

Abstract
Syphilis, a slow progressive and the third most common sexually transmitted disease found worldwide, is caused by a spirochete gram negative bacteria Treponema pallidum. Emergence of antibiotic resistant T. pallidum has led to a search for novel drugs and their targets. Subtractive genomics analyses of pathogen T. pallidum and host Homo sapiens resulted in identification of 126 proteins essential for survival and viability of the pathogen. Metabolic pathway analyses of these essential proteins led to discovery of nineteen proteins distributed among six metabolic pathways unique to T. pallidum. One hundred plant-derived terpenoids, as potential therapeutic molecules against T. pallidum, were screened for their drug likeness and ADMET (absorption, distribution, metabolism, and toxicity) properties. Subsequently the resulting nine terpenoids were docked with five unique T. pallidum targets through molecular modeling approaches. Out of five targets analyzed, D-alanine:D-alanine ligase was found to be the most promising target, while terpenoid salvicine was the most potent inhibitor. A comparison of the inhibitory potential of the best docked readily available natural compound, namely pomiferin (flavonoid) with that of the best docked terpenoid salvicine, revealed that salvicine was a more potent inhibitor than that of pomiferin. To the best of our knowledge, this is the first report of a terpenoid as a potential therapeutic molecule against T. pallidum with D-alanine:D-alanine ligase as a novel target. Further studies are warranted to evaluate and explore the potential clinical ramifications of these findings in relation to syphilis that has public health importance worldwide.
Introduction
Syphilis, caused by a spirochete gram negative bacteria Treponema pallidum subsp pallidum SS14 (T. pallidum), is the third most common and slow progressive sexually transmitted disease (STD) found worldwide. It is a multistage disease and usually transmitted through contact with active lesions of a sexual partner or from an infected pregnant woman to her fetus. Generally, antibiotics such as penicillin, erythromycin, and azithromycin are prescribed for treatment of the disease, out of which the intramuscularly administered penicillin G benzathine is the preferred treatment for syphilis, except neurosyphilis (Smith et al., 1956). Other antibiotics, including tetracyclines, macrolides, and cephalosporins, have also been used as an alternative treatment for penicillin-allergic patients (Katz and Klausner, 2008). People suffering from syphilis are at a two- to five-fold higher risk to develop HIV infection. During the past few years, the emergence of an antibiotic-resistant strain of T. pallidum (Marra et al., 2006; Mitchell et al., 2006; Stamm, 2010) has significantly hampered the treatment of syphilis. Modern approaches to the understanding of the pathogenesis of syphilis, the development of improved diagnostic tests, and the implementation of decentralized management strategies offer the promise of improving syphilis control (Hook and Peeling, 2004).
Genomics and associated high-throughput technologies provide opportunities for better understanding of infectious disease mechanism, as well as their prevention and treatment. Subtractive genomics approaches are meant for screening pathogen-specific targets which are non-human homologous genes/proteins regulating pathogen specific metabolic pathways or biological reactions. These in silico approaches reduce the time as well as the cost of target identification and have made a new paradigm shift in drug discovery research (Barh et al., 2011). Designing and virtual screening of inhibitor/drugs against the pathogen-specific unique targets may help in developing potential drugs for fatal STDs including syphilis. Thus, predictions of novel drug targets in case of a number of other pathogenic bacteria such as Mycobacterium tuberculosis, Neisseria gonorrhoeae, Staphylococcus aureus etc. have been done using bioinformatics approaches such as subtractive genomics, comparative microbial genomics, and metabolic pathway analyses of genome of the host and pathogens (Anishetty et al., 2005; Barh and Kumar, 2010; Hoseni et al., 2012; Kushwaha and Shakya, 2010; Sharma et al., 2006).
Availability of complete genome sequences for T. pallidum strain SS14 along with those of Homo sapiens (Fraser et al., 1998; Matejkova et al., 2008) has made it possible to do comparative analysis of host and pathogen genomes, proteomes as well as metabolomes. Genomic Target Database (GTD) has been developed for the target identification using subtractive genomics approach (Barh et al., 2009).
In the present study, T. pallidum-specific novel drug targets have been identified by analyzing complete genome, proteome, and metabolome of pathogen and human host through subtractive genomics approaches. Furthermore, a number of identified potential drug targets have been studied in detail by performing homology modeling and docking analyses. For docking analyses, one hundred plant-derived terpenoids were initially analyzed for their drug likeness and absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties (pharmacokinetic analyses) and such successfully screened terpenoids were docked with the identified unique and novel T. pallidum targets. In order to find out a readily available natural compound as effective inhibitor of the best identified target, a Natural Product database Set III was also screened and analyzed.
Materials and Methods
Identification of non-human homologous essential proteins in T. pallidum
The complete proteome of T. pallidum susp pallidum SS14 (ID UP000001202) was retrieved from the SWISSPROT database. The whole proteome of T. pallidum was clustered in group by using the CD-HIT (Cluster Database at High Identity with Tolerance) tool (Huang et al., 2010). CD-HIT takes FASTA format sequences and produces a set of closely related sequences, therefore reducing the database size. Sequences possessing up to 60% similarity or of less than 100 amino acids length were excluded from further analysis. The short word (identical short substrings) filtering was set to 4.The remaining set of protein sequence was subjected to Blastp analysis considering E-value of 10−4 against human proteome to identify non-human homologs. The obtained resultant set was further subjected to Blastp analysis against Database of Essential Genes (DEG) version 10.6 (Zhang and Lin, 2009). Sequences showing a bit score of higher than 100 and similarity more than 40% were screened as essential proteins (candidate drug target). The screened out proteins and corresponding gene were considered as essential for survival of pathogen.
Metabolic pathway analysis
KEGG ortholog (KO) annotation of essential proteins of T. pallidum was carried out by KASS (KEGG Automatic Annotation Server) at KEGG database (Moriya et al., 2007). KASS provides the functional annotation of genes by running Blast against its KEGG GENES database. Metabolic pathway identification corresponding to KO annotated sequences were performed by using KOBAS (KEGG Orthology-Based Annotation System) 2.0 server (Wu et al., 2006; Xie et al., 2011). KOBAS identify the frequently occurring or statistically significant enriched pathways using biological knowledge from well known pathway databases, diseases databases, and gene ontology.
Subcellular localization prediction
Protein subcellular localization prediction deals with the computational prediction of position where proteins reside in the cell. It is an important component as it deals with the prediction of protein function and genome annotation. Subcellular localization of all essential proteins was done by PSORTb tool version 2.0.4 (Gardy et al., 2005) located at http://www.psort.org/psortb/. PSORTb v.2.0 is the most precise bacterial localization prediction tool with a measured overall precision of 96%.
Functional annotation
Protein functional predictions of these essential putative uncharacterized proteins were carried out by SVMProt (Cai et al., 2003). SVMprot uses Support Vector Machine for such classifications.
