.5 mM DTT served as the mobile phase with a flow rate of 0.3 ml/min. Proteins were detected by monitoring absorbance at 214 nm and 280 nm. Proteins from low and high molecular weight calibration kits (GE Healthcare) were used to estimate apparent molecular size. Kav values were calculated as in [50].
Generation of modified-peptides and reversed-phase HPLC analysis
An unmodified peptide corresponding to the 5 residues of the N-terminus of ShhN (CGPGR) was synthesized (Anaspec Inc.). A Bzm-modified form of the peptide was generated by incubating the unmodified CGPGR peptide with a 10-fold excess of Bzm for 15 mins in the dark. Reactions were quenched with excess β-mercaptoethanol (BME).
The unmodified and Bzm-modified peptides (20 μg) were analyzed by reversed-phase HPLC on an Agilent 1200 system equipped with a photodiode array detector. A 4.6 x 150 mm Zorbax Eclipse XDB-C18 column was used and running conditions were 5 – 90% ACN w/0.1%TFA over 20 mins.
Heparin affinity chromatography assay
Unmodified and modified ShhN proteins were subjected to heparin affinity chromatography at 4°C using an Åkta FPLC system (GE Healthcare) fitted with a 1 ml HiTrap Heparin HP column (GE Healthcare). ShhN proteins (~ 50 μg) were loaded onto the column at a flow rate of 1 ml/min in 5 mM sodium phosphate pH 5.5, 150 mM NaCl, 0.5 mM DTT and eluted over 10 column volumes using a linear gradient of 0.15 to 2 M NaCl. Proteins were detected by monitoring absorbance at 280 nm.
Photocrosslinking of Bzm-ShhN to heparin
Heparin ammonium salt, MW ~13,000 Da was used for crosslinking. Bzm-ShhN (5 μg) was incubated with the heparin (5 μg) in 20 mM HEPES pH 6.5, 200 mM NaCl for 1h at room temperature in the dark. Mixtures were irradiated for 30 min at 365 nm at room temperature with the sample fixed to a handheld UV transilluminator (365 nm). After UV photolysis, samples were denatured, separated using SDS-PAGE as above and proteins transferred to a PVDF membrane for Western blotting with anti ShhN primary antibody N-19 followed by rabbit anti-Goat IgG-HRP secondary antibody. The blotted membrane was incubated with SuperSignal West Dura Extended Duration Substrate (Thermo Scientific) and visualized on X-ray film.
Results
Generation and characterization of ShhN proteins modified with thiol-specific maleimide reagents
Our strategy to generate sonic hedgehog protein probes involved labeling the N-terminal cysteine of recombinant ShhN with maleimide-activated benzophenone (Bzm) or maleimide-activated fluorescein (Flu) to generate photoactivatable and fluorescently-labeled forms respectively. N-ethyl maleimide labeling of ShhN was used as a control as this had been previously shown to not affect Shh activity [51]. Maleimide chemistry allows the formation of a stable thioether bond with the thiol moiety of N-terminal cysteine of ShhN. Labeled proteins are hereafter referred to as NEM-ShhN, Flu-ShhN and Bzm-ShhN and schematic structures of the unmodified and modified ShhN proteins are shown in Figure 1A. Modified proteins were analyzed by mass spectrometry (MS) to verify the modification and extent of modification (Fig. 1B) using unmodified ShhN as a control. Unmodified ShhN showed the expected mass of 19553 Da, while the measured experimental masses of 19679 Da, 19830 Da and 19980 Da matched with the predicted masses of the ShhN modified with NEM (Mr 125.13), Bzm (Mr 277.3) and Flu-mal (Mr 427.37) groups, respectively. The MS analysis demonstrated that the mass shift of each modified form relative to unmodified ShhN alone corresponded exactly to a single modification (Fig. 1B). No unmodified ShhN was observed in the mass spectrum of the modified forms, suggesting that the percent of ShhN coupled to NEM, Flu or Bzm approached 100%.
Fig. 1.

Structures and mass spectrometry characterization of modified human Shh proteins. (A) Schematic diagram of Shh proteins with selected modifications of the N-terminal cysteine of Shh. The N-terminal residue in processed human ShhN is cysteine 24 and the C-terminal residue is glycine 197. Unmodified ShhN (ShhN), 5-(N-ethyl-malemidyl) modified ShhN (NEM-ShhN), 4-maleimidobenzophenone modified ShhN (Bzm-ShhN) and fluorescein-maleimide modified ShhN (Flu-ShhN). (B) Whole mass spectra of modified human Shh proteins. ESI-MS spectra of modified Shh proteins with expected change in mass for each modifying group noted.
