

Abstract
Significance: Reactive oxygen and nitrogen species (ROS and RNS, respectively) can intimately control neuronal excitability and synaptic strength by regulating the function of many ion channels. In peripheral sensory neurons, such regulation contributes towards the control of somatosensory processing; therefore, understanding the mechanisms of such regulation is necessary for the development of new therapeutic strategies and for the treatment of sensory dysfunctions, such as chronic pain. Recent Advances: Tremendous progress in deciphering nitric oxide (NO) and ROS signaling in the nervous system has been made in recent decades. This includes the recognition of these molecules as important second messengers and the elucidation of their metabolic pathways and cellular targets. Mounting evidence suggests that these targets include many ion channels which can be directly or indirectly modulated by ROS and NO. However, the mechanisms specific to sensory neurons are still poorly understood. This review will therefore summarize recent findings that highlight the complex nature of the signaling pathways involved in redox/NO regulation of sensory neuron ion channels and excitability; references to redox mechanisms described in other neuron types will be made where necessary. Critical Issues: The complexity and interplay within the redox, NO, and other gasotransmitter modulation of protein function are still largely unresolved. Issues of specificity and intracellular localization of these signaling cascades will also be addressed. Future Directions: Since our understanding of ROS and RNS signaling in sensory neurons is limited, there is a multitude of future directions; one of the most important issues for further study is the establishment of the exact roles that these signaling pathways play in pain processing and the translation of this understanding into new therapeutics. Antioxid. Redox Signal. 22, 486–504.
Introduction
The mammalian peripheral somatosensory system comprises sensory fibers that innervate tissues such as skin, muscles, and viscera. These fibers sense thermal, chemical, tactile, and damage-related (“pain”) stimuli within the periphery and convey this information to the central nervous system (CNS). The fibers themselves are composed of the axons of peripheral sensory neurons whose cell bodies reside within peripheral ganglia (such as dorsal root ganglia [DRG], trigeminal ganglia [TG], or nodose ganglia). The distal ends of these axons form nerve endings within the innervated organs (i.e., skin), whereas the proximal ends synapse in the spinal cord. Peripheral sensory neurons, along with motor neurons, represent some of the longest cells within our bodies, in some cases extending axons beyond 1 m in humans (Fig. 1) and can be even longer in larger mammals. The stimulus-induced generation of action potentials within the peripheral terminals of sensory nerves and the subsequent propagation of these action potentials toward the first synapse in the spinal cord constitute peripheral somatosensory transmission. This process is mediated by the concerted action of sensory fiber plasmalemmal ion channels. The pathological deregulation of these ion channels can disturb somatosensory transmission and can result in pathological pain, which is increasingly viewed as a group of ion channel-related disorders or “channelopathies” (144). Not surprisingly, the regulation and modulation of peripheral sensory neuron ion channels, particularly in relation to pain, is a subject of intense research [see recent reviews (49, 93, 134, 144)]. Reactive oxygen species (ROS), nitric oxide (NO), and other modulators derived from soluble gaseous molecules are increasingly being recognized for their roles as intracellular second messengers and, among others, as potent regulators of ion channel activity [see, e.g., Wang (209)]. The field is, however, still in its early years and the regulation of sensory neuron excitability by ROS, NO, and gasotransmitters is a topic that we still know very little about. The aim of this article is to summarize and categorize current knowledge in this area.
FIG. 1.

Simplified anatomy of the peripheral somatosensory system. (A) Sensory fibers within the peripheral somatosensory system detect thermal, chemical, tactile, and damage-related (nociceptive) stimuli within skin, viscera, muscles, etc., and convey this information to the spinal cord. The fibers are composed of the axons of peripheral sensory neurons whose cell bodies reside within peripheral ganglia (such as DRG, TG, or nodose ganglia). The axons form nerve endings with peripheral organs and synapses in the spinal cord. (B) Peripheral sensory neurons represent some of the longest cells within our bodies. The relative size of a cell body compared with the axon of a DRG neuron innervating hindlimb skin in humans is illustrated; adapted from Devor (43) with permission. DRG, dorsal root ganglia; TG, trigeminal ganglia.
Mechanisms of ROS Generation
Molecular mechanisms of ROS metabolism are well studied in the brain with much less literature focused specifically on the peripheral nervous system. Although such mechanisms may differ between neuron types, the key steps of ROS metabolism are similar. One of the major ROS species in cells is the superoxide anion (O2•−), which is generated by one-electron reduction of O2. In mitochondria, this mainly happens at electron transport chain (ETC) complexes I and III [Fig. 2; reviewed in Murphy (118)], α-ketoglutarate dehydrogenase (175, 199), α-glycerophosphate dehydrogenase (200), and some other ETC sites. Outside of the mitochondria, O2•− can be produced by xanthine oxidase-catalyzed hypoxanthine oxidation (81), plasma membrane NADPH oxidase (87), as well as by the phospholipase A2-dependent cyclo-oxygenases and lipoxygenases (33). The O2•− is highly reactive and can oxidize many moieties; however, it is usually rapidly converted to hydrogen peroxide (H2O2) in a dismutation reaction catalyzed by the enzyme superoxide dismutase [i.e., mitochondrial manganese SOD (MnSOD) or cytosolic copper/zinc SOD (Cu/ZnSOD)] (Fig. 2) (143). H2O2 is also a potent oxidizer and it can be involved in further reactions and produce several other ROS, including the highly reactive hydroxyl radical (•OH) (in a Fenton reaction with Fe2+) (140).
FIG. 2.

Schematic representation of ROS generation in mitochondria. The main sites of ROS generation within mitochondrial ETC are ETC complexes I and III (complexes I to V are represented by gray shapes and labeled accordingly). O2•− is formed by molecular oxygen reacting with the electrons from the ETC. In the mitochondrial matrix, O2•− is converted to H2O2 by MnSOD. H2O2 can then be reduced to H2O and O2 by catalase or through glutathione/thioredoxin reduction pathways. In the intermembrane space of mitochondria, O2•− can be generated at complex III, where it is reduced to H2O2 by CuSOD and ZnSOD. Based on the generalizations provided in Refs. (118, 196). IMM, inner mitochondrial membrane; GR, gluthathione reductase; GS, glutathione synthase; TR, thioredoxin; ETC, electron transport chain; MnSOD, manganese superoxide dismutase; CuSOD, copper superoxide dismutase; ZnSOD, zinc superoxide dismutase; ROS, reactive oxygen species; H2O2, hydrogen peroxide; O2•−, superoxide anion.
Mechanisms of NO Generation
The metabolism of nitrosyl species is tightly linked with oxygen and ROS. Synthesis of NO and l-citrulline by nitric oxide synthases (NOS) occurs via a two-step conversion process involving molecular oxygen (O2), l-arginine and nicotinamide adenine dinucleotide phosphate (NADPH), as co-substrates. The reaction also involves the prosthetic groups calmodulin and haem, with flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN), and tetrahydrobiopterin (BH4) as co-factors (5) (Fig. 3). There are three isoforms of NOS in mammalian systems encoded by separate genes; two of these are termed constitutive enzymes (cNOS), encompassing neuronal NOS (nNOS, encoded by the NOS1 gene) and endothelial NOS (eNOS, encoded by NOS3). These enzymes are activated by a Ca2+/calmodulin complex, which distinguishes them from inducible NOS (iNOS, encoded by NOS2) that is Ca2+ independent and activated by inflammatory stimulation. All three isoforms of NOS can be found in the central and peripheral nervous systems. Cytosolic nNOS is expressed in neurons across a diverse range of locations, including the hippocampus and neocortex; membrane-bound eNOS is expressed in endothelial cells as well as neurons; and iNOS is expressed in astrocytes and microglia. In the peripheral somatosensory system, nNOS was detected in small- and medium-sized DRG neurons of various mammals, and it was colocalized with some nociceptive neuron markers, including calcitonin gene-related peptide (CGRP), substance P, and TRPV1 (24, 108, 116, 141, 177, 193, 203, 227). NOS expression was also reported in small- and medium-sized TG neurons (83, 88, 183).
FIG. 3.

Catalytic action of cNOS. The binding of Ca2+ to calmodulin enables the flow of electrons through the reductase domain of cNOS. NADPH donates an electron (e−) to the reductase domain of cNOS, proceeding via FAD and FMN redox carriers to the oxgyenase domain. The e− interacts with haem iron and BH4 at the active site to catalyze the reaction of O2 with l-arginine, generating NO and l-citrulline as products. Low concentrations of the substrates or co-factors result in the uncoupling of cNOS with the e− passed to O2, resulting in cNOS generating O2•− in addition to NO. The O2•− and NO can readily react and produce ONOO−, a powerful oxidant. The uncoupled pathway leading to nitrosative stress is indicated by the gray arrows. cNOS, constitutive nitric oxide synthase; NADPH, nicotinamide adenine dinucleotide phosphate; FAD, flavin adenine dinucleotide; FMN, flavin mononucleotide; O2, oxygen; NO, nitric oxide; ONOO−, peroxynitrite anion; BH4, tetrahydrobiopterin.
Interplay Between NO and ROS
The reversible binding of Ca2+ to calmodulin is the primary controller of cNOS activity, with increases in free cytosolic Ca2+ initiating the production of NO [reviewed in Schmidt et al. (162)]. Inhibition of cNOS is triggered through negative feedback of NO on the enzyme in a concentration-dependent manner. For cNOS to exclusively produce NO and l-citrulline, high concentrations of the substrates and cofactors should be present to ensure that the dimeric structure of the enzyme remains coupled. Low concentrations of any of the earlier components results in a one-electron transfer from the reductase domain of uncoupled NOS to O2, resulting in the generation of O2•− in addition to NO. NO can then react with O2•− and produce peroxynitrite anion (ONOO−), a powerful oxidant and cytotoxic agent (215). At low concentrations, ONOO− easily isomerizes into the harmless nitrate ion; however when produced at high concentrations, ONOO− may undergo symmetrical or non-symmetrical cleavage and form nitrogen dioxide (•NO2), carbonate (CO3•−), and •OH radicals, all of which are considered more reactive and toxic than NO (Fig. 4) (109). Within membranes, •NO2 is also formed from auto-oxidation of NO (101). These downstream products are more aggressive oxidants than the parent molecules, with the potential ability to induce extensive inflammation and neuronal death.
FIG. 4.

Some isomerization and cleavage pathways of protonated ONOO−. Nitrosative stress-associated pathways are indicated by the bold gray arrows.
ROS and the Peripheral Somatosensory System
Increased ROS generation is implicated in several pathological conditions related to pain; these include inflammation [i.e., chronic pancreatitis (190)] and diabetes mellitus. Diabetes is often accompanied by the peripheral neuropathies (diabetic neuropathy), and oxidative stress is a widely accepted key factor in its development (53). The incidence of neuropathy in diabetic patients can be very high (up to 50%); the major feature of this syndrome is sensory and motor fiber degeneration, with longer axons being the most affected. Diabetic neuropathy is an incapacitating condition accompanied by pain, foot ulceration, digestive abnormalities, sensory loss, erectile dysfunction, and heart arrhythmias among other conditions. In both type 1 and type 2 diabetes, there is significant accumulation of extracellular glucose, which is transported into the cytosol of cells faster than endogenous metabolic pathways can accommodate it (167). Under these conditions, glucose enters an alternative metabolic pathway where it is converted to sorbitol by the enzyme aldose reductase (112). This reaction requires NADPH, which, in turn, becomes less available for the production of the intracellular antioxidant glutathione from glutathione disulfide by the NADPH-dependent enzyme glutathione reductase. Reduced glutathione levels, in turn, make cells highly vulnerable to ROS (112, 201). Hyperglycemia may cause redox disturbances in a number of additional ways. Glucose can undergo auto-oxidation in the presence of trace amounts of free transition metals, yielding α-ketoaldehydes, H2O2 and ROS (213). Thus, reduced antioxidant capacity in combination with increased ROS production creates a cellular background for oxidative stress. As extensively described in excellent recent reviews [i.e., Refs. (112, 201)], oxidative stress and ROS are largely responsible for the neurodegeneration underlying diabetic neuropathy. Importantly, it has long been noted that the longer axons suffer the most (which is probably to do with the metabolic challenges associated with long axons).