Secondary structure prediction
The secondary structure prediction of the identified target proteins was done using PSIPRED tool version 3.3 (http://bioinf.cs.ucl.ac.uk/psipred). PSIPRED is a neural network based secondary structure prediction method that produces graphical output for the secondary structure of the given sequence (McGuffin et al., 1999).
Homology modeling
3D structures of identified target proteins of T. pallidum were generated using homology modeling module of Discovery Studio 3.1 software (DS). It involves identification of the template, building of a model, and assessing the validity of structure. The software provides a list of templates that can be selected for modeling. Templates showing similarity (positives) of greater than 40% with the target proteins were further selected for generating 3D models. After model generation, it provides a list of generated models along with their DOPE (Discrete Optimized Protein Energy) score. The DOPE score is based on statistical potential and considered as a measure of model quality. The lower the DOPE score, the better the model.
The model with the lowest DOPE score was selected and validated using Verify protein protocol. This protocol predicts the acceptability of the model on the basis of verify scores. A verify score that falls within the expected high and low score is suggestive of the model of acceptable quality. In addition to this, the generated models were also validated using Ramachandran Plot from PROCHECK (Laskowski et al., 2001) program available at SAVS server version 4 (http://services.mbi.ucla.edu/SAVES) and ProQ tools (http://www.sbc.su.se/∼bjornw/ProQ/ProQ.html) (Wallner and Elofsson, 2003). The models having maximum number of residues in the allowed region of the Ramachandran plot are considered to be acceptable. The neural network-based web tool ProQ predicts the quality of protein model based on two quality measurement scores, namely LGscore and MaxSub score. LGscore is a p-value that defines the significance level of structural similarity. MaxSub score is an automated normalized score that represents the quality of model. The range of model quality is represented as, LGscore >1.5 (correct), >3 (good) and >5 (very good) and MaxSub score >0.1 (correct), >0.5 (good), >0.8 (very good).
Pharmacokinetic analyses of one hundred plant-derived terpenoids
ADMET analyses
The in silico prediction of ADMET properties is used to assess the long-term success of a potential therapeutic molecule (Balani et al., 2005; Segall et al., 2006). Thus, the ADMET properties of one hundred plant-derived terpenoids were analyzed using DS. The toxicity prediction was done using weight of evidence (WOE) parameter of carcinogenicity of TOPKAT module of DS. WOE predictions are in accordance with animal model guidelines of U.S. Food and Drug Administration (FDA) and National Toxicology Program (NTP), USA. The ADMET screening was based on several following criteria:
a) Human intestinal absorption (HIA) and Blood brain barrier (BBB): Human intestinal absorption (HIA) is the process through which orally administered drugs are absorbed from the intestine into the bloodstream. HIA levels of 0 and 1 indicate good and moderate absorption. The blood-brain barrier (BBB) is a diffusion barrier, which hinders influx of most compounds from blood to brain (Ballabh et al., 2003). BBB levels of 1, 2, 3, and 4 indicate high, medium, low, and undefined penetration, respectively.
b) Plasma protein binding (PPB): Compounds possessing Bayesian score of −2.226 or less reflect highly bound (≥90%) to plasma protein.
c) Cytochrome P450 2D6 (CYP2D6) binding: CYP2D6 is involved in the metabolism of a wide range of metabolites in liver. Compounds having a Bayesian Score of <0.162 are non-inhibitors of CYP2D6.
d) Aqueous solubility: Solubility level of 1, 2, and 3 indicates very low, but possible and good solubility.
e) AlogP and PSA2D: AlogP98 (partition coefficient) score of <5 and PSA (polar surface area) score of <140 indicates good absorption and cell permeability (Egan et al., 2000).
f) Toxicity: WOE parameter of carcinogenicity was considered for toxicity prediction.
Drug likeness
Drug-like properties were analyzed on the basis of Lipinski's rule of five using DS. Four properties, namely H-bond donor, H-bond acceptor, molecular weight, and AlogP (partition coefficient) of the therapeutic molecule were analyzed for their drug likeness.
Molecular docking
The molecular docking analyses of modeled proteins (target) of T. pallidum and the terpenoids (ligand) were carried out using AutoDock 4.2.1 (Morris et al., 2009) software following the Lamarckian genetic algorithm (LGA) method (Morris et al. 1999). The preparation of protein was done using AutoDock Tools 4, which is the graphical user interface for AutoDock 4.2.1. The protein preparation involved the addition of polar hydrogen atoms to the macromolecule. Gasteiger charges were calculated for each atom of the macromolecule. After preparing the protein, a three-dimensional grid was positioned at the active site region of the protein. The LGA method was used for a ligand conformational search (Morris et al. 1999). The active site region for the target proteins was selected on the basis of literature evidence available for protein ligand complexes (Borrok et al., 2010; Bruning et al., 2010; Djordjevic and Stock, 1997; Eschenburg and Schonbrunn, 2000; Osborne et al., 2004). The protein ligand interaction was visualized with UCSF Chimera 1.8 (Pettersen et al., 2004).
Screening of Natural Products Database
In order to find a natural and readily available compound as effective inhibitor of the best identified target, the Natural Products Set III consisting of 117 compounds (selected from the DTP (Developmental Therapeutics Program) Open Repository collection of 140,000 compounds) were retrieved from the URL http://dtp.nci.nih.gov/branches/dscb/natprod_explanation.html and screened. For assessing the effectiveness of these compounds as potential therapeutic molecules, these 117 compounds were initially analyzed for their pharmacokinetic properties using ADMET and Lipinski's rule of five (drug likeness) analyses as described above for the terpenoids. Successfully screened compounds were subsequently docked with the best chosen target using AutoDock 4.2.1 as described for the terpenoids.
Results
Identification of non-human homologous proteins
The whole proteome (ID UP000001202) of T. pallidum, consisting of 1028 proteins, was clustered into groups by using the CD-HIT tool. Sequences possessing ≥60% similarity were included for further analysis. A total of 1015 proteins were present in the clustered group and were selected for Blastp analysis against human proteome. It was found that 530 non-human homolog proteins were present in the pathogen. The resultant set of non-human homologs was further subjected to Blastp analysis against the DEG database. Sequences showing a bit score of higher than 100 and similarity more than 40% were screened as essential proteins. Thus, out of 530 proteins, 126 proteins were found to be essential for the viability, pathogenicity, and survival of pathogen T. pallidum. These constituted 12.12% of spirochete genomes (Table 1).
Table 1.