Effect of N-terminal modifications on Shh activity
We have previously observed that adducts of the N-terminal cysteinyl thiol can result in loss of ShhN activity [49, 51]. To assess whether the ShhN proteins retained activity after modification they were assayed in the Hh-responsive cell line C3H10T1/2. In these cells, ShhN induces alkaline phosphatase (AP) activity as a marker of ShhN-mediated cell differentiation. This is a well-accepted bioassay to measure Hh activity [49] and assess Hh pathway inhibitors [49, 52]. C3H10T1/2 cells were treated with each modified ShhN over a concentration range of 0.156–25 μg/ml. A dose response curve for unmodified ShhN is included on each C3H10T1/2 assay plate as an internal control with an EC50 value range of 2.2±0.8 μg/ml (n = 17) determined. In these experiments unmodified ShhN had an EC50 of 1.6 μg/ml (Table 1) close to that previously reported [13, 49, 51]. Flu-ShhN and NEM-ShhN had activity comparable to that of the unmodified protein (Fig. 2A) with calculated EC50 values of 0.96 and 0.42 μg/ml respectively (Table 1), indicating that these modifications did not abrogate activity. Over the same concentration range Bzm-ShhN had maximal activity at all concentrations tested (data not shown). By titrating down the Bzm-ShhN concentration range to 0.0039–2 μg/ml a full dose response curve was obtained (Fig. 2B). The resulting EC50 value for Bzm-ShhN was determined to be 0.049 μg/ml (Table 1). This finding suggested that Bzm-ShhN had ~30 times the potency of ShhN in C3H10T1/2 cells. As this increase in potency was unexpected, for a comparison we then assayed a form of recombinant lipid-modified Shh (ShhNp) that was generated in human cells and reported to be 10–50-fold more active than recombinant unmodified ShhN produced in E.coli in a Gli reporter assay (StemRD product data sheet). Although, we did not analyze this protein for modifications, this lipid-modified Shh presumably represents the fully processed lipidated form with a palmitoyl on the N-terminus and cholesterol on the C-terminus (shown schematically in Fig. 2C). We first assessed the purity of this ShhNp by SDS-PAGE along with the unmodified and modified ShhN proteins (Fig. 2D). The ShhNp was ~90% pure as judged by SDS-PAGE. We have previously shown that ShhN and ShhNp migrate differently on SDS-PAGE [13] presumably due to the contribution of the lipid moiety. No significant differences were observed in the migration of the modified ShhN proteins (NEM-ShhN, Bzm-ShhN and Flu-ShhN) (Fig. 2D), nor were separate bands resolved when unmodified ShhN was mixed with equal amounts of either NEM-ShhN or Bzm-ShhN (data not shown). Gao and coworkers [53] also saw no migration shift upon palmitoylation of Shh and this may simply reflect differences in the resolving power of the various SDS-PAGE gel systems used in these different studies.
Table 1.
Activity of modified ShhN proteins in C3H10T1/2 assay.
| Protein | EC50 (μg/ml) | Relative to unmodified ShhN |
|---|---|---|
| Unmodified ShhN | 1.63±0.15 | 1 |
| NEM-ShhN | 0.42±0.05 | 3.8 |
| Flu-ShhN | 0.96 | 1.7 |
| Bzm-ShhN | 0.049±0.02 | 33 |
| ShhNp | 0.054 | 30 |
Fig 2.

Potency of modified ShhN proteins in the C3H10T1/2 cell assay. (A) Relative activities of unmodified ShhN (▲), NEM-ShhN (●) and Flu-ShhN (■). C3H10T1/2 cells were incubated with the indicated concentrations of ShhN proteins and induction of alkaline phosphatase (AP) activity measured at 405 nm using pNPP. (B) Relative activities of unmodified ShhN (▲), Bzm-ShhN (○) and dual lipidated ShhNp (□) in C3H10T1/2 cells. (C) Schematic representing fully processed dual-lipidated Shh (ShhNp). (D) SDS-PAGE analysis of modified Shh proteins. Lane M, Novex prestained protein markers. Proteins were subjected to SDS-PAGE on a 12% MOPS gel and stained with Coomassie Blue.
In the C3H10T1/2 assay the Bzm-ShhN had activity comparable to the ShhNp (Fig. 2B) with the latter having an EC50 value of 0.054 μg/ml (Table 1). Bzm-ShhN and ShhNp also induced higher maximal levels of AP than unmodified ShhN (Fig. 2B). When tested at their EC50 concentrations, unmodified ShhN (2 μg/ml), Bzm-ShhN (0.05 μg/ml) and ShhNp (0.05 μg/ml) induced the downstream markers of Hh activity, GLI1 and Ptch1, with comparable efficacy as assessed by RT-PCR (Fig. 3A). To further confirm that Bzm-ShhN was acting through the canonical Hh signaling pathway, we tested the effect of the SMO antagonist KAAD-cyclopamine (KAAD-cyc) [54] on its activity in C3H10T1/2 cells. In the presence of 300 nM KAAD-cyc, the AP induction activity of unmodified ShhN, ShhNp and Bzm-ShhN (at their EC50 concentrations) was reduced by 94, 97 and 96% respectively (Fig. 3B). Comparable inhibition data of KAAD-cyc on modified ShhN induced transcription of the Hh-pathway marker GLI1 (Fig 3C) was obtained as measured by RT-PCR.
Fig. 3.