NO and the Peripheral Somatosensory System
NO is a molecular mediator with diverse physiological roles, including vasodilation, blood clotting, and nociception. It is released from arteries, following mechanical stimuli or from dural mast cells or nerve fibers, after inflammation, contributing to pain (17). In the central and peripheral nervous systems, NO displays many properties of a neurotransmitter (55). As a gasotransmitter, NO is diffusible across membranes and its physiological effects are relatively short lived, being oxidized to nitrite in the time frame of seconds (110). NO-mediated signaling pathways include the activation of soluble guanylate cyclase (GC) and the cyclic guanosine monophosphate (cGMP)/protein kinase G (PKG) pathway (25), direct S-nitrosylation of protein thiols (addition of a nitrosyl ion NO− to generate a nitrosothiol, RS-N=O) (95) and protein tyrosine nitration (addition of NO2 to generate 3-nitrotyrosine) (16, 124). Each of these pathways has been linked to pathology of one form or another and diversion toward one of these pathways over another depends on a multitude of factors in the system, including the presence of transition metal complexes and redox status (195). With such varied and wide-ranging effects, understanding the individual signaling pathways of NO is of extreme importance, though relatively little is known about the molecular players involved in these processes. NO has been implicated in migraine, as it was reported to induce CGRP release from trigeminal afferents and CGRP levels are increased during a migraine attack in humans (51, 58). In animal models, migraines can be induced by NO donors and reduced with CGRP antagonists (85, 210) or trigeminal denervation (210). Trigeminal models show that NO increases the release of CGRP from afferent fibers (113), which is not caused by increases in CGRP mRNA (50) but which could be the result of increased neuronal activity. An injection of NO donors into the periphery is also painful and in rat paw causes hyperaemia via release of the vasodilatory CGRP from afferent nerve fibers and up-regulation of prostaglandin production (68). However, antinociceptive effects of NO have also been suggested; see “Katp channels” section and also see (134) for review.
The contribution of the individual NOS isoforms to inflammatory pain has been elucidated using isoform-specific knockout mice. In mice deficient for nNOS, thermal hyperalgesia, after an injection of complete Freund's adjuvant (CFA) was halved, mechanical hypersensitivity was absent and CFA-induced increases in CGRP in DRG neurons were reduced (21). These data suggest that nNOS plays a significant role in sensitization of DRG neurons in inflammatory pain. However, both iNOS and eNOS also contribute (21). In DRG neurons, nNOS is expressed in early developmental stages but is down-regulated with age (141, 194); both nNOS and iNOS expression was reported to be up-regulated in DRG after injury and inflammation (1, 60, 103, 104). Increased NO synthesis in microglia and astrocytes in combination with the properties that enable retrograde messenger activity has implicated astrocytes and microglia as an external source of pathogenic NO (179). Tissue inflammation thus likely leads to increases in all three isoforms (nNOS, iNOS, and eNOS) with vasodilation also increasing blood flow. Candidate mediators of this effect are the P2 purinergic receptors, as the P2 receptor antagonist pyridoxalphosphate-6-azophenyl-2′,4′-disulphonic acid (PPADS) decreased face-rubbing activity, NOS isoform expression in TG, and Fos expression in the spinal trigeminal nucleus after a subcutaneous injection of formalin (22).
An important aspect of sensory signaling by NO has recently been identified in a study that focused on the production of NO in the skin. TRPV3, a heat-sensitive ion channel (169), is expressed in keratinocytes and on activation, induces NO production (115). This is an interesting finding, as a number of the NO-modulated ion channels described next are also expressed in skin cells, including Kv7 (146), TRPV1 (20), and TRPA1 (11) channels. Characterization of the precise details of the signaling pathways between the skin and the peripheral nervous system will provide important contributions to our understanding of the role of NO and ROS in sensory processing.
While our understanding of NO signaling in the peripheral somatosensory system is still “work in progress,” roles of NO in the CNS are much better understood; several recent reviews cover these roles in processes as diverse as synaptic plasticity (26, 64, 67), excitotoxicity (29), or neurodegeneration (179) and we direct readers interested in such effects to these fine papers.
Physiological Relevance and Considerations for Future Studies
The cellular functions of NO are complex and are highly dependent on NO concentration. Broadly speaking, high concentrations of NO are cytotoxic, while low concentrations are cytoprotective. The complexities of measuring NO levels, coupled with the fact that NO freely diffuses across membranes in three-dimensional space, has made it difficult to quantify the levels that accumulate during physiological signalling in vivo. In fact, the use of the word “accumulate” may well be misleading in this context, as the presence of NO as a signaling molecule is likely highly transient. This has led to controversy and difficulties within the field, as astoundingly different physiological concentrations of NO have been reported. We also need to consider the precise biological context, with regard to both normal and pathological conditions. In this way, levels measured via various means (using electrode measurements, fluorescent probes, biosensors, and selectivity of NO for GC-coupled NO receptors) and by different groups have been reported from micromolar to picomolar concentrations [reviewed in Hall and Garthwaite (63)]. Certainly high concentrations of NO (potentially micromolar levels) can be released from inflammatory cells after their activation, while levels released from endothelial cells and neurons have been harder to measure accurately. New findings suggest that picomolar concentrations of NO can now be measured. In fact, it would appear that cells are able to detect exceedingly low concentrations of NO, even down to the picomolar level and even when the exposure is brief (e.g., subsecond range) (214).
In addition, the function of NO is tightly coupled to oxidative stress (see, e.g., Figs. 3 and 4), which makes it often difficult to separate pure NO effects from these that are more complex and require secondary mechanisms (e.g., ROS). Thus, it is still unclear whether NO is able to bind Cys directly at detectable levels or whether Cys nitrosylation requires an additional oxidative mechanism to occur (54). Certainly the biochemical tests (the “biotin switch assay” being the most prominently used method) suggest that in many proteins, cysteines are indeed nitrosylated but we will need to turn to more precise techniques in the future to confirm this.
Many of the same considerations are also true for ROS, whose precise concentrations in cells are difficult to measure, and thus the biological relevance of many of the proposed ROS mechanisms remains in question (181). Consequently, tonic intracellular H2O2 concentrations in mammalian cells were reported to vary in the range of 1–700 nM (181) and H2O2 production can increase in ischemic brain by more than 10-fold (174), suggesting that an even greater range of concentrations may potentially be achievable in vivo. Clearly, caution has to be executed when designing (and interpreting) in vitro experiments with extracellular sources of highly concentrated ROS or NO donors.
Regulation of Sensory Neuron Ion Channels by ROS and NO
M channels
Among a plethora of ion channels expressed in somatosensory (and particularly nociceptive) neurons, one subfamily is gaining increased recognition for its central role in setting and controlling background excitability, a subfamily of voltage-gated K+ channels called KCNQ (Kv7 or “M channels”). These K+ channels conduct slow, non-inactivating “delayed-rectifier” K+ currents with a threshold for activation well below −60 mV (can be as low as −80 mV) (70). These properties enable some M channels to remain active at the resting membrane potential of a sensory neuron (∼−60 mV) (6, 91, 92, 160). Since M channels have strong outwardly rectifying voltage dependence, their “background” activity represents an “intrinsic voltage-clamp” mechanism that controls the resting membrane potential, threshold for action potential (AP) firing, and accommodation within trains of AP [reviewed in Refs. (41, 49)]. Functional M channels are expressed in a variety of locations, including DRG neuron cell bodies (80, 130, 228), nerve fibers (42, 80, 129, 154), dorsal roots/central terminals (149), and nociceptive nerve endings (129). As expected from their biophysical properties, M channel activity strongly affects afferent fiber excitability both in vitro and in vivo. Thus, M channel inhibition increases and M channel enhancers decrease action potential firing in vitro [e.g., in cultured DRG neurons (91, 92, 98, 117, 130) or in skin-nerve preparations (129)]. Accordingly, a hind paw injection of the M channel blocker XE991 in rats induces moderate pain (94, 98), while peripheral injections of M channel pharmacological enhancers, such as retigabine and flupirtine, produce analgesic effects (98, 152).
In recent studies, we have found that four (Kv7.2–Kv7.5) out of five M channel subunits possess a reactive cluster of three successive cysteines in the S2–S3 linker (equivalent to the positions C156, C157, and C158 in Kv7.4; Fig. 5), which can be oxidized by physiologically relevant concentrations of H2O2 (56, 91) or S-nitrosylated by NO donors (126). Oxidation results in potent augmentation of M channel activity, while nitrosylation results in channel inhibition (Fig. 5). With regard to the H2O2 effect, we found that Kv7.2, 7.4, and 7.5 are potently augmented by extracellular H2O2 concentration as low as 5 μM. Kv7.1 and Kv7.3 are largely insensitive to H2O2 despite the fact that Kv7.3 (but not Kv7.1) possesses the conserved triple-cysteine cluster which is necessary for H2O2-induced augmentation of Kv7.2, Kv7.4, and Kv7.5 currents. The H2O2 induced a leftward shift in channel voltage dependence and induced a prominent increase in channel open probability (Po) at all voltages. Kv7 channels have highly divergent tonic maximal Po values (that is Po at saturating voltages under “control” conditions); thus, Kv7.2, 7.4 and Kv7.5 have a rather low Po in the range of 0.1–0.2, whereas Kv7.3 has a maximal Po near unity (90, 165), which is likely to explain the fact that H2O2 did not augment Kv7.3 current (as measured at saturating voltage of 0 mV). Kv7.1 differs from the rest of the family in that it has only one cysteine residue within the “triple C” region; however, reconstitution of the complete CCC sequence in Kv7.1 by substitution of R86 and S87 with cysteines bestowed some H2O2 sensitivity to this mutant, further supporting the conclusion that this triplet of cysteines mediates the redox modulation of Kv7 channels. Interestingly, it seems that oxidation of individual cysteines within the triple-C cluster has a cumulative effect on M channel activity; thus, substitution of all three cysteines was required to completely abolish channel sensitivity to H2O2, while mutation of one or even two cysteines resulted in partial reduction (but never a complete loss) of H2O2 sensitivity (56). Cysteine residues can be reversibly oxidized to cysteine sulfenic acid (Cys-SOH); this derivative can be further (irreversibly) oxidized to sulfinic acid (Cys-SO2H) and sulfonic acid (Cys-SO3H); alternatively two juxtaposed Cys-SOH can react and form intra- or intermolecular disulfide bonds with another protein or low-molecular-weight thiol such as glutathione (128). We were unable to detect the formation of intermolecular disulfide bonds specific to the triple cysteine region under non-reducing PAGE. Given the fact that the H2O2 effect on Kv7 channel activity is completely reversed by the reducing agent dithiothreitol (DTT) (56), while Cys-SO2H and Cys-SO3H modifications cannot be reduced by DTT (135), our working hypothesis is that cumulative oxidation of cysteines to cysteine sulfenic acids within the triple-C cluster mediates oxidative augmentation of M-current (Fig. 5A). Interestingly, in further experiments using patch-clamp electrophysiology and the biotin-switch technique, we found that the same triplet of cysteines can be directly S-nitrosylated in the presence of the NO donor S-nitroso-N-acetyl-DL-penicillamine (SNAP), an effect that leads to a marked decrease of M channel activity (126). This finding raises an interesting possibility that M channels, in fact, may serve a role of cellular sensors conveying intracellular ROS and NO levels into the electrical excitability of neuronal membranes. Indeed, small-diameter DRG and TG neurons respond to NO donors with (i) M channel inhibition; (ii) increased excitability; and (iii) increased CGRP release (126). Conversely, H2O2 and endogenous ROS mediate the opposite effect, that is, (i) enhancement of M channel activity; (ii) moderate hyperpolarization in small DRG neurons; and (iii) inhibition of CGRP release (91). Moreover, these anti-excitatory effects were also observed in response to endogenous O2•− release from mitochondria stimulated in small DRG neurons by substance P (91). Thus, redox/NO-sensitivity of M channels may indeed play vital regulatory and signaling roles within the somatosensory system and particularly within nociceptive fibers. However, this role of a membrane integrator of redox/NO status of the cytosol is extremely complex. NO is generated by eNOS and nNOS in the trigeminovascular system (142) and by immune cells during an inflammatory response, via cytokine-induced activation of iNOS (38), while many pathways involved in inflammatory signaling also inhibit M-current (61, 74, 75, 93, 94, 98, 224, 225). Gq-protein-coupled receptors, such as the bradykinin B2 receptor, evoke an intracellular signaling cascade that stimulates cleavage of phosphatidylinositol 4,5-bisphosphate (PIP2) into diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3), triggering Ca2+ release from IP3-sensitive stores. Both PIP2 depletion and Ca2+ release inhibit M-current directly. As previously shown, NO synthases are calmodulin dependent and elevated Ca2+ triggers NO production from nNOS by activating calmodulin (2); while NOS activity is inhibited by PIP2 (218). In this way, Gq-PCR inflammatory signaling would be able to increase NO levels by two means, the removal of PIP2-mediated inhibition of NOS and increases in Ca2+ activating NOS. This suggests that inflammatory Gq-PCR signaling and NO may work in concert to ensure robust inhibition of M-current. In addition to these effects, NO can react rapidly with endogenous O2•− and form a strong neurotoxic oxidant, ONOO−, a major cytotoxic agent produced during inflammation, sepsis, and ischemia/reperfusion (15, 95). Formation of ONOO− by O2•− and scavenging of NO leads to the inhibition of mitochondrial respiration and SOD activity, potentially establishing a feed-forward loop for ONOO− formation and oxidative modification of M channels (although the direct effect of ONOO− on M channel activity has not been demonstrated thus far). This provides for a potential difference in the cellular effects of NO, depending on redox state, by which ONOO− formation augments M-current, decreasing excitability but NO inhibits M-current, increasing excitability. As a gasotransmitter, NO and its effects are short lived and highly regionally specific, features that underlie a possibility for location- and temporal-specific regulation of ion channels. Thus, at precise loci, the redox environment will determine the ultimate effect of NO release on M-current.