Summary of Analyses for Non-human Homologous Proteins of T. pallidum
| Methods | Tools/Softwares | Number of proteins |
|---|---|---|
| Retrieval of pathogen proteins | SWISSPROT | 1028 |
| Clustering of proteome (>60%) | CD-HIT | 1015 |
| Identification of non-human homologous proteins | Blastp | 530 |
| Identification of essential proteins | DEG | 126 |
| Finding metabolic pathway of essential proteins | KASS and KOBAS | 106 |
Metabolic pathway analyses of the essential proteins in T. pallidum
Metabolic pathway analyses for 126 essential proteins of T. pallidum were done by KAAS and KOBAS server. It was revealed that, out of 126 essential proteins, 106 proteins were linked with essential metabolic and signal transduction pathway network of pathogen. Thus, out of these 106 proteins, 9 proteins were found to be involved in carbohydrate metabolism, 3 in energy metabolism (carbon fixation and nitrogen metabolism), 11 in nucleotide metabolism, 7 proteins in amino acid metabolism, 8 in cell wall biosynthesis, 8 in cofactor and vitamin biosynthesis, 4 in secondary metabolite biosynthesis, 34 in genetic processing, 13 in membrane transport, cell motility, and 2 responsible for pathogenesis. On the other hand, the analysis of metabolome of pathogens also revealed a striking lack in metabolic capabilities of pathogen with respect to TCA cycle, fatty acid and amino acid metabolic pathways (Supplementary Table S1; supplementary material is available online at www.liebertonline.com/omi).
Functional classification of uncharacterized essential proteins
Out of 126 essential proteins, functions of 20 essential proteins were unknown. For the functional annotation of these 20 proteins, the SVMProt tool was used. It was observed that out of 20 essential proteins, only 9 could be assigned function and were classified as those involved in lipid binding (2), metal binding (3), transferases (2), RNA binding (1), and transmembrane protein (1) (Supplementary Table S2).
Target identification from pathways unique to pathogen T. pallidum
In order to design highly selective drugs against T. pallidum, metabolic pathway analysis of the pathogen was carried out using KAAS and KOBAS servers. It was found that six pathways: peptidoglycan biosynthesis, D-alanine metabolism, bacterial secretion system, two component system, bacterial chemotaxis, and flagellar assembly, were unique to T. pallidum and absent in the human. Nineteen unique proteins/enzymes were identified out of the six pathways unique to T. pallidum (Table 2). These enzymes did not show homology with any of the host proteins, therefore they can be considered as putative drug targets. However, a COG (cluster of orthologous group) search (Tatusov et al., 2003) for these proteins revealed that most of the targets also exhibited homology with other pathogenic bacteria. Subcellular localization analysis of these 19 proteins, done using PSORTb tool, revealed that 10 proteins were membrane associated, 8 were cytoplasmic, while the remaining one was periplasmic (Table 2).
Table 2.
Nineteen Identified Enzymes/Proteins Out of six Pathways Unique to T. pallidum Along with Subcellular Localization
| S. No. | Pathway/Enzymes/Proteins | SWISSPROT ID | KEGG Ortholog ID | Genes | E.C. No. | Localization |
|---|---|---|---|---|---|---|
| 1. Peptidoglycan Biosynthesis | ||||||
| I. | UDP-N-acetylglucosamine 1-carboxyvinyltransferase | B2S1X7 | K00790 | murA | 2.5.1.7 | Cytoplasmic |
| II. | UDP-N-acetylmuramate dehydrogenase | B2S238 | K00075 | murB | 1.1.1.158 | Cytoplasmic |
| III. | UDP-N-acetylmuramoylalanyl-D-glutamyl-2,6-diaminopimelate–D-alanyl-D-alanine ligase | B2S2Y3 | K01929 | murF | 6.3.2.10 | Cytoplasmic |
| IV. | UDP-N-acetylglucosamine-N-acetylmuramyl-(pentapeptide) pyrophosphoryl-undecaprenol N-acetylglucosamine transferase | B2S3B6 | K02563 | murG | 2.4.1.227 | Membrane |
| V. | Penicillin-binding protein 1A | B2S3U4 | K05366 | mrcA | 2.4.1. - and 3.4.-.- | Membrane |
| VI. | Penicillin-binding protein 2 | B2S394 | K05515 | mrdA | Membrane | |
| VII. | Virulence factor | B2S3B0 | K03980 | mviN | Membrane | |
| VIII. | Penicillin-binding protein 3 | B2S329 | K03587 | ftsI | 2.4.1.129 | Membrane |
| 2. D-Alanine Metabolism | ||||||
| I. | Alanine racemase | B2S3S0 | K01775 | alr | 5.1.1.1 | Cytoplasmic |
| II. | D-alanine-D-alanine ligase | B2S3Q9 | K01921 | ddlA | 6.3.2.4 | Cytoplasmic |
| 3. Bacterial secretion system | ||||||
| I. | Preprotein translocase subunit SecD | B2S307 | K03072 | secD | Membrane | |
| II. | Preprotein translocase subunit SecY | B2S2F6 | K03076 | secY | Membrane | |
| III. | Preprotein translocase subunit SecA | B2S2X6 | K03070 | secA | Cytoplasmic | |
| IV. | Preprotein translocase subunit YidC | B2S4I6 | K03217 | yidC | Membrane | |
| V. | Preprotein translocase subunit SecF | B2S308 | K03074 | secF | Membrane | |
| 4. Two-component system | ||||||
| I. | Chemotaxis protein methyltransferase | B2S3M0 | K00575 | cheR | 2.1.1.80 | Cytoplasmic |
| II. | Sensor kinase | B2S2W0 | K03407 | cheA | 2.7.13.3 | Cytoplasmic |
| 5. Bacterial chemotaxis | ||||||
| I. | Methyl-galactoside transport system substrate-binding protein | B2S3S3 | K10540 | mglB | Periplasmic | |
| 6. Flagellar assembly | ||||||
| I. | Flagellar biosynthesis protein | B2S4J7 | K02392 | flgG1 | Membrane | |
Homology modeling of selected target proteins
Analyses of the metabolic pathways unique to T. pallidum revealed that in all six pathways a total number of 19 proteins were essential for the survival of the pathogen. Crystal structures of these proteins for T. pallidum were not available, therefore a homology modeling approach was used for generating protein 3D models for the selected proteins. Thus, one protein from each of the above mentioned six pathways, namely UDP-N-acetylglucosamine 1-carboxyvinyltransferase (UDPN), D-alanine-D-alanine ligase (DDLA), preprotein translocase subunit SecA (SECA), chemotaxis protein methyltransferase (CHER), methyl-galactoside transport system substrate-binding protein (MGLB), and flagellar biosynthesis protein were selected for molecular modeling analyses.
Homology modeling of each of these selected target proteins was done using DS software. Templates for each protein were searched using DS. Templates showing similarity (positives) of greater than 40% with the target proteins were further selected for generating 3D models. Thus, the templates, namely 1EJD (with 55% similarity, 2I87 (with 54% similarity), 1TF5 (with 59% similarity), 1AF7 (with 45% similarity), and 2FVY (with 51% similarity) were used for generating 3D models of the targets, namely UDPN, DDLA, SECA, CHER, and MGLB, respectively. In the case of flagellar biosynthesis protein, no templates were found to exhibit a similarity greater than 40%, therefore this target could not be modeled. Thus, a total number of five proteins were selected for model generation.
Among the number of generated models, the acceptable models were selected on the basis of DOPE score and validated using Verify3D, PROCHECK, ProQ tools. Results of validation are presented in Table 3 and Supplementary Figure S1. Based on all the validation scores, all the five generated models of the selected targets were found to be reliable. Results of the secondary structure prediction of all the five selected target proteins are presented in Supplementary Figures S2, S3, and S4.