Inhibition of Bzm-ShhN induced AP and transcriptional activity in C3H10T1/2 cells. (A) C3H10T1/2 cells were incubated with Shh proteins at EC50 values (2 μg/ml for unmodified ShhN, 0.02 μg/ml for ShhNp and Bzm-ShhN) for 5 days and induction of GLI and Ptch1 mRNA measured by RT-PCR relative to β–actin. (B) C3H10T1/2 cells were incubated with Shh proteins at EC50 values (2 μg/ml for unmodified ShhN, 0.02 μg/ml for ShhNp and Bzm-ShhN) for 5 days with and without 300 nM KAAD-cyclopamine (KAAD-cyc) and induction of AP activity measured as before. (C) Inhibition of ShhN induced GLI1 transcription activity in C3H10T1/2 cells. Shh proteins were incubated at EC50 values (2 μg/ml for unmodified ShhN, 0.02 μg/ml for ShhNp and Bzm-ShhN) for 5 days with and without 1 μM KAAD-cyclopamine. GLI1 mRNA expression was measured by RT-PCR relative to β–actin.
Chromatographic characterization of modified ShhN
Analytical size exclusion chromatography (SEC) had been used previously to demonstrate that unmodified ShhN exists in monomeric form whereas lipid-modified ShhN may exist as higher order oligomers [15, 55–57]. In the present study, SEC demonstrated that unmodified ShhN and Bzm-ShhN had comparable elution volumes and no higher order multimers were detected (Fig. 4A). From an analysis using molecular weight standards, both proteins had molecular sizes around 20,000 Da and hence were presumed to be monomeric (Fig. 4A). NEM-ShhN also eluted as a monomer (data not shown). Under these same (detergent free) buffer conditions the ShhNp failed to elute from the column (data not shown).
Fig. 4.
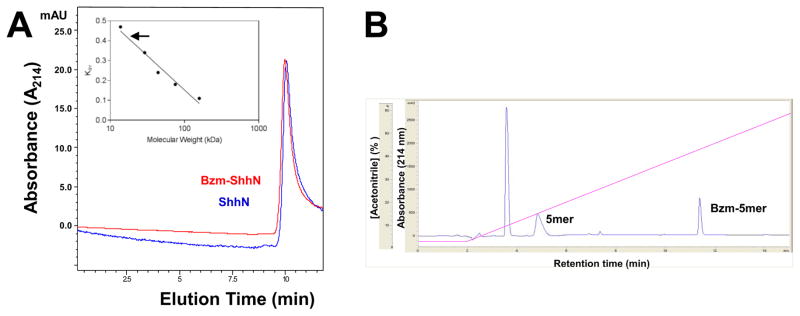
Chromatographic analysis of modified ShhN proteins. (A) Analytical size exclusion chromatography of modified ShhN proteins loaded (5 μg) on a Bio SEC-5 column at 0.3 ml/min in 5 mM potassium phosphate pH 6.5, 150 mM NaCl buffer. Inset: a calibration curve was created using the elution volumes (Ve) of the following standard proteins; adolase (Mr 158,000), conalbumin (Mr 75,000), ovalbumin (Mr 44,000), carbonic anhydrase (Mr 29,000) and ribonuclease A (Mr 13,700). Kav values were calculated [50] using a void volume (Vo) of 2.02 ml and included volume of 4.38 ml (Vi). Based on Ve of ~ 3.0 ml the molecular sizes of ShhN and Bzm-ShhN were both estimated to be ~17,000 (arrow). (B) Reversed-phase HPLC analysis of modified Shh N-terminal peptide. A peptide comprising the first five residues (5mer) of ShhN was modified on the N-terminal cysteine with acetyl-, Bz- or palmitate- using maleimide chemistry. Peptides (20 μg) were loaded on a 4.6 x 150 mm Zorbax Eclipse XDB-C18 column and eluted with a 5 – 90% ACN gradient. A representative chromatogram for a 1:1 mixture of unmodified 5mer and Bzm-5mer is shown. Earlier eluting peak is buffer component BME.
We have previously reported that ShhN potency in the C3H10T1/2 assay correlates with hydrophobicity at the N-terminus [51]. To assess the effect of the modifications on hydrophobicity and any relative differences we used reversed phase-HPLC with a synthetic unmodified peptide comprising the first 5 residues of ShhN (CGPGR). Samples of the peptide were modified with Bzm (Bzm-CGPGR), palmitate or acetyl (as a control) using maleimide chemistry. Peptides were resolved on a C18 rp-HPLC column and retention times determined. Bzm-CGPGR eluted at a higher acetonitrile (ACN) concentration (45%) than the unmodified peptide, indicating an increase in hydrophobicity (Fig. 4B). The palmitate modified peptide was more hydrophobic than the Bzm-peptide requiring 60% ACN for elution (Table 2). Predicted logP values (Table 2) for the modified peptides suggest comparable values for Bzm and fluorescein with palmitate the most non-polar.
Table 2.
