

Abstract
Bacillus azotoformans MEV2011, isolated from soil, is a microaerotolerant obligate denitrifier, which can also produce N2 by co-denitrification. Oxygen is consumed but not growth-supportive. The draft genome has a size of 4.7 Mb and contains key genes for both denitrification and dissimilatory nitrate reduction to ammonium.
Keywords: Bacillus azotoformans, Denitrification, Codenitrification, Oxygen
Introduction
Species of the genus Bacillus are characterized as Gram-positive, facultative aerobic bacteria capable of forming endospores [1]. In the absence of oxygen, many Bacillus species can respire with nitrate instead, employing either dissimilatory nitrate reduction to ammonium or denitrification [2,3]. Despite the widespread occurrence of nitrate-reducing bacilli, their molecular and genetic basis remained poorly investigated [4,5]. Only recently, genome sequencing of two denitrifying type strains, B. azotoformans LMG 9581T and B. bataviensis LMG 21883T, has yielded first insights into the genomic inventory of nitrate reduction and denitrification in Gram-positives [6].
Classification and features
B. azotoformans MEV2011 (Figure 1) was isolated at 28°C on anoxic King B plates [7] amended with KNO3 (5 g L−1) from a highly diluted top soil sample at Aarhus University, Denmark. Strain MEV2011 resembles the type strain in its chemoorganotrophic growth on short-chain fatty acids, complete denitrification, and absence of fermentation [8]. However, it differs from the type strain by its inability to grow with oxygen, even though it can tolerate and consume oxygen at atmospheric concentrations. Growth by denitrification (verified by 15N incubations; data not shown) starts at microaerobic conditions (<30 μM O2; Figure 2), yet the initial presence of oxygen in the growth medium leads to longer lag phases and no increase in final density of the culture (Figure 3); growth without nitrate was never observed. Therefore, we characterize B. azotoformans MEV2011 as microaerotolerant obligate denitrifier. In addition, B. azotoformans MEV2011 is capable of co-denitrification, a co-metabolic process, in which reduced nitrogen compounds like amino acids or hydroxylamine react with NO+ formed during denitrification to produce N2O or N2[9]; co-denitrification was verified by the mass spectrometric detection of 30 N2 + 29 N2 in cultures growing on tryptic soy broth (TSB) and 15NO3−, as suggested in [9]. B. azotoformans MEV2011 is available from the BCCM/LMG Bacteria Collection as strain LMG 28302; its general features are summarized in Table 1.
Figure 1.
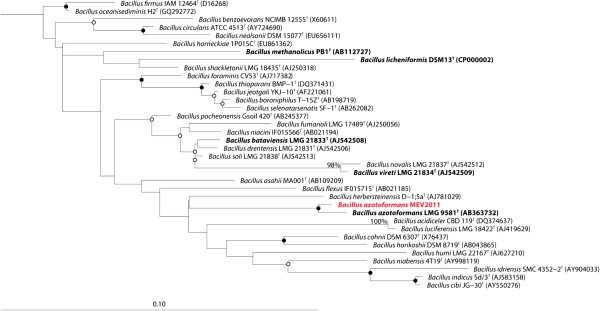
Phylogenetic tree highlighting the position of Bacillus azotoformans MEV2011 (shown in red) relative to closely related (≥95% sequence similarity) type strains within the Bacillaceae. Pre-aligned sequences were retrieved from the Ribosomal Database Project (RDP) [37]. Alignment of the B. azotoformans MEV2011 sequence as well as manual alignment optimization was performed in ARB [38]. The maximum likelihood tree was inferred from 1,478 aligned positions of 16S rRNA gene sequences and calculated based on the General Time Reversible (GTR) model with gamma rate heterogeneity using RAxML 7.4.2 [39]. Type strains with corresponding published genomes are shown in bold face. Open and closed circles indicate nodes with bootstrap support (1,000 replicates ) of 50-80% and >80%, respectively. Escherichia coli ATCC 11577T (X80725) was used to root the tree (not shown). Scale bar, 0.1 substitutions per nucleotide position.
Figure 2.