Table 3.
Summary of Post Structural Analyses of Modeled Five Target Proteins of T. pallidum
| Identified targets | ||||||
|---|---|---|---|---|---|---|
| Tools/ Software | Parameters | UDPN | DDLA | SECA | CHER | MGLB |
| DS | DOPE Score | −50885.25 | −41487.18 | −89030.57 | −31606.66 | −25578.22 |
| Verify Score | 187.06 | 152.62 | 286.20 | 113.51 | 85.67 | |
| PROCHECK (Ramachandran Plot) | Allowed region | 92.1 | 92.8 | 93.2 | 91.3 | 87.5 |
| Additionally allowed region | 6.0 | 6.0 | 5.2 | 5.2 | 11.0 | |
| Generously allowed region | 1.1 | 0.9 | 1.3 | 3.2 | 0.8 | |
| Disallowed region | 0.8 | 0.3 | 0.3 | 0.4 | 0.8 | |
| ProQ Analyses | LG Score | 5.7 | 5.3 | 2.9 | 3.7 | 1.6 |
| MaxSub Score | 0.5 | 0.5 | 0.2 | 0.3 | 0.2 | |
CHER, chemotaxis protein methyltransferase; DDLA, D-alanine-D-alanine ligase; MGLB, methyl-galactoside transport system substrate-binding protein; SECA, preprotein translocase subunit SecA; UDPN, UDP-N-acetylglucosamine 1-carboxyvinyltransferase
Pharmacokinetic analyses of one hundred plant-derived terpenoids for therapeutic applications
Before subjecting the selected plant-derived terpenoids for docking analyses, one hundred terpenoids were screened through ADMET and Lipinski's rule of five analyses using DS software. Results are presented in Tables 4 and 5. It is noteworthy that out of 100 selected terpenoids, only 9 could successfully fulfill the ADMET and Lipinski's rule of five criteria.
Table 4.
List of Successfully Screened Terpenoids Fulfilling ADMET Crteria
| S. No. | Compound Name | AlogP98* | PSA* 2D | HIA* | BBB* level | Solubility Level | CYP2d6* | PPB | PPB* prediction | Toxicity WOE* Prediction |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Tanshinone | 4.661 | 47.155 | 0 | 1 | 1 | −3.0869 | 2.266 | true | NC |
| 2 | Sclareol | 4.27 | 41.631 | 0 | 1 | 2 | −2.5333 | −0.235 | true | NC |
| 3 | Sarsasapoge-nin | 4.883 | 38.675 | 0 | 0 | 1 | −3.2867 | −1.084 | true | NC |
| 4 | Salvicine | 3.633 | 76.232 | 0 | 2 | 3 | −3.5442 | 2.181 | true | NC |
| 5 | Menthol | 2.779 | 20.815 | 0 | 1 | 3 | −3.0633 | −1.260 | true | NC |
| 6 | Mansonone E | 2.762 | 43.531 | 0 | 1 | 2 | −3.0492 | 0.434 | true | NC |
| 7 | Limonene | 3.502 | 0 | 0 | 0 | 3 | −2.3187 | 0.577 | true | NC |
| 8 | Cynaropicrin | 1.381 | 94.092 | 0 | 3 | 3 | −4.6654 | −1.184 | true | NC |
| 9 | Carnosol | 4.387 | 67.861 | 0 | 1 | 2 | −1.9658 | 0.435 | true | NC |
AlogP98, partition coefficient; BBB, blood brain barrier; CYP2D6, cytochrome P450 2D6 binding; HIA, human intestinal absorption; PPB, plasma protein binding; PSA, polar surface area; WOE, weight of evidence.
Table 5.
Analyses of Drug Like Properties of Successfully Screened Terpenoids, Fulfilling ADMET Criteria, on Basis of Lipinsiki's Rule of Five
| S. No. | Compound | H-donor | H-acceptor | Mol. Wt. | AlogP |
|---|---|---|---|---|---|
| 1 | Tanshinone | 0 | 3 | 294.344 | 4.66 |
| 2 | Sclareol | 2 | 2 | 308.499 | 4.27 |
| 3 | Sarsasapogenin | 1 | 3 | 416.636 | 4.883 |
| 4 | Salvicine | 2 | 4 | 330.418 | 3.633 |
| 5 | Menthol | 1 | 1 | 156.265 | 2.779 |
| 6 | Mansonone E | 0 | 3 | 242.27 | 2.762 |
| 7 | Limonene | 0 | 0 | 136.234 | 3.502 |
| 8 | Cynaropicrin | 2 | 6 | 346.374 | 1.381 |
| 9 | Carnosol | 2 | 4 | 330.418 | 4.387 |
Docking analyses
Based on the results of pharmacokinetic analyses, nine successfully screened terpenoids were subjected to docking with all five target proteins, namely UDPN, DDLA, SECA, CHER, and MGLB. Results of the docking analyses in terms of binding free energy and predicted Ki (inhibition constant) are presented in Table 6. The complexes of each of the five selected targets with their best docked terpenoids have been depicted in Figure 1. It was observed that out of nine terpenoids, four, namely salvicine, tanshinone, menthol, and limonene, showed best docking results (having minimum binding energy and Ki value) in the case of the target DDLA. Terpenoids, namely sclerol, carnasol, and mansonone E, showed best docking results in the case of target CHER; terpenoid sarasasapogenin showed best docking result in the case of target UDPN, while that of cynaropicrin showed best docking result in the case of target MGLB. From the results of docking analyses, it is evident that DDLA is the most promising and SECA is the least favorable drug target among the selected five targets. Thus, it is evident that DDLA showed best docking results with the maximum number of terpenoids (four out of nine) among all the five docked targets. In addition to this, the terpenoid, salvicine, was found to be the most potent inhibitor with respect to binding energy (-8.52 kcal/mole) and least inhibition constant, Ki, (0.5 μM).
Table 6.