Relative hydrophobicity of modifications.
| CGPGR Peptide | Acetonitrile (% to elute) | logP of modificationa |
|---|---|---|
| No modification | 17% | N/A |
| Acetyl- | 20% | −0.4 |
| NEM- | ND | −0.06 |
| Bzm- | 45% | 2.71 |
| Flu(m)- | ND | 2.5 |
| Palmitate- | 60% | 6.26 |
Calculated logP values from www.chemicalize.org
Binding and photocrosslinking between Bzm-ShhN and heparin
ShhN possesses a heparin-binding site near the N-terminus and ShhN proteins have been shown to bind tightly to heparin [32, 37]. The effect of the N-terminal Bzm modification on heparin binding was assessed by heparin affinity-chromatography [55, 56, 58]. Unmodified ShhN, NEM-ShhN and Bzm-ShhN had comparable binding to heparin, with all forms eluting at ~1 M NaCl (Fig. 5A).
Fig. 5.

Binding and UV crosslinking of Bzm-ShhN in the presence of heparin. (A) Binding of modified ShhN proteins (50 μg) to a heparin-affinity column and elution with a linear gradient of 0 – 2.0 M NaCl in 5 mM sodium phosphate pH 5.5, 150 mM NaCl, 0.5 mM DTT buffer. (B) Bzm-ShhN was incubated with heparin and subjected to UV light (30 min, dark, 365 nm). Controls included ShhN + heparin, Bzm-ShhN alone or no UV. Crosslinking, SDS-PAGE and Western blotting carried out as described in Methods. Western blot with anti-ShhN polyclonal Ab. Cross-linked bands are indicated by vertical bar.
Bzm-ShhN was tested for its ability to cross-link with a known Hh binder, heparin. For cross-linking, equal amounts of Bzm-ShhN was incubated with or without heparin (MW~13,000 Da) and exposed to UV irradiation to test whether the photoactivatable property of Bzm could be used to induce covalent crosslinks in the presence of heparin. Negative controls for UV treatment included Bzm-ShhN alone and ShhN + heparin. Sample and control reactions were incubated in either the presence or absence of UV irradiation and evidence of crosslinking was assessed by ShhN migration shifts on SDS-PAGE. After UV irradiation to induce cross-linking, the samples were analyzed by reducing SDS-PAGE followed by Western blotting using a ShhN specific primary antibody (Fig. 5B). This approach detected ShhN in each sample and control with no additional bands indicative of cross-linked species observable in the non UV irradiated (−UV) samples. Several bands of higher molecular weight than ShhN were evident only in the UV irradiated (+UV) Bzm-ShhN + heparin sample indicated by a vertical bar (Fig. 5B). Higher mass bands were not observed in the Bzm-ShhN alone (Fig. 5B) or ShhN+heparin (not shown) UV treated samples.
Discussion
In this study our aim was to generate a photoactivatable form of ShhN to probe potential binding partners both in vitro and in cell-based models. Herein, we show that not only could ShhN be specifically modified with the photoactivatable group benzophenone (Bz) and retain signaling activity, but also that its activity was comparable to that of the dual-lipidated form of ShhN that is presumed to be the fully active endogenous form. Analysis of the Bzm modified protein suggested that it was monomeric and that the hydrophobic nature of the benzophenone group may contribute in part to the increase in Hh signaling activity. Further we showed that Bzm-ShhN was able to crosslink in the presence of a known binding partner, heparin.
We [45, 46, 59] and others (e.g. [60]) have used the photolabile group benzophenone as a photoaffinity probe to target and map ligand interactions. The presence of an N-terminal cysteine residue on ShhN facilitated the use of maleimide chemistry to modify ShhN with activated benzophenone-maleimide (Bzm). We demonstrate that Bzm labeling of ShhN results in a modified form with the Bzm group located at a single site and with a labeling efficiency close to 100%.
We were cognizant that modifications to the N-terminal cysteine of ShhN could lead to loss of activity [49]. In the present study, the Bzm modification did not abrogate activity in the C3H10T1/2 Hh functional assay and in fact substantially increased the potency of the Bzm-ShhN (>30-fold) compared to the unmodified or NEM-modified ShhN. The Bzm-ShhN also showed comparable activity to a form of ShhN presumed to represent the fully active dual-lipidated form (ShhNp; as it was expressed as a full length construct in a mammalian cell system). Further, the activity of Bzm-ShhN was close to that of myristolated-ShhN, the most potent form of modified ShhN reported [51]. The activity of Bzm-ShhN like that of ShhN and ShhNp was fully inhibited by the Hh pathway inhibitor KAAD-cyclopamine indicating that these proteins were all acting through the same canonical Hh signaling pathway.
We investigated potential mechanisms that could cause the increased activity of Bzm-ShhN; formation of multimers and/or an increase in hydrophobicity. We have previously shown that increases in ShhN activity in the C3H10T1/2 assay are correlated with increases in hydrophobicity on the N-terminus of the protein [51]. The mechanism imparting this increase in activity with increasing hydrophobicity was not apparent but was presumed to involve some form of membrane interaction [51]. Labeling of cells stably expressing Shh with an alkynyl palmitic acid probe demonstrated that palmitoylated Shh localized to membrane fractions and that this modification did not affect activity [53]. In our present study, rp-HPLC analysis along with calculated logP values suggests that the Bzm modification is less non-polar than palmitate. Our observation that Bzm-ShhN has comparable activity to ShhNp while the Bzm group may be less hydrophobic than palmitate suggests that additional factors other than hydrophobicity may dictate ShhN potency, or that a hydrophobicity threshold exists. However, ShhN modified with fluorescein (Flu), a group with comparable logP to Bzm, showed no increased activity, favoring the suggestion that additional factors other than hydrophobicity promote the increased activity of Bzm-ShhN. The differences in observed activity of Flu-ShhN and Bzm-ShhN in the C3H10T1/2 assays despite comparable hydrophobic modifications may be due to the ability of Bzm-ShhN to interact with other binding partners or due to the larger size and steric constraints of the fluorescein group.