FIG. 5.

Modulation of M-type K+ channels by ROS and NO. (A) Schematic showing the domain structure and location of triplets of reactive cysteines within M channel subunits Kv7.2–7.5. Putative chemical modifications of the cysteines within the triplet are indicated with “SOH” denoting cysteine sulfenic acid formation and “SNO” denoting cysteine S-nitrosylaton. (B) Modification by ROS increases M-current, which leads to the inhibition of neuronal excitability (green); whereas NO-mediated modification suppresses M channel activity, leading to increased excitability (red).
KATP channels
ATP-sensitive K+ channels (KATP) are heteromeric channels formed by Kir6.1 or Kir6.2 subunits of inward-rectifier K+ channels and regulatory sulfonylurea receptor subunits SUR1 or SUR2 (65). Physiologically, KATP channels are inhibited by intracellular ATP and activated by cytosolic ATP depletion, thus serving the role of cellular metabolic sensors. KATP channels have been suggested to play a neuroprotective role in parts of the CNS in some pathological conditions, such as ischemia (216). In the peripheral nervous system, KATP channels (i.e., Kir6.2, SUR1, and SUR2) are expressed in a subpopulation of DRG neurons of various sizes (229). KATP currents in small DRG neurons are relatively small, but KATP channel enhancers (diazoxide and pinacidil) hyperpolarize nociceptive neurons and reduce bradykinin-induced pain (48). Similar to M channels, KATP channels are augmented by endogenous H2O2, as has been shown in dorsal striatal neurons (12), and the resulting hyperpolarization of neurons has been suggested as a mechanism of neuroprotective silencing in hypoxia (198). The molecular mechanism of the KATP channel sensitivity to ROS remains to be established; thus far, it has been reported that the effect is probably direct and is mediated by decreasing channel sensitivity to ATP (71); SUR1-containing KATP channels are more sensitive to H2O2 (147). It is not yet tested whether ROS sensitivity of KATP channels plays any role in sensory neurons.
KATP channels are also sensitive to NO; however, in contrast to M channels, NO, similar to ROS, activate KATP channels in DRG neurons (as shown using excised-patch electrophysiology); the effect is possibly mediated via the direct S-nitrosylation of the SUR1 subunit of the channel complex (77). However, an indirect activation of KATP channels in DRG neurons by NO acting via the classical NO/sGC/cGMP/PKG pathway has also been suggested in other studies (170, 171) (Fig. 6). After peripheral nerve injury, the nociceptive effects of NO are exacerbated, potentially due to inflammation and associated increases in neuronal excitability. The NO donor SNAP caused activation of KATP channels, resulting in decreased neuronal excitation of large DRG neurons after axotomy (77). Peripheral injections of NO donors showed antihyperalgesic effects in some studies (although pro-algesic effects were also demonstrated in others and these were suggested to be mediated by KATP channel opening via the NO/sGC/cGMP/PKG pathway (156).
FIG. 6.

Putative mechanisms of regulation of KATP channels by NO. NBD, nucleotide binding domain; NEM, N-ethylmaleimide; DTT, dithiothreitol; SNAP, S-nitroso-N-acetyl-DL-penicillamine; GC, guanylate cyclase; cGMP, cyclic guanosine monophosphate; PKG, protein kinase G. Based on the data from Refs. (77, 122, 134).
As per examples given earlier, it appears that peripheral somatosensory actions of NO can be either pro-nociceptive or antinociceptive depending on the experimental conditions and models used [for review see Refs. (40, 134)]. For instance, a dual action of locally applied NO donors on tactile allodynia induced by surgical incision in rats has been noted (136); lower concentrations of SNAP reduced allodynia in a GC-dependent manner, while higher concentrations enhanced allodynia, independently of GC. Since NO has opposing effects on M channels and KATP channels, it is conceivable that at least some pro-algesic effects can be mediated by M channel inhibition, while analgesic effects can potentially be mediated by KATP activation. In such a scenario, the exact outcome of the NO effect would depend on the relative expression and activity of M channels and KATP channels in the affected neurons and the spatial correlation of the NO release events with the locality of these channels within the neuronal plasma membrane. It should be noted that KATP channels appear to be slightly more sensitive to NO as compared with M channels; the activating effect of SNAP on KATP channels reached saturation at 100 μM (77), while for M channels the SNAP IC50 has been determined at 370 μM (126).
BK channels
Large-conductance Ca2+-activated K+ channels (Slo1, BK, and Maxi-K) are expressed in DRG neurons of various sizes as demonstrated with correlative patch clamp analysis and RT-PCR (89, 163); the auxiliary subunit β2 was suggested to participate in the BK channel complex in DRG (89, 163). The major functions of BK channels in sensory neurons were suggested to include shortening of the action potential, acceleration of repolarization, and fast after-hyperpolarization (89, 163). These effects may both limit neuronal excitability/output or, somewhat paradoxically, increase it as fast repolarization and more prominent after hyperpolarization may result in an increased firing rate due to improved recovery of voltage-gated Na+ channels (VGNC) from the inactivation (82). The BK channel protein contains multiple cysteine and methionine residues that potentially can be oxidized to modulate channel function (i.e., human Slo1 protein AAB65837 has 29 cysteines and 30 methionines) and BK channels are well established as O2 sensors, responsive to gasotransmitter modulation (78, 157). The general mechanisms of redox modulation of BK channels were recently reviewed in depth in an excellent review (157); therefore, here, we only briefly consider some key facts. The presence of multiple cysteines and methionines within Slo1 proteins results in a complex response to oxidizers and cysteine-modifying reagents. Thus, heterologousely expressed Slo1 channels are inhibited by H2O2 and activated by DTT (45), presumably via cysteine-oxidation mechanisms. The application of various cysteine-modifying agents such as 2-(trimethylammonium)ethyl methanethiosulfonate (MTSET), (2-sulfonatoethyl)methanethiosulfonate (MTSES), or N-ethylmaleimide (NEM) to Slo1 changed channel voltage- and Ca2+ dependence of steady-state activation. Moreover these agents, while acting on the same cysteines, had different effects on channel function. For instance, MTSET increased Slo1 K+ conductance and shifted voltage dependence to more negative voltages, while MTSES and NEM had the opposite effects (226). There is also evidence that methionine oxidation and cysteine oxidation may have opposing effects on channel activity (191). Thus, redox modulation of BK channels is incredibly complex and the outcome of such modulation is likely to depend on the nature of the redox changes and local environment. The significance of redox modulation of BK channels for sensory neurons has not yet been elucidated.
Literature on the regulation of BK channels by NO is more coherent, as there is a broad agreement that in neurons and smooth muscle cells, BK channel activity can be augmented via the classical NO/sGC/cGMP/PKG pathway (10, 82, 102, 164, 217), by which the activation of PKG by cGMP leads to phosphorylation of the Slo1 channel protein at three specific PKG phosphorylation sites, an effect that results in enhancement of channel activity (86).
While redox/NO modulation of BK channels can be complex, there is possibly a pattern for reciprocal effects; however, in contrast to M channels, ROS, such as H2O2, are rather inhibitory, while NO enhances channel activity. It would be interesting to investigate how (and if ) the redox/NO modulation of BK channels affects peripheral somatosensory transmission.
A-type K+ channels
Somatosensory neurons express robust A currents (IKA, inactivating voltage-gated K+ current) that can be further pharmacologically and electrophysiologically separated into two or three subtypes which are distinguishable by different inactivation kinetics (i.e., slow- and fast-inactivating IKA) and sensitivity to tetraethylammonium (TEA) (3, 52, 59, 111, 173, 219); see Du and Gamper (49) for review. The IKA with slower inactivation kinetics is predominantly found in small-diameter nociceptors that also express TTX-resistant Na+channels and TRPV1 (59, 223). IKA is conducted by Kvs, several members of which are expressed in sensory neurons. Thus, the expression of several members of the Kv1.1 channel family has been reported with Kv1.1, Kv1.2, and Kv1.4 being the most abundantly expressed subunits (79, 145, 204, 219). Kv1.1 and Kv1.2 are predominantly found in non-nociceptive, large-diameter DRG neurons. In small-diameter nociceptive DRG neurons, the predominant Kv1 α-subunit is Kv1.4 (145, 204), a rapidly inactivating A-type K+ channel (37). Inactivating subunits of Kv3 (Kv3.4) and Kv4 (Kv4.3) channel families are also expressed in sensory neurons and contribute to IKA, particularly in nociceptors (32).
Several A-type forming Kv subunits have been found to be sensitive to ROS. Thus, inactivation of Kv1.4 and Kv3.4 is slowed by oxidation, an effect reversed by reducing agents (155). Cysteine C13 was found to be responsible for this effect in Kv1.4 (155) [a detailed discussion on the regulation of Kv channel inactivation by ROS can be found in Sahoo et al. (157)]. Interestingly, the modulation of native IKA by ROS has been reported in small-diameter DRG neurons (69), suggesting that the redox sensitivity of Kv channels may have a physiological role in peripheral somatosensory processing. Thus, the oxidizing agents DTBNP and chloramine-T reduced fast inactivation of IKA in cultured small DRG neurons, while the reducing agent DTT reversed this effect (69). If the fast inactivation of IKA being reduced by oxidation, the underlying Kv channels would become less susceptible for cumulative inactivation during repetitive firing, which, in turn, may affect firing rate and thus modulate the sensitivity of nociceptive fibers. It, however, should be noted that in the study (69), the IKA has been pharmacologically isolated in DRG neurons by the addition of 5 mM TEA to inhibit the delayed-rectifier K+ channel current. However, at this concentration, TEA would only partially block M channels; only Kv7.2 is sensitive to TEA in a low millimolar range, while other M channel subunits expressed in DRG, Kv7.3 and Kv7.5 are poorly TEA insensitive (62, 166, 221). The classical M channel, which is a Kv7.2/Kv7.3 heteromer, has a TEA IC50 in the range of 5 mM (62, 166, 208, 221) and, thus, will only be 50% blocked at this concentration. M channels have slow kinetics and as discussed earlier are potently augmented by oxidizing agents [and these effects are reversed by DTT (56, 91, 92)]. Therefore, the increase in steady-state K+ current on application of oxidizing agents that was interpreted as reduction of inactivation (69) is also consistent with an increased contribution of M-current to the total K+ current recorded. Clearly, further research is needed to elucidate the exact molecular mechanisms of sensory neuron K+ current modulation by redox signaling.