Summary of Results of Docking of Selected Terpenoids with Five Unique Target Proteins of T. pallidum*
| Terpenoids | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Target | Docking Result | Salvicine | Tanshinone | Sclerol | Sarasasapogenin | Carnasol | Menthol | Limonene | ManasononeE | Cynaropicrin |
| DDLA | Binding energy(kcal/mole) | −8.52 | −7.87 | −7.25 | −6.86 | −6.56 | −6.52 | −5.98 | −5.60 | −5.56 |
| Ki (μm) | 0.56 | 1.70 | 4.83 | 9.93 | 15.48 | 16.54 | 41.10 | 48.30 | 84.43 | |
| UDPN | Binding energy(kcal/mole) | −6.02 | −6.41 | −5.53 | −8.58 | −5.80 | −4.57 | −4.87 | −5.36 | −5.95 |
| Ki(μm) | 38.79 | 20.02 | 88.68 | 513.29 | 56.11 | 446.50 | 267.49 | 117.03 | 43.47 | |
| SECA | Binding energy(kcal/mole) | −4.02 | −4.77 | −4.05 | −3.70 | −3.99 | −4.22 | −3.94 | −4.57 | −4.24 |
| Ki(μm) | 1120.00 | 321.08 | 1080.00 | 1900.0 | 1200 | 804.14 | 1300 | 447.37 | 776.01 | |
| CHER | Binding energy(kcal/mole) | −6.64 | −7.67 | −7.79 | +21.61 | −7.49 | −6.06 | −5.17 | −6.86 | −6.79 |
| Ki(μm) | 13.48 | 2.37 | 1.94 | - | 3.23 | 36.23 | 161.46 | 9.32 | 10.52 | |
| MGLB | Binding energy(kcal/mole) | −6.00 | −6.37 | −7.15 | −7.54 | −7.11 | −5.06 | −4.29 | −5.94 | −6.95 |
| Ki(μm) | 40.06 | 21.43 | 5.76 | 2.49 | 6.19 | 194.59 | 711.47 | 44.56 | 8.00 | |
CHER, chemotaxis protein methyltransferase; DDLA, D-alanine-D-alanine ligase; MGLB, methyl-galactoside transport system substrate-binding protein; SECA, preprotein translocase subunit SecA; UDPN, UDP-N-acetylglucosamine 1-carboxyvinyltransferase.
FIG. 1.

Modeled 3D structures of five identified target proteins of T. pallidum along with the respective best docked terpenoids (red). (A) UDPN with tanshinone; (B) DDLA with salvicine; (C) SECA with tanshinone; (D) CHER with sclerol; (E) MGLB with sarasasapogenin.
While considering DDLA as the best target protein and salvicine as the best docked terpenoid, a comparative analysis was also done with D-cycloserine, which is a commonly used drug against DDLA. The docking analyses revealed that the best docked terpenoid, salvicine, exhibited four-fold lower binding energy and approximately A thousand-fold lower inhibition constant as compared to the drug D-cycloserine.
The regiospecific interactions of terpenes, as well as that of the drug, D-cycloserine, were analyzed within a zone of 6 Å at the active site of DDLA of the respective docked complexes. Results are presented in Table 7. It is noteworthy that the most potent inhibitor terpenoid, namely salvicine, interacted with a total of 11 residues, namely Glu14, Phe99, Pro100, Val101, Leu102, His103, Thr317, Met318, Pro319, Gly320, and Phe321 at the active site of DDLA. Furthermore, binding of salvicine involved H-bond interaction with the residues His103 and Met318 (Fig. 2). Regiospecific interaction studies also revealed that all the interacting residues at the active site region were present in the allowed region of the Ramachandran plot of DDLA. However the residues in the disallowed region, namely Leu64, Phe86, Asn235, and Ala265, were not present at the active site. A comparison of the regiospecific interactions of other 8 terpenes with that of salvicine revealed that, among the 11 interacting residues, 8 residues namely, Glu14, Val101, Leu102, His103, Met318, Pro319, Gly320, and Phe 321, were found to be common in all the terpenoids with the exception of limonene and sarasasapogenin in which Glu14, His 103 and Phe321, respectively, were absent (Table 7). The analyses of the regiospecific interactions of the drug D-cycloserine revealed the participation of eight residues, namely His 103, Asp109, Ala134, Met 135, Val 315, Asn 316, Thr317, and Met318, of DDLA at the active site.
Table 7.
Summary of Results of Docking Analyses of Selected Terpenoids and Commonly Used Drug D-Cycloserine with Regiospecific Interactions with DDLA as Target
| Compound | Binding energy (kcal/mole) | Ki (μM) | Regiospecific interacting residues |
|---|---|---|---|
| Salvicine | −8.52 | 0.5 | Glu14, Phe99, Pro100, Val101, Leu102, His103, Thr317, Met318, Pro319, Gly320, Phe321(11) |
| Tanshinone | −7.87 | 1.7 | Glu14, Val17, Phe99, Pro100, Val101, Leu102, His103, Thr317, Met318, Pro319, Gly320, Phe321(12) |
| Sclerol | −7.25 | 4.83 | Glu14, Phe99, Pro100, Val101, Leu102, His103, Thr317, Met318, Pro319, Gly320, Phe321(11) |
| Sarasasapogenin | −6.86 | 9.93 | Glu14, His15, Val17, Ser18, Val101, Leu102, His103, Arg299, Met318, Pro319, Gly320(11) |
| Carnasol | −6.56 | 15.48 | Glu14, Pro100, Val101, Leu102, His103, Met318, Pro319, Gly320, Phe321(9) |
| Menthol | −6.52 | 16.54 | Glu14, Phe99, Pro100, Val101, Leu102, His103, Met318, Pro319, GLy320, Phe321 (10) |
| Limonene | −5.98 | 41.10 | Pro100, Val101, Leu102, Val124, Gly125, Cys126, Thr317, Met318, Pro319, Gly320, Phe321 (11) |
| Mansonone E | −5.60 | 78.30 | Glu14, His15, Val17, Ser18, Val101, Leu102, His103, Asn316, Met318, Pro319, Gly320, Phe321(12) |
| Cynaropicrin | −5.56 | 84.43 | Glu14, His15, Val17, Ser18, Val101, Leu102, His103, Arg299, Asn316, Met318, Pro319, Glu320, Phe321(13) |
| D-cycloserine | −4.80 | 3.0 | His 103, Asp109, Ala134, Met 135, Val 315, Asn 316, Thr317, Met 318 (8) |
FIG. 2.
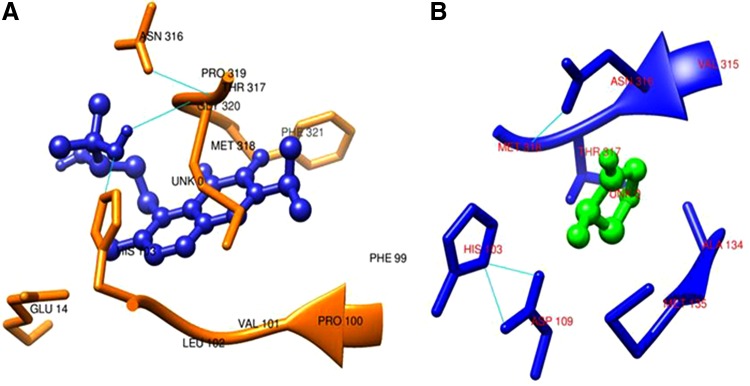
3D view showing the interacting residues of DDLA at the active site along with salvicine (A) and D-cycloserine (B).
Mode of action of salvicine, the most potent inhibitor of DDLA
In order to investigate the mechanism of inhibition of the DDLA by salvicine, the inhibitor salvicine was docked with the target DDLA complexed with substrate D-alanine and the co-factor ADP. Results of the docking revealed that salvicine binds with DDLA at a site different to those of ADP site and the D-alanine binding sites (Fig. 3), suggesting that salvicine did not inhibit DDLA by competing with either ADP or D-alanine binding sites.