Our previous study correlating hydrophobicity with increasing activity indicated no oligomerization of ShhN and unmodified- and myristoylated-modified ShhN proteins were monomeric as assessed by analytical ultracentrifugation and dynamic light scattering [51]. Lipidated forms of ShhN have been shown to form higher ordered structures or multimers that facilitate its association with lipid rafts, and its release, diffusion and reception [20, 57, 61–63], with more recent studies demonstrating that expression of full length Shh in mammalian cells yielded multimers of ShhNp [55, 56]. Our analytical SEC analysis indicated that Bzm-ShhN like unmodified ShhN does not form stable multimers under the conditions used, suggesting that the increased signaling potency of Bzm-ShhN is not due to multimer formation. It may be that the ShhN forms, modified chemically with Bzm or lipid do not form multimers spontaneously.
Heparin, a highly sulfated glycosaminoglycan, is a known binding partner of ShhN [7, 15, 37–39, 55, 56, 61]. Heparin has been reported to promote multimerization of ShhNp [55], and of unmodified ShhN proteins [61], although incubation of Hh with a heparin oligosaccharide failed to promote this oligomerization [39]. Using heparin affinity chromatography we did not observe any differences in Bzm-ShhN binding to heparin compared to unmodified ShhN indicating that the Bzm modification does not appear to cause steric hindrance for heparin binding nor increase binding affinity. Incubation of Bzm-ShhN or unmodified ShhN with heparin did not result in the observation of multimers as assessed by SDS-PAGE under reducing conditions followed by Western blotting. Under these conditions either multimers did not form at all or they were labile to the SDS-PAGE/DTT gel conditions. Interaction with heparin or other constituents present in the C3H10T1/2 assay may promote multimer formation of Bzm-ShhN not observed in vitro.
The conserved N-terminus of ShhN extends away from the core structure [58] and provides a somewhat unstructured and flexible arm to probe for binding interactions in close proximity. When Bzm-ShhN was UV irradiated alone, no crosslinked products were observed by SDS-PAGE/Western blotting. In the presence of heparin, however, crosslinked products were detected in the range of ~50–80 kDa. This distribution of crosslinked products may represent UV crosslinks between just Bzm-ShhN molecules where heparin is not incorporated in the final crosslinked product but rather serves as a template to promote crosslinking between Bzm-ShhN molecules. Hence, the bands at ~ 60 kDa and 80 kDa may represent Bzm-ShhN trimers and tetramers, respectively. Such heparan sulfate dependent oligomers of ShhNp and ShhN have been previously reported [61]. Alternatively, the number and molecular weight range of the UV-crosslinked products may suggest that heparin is incorporated into some of the crosslinked species, with multiple Bzm-ShhN molecules attached to the same heparin chain, e.g. heparin plus two Bzm-ShhN molecules would present as a ~53kDa band. Support for this model comes from the proximity of the Bzm-labeled N-terminal Cys to the Cardin-Weintraub heparin binding motif on ShhN [32], potentially within the distance constraint of 10 Å proposed for crosslinking to the reactive carbonyl in benzophenone [57], and from previous observations that heparin can bind multiple ShhN molecules [38]. Confirmation that these bands represent Bzm-ShhN multimers and/or Bzm-ShhN-heparin cross-linked species would require extensive mass spectroscopy analysis. The heparan sulfate dependent oligomers of ShhNp and ShhN that are resistant to heat, SDS and reducing conditions have been termed undisruptable (UD) [61]. One possibility in our study is that, rather than due to UV crosslinking, the multiple, migrated bands represent UD oligomers of Bzm-ShhN that are favored compared to ShhN. However, the lack of higher molecular weight species for Bzm-ShhN incubated with heparin in the absence of UV irradiation suggests that direct cross-linking is occurring in the presence of UV irradiation rather than multimerization and formation of UD Bzm-ShhN. The absence of higher molecular-weight species when unmodified ShhN was incubated with heparin would also indicate the absence of stable multimers.
Although lipid modifications are important for ShhN function, it is challenging to isolate and purify fully lipidated, processed ShhN (ShhNp) in sufficient quantities to sustain in vitro studies required to fully characterize ShhN function – even when using cultured cell lines, such as stably transfected HEK293 cells [64]. This is also a particular problem for large scale studies or high-throughput screens aiming to identify direct inhibitors of ShhN function. Furthermore, lipid modifications impair ShhNp solubility as suggested by the failure of ShhNp to pass through the SEC column in the absence of detergent as shown in our current and previous studies [51]. Unlike the ShhNp used in the present studies that is formulated with 1% CHAPS detergent, we found no requirement for detergent in the buffer to maintain Bzm-ShhN solubility. Bzm-ShhN was able to pass through the SEC column and also bind to heparin-Sepharose in the absence of added detergent. Due to the insolubility of ShhNp, in vitro assays have tended to substitute ShhNp with unmodified ShhN free of lipid modifications (unmodified ShhN). Results from these studies may not always be equivalent to results obtained using ShhNp. Utilizing Bzm-ShhN in screens may improve the physiological relevance of in vitro assays.