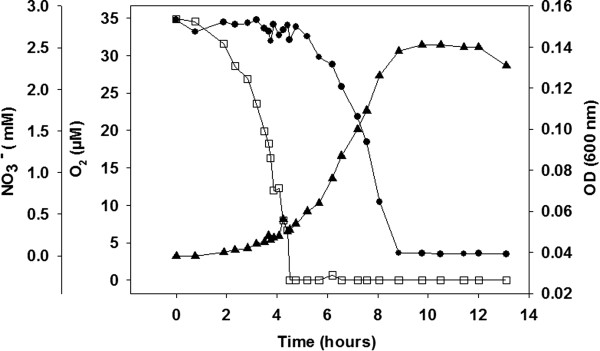
Consumption of oxygen (□; measured online with an oxygen microsensor) and nitrate (●; measured by HPLC) during growth (▲; OD600) of B. azotoformans MEV2011. No growth was observed at oxygen concentrations >30-35 μM, and the initiation of growth coincided with the first detection of 30 N2 from 15NO3− (data not shown), indicating that growth was coupled to denitrification.
Figure 3.
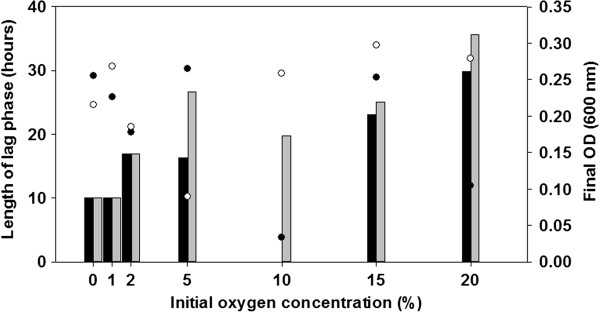
Length of lag phase (h; bars), and final biomass (OD600; circles) of B. azotoformans MEV2011 as function of the initial oxygen concentration in the culture. Cultures were grown in TSB (10 g L−1, Scharlau®) amended with 3 mM KNO3. Black and grey bars and circles represent data from replicate incubations. Growth was first detected when oxygen had been consumed to <30–35 μM (see Figure 2), explaining the increasing lag time with increasing oxygen concentrations. The final OD was almost identical in all incubations and unrelated to the initial oxygen concentration, indicating that oxygen did not contribute to biomass production.
Table 1.
Classification and general features of Bacillus azotoformans MEV2011[27]
| MIGS ID | Property | Term | Evidence code a |
|---|---|---|---|
| |
Classification |
Domain Bacteria |
TAS [28] |
| Phylum Firmicutes |
TAS [29]–[31] |
||
| Class Bacilli |
TAS [32,33] |
||
| Order Bacillales |
TAS [29,34] |
||
| Family Bacillaceae |
TAS [29,35] |
||
| Genus Bacillus |
TAS [29,36] |
||
| Species Bacillus azotoformans |
TAS [8] |
||
| Strain: MEV2011 (LMG 28302) |
IDA |
||
| |
Gram stain |
Variable |
IDA |
| |
Cell shape |
Rod |
IDA |
| |
Motility |
Motile |
IDA |
| |
Sporulation |
Endospore-forming |
IDA |
| |
Temperature range |
15 – 42°C |
IDA |
| |
Optimum temperature |
39 – 42°C |
IDA |
| |
pH range; Optimum |
4–9; 7 |
IDA |
| |
Carbon source |
Malate, acetate, lactate, citrate, succinate, yeast extract |
IDA |
| |
Terminal electron acceptor |
Nitrate, nitrite, NO, N2O (O2 is reduced but does not support growth) |
IDA |
| MIGS-6 |
Habitat |
Soil |
IDA |
| MIGS-6.3 |
Salinity |
0–3% NaCl (w/v) |
IDA |
| MIGS-22 |
Oxygen requirement |
Anaerobic, microaerotolerant |
IDA |
| MIGS-15 |
Biotic relationship |
Free-living |
IDA |
| MIGS-14 |
Pathogenicity |
Non-pathogen |
IDA |
| MIGS-4 |
Geographic location |
Denmark/Aarhus University campus, Aarhus |
IDA |
| MIGS-5 |
Sample collection |
2011-02-01 |
IDA |
| MIGS-4.1 |
Latitude |
56° 10’ 0.12” N |
IDA |
| MIGS-4.2 |
Longitude |
10° 12’ 6.12” E |
IDA |
| MIGS-4.4 | Altitude | 38.6 m | IDA |
aEvidence codes - IDA: Inferred from Direct Assay; TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [10].