FIG. 3.

3D view showing three distinct binding sites for each of salvicine (red), d-alanine (blue), and ADP (brown) in DDLA.
Screening of natural products with best identified target DDLA
In order to find a natural compound as effective inhibitor of the best target identified from the present study, namely DDLA, a Natural Products database Set III consisting of 117 readily available compounds (for further experimental validation), was analyzed for pharmacokinetic properties. The results of ADMET and Lipinski rule of five analyses are presented in Tables 8 and 9, respectively. The data revealed that out of 117, only 13 compounds could successfully pass the ADMET and Lipinski rule of five criteria. These 13 compounds were further subjected for docking analyses with DDLA as target using AutoDock 4.2.1 software. Results are presented in Table 10. It is noteworthy that compound pomiferin (a flavonoid) was found to be the most potent inhibitor of DDLA with Ki value of 3.09 μM. A comparison of the inhibitory potential of the best docked readily available natural compound, namely pomiferin, with that of the best docked terpenoid from the present study, namely salvicine, revealed that the salvicine was more potent inhibitor (about six-fold stronger) than that of the pomiferin. At the level of binding energy also, salvicine was found to be better than that of pomiferin.
Table 8.
List of Successfully Screened Compounds from Natural Product Set III of DTP Repository Fulfilling ADMET Criteria
| S. No. | Compound ID | AlogP98 | PSA2D | HIA* | BBB* level | Solubility level | CYP2D6* | PPB* | PPB prediction | Toxicity WOE* Prediction |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 7668 | 2.963 | 100.562 | 0 | 3 | 4 | −3.87787 | 1.61957 | TRUE | NC |
| 2 | 26258 | 3.93 | 61.951 | 0 | 1 | 2 | −3.08797 | 2.03212 | TRUE | NC |
| 3 | 32192 | 2.829 | 65.303 | 0 | 2 | 2 | −4.5736 | 0.694091 | TRUE | NC |
| 4 | 32979 | 3.308 | 50.958 | 0 | 1 | 2 | −0.6143 | −1.88296 | TRUE | NC |
| 5 | 5113 | 4.784 | 97.607 | 1 | 4 | 2 | −1.40136 | −0.52834 | TRUE | NC |
| 6 | 9665 | 1.395 | 47.605 | 0 | 2 | 3 | −5.86162 | −0.90384 | TRUE | NC |
| 7 | 32982 | 3.554 | 94.092 | 0 | 3 | 3 | −4.36961 | 3.38794 | TRUE | NC |
| 8 | 11440 | 2.953 | 56.373 | 0 | 2 | 2 | −0.12488 | 2.59889 | TRUE | NC |
| 9 | 5366 | 3.012 | 74.233 | 0 | 2 | 2 | −2.91479 | −1.55838 | TRUE | NC |
| 10 | 36351 | 3.4 | 39.072 | 0 | 1 | 2 | −1.82962 | −0.96727 | TRUE | NC |
| 11 | 58368 | 3.421 | 91.137 | 0 | 3 | 3 | −5.48094 | −1.44069 | TRUE | NC |
| 12 | 76022 | 2.914 | 73.674 | 0 | 2 | 2 | −5.5384 | 0.322006 | TRUE | NC |
| 13 | 267461 | 1.45 | 102.463 | 0 | 3 | 3 | −9.79909 | −1.10165 | TRUE | NC |
AlogP98, partition coefficient; BBB, blood brain barrier; CYP2D6, cytochrome P450 2D6 binding; HIA, human intestinal absorption; NC, noncarcinogenic; PPB, plasma protein binding; PSA, polar surface area; WOE, weight of evidence.
Table 9.
Analyses of Drug Like Properties of Successfully Screened Compounds from Natural Product Set III of DTP Repository Fulfilling ADMET Criteria, on Basis of Lipinski's Rule of Five
| S No. | Compound ID | H donor | H Acceptor | Molecular weight | AlogP |
|---|---|---|---|---|---|
| 1 | 7668 | 4 | 5 | 304.422 | 2.963 |
| 2 | 26258 | 0 | 6 | 394.417 | 3.93 |
| 3 | 32192 | 0 | 7 | 367.352 | 2.829 |
| 4 | 32979 | 1 | 5 | 341.401 | 3.308 |
| 5 | 5113 | 3 | 6 | 420.454 | 4.784 |
| 6 | 9665 | 1 | 4 | 282.375 | 1.395 |
| 7 | 32982 | 2 | 6 | 368.38 | 3.554 |
| 8 | 11440 | 0 | 6 | 389.829 | 3.335 |
| 9 | 5366 | 0 | 8 | 413.421 | 3.012 |
| 10 | 36351 | 0 | 5 | 339.385 | 3.4 |
| 11 | 58368 | 2 | 8 | 639.862 | 4.217 |
| 12 | 76022 | 1 | 9 | 429.42 | −0.325 |
| 13 | 267461 | 2 | 6 | 302.279 | 1.45 |
Table 10.
Results of Docking of Natural Products Set III Compounds (Selected from the DTP Open Repository Collection) Successfully Passing ADMET and Drug Likeness Parameters
| S. No. | Name and compound ID | Binding Energy (kcal/mol) | Ki (μM) |
|---|---|---|---|
| 1 | Pomiferin (5113) | −7.52 | 3.09 |
| 2 | Curcumin (32982) | −7.40 | 3.79 |
| 3 | Nanaomycin (267461) | −7.38 | 3.86 |
| 4 | Aleuritic acid (7668) | −6.69 | 12.40 |
| 5 | Bicuculline (32192) | −6.42 | 19.84 |
| 6 | Canadine (36351) | −5.40 | 110.60 |
| 7 | Gelcohol (9665) | −5.21 | 151.85 |
| 8 | Isocorydine (32979) | −5.00 | 217.02 |
| 9 | Protopine (11440) | −4.07 | 1040.00 |
| 10 | Rotenone (26258) | −3.55 | 2520.00 |
| 11 | Noscapine (5366) | −3.30 | 3830.00 |
| 12 | Thaspine (76022) | −1.93 | 38400.00 |
| 13 | Fugilin dicyclohexylamine salt (58368) | −1.72 | 54750.00 |
Discussion
In recent years, due to increasing cases of antibiotic resistance in T. pallidum, syphilis has drawn more attention by necessitating the identification of novel and specific drug targets (Stamm, 2010). With the availability of complete genome sequences for the host H. sapiens and pathogen T. pallidum, it has become possible to identify novel and specific drug targets through subtractive genomics/proteomics approaches (Fraser et al., 1998; Matejkova et al., 2008, Michael and Eugene, 1999). Thus, in the present study, a large scale analysis of T. pallidum proteome led to identification of 126 proteins essential for the survival of pathogen. Metabolic pathway analysis led to identification of 6 unique pathways (Table 2) with 19 non-human homologous essential proteins. Molecular modeling analyses of the each of the five target enzymes/proteins, among the identified six unique pathways, serving as potential drug targets, led to emergence of DDLA as the most promising target and salvicine as the most promising inhibitor of DDLA. These enzyme/proteins were selected on the basis of their role in the survival of the pathogen. Many of these targets have been reported as potential drug targets in several other bacteria [e.g, Neisseria gonorrhoeae (Barh and Kumar, 2009), Mycobacterium tuberculosis (Bruning et al., 2010), Staphylococcus aureus (Liu et al., 2006)]. Thus, the enzyme protein UDPN of the peptidoglycan biosynthesis pathway has been analyzed as an antibacterial drug target (Gu et al., 2004; Lovering et al., 2012; Steven, 2002). The DDLA enzyme protein of the D-alanine metabolic pathway has also been used as a potential drug target in M. tuberculosis, S. aureus, Helicobacter pylori, and Escherichia coli (Bruning et al., 2010; Wu et al., 2008). Similarly, the SECA protein of bacterial secretion system has also been analyzed as a drug target in E. coli and S. aureus (Jang et al., 2011; Li et al., 2008). Though the CHER of the two component system and MGLB protein of the bacterial chemotaxis pathways have not yet been reported as potential drug targets, they have been suggested to be the significant ones (Djordjevic and Stock, 1997; Robins and Rotman, 1972). To the best of our knowledge, there are no reports available in literature regarding exploitation of any of the identified unique targets of T. palllidum, from the present study, as potential drug targets.