In conclusion, in utilizing maleimide chemistry to covalently attach a photoactivatable crosslinker benzophenone to the thiol on the N-terminal cysteine of ShhN, we have generated a modified recombinant form of ShhN that can be used to probe direct binding partners of the protein in vitro, but also possesses comparable activity to the fully lipidated natural form. This may be a first step towards improving the physiological relevance of in vitro assays to assess ShhNp function and to probe potential novel interactions between sonic hedgehog and extracellular binding partners. How lipidated hedgehog protein is trafficked and/or diffuses to target cells is still an intriguing question. The availability of the ShhNp mimetic Bzm modified-ShhN will allow us to probe potential mechanism of reception of the lipidated hedgehog morphogen. Our results indicate the feasibility of using photo-activatable forms of ShhN protein to probe for binding interactions.
Acknowledgments
This work was supported by the National Institute on Alcohol Abuse and Alcoholism and National Cancer Institute of the NIH under award numbers U54AA019765, R15CA159209 and U54CA156735 (KPW). Additional funding was from Golden LEAF Foundation and the BIOIMPACT Initiative of the State of North Carolina through the Biomanufacturing Research Institute & Technology Enterprise (BRITE) Center for Excellence at North Carolina Central University. The authors thank Robert Ndoyi, Willietta Gibson, Khalia Williams and Shalonda Ingram (BRITE, NCCU) for technical assistance, and acknowledge Blake Pepinsky for initial discussions.
Abbreviations used
- ShhN
Sonic Hedgehog N-terminal domain
- Bzm
4-maleimidobenzophenone
- HSPG
heparan sulfate proteoglycan
- Ptc
Patched
- NEM
N-ethylmaleimide
- Flu
fluorescein
- AP
alkaline phosphatase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ingham P, McMahon A. Genes & Development. 2001;15:3059–3087. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- 2.Rubin LL, de Sauvage FJ. Nat Rev Drug Discov. 2006;5:1026–1033. doi: 10.1038/nrd2086. [DOI] [PubMed] [Google Scholar]
- 3.Pathi S, Pagan-Westphal S, Baker D, Garber E, Rayhorn P, Bumcrot D, Tabin C, Pepinsky RB, Williams K. Mechanisms of Development. 2001;106:107–117. doi: 10.1016/s0925-4773(01)00427-0. [DOI] [PubMed] [Google Scholar]
- 4.Johnson R, Riddle R, Laufer E, Tabin C. Biochemical Society Transactions. 1994;22:569–574. doi: 10.1042/bst0220569. [DOI] [PubMed] [Google Scholar]
- 5.Chiang C, Litingtung Y, Lee E, Young K, Corden J, Westphal H, Beachy P. Nature. 1996;383:407–413. doi: 10.1038/383407a0. [DOI] [PubMed] [Google Scholar]
- 6.Hu D, Helms JA. Development. 1999;126:4873–4884. doi: 10.1242/dev.126.21.4873. [DOI] [PubMed] [Google Scholar]
- 7.Bumcrot D, Takada R, McMahon A. Molecular and Cellular Biology. 1995;15:2294. doi: 10.1128/mcb.15.4.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patten I, Placzek M. Cellular and Molecular Life Sciences CMLS. 2000;57:1695–1708. doi: 10.1007/PL00000652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roelink H, Augsburger A, Heemskerk J, Korzh V, Norlin S, Ruiz A, Tanabe Y, Placzek M, Edlund T, Jessell T. Cell. 1994;76:761–775. doi: 10.1016/0092-8674(94)90514-2. [DOI] [PubMed] [Google Scholar]
- 10.Riddle R, Johnson R, Laufer E, Tabin C. Cell(Cambridge) 1993;75:1401–1416. doi: 10.1016/0092-8674(93)90626-2. [DOI] [PubMed] [Google Scholar]
- 11.Marti E, Bumcrot DA, Takada R, McMahon AP. Nature. 1995;375:322–325. doi: 10.1038/375322a0. [DOI] [PubMed] [Google Scholar]
- 12.Porter J, Young K, Beachy P. Science. 1996;274:255–259. doi: 10.1126/science.274.5285.255. [DOI] [PubMed] [Google Scholar]
- 13.Pepinsky R, Zeng C, Wen D, Rayhorn P, Baker D, Williams K, Bixler S, Ambrose C, Garber E, Miatkowski K, Taylor FR, Wang E, Galdes A. Journal of Biological Chemistry. 1998;273:14037–14045. doi: 10.1074/jbc.273.22.14037. [DOI] [PubMed] [Google Scholar]
- 14.Chamoun Z, Mann RK, Nellen D, von Kessler DP, Bellotto M, Beachy PA, Basler K. Science. 2001;293:2080–2084. doi: 10.1126/science.1064437. [DOI] [PubMed] [Google Scholar]