Genome sequencing and annotation
Genome project history
Bacillus azotoformans MEV2011 was selected for whole genome sequencing based on its unusual “obligate” denitrifying phenotype, i.e. its inability to grow under oxic conditions, together with its co-denitrifying capacity. Comparing the genome of strain MEV2011 to that of the oxygen-respiring and conventionally denitrifying type strain [8] may provide insights into the molecular basis of its metabolic features. The draft genome sequence was completed on July 20, 2013. The genome project is deposited in the Genomes OnLine Database (GOLD) as project Gp0043190. Raw sequencing reads have been deposited at the NCBI Sequence Read Archive (SRA) under the experiment numbers SRX527325 (100 bp library) and SRX527326 (400 bp library). This Whole Genome Shotgun project has been deposited at GenBank under the accession number JJRY00000000. The version described in this paper is version 1. Table 2 presents the project information and its association with MIGS version 2.0 compliance [27].
Table 2.
Project information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 |
Finishing quality |
High quality draft |
| MIGS-28 |
Libraries used |
IonTorrent 100 bp and 400 bp single end reads |
| MIGS-29 |
Sequencing platforms |
IonTorrent PGM |
| MIGS-31.2 |
Fold coverage |
110× |
| MIGS-30 |
Assemblers |
Newbler 2.6, MIRA 3.9.18, Sequencher 5.0.1 |
| MIGS-32 |
Gene calling method |
Prodigal |
| |
Locus Tag |
M670 |
| |
Genbank ID |
JJRY00000000 |
| |
Genbank Date of Release |
2014-06-16 |
| |
GOLD ID |
Gi0050495 |
| |
BIOPROJECT |
PRJNA209301 |
| |
Project relevance |
Environmental, co-denitrification |
| MIGS 13 | Source Material Identifier | LMG 28302 |
Growth conditions and genomic DNA preparation
B. azotoformans MEV2011 was grown at 28°C in N2-flushed TSB (10 g L−1, Scharlau®) amended with KNO3 (3 g L−1). DNA was extracted using the DNeasy Blood & Tissue kit (Qiagen®).
Genome sequencing and assembly
Sequencing of the B. azotoformans MEV2011 genome was performed with an Ion Torrent PGM sequencer (Life Sciences) using 100 and 400 bp sequencing chemistries. Sequencing libraries were prepared using Ion Xpress™ Plus Fragment Library Kits (Life Sciences), and Ion OneTouch™ Template Kits (Life Sciences). Sequencing of the 100 bp library generated 442,853 reads (representing 42 Mbp of sequence information), while sequencing of the 400 bp library generated 2,401,947 reads (477 Mbp). Together, both libraries achieved a genome coverage of c. 110× for an estimated genome size of 4.7 Mbp. The reads were quality trimmed using the prinseq-lite.pl script [11] with the following parameters; reads generated with 100 bp chemistry: -min_len 50 -trim_to_len 110 -trim_left 15 -trim_qual_right 20 -trim_qual_window 4 -trim_qual_type mean; reads generated with 400 bp chemistry: -min_len 50 -trim_to_len 400 -trim_left 15 -trim_qual_right 20 -trim_qual_window 4 -trim_qual_type mean. The trimmed reads (2,491,456 reads representing 444 Mbp) were assembled using MIRA 3.9.18 [12] with the following parameters: job = genome,denovo,accurate; technology = iontor. In parallel, the reads were also assembled using Newbler 2.6 (Roche) with the following parameters: -mi 96 –ml 50 (i.e. 96% minimum sequence similarity and 50 bp minimum overlap). Contigs shorter than 1,000 bp were removed from both assemblies. All remaining contigs were trimmed by 50 bp from the 5’ and the 3’ ends using the prinseq-lite.pl script in order to remove error-prone contig ends. The two assemblies were merged and manually inspected using Sequencher 5.0.1 (Genecodes). In cases where the bases of the two assemblies disagreed, the Newbler variant was preferred. Contigs not contained in both assemblies were removed from the data set. The final assembly yielded 56 contigs representing 4.7 Mbp of sequence information.