T-type Ca2+ channels
T-type Ca2+ channels (encoded by the CACNA1G, CACNA1H, and CACNA1I genes, which code for Cav3.1–3.3 channel α-subunits, respectively) are expressed in a subpopulation of DRG neurons of various types [from small/medium-diameter nociceptors to some large-diameter, mechanosensitive neurons (39, 73, 153)]. These channels differ from other voltage-gated Ca2+ channels (VGCCs) in several important features; thus, they are activated at voltages below −60 mV (hence, they are also termed “low-voltage activated” Ca2+ channels [LVA]); T-type channels activate and inactivate rapidly and have a smaller single-channel conductance than high-voltage-activated VGCCs (30, 72, 133). In the CNS, T-type channels are responsible for pacemaker activity and low threshold spikes; they also may display a significant window current (i.e., they can be tonically active) at membrane potentials close to the resting membrane potential and consequently can contribute to tonic Ca2+ influx (30, 72, 133). In DRG neurons, Cav3.2 is the predominantly expressed T-type channel subunit (168, 189). It is hypothesized that in sensory fibers, T-type channels can participate in setting the threshold for action potential firing and control burst firing (119, 121, 211). It has been shown that in nociceptive neurons, T-type current amplitudes can be augmented by reducing agents such as DTT or l-cysteine and inhibited by the oxidizing agent 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) (197). In accord with its role in nociception, plantar injections of DTT or l-cysteine into rat hind paws induced thermal and mechanical hyperalgesia (197). These effects were prevented by the T-type inhibitor mibefradil; moreover, a peripheral injection of DTNB had an anti-nociceptive effect. Currents mediated by Cav3.2 are also strongly inhibited by ascorbate in a mechanism that is unique to Cav3.2. Ascorbate is best known for its “antioxidant” properties. However, ascorbate can catalyze the formation of several ROS, including •OH, O2•−, and H2O2 by reducing Fe3+ and Cu2+ to Fe2+ and Cu+ and by reducing molecular oxygen (ascorbate-metal ion catalyzed oxidation [MCO]) (172). It has been suggested that ascorbate can inhibit Cav3.2 (but not the other T-type α subunits) by an MCO of a unique metal-binding histidine residue, His191 (in human Cav3.2) in the extracellular S3–S4 linker of domain I (120). The same histidine residue apparently mediates augmentation of Cav3.2 currents by l-cysteine (121). l-cysteine was shown to prevent tonic channel inhibition by endogenous zinc ions.
Carbon monoxide (CO) is increasingly being recognized as a potent intracellular second messenger or gasotransmitter [for review see Peers and Steele (131)]. A recent study reported CO-mediated inhibition of Cav3.2 in DRG neurons, an effect which was based on a redox mechanism distinct from that involving the MCO of His191 (23). CO is produced in cells by the heme oxygenase (HO) enzyme, which degrades heme and produces biliverdin, free iron (Fe2+), and CO. Growing evidence suggests that HOs can modulate nociception; thus, HO-1 is induced in DRG neurons in response to heat stress (132) or axonal damage (107). Accordingly, local endogenous CO has been suggested to suppress inflammatory hyperalgesia in the carrageenan model of pain (178). Moreover, the chemical induction of HO-1 inhibited the nociceptive response to formalin, while the transcriptional inhibition of HO-1 production prevented this effect (151). Boycott et al. (23) have shown that heterologousely expressed Cav3.1, Cav3.2, and Cav3.3 as well as native T-type current in cultured DRG neurons is potently inhibited by the CO donor CORM-2, an effect that was significantly reversed by DTT. CO sensitivity of Cav3.2 (but not the other two subunits) was mediated by an extracellular redox-sensitive site, which was also highly sensitive to thioredoxin (TR) but was distinct from His191. It has been suggested that TR acts as a tonic, endogenous regulator of Cav3.2 channels; while HO-1-derived CO disrupts this regulation, causing channel inhibition. Taken together, these studies emphasize the importance of T-type channels and their redox sensitivity to peripheral somatosensory (and particularly nociceptive) transmission.
TRP channels
Transient receptor potential (TRP) channels are universal cellular sensors that are capable of responding to versatile external stimuli such as temperature, pH, and changes in the chemical milieu. Several TRP channels are preferentially expressed in peripheral somatosensory neurons where these mediate neuronal responses to the environment. Probably the best studied sensory TRP channels within peripheral sensory nerves are TRPV1 and TRPA1 [for a review, see e.g., Vay et al. (202)]. TRPV1 is a member of the vanilloid subfamily of TRP channels; it is expressed in subpopulations of all the major types of nociceptive neurons (nociceptive Aδ fibers, peptidergic and non-peptidergic C-fibers). TRPV1 is a versatile sensor that is activated by a wide range of stimuli such as capsaicin, noxious heat, protons (acidification), endocannabinoids, ethanol, camphor, allicin, 2-APB, lidocaine, ω-3 fatty acids, and many others. The mechanisms of TRPV1 activation and its role in thermosensation and pain have been extensively reviewed [e.g., Refs. (31, 44, 202)]; therefore, here, we will only consider mechanisms relevant to the topic of this article. TRPV1 (together with several other TRP channels, for example, TRPC1, TRPC4, TRPC5, TRPV3, and TRPV4) is activated by NO in a process that is likely mediated by direct S-nitrosylation of cysteines (222). Likewise, TRPV1 is also activated by H2O2. In TRPC5, the action of these cysteine-modifying agents was localized to C553 and C558 within the extracellular S5–S6 linker (Fig. 7A); TRPV1 also has two cysteines in this region (C616 and C621), accordingly it was suggested that TRPV1 is regulated by the same mechanism as the substitution of these two residues reduced sensitivity to H2O2 and the cysteine-modifying reagent 5-nitro-2-PDS (222). Surprisingly, the sensitization of heat- and capsaicin sensitivity of TRPV1 by both oxidizing (diamide, H2O2 and chloramine-T) and reducing (DTT and reduced glutathione) agents has also been reported (186, 205). Interestingly, in the latter study, the effect of the reducing agents was linked to the same extracellular cysteines C616 and C621 (with the additional C634 that was also considered important for the effect in this study); while the sensitizing effect of the oxidizing compounds was found to be dependent on intracellularly facing residues. Chuang and Lin have identified the C-terminal cysteines C772 and C783 as being necessary for the sensitizing action of oxidizing agents (36). TRPV1 can also be activated by pungent compounds from onion and garlic, such as allicin (106). The action of allicin has been localized to a single cysteine C157 within the TRPV1 N-terminus (159). This is a physiologically relevant mechanism, as it underlies the responses of sensory neurons that express TRPV1 (and TRPA1, which is also sensitive to allicin via a similar mechanism) to these pungent stimuli. While the precise mechanics of redox/NO regulation of TRPV1 are still emerging, it is clear that the channel is very sensitive to such modulation and that multiple mechanisms exist. Since tissue damage and inflammation are linked to ROS and RNS production, it is likely that redox/NO modulation of TRPV1 has a prominent role in inflammatory pain mechanisms.
FIG. 7.
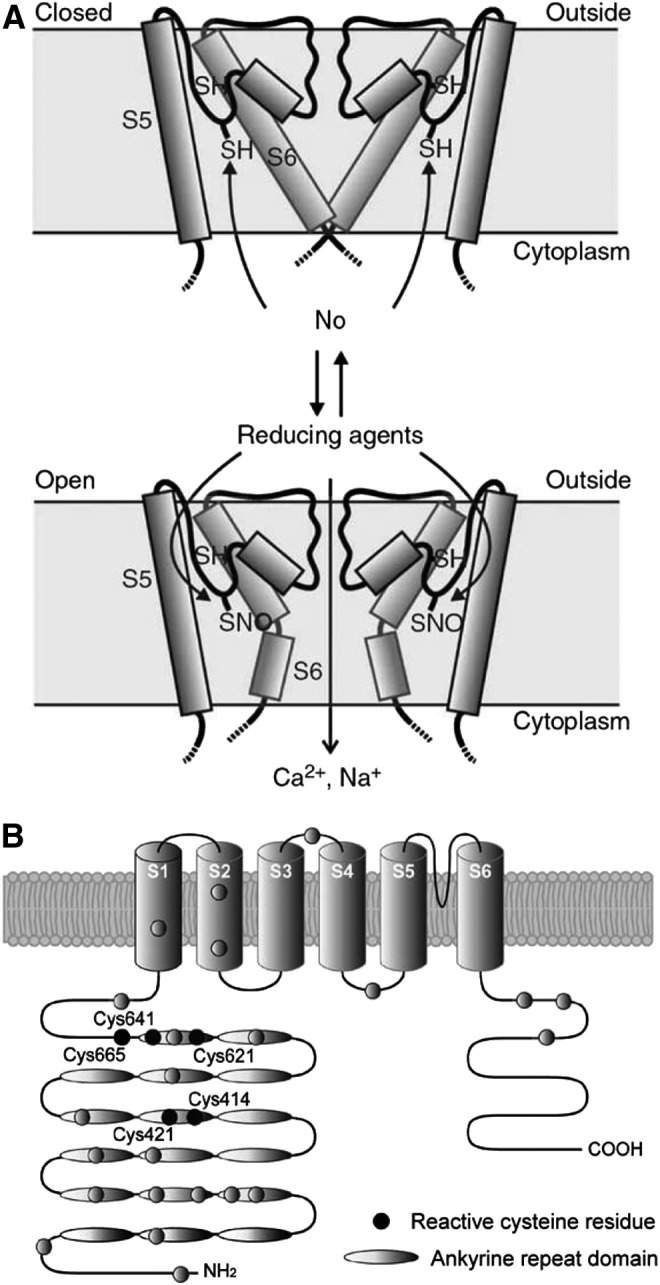
Modulation of TRP channels by ROS and nitrosylation. (A) Model for TRP channel activation by NO and reactive disulfides (based on TRPC5 and proposed to be applicable to other NO-sensitive TRP channels, including TRPV1). Adapted from Yoshida et al. (222) with permission. (B) Reactive cysteine residues (black circles) of human TRPA1; adapted from Takahashi and Mori (188) with permission. TRP, transient receptor potential.
TRPA1 is the only member of the ankyrin subfamily of TRP channels in mammals; similar to TRPV1, TRPA1 is also highly expressed in peripheral somatosensory fibers, particularly in those that are activated by both noxious cold and a wide range of environmental irritants (13, 19, 182). Similar to TRPV1, TRPA1 is a “promiscuous” sensor that is activated by a variety of stimuli, including temperatures below 15°C, cinnamaldehyde, endocannabinoids, acrolein, allicin, icilin, gingerol chlorine, ROS, nicotine, and many others [for review see Refs. (14, 123, 202)]. Some of the compounds that activate TRPV1 can also activate TRPA1; moreover, both channels are often co-expressed in the same sensory neurons and possibly interact physically (4, 158, 176). Similar to the case of TRPV1, both NO donors (SNAP, NOR3) and ROS (H2O2) activate TRPA1 (both the native channel in sensory neurons and heterologously overexpressed TRPA1) (7, 18, 114, 161, 187). Again, similar to TRPV1, a common theme in the action of ROS, NO, and irritants is multiple modifications of channel cysteines. Thus, two groups have identified five cysteines (all in the channel N-terminus) to be responsible for TRPA1 activation by irritants but only one of them (C622 in mouse TRPA1) was identified by both groups (66, 105). Covalent modification of lysine 708 (in human TRPA1) by isothiocyanates was also reported to contribute to the activation of TRPA1 (66). In another study, C421, C641, and C665 of human TRPA1 (all within the N-terminus) were found to be mostly responsible for channel TRPA1 activation by NO donors and H2O2 (187); positions of the reactive cysteines in the human TRPA1 N-terminus are indicated in Figure 7B. Thus, while having similar functional outcomes, the sensitivity of the TRPA1 channel to redox/NO modulation may be underwritten by somewhat different structural mechanisms as compared with TRPV1 in which both the N-terminus and the pore region were implicated. Modulation of TRPA1 via modification of reactive cysteines has obvious physiological importance, as it underlies sensory responses to airborne irritants (13, 18) and is likely to contribute to disorders such as asthma (27) and chronic itch (97, 212).