Salvicine, the most potent inhibitor of DDLA among the screened terpenoids, was found to inhibit DDLA in an allosteric manner by binding to DDLA at a site different to those of substrate (D-alanine) and cofactor (ATP/ADP) binding sites (Fig. 3). Thus, the mode of inhibition of DDLA by salvicine was different from those previously reported inhibitors of DDLA such as phosphinate, phosphonate, and D-cycloserine where these inhibitors have been reported to act as competitive inhibitors for either substrate or cofactor (Liu et al., 2006). In agreement to our finding, Liu, et al., (2006), based on crystallographic analyses, have proposed an allosteric inhibition model for Staphylococcus aureus DDLA by the inhibitor 3-chloro-2,2-dimethyl-N-(4(trifluoromethyl)phenyl) propanamide. Although based on the screening of a natural products database of readily available compounds, pomiferin, a flavonoid was found to be the best, but on comparison, the terpenoid salvicine was found to be an even more efficient inhibitor of DDLA than pomiferin. Thus, based on present studies, the DDLA could be suggested as a promising drug target and the terpenoid, salvicine, as a potent inhibitor for further experimental validation as a drug against syphilis.
Conclusions
Syphilis is still a major concern among STD, especially in view of increasing cases of antibiotic resistance, making a call for the identification and validation of newer and novel putative drug targets in T. pallidum, the causative agent for the disease. With this objective, the present study was initiated. Thus, using a subtractive genomics approach covering comparative analysis of complete genome, proteome, and metabolome of pathogen T. pallidum and the host H. sapiens, nineteen proteins/enzymes, distributed among 6 metabolic pathways unique to the T. pallidum, were identified. Out of these nineteen proteins, five, namely, UDPN, DDLA, SECA, CHER, and MGLB, which have been extensively studied as potent drug targets for a number of pathogenic bacteria but not yet for T. pallidum, were studied in detail by performing molecular modeling analyses with terpenoids as potential anti-syphilis drugs. The molecular modeling analyses revealed DDLA as a putative drug target as a maximum number of terpenoids showed best docking results with DDLA in comparison to other four targets. Along with this, the salvicine was found to be a relatively more potent inhibitor as compared to that of the commonly used synthetic drug D-cycloserine, as well as to that of pomiferin, a flavonoid coming out of the screening of a database of readily available natural compounds. Therefore, DDLA as a promising drug target and salvicine as a potent inhibitor can be considered for further experimental validation for treating syphilis.
Supplementary Material
Acknowledgments
Financial supports from Department of Biotechnology (DBT), New Delhi under Bioinformatics Infrastructure Facility, Department of Higher Education, Government of U.P., Lucknow under Centre of Excellence Grant and Department of Science and Technology, New Delhi under Promotion of University Research and Scientific Excellence (DST-PURSE) program are gratefully acknowledged.
Author Disclosure Statement
No conflict of financial interests exists.
References
- Anishetty S, Pulimi M, and Pennathur G. (2005). Potential drug targets in Mycobacterium tuberculosis through metabolic pathway analysis. Comp Biol Chem 29, 368–378 [DOI] [PubMed] [Google Scholar]
- Balani SK, Mewa GT, Gan LS, Wu JT, and Lee FW. (2005). Strategy of utilizing in vitro and in vivo ADME tools for lead optimization and drug candidate selection. Curr Top Med Chem 5, 1033–1038 [DOI] [PubMed] [Google Scholar]
- Barh D, and Kumar A. (2009). In silico identification of candidate drug and vaccine targets from various pathways in Neisseria gonorrhoeae. In Silico Biol 9, 225–231 [PubMed] [Google Scholar]
- Barh D, Kumar A, and Misra AN. (2009). Genomic Target Database (GTD): A database of potential targets in human pathogenic bacteria, Bioinformation 4, 50–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barh D, Tiwari S, Jain N, et al. (2011). In Silico subtractive genomics for target identification in human bacterial pathogens. Drug Dev Res 72, 162–177 [Google Scholar]
- Borrock MJ, Kiessling LL, and Forest KT. (2007). Conformational changes of glucose/galactose-binding protein illuminated by open, unliganded, and ultra-high-resolution ligand-bound structures. Protein Sci 16, 1032–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruning JB, Murillo AC, Chacon O, Barletta RG, and Sacchettini JC. (2010). Structure of the Mycobacterium tuberculosis D-Alanine:D-Alanine ligase, a target of the antituberculosis drug D-Cycloserine. Antimicrob Agents Chemother 55, 291–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai CZ, Han LY, Ji ZL, Chen X, and Chen YZ. (2003). SVM-Prot: Web-based support vector machine software for functional classification of a protein from its primary sequence. Nucleic Acids Res 31,3692–3697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djorjevic S, and Stock AM. (1997). Crystal structure of the chemotaxis receptor methyltransferase CheR suggests a conserved structural motif for binding S-adenosylmethionine. Structure 5, 545–558 [DOI] [PubMed] [Google Scholar]
- Echenburg S, and Scronbrunn E. (2000). Comparative X-ray analyses of un-liganded fosfomycin-target MurA. Proteins: Struct Funct Genet 40, 290–298 [DOI] [PubMed] [Google Scholar]
- Fraser CM, Norris SJ, Wienstock GM, et al. (1998). Complete genome sequence of Treponema pallidum, the syphilis spirochete. Science 281, 375. [DOI] [PubMed] [Google Scholar]
- Egan WJ, Merz KM, Jr, and Baldwin JJ. (2000). Prediction of drug absorption using multivariate statistics. J Med Chem 43, 3867–3877 [DOI] [PubMed] [Google Scholar]
- Gardy JL, Laird MR, Chen F, et al. (2005). PSORTb v.2.0: Expanded prediction of bacterial protein subcellular localization and insights gained from comparative proteome analysis. Bioinformatics 21, 617–623 [DOI] [PubMed] [Google Scholar]
- Gu YG, Florjancic AS, Clark RF, et al. (2004). Structure-activity relationships of novel potent MurF inhibitors. Bio Med Chem Lett 14, 267–270 [DOI] [PubMed] [Google Scholar]
- Hook EW, III, and Peeling RW. (2004). Syphilis control: A continuing challenge. N Engl J Med 351, 122–124 [DOI] [PubMed] [Google Scholar]