Genome annotation
The draft genome was auto-annotated using the standard operation procedure of the Integrated Microbial Genomes Expert Review (IMG-ER) platform developed by the Joint Genome Institute, Walnut Creek, CA, USA [13]. In short, CRISPR regions were identified by CRT [14] and PILERCR [15], tRNAs were identified by tRNAScan-SE-1.23 [16], rRNAs were identified by RNAmmer [17], and finally all other genes were identified by Prodigal [18]. Functional annotation was based on gene comparisons with the KEGG database (release 63.0, July 1, 2012) [19], the PFAM database (version 25.0, March 30, 2011) [20], the cluster of orthologous groups (COG) [21] database, and the TIGRfam database (release 11.0, August 3, 2011) [22].
Genome properties
The MEV2011 draft genome is 4,703,886 bp long and comprises 56 contigs ranging in size from 1,773 to 525,568 bp, with an overall GC content of 37.49% (Table 3). Of the 4,986 predicted genes, 4,809 (96.45%) are protein-coding genes, and 177 are RNAs. Of the RNAs, 94 are tRNAs, and 37 are rRNAs. The number of 5S rRNAs as well as the number of partial 16S and 23S rRNA genes indicates a total of 11 rRNA operons. Most (75.3%) protein-coding genes were assigned to putative functions. The distribution of genes into COG functional categories is presented in Table 4.
Table 3.
Nucleotide content and gene count levels of the genome
| Attribute | Value | % of total a |
|---|---|---|
| Genome size (bp) |
4,703,886 |
100 |
| DNA coding (bp) |
4,075,859 |
86.7 |
| DNA G + C (bp) |
1,763,498 |
37.5 |
| DNA scaffolds |
56 |
100 |
| Total genes |
4,986 |
100 |
| Protein coding genes |
4,809 |
96.5 |
| RNA genes |
177 |
3.6 |
| Pseudo genes |
0 |
0 |
| Genes in internal clusters |
3,448 |
69.1 |
| Genes with function prediction |
3,755 |
75.3 |
| Genes assigned to COGs |
2,809 |
56.3 |
| Genes with Pfam domains |
3,890 |
78.0 |
| Genes with signal peptides |
182 |
3.7 |
| Genes with transmembrane helices |
1,233 |
24.7 |
| CRISPR repeats | 4 | - |
a)The total is based on either the size of the genome in base pairs or the total number of protein coding genes in the annotated genome.
Table 4.
Number of genes associated with general COG functional categories
| Code | Value | % age a | Description |
|---|---|---|---|
| A |
0 |
0 |
RNA processing and modification |
| J |
160 |
3.32 |
Translation, ribosomal structure and biogenesis |
| K |
246 |
5.11 |
Transcription |
| L |
185 |
3.85 |
Replication, recombination and repair |
| B |
1 |
0.02 |
Chromatin structure and dynamics |
| D |
34 |
0.70 |
Cell cycle control, Cell division, chromosome partitioning |
| V |
45 |
0.94 |
Defense mechanisms |
| T |
207 |
4.30 |
Signal transduction mechanisms |
| M |
123 |
2.56 |
Cell wall/membrane biogenesis |
| N |
77 |
1.60 |
Cell motility |
| U |
50 |
1.04 |
Intracellular trafficking and secretion |
| O |
106 |
2.20 |
Posttranslational modification, protein turnover, chaperones |
| C |
216 |
4.49 |
Energy production and conversion |
| G |
136 |
2.83 |
Carbohydrate transport and metabolism |
| E |
299 |
6.22 |
Amino acid transport and metabolism |
| F |
67 |
1.39 |
Nucleotide transport and metabolism |
| H |
144 |
2.99 |
Coenzyme transport and metabolism |
| I |
135 |
2.81 |
Lipid transport and metabolism |
| P |
173 |
3.60 |
Inorganic ion transport and metabolism |
| Q |
81 |
1.68 |
Secondary metabolites biosynthesis, transport and catabolism |
| R |
350 |
7.28 |
General function prediction only |
| S |
291 |
6.05 |
Function unknown |
| - | 2,177 | 45.27 | Not in COGs |
a)The total is based on the total number of protein coding genes in the annotated genome.