Other channels
The number of sensory ion channels being identified as NO- and ROS-regulated is growing rapidly, indicating the importance of these signaling molecules in controlling excitability and pain processing. In DRG neurons, acid-sensing ion channels are potentiated by NO, and NO donors increased the pain of acid injections into the skin in humans (28). Potentiation of VGNC by endogenous ROS in small- and medium-sized DRG neurons has been recently suggested, as VGNC current density decreased after application of ROS scavengers (207). In addition, another study reported that in vitro hyperglycemia produced both increased ROS production and augmented VGNC currents in cultured DRG neurons. These are, however, indirect evidence and the sensitivity of VGNC to ROS needs to be tested further. Peripheral sensory neurons use glutamate as their main neurotransmitter; interestingly, DRG neurons also abundantly express somatic N-methyl-d-aspartate (NMDA) receptors (84), and there is evidence that there is also extrasynaptic (i.e., somatic) glutamate release in DRG (84). While the function of extrasynaptic glutamatergic circuitry in DRG remains unknown, a recent report somewhat paradoxically demonstrated that DRG-specific genetic deletion of the NR1 subunit of NMDA receptors in mice resulted in DRG hyperexcitability and pain hypersensitivity (127). NMDA receptors represent another ion channel type which is robustly modulated by NO and ROS through mechanisms that are relatively well understood [see Lipton et al. (96) for review]. Thus, six cysteine residues (Fig. 8) are involved in redox modulation of the channel by oxidizing agents, such as DTNB, inhibiting NMDA receptor current while reducing agents such as DTT or dihydrolipoic acid increase channel activity (34, 35, 127, 184). Four of these six cysteines are located within NR1 subunit (C79, C308, C744, and C798), while the final pair is in the NR2A subunit (C87 and C320). Five cysteines can be directly S-nitrosylated: C744 and C798 of NR1, and C87, C320, and C399 of NR2A (35). S-nitrosylation of all these residues is inhibitory. Four residues (C744, C798 of NR1, C87, and C320 of NR2A) are susceptible to both redox and NO modulation (Fig. 8). While it is not yet clear whether redox/NO modulation of NMDA receptors plays any role in peripheral somatosensory transduction, the fact that genetic deletion of NR1 specifically in peripheral somatosensory neurons results in distinct pain hypersensitivity suggests that this is likely to be the case.
FIG. 8.

Cysteines involved in redox/NO modulation of NMDA receptors. Residues involved in redox and NO modulation are shown within the darker and lighter gray circles, respectively; residues that were implicated in both types of modulation are shown within the overlap of two circles. Based on data from Refs. (34, 35, 127, 184). NMDA, N-methyl-d-aspartate.
Role for Lipids in Redox and NO Modulatory Pathways
When considering the regulation of channel proteins by ROS and NO, the contribution of modifications to the lipid membrane in which these channels are located should not be overlooked. Many of the channels mentioned earlier display a complex relationship not only with ROS and NO but also with the molecular composition of the lipid bilayer. Oxidative stress is the physiological state in which the formation of ROS is favored and oxidative damage to a cell or tissue can subsequently result. A large proportion of ROS are free radicals that possess one unpaired electron, giving them a high degree of reactivity. These species are produced during normal cellular metabolism, particularly cellular respiration that occurs in the mitochondria. Cells have a protective defence system made up of enzymatic and non-enzymatic antioxidants that decrease levels of ROS on their generation (see, e.g., Fig. 2). Consequently cells exist in a state of equilibrium with a fine balance between the production and sequestration of ROS (47). Neurons have an inherent susceptibility to oxidative stress for a number of reasons, including a deficiency in antioxidants compared with other tissues of the body, meaning a reduced ability to defend against ROS attack (9). In addition, there is a greater production of ROS in neurons due to their high rate of metabolic activity and a high content of transition metals that are actively redistributed within the cell (46). Finally, the brain is composed of a high proportion of polyunsaturated fatty acids (PUFA), which are susceptible to free radical attack, causing changes in membrane permeability, fluidity, and function (148).
Higher concentrations of cellular ROS arise due to dysregulation of cellular H2O2, NO, and •OH processing. The increase in cellular ROS is a self-perpetuating mechanism, as ROS damage lipids, which, in turn, also become radical species. Lipid peroxidation occurs via a ROS mediated bis-allylic reaction. The ROS removes a hydrogen atom from the carbon adjacent to the double bond of PUFA, generating species that are highly reactive electrophilic aldehydes (206). This lipid peroxidation impairs the normal function of the cell as lipid radicals and ROS are highly reactive and thus induce lipid–lipid and lipid–protein cross-linking, which leads to an increase in membrane rigidity, a decrease in membrane enzyme activity, as well as alterations to membrane permeability and ion channel activity (8, 185, 220).
Phospholipids, sphingolipids, and sterols are known to regulate a number of channels, including M channels (192), inwardly rectifying Kir channels (150), TRPV1 (100, 138), and TRPM8 (99). These lipid species are particularly susceptible to peroxidative damage due to higher levels of PUFAs, which are thought to be targeted by oxidative radicals in disease (137). Lipid rafts are membrane microdomains that are rich in cholesterol and sphingolipids, which further control ion channel distribution and function. These lipid rafts control the function of a number of ion channels in sensory neurons, including M channels (125), Ca2+-activated Cl− channel ANO1/TMEM16A (76) purinergic P2 receptors (57), and voltage-gated sodium channels (139). These papers show that the manipulation of cholesterol levels within the membrane has profound effects on ion channel function. Therefore, we can consider lipids as playing an important role in signaling by ROS and NO. However, due to the inherent difficulties in manipulating levels of individual lipid species in sensory neurons, their precise contribution to changes in ion channel activity by ROS and RNS remains to be determined.
Concluding Remarks
The picture being assembled in the regulation of ion channels by ROS and RNS is one of extraordinary complexity. These species are able to regulate channels by direct modification of amino acid residues, affecting signaling pathways and/or altering redox state. ROS and RNS have interconnected modes of action and important physiological roles. However, the exact mechanisms of ROS- and RNS-mediated regulation of peripheral sensory neuron excitability are, at best, fragmented and incomplete. We know that many ion channels which are expressed in sensory fibers are sensitive to ROS and RNS, and we also know that these reactive species are being generated at increased levels in many pain states (i.e., in inflammation, injury, etc.); nevertheless, a consistent and inclusive picture is yet to be produced. Obviously, intensive future research is needed; moreover, these future studies will need to consider the system, as a whole, rather than individual reactions and responses, in order to decipher the most important effects. Evidently, a focus on ROS and NO signaling in sensory processing is crucial not only for understanding such processing (particularly in the disease states where these transmitters are implicated) but also for the development of future therapeutics.
Abbreviations Used
- ANO1
anoctamin, member 1
- BK
big conductance K+
- BH4
tetrahydrobiopterin
- Cav, VGCC
voltage-gated Ca2+ channel
- CFA
complete Freund's adjuvant
- cGMP
cyclic guanosine monophosphate
- CGRP
calcitonin gene-related peptide
- cNOS
constitutive NOS
- CNS
central nervous system
- CO
carbon monoxide
- CO3•−
carbonate
- CORM-2
tricarbonyldichlororuthenium(II) dimer
- CuSOD
copper superoxide dismutase
- DAG
diacylglycerol
- DRG
dorsal root ganglia
- DTNB
5,5′-dithiobis-(2-nitrobenzoic acid)
- DTT
dithiothreitol
- eNOS
endothelial NOS
- ETC
electron transport chain
- FAD
flavin adenine dinucleotide
- FMN
flavin mononucleotide
- GC
guanylate cyclase
- GR
gluthathione reductase
- GS
glutathione synthase
- H2O2
hydrogen peroxide
- HO
heme oxygenase
- iNOS
inducible NOS
- IP3
inositol 1,4,5-trisphosphate
- Kv
voltage-gated K+ channel
- LVA
low-voltage activated Ca2+ channels
- MCO
ascorbate-metal ion catalyzed oxidation
- MnSOD
manganese superoxide dismutase
- MTSES
(2-sulfonatoethyl)methanethiosulfonate
- MTSET
2-(trimethylammonium)ethyl methanethiosulfonate
- NEM
N-ethylmaleimide
- NMDA
N-methyl-d-aspartate
- nNOS
neuronal NOS
- NO
nitric oxide
- •NO2
nitrogen dioxide
- NOS
nitric oxide synthase
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PKG
protein kinase G
- PPADS
pyridoxalphosphate-6-azophenyl-2′,4′-disulphonic acid
- PUFA
polyunsaturated fatty acids
- O2•−
superoxide anion
- •OH
hydroxyl radical
- ONOO−
peroxynitrite anion
- ROS
reactive oxygen species
- RNS
reactive nitrogen species
- SNAP
S-nitroso-N-acetyl-DL-penicillamine
- SOD
superoxide dismutase
- TEA
tetraethylammonium
- TG
trigeminal ganglia
- TR
thioredoxin
- TRP
transient receptor potential
- TRPA1
transient receptor potential cation channel, subfamily A, member 1
- TRPC
transient receptor potential cation channel, canonical
- TRPM8
transient receptor potential cation channel subfamily M member 8
- TRPV1
transient receptor potential cation channel, subfamily V, member 1
- VGNC
voltage-gated Na+ channels
- ZnSOD
zinc superoxide dismutase
Acknowledgments
The work that has led up to this review has been supported by MRC, BBSRC, and the Wellcome Trust.