- Ismail H, Arif T, Deena-Al M, et al. (2012). Application of a subtractive genomics approach for in silico identification and characterization of novel drug targets in Mycobacterium tuberculosis F11. Interdiscip Sci Comput Life Sci 6, 48–56 [DOI] [PubMed] [Google Scholar]
- Jang MY, Jonghe SD, Segers K, Anne J, and Herdwijin P. (2010). Synthesis of novel 5-amino-thiazolo(4,5-d)pyrimidines as E. coli and S. Aureus SecA inhibitors. Bioorg Med Chem 19, 702–714 [DOI] [PubMed] [Google Scholar]
- Katz KA, and Klausner D. (2008). Azithromycin resistance in Treponema pallidum. Curr Opin Infec Dis 21, 83–91 [DOI] [PubMed] [Google Scholar]
- Kushwaha SK, and Shakya M. (2010). Protein interaction network analysis—Approach for potential drug target identification in Mycobacterium tuberculosis. J Theor Biol 262, 284–294 [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, and Thornton JM. (1993). PROCHECK: A program to check the steriochemical quality of protein structure. J App Cryst 26, 283–291 [Google Scholar]
- Li M, Huang YJ, Tai PC, and Wang B. (2008). Discovery of the first SecA inhibitors using structure-based virtual screening. Biochem Biophy Res Comm 368, 839–845 [DOI] [PubMed] [Google Scholar]
- Liu S, Chang JS, Herberg JT, et al. (2006). Allosteric inhibition of Staphylococcus aureus D-alanine:D-alanine ligase revealed by crystallographic studies. Proc Natl Acad Sci USA 103,15178–15183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovering AL, Safadi SS, and Strynadka NCJ. (2012). Structural perspective of peptidoglycan biosynthesis and assembly. Annu Rev Biochem 81, 451–478 [DOI] [PubMed] [Google Scholar]
- Marra CM, Colina AP, Godornes C, et al. (2006). Antibiotic selection may contribute to increases in macrolide-resistant Treponema pallidum. J Infect Dis 194, 771–1773 [DOI] [PubMed] [Google Scholar]
- Matejkova P, Strouhal M, Smajs D, et al. (2008). Complete genome sequence of Treponema pallidum ssp. Pallidum strain SS14 determined with oligonucleotide arrays. BMC Microbiol 8, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffin LJ, Bryson K, and Jones DT. (1999). The PSIPRED protein structure prediction server. Bioinformat App Note 16, 404–405 [DOI] [PubMed] [Google Scholar]
- Michael YG, and Eugene VK. (1999). Searching for drug targets in microbial genomes. Curr Opin Biotech 10, 571–578 [DOI] [PubMed] [Google Scholar]
- Mitchell SJ, Engelman J, Kent CK, Lukehart SA, Godornes C, and Klausner JD. (2006). Azithromycin-resistant syphilis infection: San Francisco, California, 2000–2004. Clin Infec Dis 42, 337–345 [DOI] [PubMed] [Google Scholar]
- Moriya Y, Itoh M, Okuda S, Yoshizawa AC, and Kanehisa M. (2007). KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 35, 182–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris GM, Goodsell DS, Halliday RS, et al. (1999). Automated docking using a Lamarkian genetic algorithm and an empirical binding free energy function. J Comput Chem 19, 1639–1662 [Google Scholar]
- Morris GM, Huey R, Lindstrom W, et al. (2009). AutoDock4 and AutoDock tools4: Automated docking with selective receptor flexibility J Comput Chem 30, 2785–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris SJ. and The Treponema pallidum Polypeptide Research Group. (1993). Polypeptides of Treponema pallidum: Progress towards understanding their structural, functional and immunologic roles. Microbiol Rev 57, 750–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris SJ, Charon NW, Cook RG, Fuentes ND. and Limberger RJ. (1988). Antigenic relatedness and N-terminal homology define two classes of periplasmic flagellar proteins of Treponema pallidum subsp. pallidum and Treponema phagedenis. J Bacteriol 170, 4072–4082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne AR, Clemons WM, Jr, and Rapoport TA. (2004). A large conformational change of translocation ATPase SecA. Proc Natl Acad Sci USA 101, 10937–10942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, et al. (2004). UCSF Chimera—A visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- Segall MD, Beresford AP, Gola JM, Hawksley D, and Tarbit MH. (2006). Focus on success: Using a probabilistic approach to achieve an optimal balance of compound properties in drug discovery. Expert Opin Drug Metab Toxicol 2, 325–337 [DOI] [PubMed] [Google Scholar]
- Sharma V, Gupta P, and Dixit A. (2006). In silico identification of putative drug targets from different metabolic pathways of Aeromonas hydrophila. In Silico Biol 8, 331–338 [PubMed] [Google Scholar]
- Smith CA, Kamp M, Olansky S, and Price EV. (1956). Benzathine penicillin G in the treatment of syphilis. Bull World Health Organ 15, 1087–1096 [PMC free article] [PubMed] [Google Scholar]
- Stamm LV. (2010). Global challenge of antibiotic resistant Treponema pallidum. Antimicrob Agents Chemother 54, 583–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steven JP. (2002). New (and not so new) antibacterial targets—From where and when will the novel drugs come. Curr Opin Pharmaco 2, 513–522 [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Fedorova ND, Jackson JD, et al. (2003). The COG database: An updated version includes eukaryotes. BMC Bioinformatics 4, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallner B, and Elofsson A. (2003). Can correct protein models be identified? Protein Sci 12, 1073–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh CT. (1989). Enzymes in the D-alanine branch of bacterial cell wall peptidoglycan assembly. J Biol Chem 264, 393–396 [PubMed] [Google Scholar]
- Wu D, Kong Y, Han C, Chen J, Hu L, Jiang H, and Shen X. (2008). D-alanine:D-alanine ligase as a new target for flavanoids quercetin and apigenin. Int J Antimicrob Agents 32, 421–426 [DOI] [PubMed] [Google Scholar]
- Wu JM, Mao XZ, Cai T, Luo JC, and Wei LP. (2006). KOBAS server: A web-based platform for automated annotation and pathway identification. Nucleic Acids Res 34, W720–W724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C, Mao X, Huang J, et al. (2011) KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res 39, 316–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, and Lin Y. (2009). DEG 5.0 a database of essential genes in both prokaryotes and eukaryotes. Nucleic Acids Res 37, 455–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.