- 15.Ohlig S, Farshi P, Pickhinke U, van den Boom J, Höing S, Jakuschev S, Hoffmann D, Dreier R, Schöler HR, Dierker T. Developmental Cell. 2011;20:764–774. doi: 10.1016/j.devcel.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 16.Dawber R, Hebbes S, Herpers B, Docquier F, van den Heuvel M. BMC Dev Biol. 2005;5:21. doi: 10.1186/1471-213X-5-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grover VK, Valadez JG, Bowman AB, Cooper MK. PloS one. 2011;6:e21353. doi: 10.1371/journal.pone.0021353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kornberg TB. Developmental Biology. 2014;394:1–5. doi: 10.1016/j.ydbio.2014.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kohtz JD, Lee HY, Gaiano N, Segal J, Ng E, Larson T, Baker DP, Garber EA, Williams KP, Fishell G. Development. 2001;128:2351–2363. doi: 10.1242/dev.128.12.2351. [DOI] [PubMed] [Google Scholar]
- 20.Chen MH, Li YJ, Kawakami T, Xu SM, Chuang PT. Genes & Development. 2004;18:641–659. doi: 10.1101/gad.1185804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Yang H, Zdanowicz M, Sicklick J, Qi Y, Camp T, Diehl A. Laboratory investigation. 2007;87:231–240. doi: 10.1038/labinvest.3700516. [DOI] [PubMed] [Google Scholar]
- 22.Mao H, Diehl AM, Li YX. Lab Invest. 2009;89:290–300. doi: 10.1038/labinvest.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Y, Struhl G. Cell(Cambridge) 1996;87:553–563. doi: 10.1016/s0092-8674(00)81374-4. [DOI] [PubMed] [Google Scholar]
- 24.Marigo V, Davey R, Zuo Y, Cunningham J, Tabin C. Nature. 1996;384:176–179. doi: 10.1038/384176a0. [DOI] [PubMed] [Google Scholar]
- 25.Beachy PA, Hymowitz SG, Lazarus RA, Leahy DJ, Siebold C. Genes & Development. 2010;24:2001–2012. doi: 10.1101/gad.1951710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang JS, Zhang W, Krauss RS. Science Signaling. 2007;2007:pe50. doi: 10.1126/stke.4032007pe50. [DOI] [PubMed] [Google Scholar]
- 27.Izzi L, Lévesque M, Morin S, Laniel D, Wilkes BC, Mille F, Krauss RS, McMahon AP, Allen BL, Charron F. Developmental Cell. 2011;20:788–801. doi: 10.1016/j.devcel.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kavran JM, Ward MD, Oladosu OO, Mulepati S, Leahy DJ. Journal of Biological Chemistry. 2010;285:24584–24590. doi: 10.1074/jbc.M110.131680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tenzen T, Allen BL, Cole F, Kang JS, Krauss RS, McMahon AP. Developmental Cell. 2006;10:647–656. doi: 10.1016/j.devcel.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 30.Sanders TA, Llagostera E, Barna M. Nature. 2013;497:628–632. doi: 10.1038/nature12157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Varjosalo M, Taipale J. Journal of Cell Science. 2007;120:3–6. doi: 10.1242/jcs.03309. [DOI] [PubMed] [Google Scholar]
- 32.Rubin J, Choi Y, Segal R. Development. 2002;129:2223–2232. doi: 10.1242/dev.129.9.2223. [DOI] [PubMed] [Google Scholar]
- 33.Callejo A, Torroja C, Quijada L, Guerrero I. Development. 2006;133:471–483. doi: 10.1242/dev.02217. [DOI] [PubMed] [Google Scholar]
- 34.Capurro MI, Xu P, Shi W, Li F, Jia A, Filmus J. Developmental Cell. 2008;14:700–711. doi: 10.1016/j.devcel.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 35.Williams E, Pappano W, Saunders A, Kim M, Leahy D, Beachy P. Proceedings of the National Academy of Sciences. 2010;107:5869–5874. doi: 10.1073/pnas.1001777107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wojcinski A, Nakato H, Soula C, Glise B. Developmental Biology. 2011;358:168–180. doi: 10.1016/j.ydbio.2011.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang F, McLellan J, Ayala A, Leahy D, Linhardt R. Biochemistry. 2007;46:3933–3941. doi: 10.1021/bi6025424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Whalen DM, Malinauskas T, Gilbert RJ, Siebold C. Proceedings of the National Academy of Sciences. 2013;110:16420–16425. doi: 10.1073/pnas.1310097110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McLellan JS, Yao S, Zheng X, Geisbrecht BV, Ghirlando R, Beachy PA, Leahy DJ. Proceedings of the National Academy of Sciences. 2006;103:17208–17213. doi: 10.1073/pnas.0606738103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bishop B, Aricescu A, Harlos K, O'Callaghan C, Jones E, Siebold C. Nature Structural & Molecular Biology. 2009;16:698–703. doi: 10.1038/nsmb.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bosanac I, Maun H, Scales S, Wen X, Lingel A, Bazan J, de Sauvage F, Hymowitz S, Lazarus R. Nature Structural & Molecular Biology. 2009;16:691–697. doi: 10.1038/nsmb.1632. [DOI] [PubMed] [Google Scholar]