References
- 1.Abe S, Mizusawa I, Kanno K, Yabashi A, Suto M, Kuraya M, Honda T, and Hiraiwa K. Nitric oxide synthase expressions in rat dorsal root ganglion after a hind limb tourniquet. Neuroreport 14: 2267–2270, 2003 [DOI] [PubMed] [Google Scholar]
- 2.Abu-Soud HM. and Stuehr DJ. Nitric oxide synthases reveal a role for calmodulin in controlling electron transfer. Proc Natl Acad Sci U S A 90: 10769–10772, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akins PT. and McCleskey EW. Characterization of potassium currents in adult rat sensory neurons and modulation by opioids and cyclic AMP. Neuroscience 56: 759–769, 1993 [DOI] [PubMed] [Google Scholar]
- 4.Akopian AN. Regulation of nociceptive transmission at the periphery via TRPA1-TRPV1 interactions. Curr Pharm Biotechnol 12: 89–94, 2011 [DOI] [PubMed] [Google Scholar]
- 5.Alderton WK, Cooper CE, and Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J 357: 593–615, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amir R. and Devor M. Spike-evoked suppression and burst patterning in dorsal root ganglion neurons of the rat. J Physiol 501: 183–196, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andersson DA, Gentry C, Moss S, and Bevan S. Transient receptor potential A1 is a sensory receptor for multiple products of oxidative stress. J Neurosci 28: 2485–2494, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anzai K, Ogawa K, Goto Y, Senzaki Y, Ozawa T, and Yamamoto H. Oxidation-dependent changes in the stability and permeability of lipid bilayers. Antioxid Redox Signal 1: 339–347, 1999 [DOI] [PubMed] [Google Scholar]
- 9.Aoyama K, Suh SW, Hamby AM, Liu J, Chan WY, Chen Y, and Swanson RA. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat Neurosci 9: 119–126, 2006 [DOI] [PubMed] [Google Scholar]
- 10.Archer SL, Huang JM, Hampl V, Nelson DP, Shultz PJ, and Weir EK. Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proc Natl Acad Sci U S A 91: 7583–7587, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Atoyan R, Shander D, and Botchkareva NV. Non-neuronal expression of transient receptor potential type A1 (TRPA1) in human skin. J Invest Dermatol 129: 2312–2315, 2009 [DOI] [PubMed] [Google Scholar]
- 12.Avshalumov MV. and Rice ME. Activation of ATP-sensitive K+ (KATP) channels by H2O2 underlies glutamate-dependent inhibition of striatal dopamine release. Proc Natl Acad Sci U S A 100: 11729–11734, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bautista DM, Jordt SE, Nikai T, Tsuruda PR, Read AJ, Poblete J, Yamoah EN, Basbaum AI, and Julius D. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 124: 1269–1282, 2006 [DOI] [PubMed] [Google Scholar]
- 14.Bautista DM, Pellegrino M, and Tsunozaki M. TRPA1: A gatekeeper for inflammation. Annu Rev Physiol 75: 181–200, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beckman JS, Beckman TW, Chen J, Marshall PA, and Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A 87: 1620–1624, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beckman JS, Ischiropoulos H, Zhu L, van der Woerd M, Smith C, Chen J, Harrison J, Martin JC, and Tsai M. Kinetics of superoxide dismutase- and iron-catalyzed nitration of phenolics by peroxynitrite. Arch Biochem Biophys 298: 438–445, 1992 [DOI] [PubMed] [Google Scholar]
- 17.Berger RJ, Zuccarello M, and Keller JT. Nitric oxide synthase immunoreactivity in the rat dura mater. Neuroreport 5: 519–521, 1994 [DOI] [PubMed] [Google Scholar]
- 18.Bessac BF, Sivula M, von Hehn CA, Caceres AI, Escalera J, and Jordt SE. Transient receptor potential ankyrin 1 antagonists block the noxious effects of toxic industrial isocyanates and tear gases. FASEB J 23: 1102–1114, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bessac BF, Sivula M, von Hehn CA, Escalera J, Cohn L, and Jordt SE. TRPA1 is a major oxidant sensor in murine airway sensory neurons. J Clin Invest 118: 1899–1910, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bodo E, Kovacs I, Telek A, Varga A, Paus R, Kovacs L, and Biro T. Vanilloid receptor-1 (VR1) is widely expressed on various epithelial and mesenchymal cell types of human skin. J Invest Dermatol 123: 410–413, 2004 [DOI] [PubMed] [Google Scholar]
- 21.Boettger MK, Üceyler N, Zelenka M, Schmitt A, Reif A, Chen Y, and Sommer C. Differences in inflammatory pain in nNOS-, iNOS- and eNOS-deficient mice. Eur J Pain 11: 810–818, 2007 [DOI] [PubMed] [Google Scholar]
- 22.Borsani E, Albertini R, Labanca M, Lonati C, Rezzani R, and Rodella LF. Peripheral purinergic receptor modulation influences the trigeminal ganglia nitroxidergic system in an experimental murine model of inflammatory orofacial pain. J Neurosci Res 88: 2715–2726, 2010 [DOI] [PubMed] [Google Scholar]
- 23.Boycott HE, Dallas ML, Elies J, Pettinger L, Boyle JP, Scragg JL, Gamper N, and Peers C. Carbon monoxide inhibition of Cav3.2 T-type Ca2+ channels reveals tonic modulation by thioredoxin. FASEB J 27: 3395–3407, 2013 [DOI] [PubMed] [Google Scholar]
- 24.Bredt DS, Glatt CE, Hwang PM, Fotuhi M, Dawson TM, and Snyder SH. Nitric oxide synthase protein and mRNA are discretely localized in neuronal populations of the mammalian CNS together with NADPH diaphorase. Neuron 7: 615–624, 1991 [DOI] [PubMed] [Google Scholar]
- 25.Bredt DS. and Snyder SH. Nitric oxide mediates glutamate-linked enhancement of cGMP levels in the cerebellum. Proc Natl Acad Sci U S A 86: 9030–9033, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brenman JE. and Bredt DS. Synaptic signaling by nitric oxide. Curr Opin Neurobiol 7: 374–378, 1997 [DOI] [PubMed] [Google Scholar]
- 27.Caceres AI, Brackmann M, Elia MD, Bessac BF, del Camino D, D'Amours M, Witek JS, Fanger CM, Chong JA, Hayward NJ, Homer RJ, Cohn L, Huang X, Moran MM, and Jordt SE. A sensory neuronal ion channel essential for airway inflammation and hyperreactivity in asthma. Proc Natl Acad Sci U S A 106: 9099–9104, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cadiou H, Studer M, Jones NG, Smith ES, Ballard A, McMahon SB, and McNaughton PA. Modulation of acid-sensing ion channel activity by nitric oxide. J Neurosci 27: 13251–13260, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, and Stella AM. Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci 8: 766–775, 2007 [DOI] [PubMed] [Google Scholar]
- 30.Carbone E. and Lux HD. A low voltage-activated, fully inactivating Ca channel in vertebrate sensory neurones. Nature 310: 501–502, 1984 [DOI] [PubMed] [Google Scholar]
- 31.Caterina MJ. and Julius D. The vanilloid receptor: a molecular gateway to the pain pathway. Annu Rev Neurosci 24: 487–517, 2001 [DOI] [PubMed] [Google Scholar]
- 32.Chien LY, Cheng JK, Chu D, Cheng CF, and Tsaur ML. Reduced expression of A-type potassium channels in primary sensory neurons induces mechanical hypersensitivity. J Neurosci 27: 9855–9865, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cho KJ, Seo JM, and Kim JH. Bioactive lipoxygenase metabolites stimulation of NADPH oxidases and reactive oxygen species. Mol Cells 32: 1–5, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi Y, Chen HV, and Lipton SA. Three pairs of cysteine residues mediate both redox and zn2+modulation of the nmda receptor. J Neurosci 21: 392–400, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi YB, Tenneti L, Le DA, Ortiz J, Bai G, Chen HS, and Lipton SA. Molecular basis of NMDA receptor-coupled ion channel modulation by S-nitrosylation. Nat Neurosci 3: 15–21, 2000 [DOI] [PubMed] [Google Scholar]
- 36.Chuang HH. and Lin S. Oxidative challenges sensitize the capsaicin receptor by covalent cysteine modification. Proc Natl Acad Sci U S A 106: 20097–20102, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coetzee WA, Amarillo Y, Chiu J, Chow A, Lau D, McCormack T, Moreno H, Nadal MS, Ozaita A, Pountney D, Saganich M, Vega-Saenz de Miera E, and Rudy B. Molecular diversity of K+ channels. Ann N Y Acad Sci 868: 233–285, 1999 [DOI] [PubMed] [Google Scholar]
- 38.Corbett JA, Kwon G, Turk J, and McDaniel ML. IL-1 beta induces the coexpression of both nitric oxide synthase and cyclooxygenase by islets of Langerhans: activation of cyclooxygenase by nitric oxide. Biochemistry 32: 13767–13770, 1993 [DOI] [PubMed] [Google Scholar]
- 39.Coste B, Crest M, and Delmas P. Pharmacological dissection and distribution of NaN/Nav1.9, T-type Ca2+ currents, and mechanically activated cation currents in different populations of DRG neurons. J Gen Physiol 129: 57–77, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cury Y, Picolo G, Gutierrez VP, and Ferreira SH. Pain and analgesia: the dual effect of nitric oxide in the nociceptive system. Nitric Oxide 25: 243–254, 2011 [DOI] [PubMed] [Google Scholar]
- 41.Delmas P. and Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci 6: 850–862, 2005 [DOI] [PubMed] [Google Scholar]
- 42.Devaux JJ, Kleopa KA, Cooper EC, and Scherer SS. KCNQ2 is a nodal K+ channel. J Neurosci 24: 1236–1244, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Devor M. Unexplained peculiarities of the dorsal root ganglion. Pain Suppl 6: S27–S35, 1999 [DOI] [PubMed] [Google Scholar]
- 44.Dhaka A, Viswanath V, and Patapoutian A. TRP ion channels and temperature sensation. Annu Rev Neurosci 29: 135–161, 2006 [DOI] [PubMed] [Google Scholar]
- 45.DiChiara TJ. and Reinhart PH. Redox modulation of hslo Ca2+-activated K+ channels. J Neurosci 17: 4942–4955, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dodani SC, Domaille DW, Nam CI, Miller EW, Finney LA, Vogt S, and Chang CJ. Calcium-dependent copper redistributions in neuronal cells revealed by a fluorescent copper sensor and X-ray fluorescence microscopy. Proc Natl Acad Sci U S A 108: 5980–5985, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Droge W. Free radicals in the physiological control of cell function. Physiol Rev 82: 47–95, 2002 [DOI] [PubMed] [Google Scholar]
- 48.Du X, Wang C, and Zhang H. Activation of ATP-sensitive potassium channels antagonize nociceptive behavior and hyperexcitability of DRG neurons from rats. Mol Pain 7: 35, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Du X. and Gamper N. Potassium channels in peripheral pain pathways: expression, function and therapeutic potential. Curr Neuropharmacol 11: 621–640, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eberhardt M, Neeb L, Vogel E-M, Tiegs G, Reuter U, Messlinger K, and Fischer MJM. Glyceroltrinitrate facilitates stimulated CGRP release but not gene expression of CGRP or its receptor components in rat trigeminal ganglia. Neuropeptides 43: 483–489, 2009 [DOI] [PubMed] [Google Scholar]
- 51.Edvinsson L. and Goadsby PJ. Extracerebral manifestations in migraine. A peptidergic involvement? J Intern Med 228: 299–304, 1990 [DOI] [PubMed] [Google Scholar]
- 52.Everill B, Rizzo MA, and Kocsis JD. Morphologically identified cutaneous afferent DRG neurons express three different potassium currents in varying proportions. J Neurophysiol 79: 1814–1824, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fernyhough P, Roy Chowdhury SK, and Schmidt RE. Mitochondrial stress and the pathogenesis of diabetic neuropathy. Expert Rev Endocrinol Metab 5: 39–49, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Forstermann U. Nitric oxide and oxidative stress in vascular disease. Pflugers Arch 459: 923–939, 2010 [DOI] [PubMed] [Google Scholar]
- 55.Gally JA, Montague PR, Reeke GN, Jr., and Edelman GM. The NO hypothesis: possible effects of a short-lived, rapidly diffusible signal in the development and function of the nervous system. Proc Natl Acad Sci U S A 87: 3547–3551, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gamper N, Zaika O, Li Y, Martin P, Hernandez CC, Perez MR, Wang AY, Jaffe DB, and Shapiro MS. Oxidative modification of M-type K+ channels as a mechanism of cytoprotective neuronal silencing. EMBO J 25: 4996–5004, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gnanasekaran A, Sundukova M, van den Maagdenberg AM, Fabbretti E, and Nistri A. Lipid rafts control P2X3 receptor distribution and function in trigeminal sensory neurons of a transgenic migraine mouse model. Mol Pain 7: 77, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goadsby P, Edvinsson L, and Ekman R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol 28: 183–187, 1990 [DOI] [PubMed] [Google Scholar]
- 59.Gold MS, Shuster MJ, and Levine JD. Characterization of six voltage-gated K+ currents in adult rat sensory neurons. J Neurophysiol 75: 2629–2646, 1996 [DOI] [PubMed] [Google Scholar]