Insights from the genome sequence
Overall, the genome of the novel strain MEV2011 appeared highly similar to that of the B. azotoformans type strain LMG 9581T[8]. In silico DNA–DNA hybridization (DDH) was performed for the assembled MEV2011 genome against the published genome of LMG 9581T (Acc. number NZ_AJLR00000000); the contigs of B. azotoformans LMG 9581T were assembled into one FASTA file before uploading to the online genome-to-genome calculator provided by the DSMZ [23]. Using the GGDC 2.0 model, DHH estimates were always >70%, irrespective of the formula used for computing DHH, and with probabilities between 78 and 87%. These results confirm that MEV2011 is a novel strain of the species B. azotoformans.
Just as B. azotoformans LMG 9581T, strain MEV2011 carries multiple copies of key denitrification genes, encodes both membrane-bound and periplasmic nitrate reductases, and the key genes for nitrite reduction to both NO (in denitrification) and ammonium (in DNRA); see (Additional file 1: Table S1) and reference [6] for details. Modularity and redundancy in nitrate reduction pathways has also been observed in other Bacillus species (e.g. B. bataviensis[6], Bacillus sp. strain ZYK [24], Bacillus sp. strain 1NLA3E [25]), and may be a general feature of nitrate-reducing members of this genus.
All genes essential for aerobic respiration were identified, including those for terminal oxidases (see Additional file 1: Table S2) and for detoxifying reactive oxygen species (see Additional file 1: Table S3). Therefore, the inability of B. azotoformans MEV2011 to grow with oxygen remains a conundrum and in some way resembles that of various sulfate-reducing bacteria, which also consume oxygen and even produce ATP during oxic respiration but are unable to grow in the presence of oxygen [26].
Conclusion
Based on our whole genome comparison, the microaerotolerant obligate (co-) denitrifying Bacillus sp. MEV2011 (LMG 28302) is a novel strain of Bacillus azotoformans, with similar redundancy in its nitrate reduction pathways, including the potential for DNRA, and a complete set of genes for oxic respiration and oxygen detoxification; its inability to grow with oxygen remains enigmatic.
Abbreviations
DNRA: Dissimilatory nitrate reduction to ammonium.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KF and AS designed research, MN isolated and characterized strain MEV2011 and carried out the genome sequencing, LS performed bioinformatics analyses, all authors analyzed data, MN and LS wrote the manuscript with help of AS and KF, all authors read and approved the final manuscript.
Supplementary Material
Overview of the genomic inventory for dissimilatory nitrogen transformations in Bacillus azotoformans MEV2011. Table S2. Overview of the genomic inventory for enzymatic reduction of O2 and ATP synthase in Bacillus azotoformans MEV2011. Table S3. Overview of the genomic inventory for the detoxification of reactive oxygen species in Bacillus azotoformans MEV2011.
Contributor Information
Maja Nielsen, Email: mnimajanielsen@hotmail.com.
Lars Schreiber, Email: lars.schreiber@biology.au.dk.
Kai Finster, Email: Kai.finster@biology.au.dk.
Andreas Schramm, Email: andreas.schramm@biology.au.dk.
Acknowledgements
We thank Anne Stentebjerg and Britta Poulsen for their excellent technical assistance. Eline Palm Hansen is acknowledged for the isolation and initial characterization of strain MEV2011 (together with Maja Nielsen). This work was supported by the Technology Transfer Office (TTO), Aarhus University, Denmark. Funding for the Stellar Astrophysics Centre is provided by The Danish National Research Foundation (Grant agreement no.: DNRF106).