- 42.McLellan J, Zheng X, Hauk G, Ghirlando R, Beachy P, Leahy D. Nature. 2008;455:979–983. doi: 10.1038/nature07358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim MS, Saunders AM, Hamaoka BY, Beachy PA, Leahy DJ. Proceedings of the National Academy of Sciences. 2011;108:13112–13117. doi: 10.1073/pnas.1109877108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cleveland TE, Iv, McCabe JM, Leahy DJ. Protein Expression and Purification. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Olszewski S, Deeney J, Schuppin G, Williams K, Corkey B, Rhodes C. Journal of Biological Chemistry. 1994;269:27987–27991. [PubMed] [Google Scholar]
- 46.Williams K, Shoelson S. Journal of Biological Chemistry. 1993;268:5361–5364. [PubMed] [Google Scholar]
- 47.Wittelsberger A, Mierke DF, Rosenblatt M. Chemical Biology & Drug Design. 2008;71:380–383. doi: 10.1111/j.1747-0285.2008.00646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Daye L, Gibson W, Williams K. International Journal of High Throughput Screening. 2010;2010:69–80. [Google Scholar]
- 49.Williams KP, Rayhorn P, Chi-Rosso G, Garber EA, Strauch KL, Horan GS, Reilly JO, Baker DP, Taylor FR, Koteliansky V, Pepinsky RB. J Cell Sci. 1999;112(Pt 23):4405–4414. doi: 10.1242/jcs.112.23.4405. [DOI] [PubMed] [Google Scholar]
- 50.Rhodes D, Bossio R, Laue T. Methods in Enzymology. 2009;463:691–723. doi: 10.1016/S0076-6879(09)63039-1. [DOI] [PubMed] [Google Scholar]
- 51.Taylor F, Wen D, Garber E, Carmillo A, Baker D, Arduini R, Williams K, Weinreb P, Rayhorn P, Hronowski X, Whitty A, Day E, Boriack-Sjodin A, Shapiro R, Glades A, Pepinsky B. Biochemistry. 2001;40:4359–4371. doi: 10.1021/bi002487u. [DOI] [PubMed] [Google Scholar]
- 52.Wang L, Liu Z, Gambardella L, Delacour A, Shapiro R, Yang J, Sizing I, Rayhorn P, Garber E, Benjamin C, Williams K, Taylor F, Barrandon Y, Ling LE, Burkly L. J Invest Dermatol. 2000;114:901–908. doi: 10.1046/j.1523-1747.2000.00951.x. [DOI] [PubMed] [Google Scholar]
- 53.Gao X, Arenas-Ramirez N, Scales SJ, Hannoush RN. FEBS Letters. 2011;585:2501–2506. doi: 10.1016/j.febslet.2011.06.033. [DOI] [PubMed] [Google Scholar]
- 54.Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, Scott MP, Beachy PA. Nature. 2000;406:1005–1009. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- 55.Chang SC, Mulloy B, Magee AI, Couchman JR. Journal of Biological Chemistry. 2011;286:44391–44402. doi: 10.1074/jbc.M111.285361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Farshi P, Ohlig S, Pickhinke U, Hoing S, Jochmann K, Lawrence R, Dreier R, Dierker T, Grobe K. Journal of Biological Chemistry. 2011;286:23608–23619. doi: 10.1074/jbc.M110.206474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goetz J, Singh S, Suber L, Kull F, Robbins D. Journal of Biological Chemistry. 2006;281:4087–4093. doi: 10.1074/jbc.M511427200. [DOI] [PubMed] [Google Scholar]
- 58.Ohlig S, Pickhinke U, Sirko S, Bandari S, Hoffmann D, Dreier R, Farshi P, Götz M, Grobe K. Journal of Biological Chemistry. 2012;287:43708–43719. doi: 10.1074/jbc.M112.356667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Digard P, Williams K, Hensley P, Brooks I, Dahl C, Coen D. Proceedings of the National Academy of Sciences. 1995;92:1456–1460. doi: 10.1073/pnas.92.5.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Toedt G, Krishnan R, Friedhoff P. Nucleic Acids Research. 2003;31:819–825. doi: 10.1093/nar/gkg191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dierker T, Dreier R, Migone M, Hamer S, Grobe K. Journal of Biological Chemistry. 2009;284:32562–32571. doi: 10.1074/jbc.M109.044867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vyas N, Goswami D, Manonmani A, Sharma P, Ranganath H, VijayRaghavan K, Shashidhara L, Sowdhamini R, Mayor S. Cell. 2008;133:1214–1227. doi: 10.1016/j.cell.2008.05.026. [DOI] [PubMed] [Google Scholar]
- 63.Kornberg TB. Science Signaling. 2011;4:pe44. doi: 10.1126/scisignal.2002447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cooper MK, Porter JA, Young KE, Beachy PA. Science. 1998;280:1603–1607. doi: 10.1126/science.280.5369.1603. [DOI] [PubMed] [Google Scholar]