- 60.Guan J-S, Haggarty SJ, Giacometti E, Dannenberg J-H, Joseph N, Gao J, Nieland TJF, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, and Tsai L-H. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459: 55–60, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guo J. and Schofield GG. Activation of muscarinic m5 receptors inhibits recombinant KCNQ2/KCNQ3 K+ channels expressed in HEK293T cells. Eur J Pharmacol 462: 25–32, 2003 [DOI] [PubMed] [Google Scholar]
- 62.Hadley JK, Passmore GM, Tatulian L, Al-Qatari M, Ye F, Wickenden AD, and Brown DA. Stoichiometry of expressed KCNQ2/KCNQ3 potassium channels and subunit composition of native ganglionic M channels deduced from block by tetraethylammonium. J Neurosci 23: 5012–5019, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hall CN. and Garthwaite J. What is the real physiological NO concentration in vivo? Nitric Oxide 21: 92–103, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hardingham N, Dachtler J, and Fox K. The role of nitric oxide in pre-synaptic plasticity and homeostasis. Front Cell Neurosci 7: 190, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, and Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90: 291–366, 2010 [DOI] [PubMed] [Google Scholar]
- 66.Hinman A, Chuang HH, Bautista DM, and Julius D. TRP channel activation by reversible covalent modification. Proc Natl Acad Sci U S A 103: 19564–19568, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Holscher C. Nitric oxide, the enigmatic neuronal messenger: its role in synaptic plasticity. Trends Neurosci 20: 298–303, 1997 [DOI] [PubMed] [Google Scholar]
- 68.Holzer P, Jocic M, and Peskar BA. Mediation by prostaglandins of the nitric oxide-induced neurogenic vasodilatation in rat skin. Br J Pharmacol 116: 2365–2370, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hsieh CP. Redox modulation of A-type K+ currents in pain-sensing dorsal root ganglion neurons. Biochem Biophys Res Commun 370: 445–449, 2008 [DOI] [PubMed] [Google Scholar]
- 70.Huang H. and Trussell LO. KCNQ5 channels control resting properties and release probability of a synapse. Nat Neurosci 14: 840–847, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ichinari K, Kakei M, Matsuoka T, Nakashima H, and Tanaka H. Direct activation of the ATP-sensitive potassium channel by oxygen free radicals in guinea-pig ventricular cells: its potentiation by MgADP. J Mol Cell Cardiol 28: 1867–1877, 1996 [DOI] [PubMed] [Google Scholar]
- 72.Iftinca MC. and Zamponi GW. Regulation of neuronal T-type calcium channels. Trends Pharmacol Sci 30: 32–40, 2009 [DOI] [PubMed] [Google Scholar]
- 73.Jevtovic-Todorovic V. and Todorovic SM. The role of peripheral T-type calcium channels in pain transmission. Cell Calcium 40: 197–203, 2006 [DOI] [PubMed] [Google Scholar]
- 74.Jia Q, Jia Z, Zhao Z, Liu B, Liang H, and Zhang H. Activation of epidermal growth factor receptor inhibits KCNQ2/3 current through two distinct pathways: membrane PtdIns(4,5)P2 hydrolysis and channel phosphorylation. J Neurosci 27: 2503–2512, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jia Z, Bei J, Rodat-Despoix L, Liu B, Jia Q, Delmas P, and Zhang H. NGF inhibits M/KCNQ currents and selectively alters neuronal excitability in subsets of sympathetic neurons depending on their M/KCNQ current background. J Gen Physiol 131: 575–587, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jin X, Shah S, Liu Y, Zhang H, Lees M, Fu Z, Lippiat JD, Beech DJ, Sivaprasadarao A, Baldwin SA, Zhang H, and Gamper N. Activation of the Cl− channel ANO1 by localized calcium signals in nociceptive sensory neurons requires coupling with the IP3 receptor. Sci Signal 6: ra73, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kawano T, Zoga V, Kimura M, Liang MY, Wu HE, Gemes G, McCallum JB, Kwok WM, Hogan QH, and Sarantopoulos CD. Nitric oxide activates ATP-sensitive potassium channels in mammalian sensory neurons: action by direct S-nitrosylation. Mol Pain 5: 12, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kemp PJ. and Peers C. Oxygen sensing by ion channels. Essays Biochem 43: 77–90, 2007 [DOI] [PubMed] [Google Scholar]
- 79.Kim DS, Choi JO, Rim HD, and Cho HJ. Downregulation of voltage-gated potassium channel alpha gene expression in dorsal root ganglia following chronic constriction injury of the rat sciatic nerve. Brain Res Mol Brain Res 105: 146–152, 2002 [DOI] [PubMed] [Google Scholar]
- 80.King CH. and Scherer SS. Kv7.5 is the primary Kv7 subunit expressed in C-fibers. J Comp Neurol 520: 1940–1950, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kinuta Y, Kimura M, Itokawa Y, Ishikawa M, and Kikuchi H. Changes in xanthine oxidase in ischemic rat brain. J Neurosurg 71: 417–420, 1989 [DOI] [PubMed] [Google Scholar]
- 82.Klyachko VA, Ahern GP, and Jackson MB. cGMP-mediated facilitation in nerve terminals by enhancement of the spike afterhyperpolarization. Neuron 31: 1015–1025, 2001 [DOI] [PubMed] [Google Scholar]
- 83.Kolesar D, Kolesarova M, Schreiberova A, Lackova M, and Marsala J. Distribution of NADPH diaphorase-exhibiting primary afferent neurons in the trigeminal ganglion and mesencephalic trigeminal nucleus of the rabbit. Cell Mol Neurobiol 26: 1265–1279, 2006 [DOI] [PubMed] [Google Scholar]
- 84.Kung LH, Gong K, Adedoyin M, Ng J, Bhargava A, Ohara PT, and Jasmin L. Evidence for glutamate as a neuroglial transmitter within sensory ganglia. PLoS One 8: e68312, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kurosawa M, Messlinger K, Pawlak M, and Schmidt RF. Increase of meningeal blood flow after electrical stimulation of rat dura mater encephali: mediation by calcitonin gene-related peptide. Br J Pharmacol 114: 1397–1402, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kyle BD, Hurst S, Swayze RD, Sheng J, and Braun AP. Specific phosphorylation sites underlie the stimulation of a large conductance, Ca2+-activated K+ channel by cGMP-dependent protein kinase. FASEB J 27: 2027–2038, 2013 [DOI] [PubMed] [Google Scholar]
- 87.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 4: 181–189, 2004 [DOI] [PubMed] [Google Scholar]
- 88.Lazarov N. and Dandov A. Distribution of NADPH-diaphorase and nitric oxide synthase in the trigeminal ganglion and mesencephalic trigeminal nucleus of the cat. A histochemical and immunohistochemical study. Acta Anat (Basel) 163: 191–200, 1998 [DOI] [PubMed] [Google Scholar]
- 89.Li W, Gao SB, Lv CX, Wu Y, Guo ZH, Ding JP, and Xu T. Characterization of voltage-and Ca2+-activated K+ channels in rat dorsal root ganglion neurons. J Cell Physiol 212: 348–357, 2007 [DOI] [PubMed] [Google Scholar]
- 90.Li Y, Gamper N, and Shapiro MS. Single-channel analysis of KCNQ K+ channels reveals the mechanism of augmentation by a cysteine-modifying reagent. J Neurosci 24: 5079–5090, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Linley JE, Ooi L, Pettinger L, Kirton H, Boyle JP, Peers C, and Gamper N. Reactive oxygen species are second messengers of neurokinin signaling in peripheral sensory neurons. Proc Natl Acad Sci U S A 109: E1578–E1586, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Linley JE, Pettinger L, Huang D, and Gamper N. M channel enhancers and physiological M channel block. J Physiol 590: 793–807, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Linley JE, Rose K, Ooi L, and Gamper N. Understanding inflammatory pain: ion channels contributing to acute and chronic nociception. Pflugers Arch 459: 657–669, 2010 [DOI] [PubMed] [Google Scholar]
- 94.Linley JE, Rose K, Patil M, Robertson B, Akopian AN, and Gamper N. Inhibition of M current in sensory neurons by exogenous proteases: a signaling pathway mediating inflammatory nociception. J Neurosci 28: 11240–11249, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lipton SA, Choi YB, Pan ZH, Lei SZ, Chen HS, Sucher NJ, Loscalzo J, Singel DJ, and Stamler JS. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature 364: 626–632, 1993 [DOI] [PubMed] [Google Scholar]
- 96.Lipton SA, Choi YB, Takahashi H, Zhang D, Li W, Godzik A, and Bankston LA. Cysteine regulation of protein function—as exemplified by NMDA-receptor modulation. Trends Neurosci 25: 474–480, 2002 [DOI] [PubMed] [Google Scholar]
- 97.Liu B, Escalera J, Balakrishna S, Fan L, Caceres AI, Robinson E, Sui A, McKay MC, McAlexander MA, Herrick CA, and Jordt SE. TRPA1 controls inflammation and pruritogen responses in allergic contact dermatitis. FASEB J 27: 3549–3563, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu B, Linley JE, Du X, Zhang X, Ooi L, Zhang H, and Gamper N. The acute nociceptive signals induced by bradykinin in rat sensory neurons are mediated by inhibition of M-type K+ channels and activation of Ca2+-activated Cl− channels. J Clin Invest 120: 1240–1252, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu B. and Qin F. Functional control of cold- and menthol-sensitive TRPM8 ion channels by phosphatidylinositol 4,5-bisphosphate. J Neurosci 25: 1674–1681, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu M, Huang W, Wu D, and Priestley JV. TRPV1, but not P2×, requires cholesterol for its function and membrane expression in rat nociceptors. Eur J Neurosci 24: 1–6, 2006 [DOI] [PubMed] [Google Scholar]
- 101.Liu X, Miller MJ, Joshi MS, Thomas DD, and Lancaster JR., Jr.Accelerated reaction of nitric oxide with O2 within the hydrophobic interior of biological membranes. Proc Natl Acad Sci U S A 95: 2175–2179, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lu G, Mazet B, Sarr MG, and Szurszewski JH. Effect of nitric oxide on calcium-activated potassium channels in colonic smooth muscle of rabbits. Am J Physiol 274: G848–G856, 1998 [DOI] [PubMed] [Google Scholar]
- 103.Luo ZD, Chaplan SR, Scott BP, Cizkova D, Calcutt NA, and Yaksh TL. Neuronal nitric oxide synthase mRNA upregulation in rat sensory neurons after spinal nerve ligation: lack of a role in allodynia development. J Neurosci 19: 9201–9208, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ma W. and Eisenach JC. Neuronal nitric oxide synthase is upregulated in a subset of primary sensory afferents after nerve injury which are necessary for analgesia from α2-adrenoceptor stimulation. Brain Res 1127: 52–58, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Macpherson LJ, Dubin AE, Evans MJ, Marr F, Schultz PG, Cravatt BF, and Patapoutian A. Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature 445: 541–545, 2007 [DOI] [PubMed] [Google Scholar]
- 106.Macpherson LJ, Geierstanger BH, Viswanath V, Bandell M, Eid SR, Hwang S, and Patapoutian A. The pungency of garlic: activation of TRPA1 and TRPV1 in response to allicin. Curr Biol 15: 929–934, 2005 [DOI] [PubMed] [Google Scholar]
- 107.Magnusson S, Ekstrom TJ, Elmer E, Kanje M, Ny L, and Alm P. Heme oxygenase-1, heme oxygenase-2 and biliverdin reductase in peripheral ganglia from rat, expression and plasticity. Neuroscience 95: 821–829, 2000 [DOI] [PubMed] [Google Scholar]
- 108.Majewski M, Sienkiewicz W, Kaleczyc J, Mayer B, Czaja K, and Lakomy M. The distribution and co-localization of immunoreactivity to nitric oxide synthase, vasoactive intestinal polypeptide and substance P within nerve fibers supplying bovine and porcine female genital organs. Cell Tissue Res 281: 445–464, 1995 [DOI] [PubMed] [Google Scholar]
- 109.Malinski T. Nitric oxide and nitroxidative stress in Alzheimer's disease. J Alzheimers Dis 11: 207–218, 2007 [DOI] [PubMed] [Google Scholar]
- 110.Marletta MA, Yoon PS, Iyengar R, Leaf CD, and Wishnok JS. Macrophage oxidation of L-arginine to nitrite and nitrate: nitric oxide is an intermediate. Biochemistry 27: 8706–8711, 1988 [DOI] [PubMed] [Google Scholar]
- 111.McFarlane S. and Cooper E. Kinetics and voltage dependence of A-type currents on neonatal rat sensory neurons. J Neurophysiol 66: 1380–1391, 1991 [DOI] [PubMed] [Google Scholar]
- 112.McHugh JM. and McHugh WB. Diabetes and peripheral sensory neurons: what we don't know and how it can hurt us. AACN Clin Issues 15: 136–149, 2004 [DOI] [PubMed] [Google Scholar]