References
- Fritze D. Taxonomy of the genus Bacillus and related genera: the aerobic endospore-forming bacteria. Phytopathology. 2004;94:1245–1248. doi: 10.1094/PHYTO.2004.94.11.1245. [DOI] [PubMed] [Google Scholar]
- Tiedje JM. In: Biology of anaerobic microorganisms. Zehnder AJB, editor. New York, NJ: John Wiley and Sons; 1988. Ecology of denitrification and dissimilatory nitrate reduction to ammonium; pp. 179–244. [Google Scholar]
- Verbaendert I, Boon N, De Vos P, Heylen K. Denitrification is a common feature among members of the genus Bacillus. System Appl Microbiol. 2011;34:385–391. doi: 10.1016/j.syapm.2011.02.003. [DOI] [PubMed] [Google Scholar]
- Jones CM, Welsh A, Throbäck IN, Dörsch P, Bakken LR, Hallin S. Phenotypic and genotypic heterogeneity among closely related soil‒borne N2- and N2O-producing Bacillus isolates harboring the nosZ gene. FEMS Microbiol Ecol. 2011;76:541–552. doi: 10.1111/j.1574-6941.2011.01071.x. [DOI] [PubMed] [Google Scholar]
- Verbaendert I, De Vos P, Boon N, Heylen K. Denitrification in Gram-positive bacteria: an underexplored trait. Biochem Soc Transac. 2011;39:254–258. doi: 10.1042/BST0390254. [DOI] [PubMed] [Google Scholar]
- Heylen K, Keltjens J. Redundancy and modularity in membrane-associated dissimilatory nitrate reduction in Bacillus. Frontiers Microbiol. 2012;3:371. doi: 10.3389/fmicb.2012.00371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King E, Ward MK, Raney DE. Two simple media for the demonstration of pyocyanin and fluorescein. J Laborat Clin Med. 1955;44:301–307. [PubMed] [Google Scholar]
- Pichinoty F, De Barjac H, Mandel M, Asselineau J. Description of Bacillus azotoformans sp. nov. Int J Syst Bac. 1983;33:660–662. doi: 10.1099/00207713-33-3-660. [DOI] [Google Scholar]
- Spott O, Russow R, Stange CF. Formation of hybrid N2O and hybrid N2 due to codenitrification: First review of a barely considered process of microbially mediated N-nitrosation. Soil Biol Biochem. 2011;43:1995–2011. doi: 10.1016/j.soilbio.2011.06.014. [DOI] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevreux B, Wetter T, Suhai S. Computer Science and Biology. Proceedings of the German Conference on Bioinformatics. Hannover, Germany: GCB; 1999. Genome sequence assembly using trace signals and additional sequence information; pp. 45–56. [Google Scholar]
- Markowitz VM, Mavromatis K, Ivanova NN, Chen I-MA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics. 2009;25:2271–2278. doi: 10.1093/bioinformatics/btp393. [DOI] [PubMed] [Google Scholar]
- Bland C, Ramsey T, Sabree F, Lowe M, Brown K, Kyrpides N, Hugenholtz P. CRISPR Recognition Tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics. 2007;8:209. doi: 10.1186/1471-2105-8-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. PILER-CR: Fast and accurate identification of CRISPR repeats. BMC Bioinformatics. 2007;8:18. doi: 10.1186/1471-2105-8-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: A Program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagesen K, Hallin P, Rødland EA, Stærfeldt H-H, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D, Chen G-L, LoCascio P, Land M, Larimer F, Hauser L. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer ELL, Eddy SR, Bateman A, Finn RD. The Pfam protein families database. Nucleic Acids Res. 2004;32(suppl 1):D138–D141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov R, Fedorova N, Jackson J, Jacobs A, Kiryutin B, Koonin E, Krylov D, Mazumder R, Mekhedov S, Nikolskaya A, Rao BS, Smirnov S, Sverdlov A, Vasudevan S, Wolf Y, Yin J, Natale D. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft DH, Selengut JD, White O. The TIGRFAMs database of protein families. Nucleic Acid Res. 2003;31:371–373. doi: 10.1093/nar/gkg128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auch AF, von Jan M, Klenk H-P, Göker M. Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand Genomic Sci. 2010;2:117–134. doi: 10.4056/sigs.531120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao P, Su J, Hu Z, Häggblom M, Zhu Y. Genome sequence of the facultative anaerobic bacterium Bacillus sp. strain ZYK, a selenite and nitrate reducer from paddy soil. Stand Genomic Sci. 2014;9:646–54. doi: 10.4056/sigs.3817480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatramanan R, Prakash O, Woyke T, Chain P, Goodwin LA, Watson D, Brooks S, Kostka JE, Green SJ. Genome sequences for three denitrifying bacterial strains isolated from a uranium- and nitrate-contaminated subsurface environment. Genome Announc. 2013;1(4):e00449–13. doi: 10.1128/genomeA.00449-13. 10.1128/genomeA. 00449-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilling W, Cypionka H. Aerobic respiration in sulfate-reducing bacteria. FEMS Microbiol Letters. 1990;71:123–127. [Google Scholar]
- Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, Ashburner M, Axelrod N, Baldauf S, Ballard S, Boore J, Cochrane G, Cole J, Dawyndt P, De Vos P, dePamphilis C, Edwards R, Faruque N, Feldman R, Gilbert J, Gilna P, Glöckner FO, Goldstein P, Guralnick R, Haft D, Hancock D. The minimum information about a genome sequence (MIGS) specification. Nature Biotechnol. 2008;26:541–547. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skerman VBD, McGowan V, Sneath PHA. Approved Lists of Bacterial Names. Int J Syst Bacteriol. 1980;30:225–420. doi: 10.1099/00207713-30-1-225. [DOI] [PubMed] [Google Scholar]
- Gibbons NE, Murray RGE. Proposals Concerning the Higher Taxa of Bacteria. Int J Syst Bacteriol. 1978;28:1–6. doi: 10.1099/00207713-28-1-1. [DOI] [Google Scholar]
- Murray RGE. In: Bergey's Manual of Systematic Bacteriology, Volume 1. 1. Holt JG, editor. Baltimore: The Williams and Wilkins Co.; 1984. The Higher Taxa, or, a Place for Everything…? pp. 31–34. [Google Scholar]
- List no. 132. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol. 2010;60:469–472. doi: 10.1099/ijs.0.024562-0. [DOI] [PubMed] [Google Scholar]
- Ludwig W, Schleifer KH, Whitman WB. In: Bergey's Manual of Systematic Bacteriology, Volume 3. 2. De Vos P, Garrity G, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer KH, Whitman WB, editor. New York: Springer-Verlag; 2009. Class I. Bacilli class nov; pp. 19–20. [Google Scholar]
- Prévot AR, Hauderoy P, Ehringer G, Guillot G, Magrou J, Prevot AR, Rosset D, Urbain A, editor. Dictionnaire des Bactéries Pathogènes. 2. Paris: Masson et Cie; 1953. p. 1–692. [Google Scholar]
- Fischer A. Untersuchungen über Bakterien. Jahrbücher für Wissenschaftliche Botanik. 1895;27:1–163. [Google Scholar]
- Cohn F. Untersuchungen über Bakterien. Beitr Biol Pflanz. 1872;1:127–224. [Google Scholar]
- Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37(suppl 1):D141–D145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig W, Strunk O, Westram R, Richter L, Meier H, Kumar Y, Buchner A, Lai T, Steppi S, Jobb G, Förster W, Brettske I, Gerber S, Ginhart AW, Gross O, Grumann S, Hermann S, Jost R, König A, Liss T, Lüßmann R, May M, Nonhoff B, Reichel B, Strehlow R, Stamatakis A, Stuckmann N, Vilbig A, Lenke M, Ludwig T. ARB: a software environment for sequence data. Nucleic Acids Res. 2004;32:1363–1371. doi: 10.1093/nar/gkh293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML web servers. System Biol. 2008;57:758–771. doi: 10.1080/10635150802429642. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Overview of the genomic inventory for dissimilatory nitrogen transformations in Bacillus azotoformans MEV2011. Table S2. Overview of the genomic inventory for enzymatic reduction of O2 and ATP synthase in Bacillus azotoformans MEV2011. Table S3. Overview of the genomic inventory for the detoxification of reactive oxygen species in Bacillus azotoformans MEV2011.