- 113.Messlinger K, Suzuki A, Pawlak M, Zehnter A, and Schmidt RF. Involvement of nitric oxide in the modulation of dural arterial blood flow in the rat. Br J Pharmacol 129: 1397–1404, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Miyamoto T, Dubin AE, Petrus MJ, and Patapoutian A. TRPV1 and TRPA1 mediate peripheral nitric oxide-induced nociception in mice. PLoS One 4: e7596, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Miyamoto T, Petrus MJ, Dubin AE, and Patapoutian A. TRPV3 regulates nitric oxide synthase-independent nitric oxide synthesis in the skin. Nat Commun 2: 369, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Morris R, Southam E, Braid DJ, and Garthwaite J. Nitric oxide may act as a messenger between dorsal root ganglion neurones and their satellite cells. Neurosci Lett 137: 29–32, 1992 [DOI] [PubMed] [Google Scholar]
- 117.Mucha M, Ooi L, Linley JE, Mordaka P, Dalle C, Robertson B, Gamper N, and Wood IC. Transcriptional control of KCNQ channel genes and the regulation of neuronal excitability. J Neurosci 30: 13235–13245, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 417: 1–13, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nelson MT, Joksovic PM, Perez-Reyes E, and Todorovic SM. The endogenous redox agent L-cysteine induces T-type Ca2+ channel-dependent sensitization of a novel subpopulation of rat peripheral nociceptors. J Neurosci 25: 8766–8775, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nelson MT, Joksovic PM, Su P, Kang HW, Van Deusen A, Baumgart JP, David LS, Snutch TP, Barrett PQ, Lee JH, Zorumski CF, Perez-Reyes E, and Todorovic SM. Molecular mechanisms of subtype-specific inhibition of neuronal T-type calcium channels by ascorbate. J Neurosci 27: 12577–12583, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nelson MT, Woo J, Kang HW, Vitko I, Barrett PQ, Perez-Reyes E, Lee JH, Shin HS, and Todorovic SM. Reducing agents sensitize C-type nociceptors by relieving high-affinity zinc inhibition of T-type calcium channels. J Neurosci 27: 8250–8260, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature 440: 470–476, 2006 [DOI] [PubMed] [Google Scholar]
- 123.Nilius B, Appendino G, and Owsianik G. The transient receptor potential channel TRPA1: from gene to pathophysiology. Pflugers Arch 464: 425–458, 2012 [DOI] [PubMed] [Google Scholar]
- 124.Ohshima H, Friesen M, Brouet I, and Bartsch H. Nitrotyrosine as a new marker for endogenous nitrosation and nitration of proteins. Food Chem Toxicol 28: 647–652, 1990 [DOI] [PubMed] [Google Scholar]
- 125.Oldfield S, Hancock J, Mason A, Hobson SA, Wynick D, Kelly E, Randall AD, and Marrion NV. Receptor-mediated suppression of potassium currents requires colocalization within lipid rafts. Mol Pharmacol 76: 1279–1289, 2009 [DOI] [PubMed] [Google Scholar]
- 126.Ooi L, Gigout S, Pettinger L, and Gamper N. Triple cysteine module within M-type K+ channels mediates reciprocal channel modulation by nitric oxide and reactive oxygen species. J Neurosci 33: 6041–6046, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pagadala P, Park CK, Bang S, Xu ZZ, Xie RG, Liu T, Han BX, Tracey WD, Jr., Wang F, and Ji RR. Loss of NR1 subunit of NMDARs in primary sensory neurons leads to hyperexcitability and pain hypersensitivity: involvement of Ca2+-activated small conductance potassium channels. J Neurosci 33: 13425–13430, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Paget MS. and Buttner MJ. Thiol-based regulatory switches. Annu Rev Genet 37: 91–121, 2003 [DOI] [PubMed] [Google Scholar]
- 129.Passmore GM, Reilly JM, Thakur M, Keasberry VN, Marsh SJ, Dickenson AH, and Brown DA. Functional significance of M-type potassium channels in nociceptive cutaneous sensory endings. Front Mol Neurosci 5: 63, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Passmore GM, Selyanko AA, Mistry M, Al-Qatari M, Marsh SJ, Matthews EA, Dickenson AH, Brown TA, Burbidge SA, Main M, and Brown DA. KCNQ/M currents in sensory neurons: significance for pain therapy. J Neurosci 23: 7227–7236, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Peers C. and Steele DS. Carbon monoxide: a vital signalling molecule and potent toxin in the myocardium. J Mol Cell Cardiol 52: 359–365, 2012 [DOI] [PubMed] [Google Scholar]
- 132.Peng J, Lu R, Ye F, Deng HW, and Li YJ. Induction of alpha-calcitonin gene-related peptide mRNA expression in rat dorsal root ganglia by heat stress involves the heme oxygenase-1/carbon monoxide pathway. Neuropeptides 35: 297–302, 2001 [DOI] [PubMed] [Google Scholar]
- 133.Perez-Reyes E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev 83: 117–161, 2003 [DOI] [PubMed] [Google Scholar]
- 134.Petho G. and Reeh PW. Sensory and signaling mechanisms of bradykinin, eicosanoids, platelet-activating factor, and nitric oxide in peripheral nociceptors. Physiol Rev 92: 1699–1775, 2012 [DOI] [PubMed] [Google Scholar]
- 135.Poole LB, Karplus PA, and Claiborne A. Protein sulfenic acids in redox signaling. Annu Rev Pharmacol Toxicol 44: 325–347, 2004 [DOI] [PubMed] [Google Scholar]
- 136.Prado WA, Schiavon VF, and Cunha FQ. Dual effect of local application of nitric oxide donors in a model of incision pain in rats. Eur J Pharmacol 441: 57–65, 2002 [DOI] [PubMed] [Google Scholar]
- 137.Prasad MR, Lovell MA, Yatin M, Dhillon H, and Markesbery WR. Regional membrane phospholipid alterations in Alzheimer's disease. Neurochem Res 23: 81–88, 1998 [DOI] [PubMed] [Google Scholar]
- 138.Prescott ED. and Julius D. A modular PIP2 binding site as a determinant of capsaicin receptor sensitivity. Science 300: 1284–1288, 2003 [DOI] [PubMed] [Google Scholar]
- 139.Pristera A, Baker MD, and Okuse K. Association between tetrodotoxin resistant channels and lipid rafts regulates sensory neuron excitability. PLoS One 7: e40079, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Prousek J. Fenton chemistry in biology and medicine. Pure Appl Chem 79: 2325–2338, 2007 [Google Scholar]
- 141.Qian Y, Chao DS, Santillano DR, Cornwell TL, Nairn AC, Greengard P, Lincoln TM, and Bredt DS. cGMP-dependent protein kinase in dorsal root ganglion: relationship with nitric oxide synthase and nociceptive neurons. J Neurosci 16: 3130–3138, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ramachandran R, Ploug KB, Hay-Schmidt A, Olesen J, Jansen-Olesen I, and Gupta S. Nitric oxide synthase (NOS) in the trigeminal vascular system and other brain structures related to pain in rats. Neurosci Lett 484: 192–196, 2010 [DOI] [PubMed] [Google Scholar]
- 143.Ramasarma T. In praise of H2O2, the versatile ROS, and its vanadium complexes. Toxicol Mech Methods 22: 336–346, 2012 [DOI] [PubMed] [Google Scholar]
- 144.Raouf R, Quick K, and Wood JN. Pain as a channelopathy. J Clin Invest 120: 3745–3752, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rasband MN, Park EW, Vanderah TW, Lai J, Porreca F, and Trimmer JS. Distinct potassium channels on pain-sensing neurons. Proc Natl Acad Sci U S A 98: 13373–13378, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Reilly JM, Telezhkin V, Passmore GM, Marsh SJ, and Brown DA. Kv7/M-type potassium channels in rat skin keratinocytes. Pflugers Arch 465: 1371–1381, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Rice ME, Avshalumov MV, and Patel JC. Hydrogen peroxide as a diffusible messenger: evidence from voltammetric studies of dopamine release in brain slices. In: Electrochemical Methods for Neuroscience, edited by Michael AC. and Borland LM. Boca Raton, FL: CRC Press, 2007 [PubMed] [Google Scholar]
- 148.Richter C. Biophysical consequences of lipid peroxidation in membranes. Chem Phys Lipids 44: 175–189, 1987 [DOI] [PubMed] [Google Scholar]
- 149.Rivera-Arconada I. and Lopez-Garcia JA. Retigabine-induced population primary afferent hyperpolarisation in vitro. Neuropharmacology 51: 756–763, 2006 [DOI] [PubMed] [Google Scholar]
- 150.Rohacs T, Lopes CM, Jin T, Ramdya PP, Molnar Z, and Logothetis DE. Specificity of activation by phosphoinositides determines lipid regulation of Kir channels. Proc Natl Acad Sci U S A 100: 745–750, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rosa AO, Egea J, Lorrio S, Rojo AI, Cuadrado A, and Lopez MG. Nrf2-mediated haeme oxygenase-1 up-regulation induced by cobalt protoporphyrin has antinociceptive effects against inflammatory pain in the formalin test in mice. Pain 137: 332–339, 2008 [DOI] [PubMed] [Google Scholar]
- 152.Rose K, Ooi L, Dalle C, Robertson B, Wood IC, and Gamper N. Transcriptional repression of the M channel subunit Kv7.2 in chronic nerve injury. Pain 152: 742–754, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Rose KE, Lunardi N, Boscolo A, Dong X, Erisir A, Jevtovic-Todorovic V, and Todorovic SM. Immunohistological demonstration of Ca3.2 T-type voltage-gated calcium channel expression in soma of dorsal root ganglion neurons and peripheral axons of rat and mouse. Neuroscience 250C: 263–274, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Roza C, Castillejo S, and Lopez-Garcia JA. Accumulation of Kv7.2 channels in putative ectopic transduction zones of mice nerve-end neuromas. Mol Pain 7: 58, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ruppersberg JP, Stocker M, Pongs O, Heinemann SH, Frank R, and Koenen M. Regulation of fast inactivation of cloned mammalian IK(A) channels by cysteine oxidation. Nature 352: 711–714, 1991 [DOI] [PubMed] [Google Scholar]
- 156.Sachs D, Cunha FQ, and Ferreira SH. Peripheral analgesic blockade of hypernociception: activation of arginine/NO/cGMP/protein kinase G/ATP-sensitive K+ channel pathway. Proc Natl Acad Sci U S A 101: 3680–3685, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sahoo N, Hoshi T, and Heinemann SH. Oxidative modulation of voltage-gated potassium channels. Antioxid Redox Signal 21: 933–952, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Salas MM, Hargreaves KM, and Akopian AN. TRPA1-mediated responses in trigeminal sensory neurons: interaction between TRPA1 and TRPV1. Eur J Neurosci 29: 1568–1578, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Salazar H, Llorente I, Jara-Oseguera A, Garcia-Villegas R, Munari M, Gordon SE, Islas LD, and Rosenbaum T. A single N-terminal cysteine in TRPV1 determines activation by pungent compounds from onion and garlic. Nat Neurosci 11: 255–261, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sapunar D, Ljubkovic M, Lirk P, McCallum JB, and Hogan QH. Distinct membrane effects of spinal nerve ligation on injured and adjacent dorsal root ganglion neurons in rats. Anesthesiology 103: 360–376, 2005 [DOI] [PubMed] [Google Scholar]
- 161.Sawada Y, Hosokawa H, Matsumura K, and Kobayashi S. Activation of transient receptor potential ankyrin 1 by hydrogen peroxide. Eur J Neurosci 27: 1131–1142, 2008 [DOI] [PubMed] [Google Scholar]
- 162.Schmidt HH, Pollock JS, Nakane M, Forstermann U, and Murad F. Ca2+/calmodulin-regulated nitric oxide synthases. Cell Calcium 13: 427–434, 1992 [DOI] [PubMed] [Google Scholar]
- 163.Scholz A, Gruss M, and Vogel W. Properties and functions of calcium-activated K+ channels in small neurones of rat dorsal root ganglion studied in a thin slice preparation. J Physiol 513: 55–69, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Schubert R. and Nelson MT. Protein kinases: tuners of the BKCa channel in smooth muscle. Trends Pharmacol Sci 22: 505–512, 2001 [DOI] [PubMed] [Google Scholar]
- 165.Selyanko AA, Hadley JK, and Brown DA. Properties of single M-type KCNQ2/KCNQ3 potassium channels expressed in mammalian cells. J Physiol 534: 15–24, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Shapiro MS, Roche JP, Kaftan EJ, Cruzblanca H, Mackie K, and Hille B. Reconstitution of muscarinic modulation of the KCNQ2/KCNQ3 K+ channels that underlie the neuronal M current. J Neurosci 20: 1710–1721, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sheetz MJ. and King GL. Molecular understanding of hyperglycemia's adverse effects for diabetic complications. JAMA 288: 2579–2588, 2002 [DOI] [PubMed] [Google Scholar]
- 168.Shin JB, Martinez-Salgado C, Heppenstall PA, and Lewin GR. A T-type calcium channel required for normal function of a mammalian mechanoreceptor. Nat Neurosci 6: 724–730, 2003 [DOI] [PubMed] [Google Scholar]
- 169.Smith GD, Gunthorpe MJ, Kelsell RE, Hayes PD, Reilly P, Facer P, Wright JE, Jerman JC, Walhin JP, Ooi L, Egerton J, Charles KJ, Smart D, Randall AD, Anand P, and Davis JB. TRPV3 is a temperature-sensitive vanilloid receptor-like protein. Nature 418: 186–190, 2002 [DOI] [PubMed] [Google Scholar]
- 170.Soares AC. and Duarte IDG. Dibutyryl-cyclic GMP induces peripheral antinociception via activation of ATP-sensitive K+ channels in the rat PGE2-induced hyperalgesic paw. Br J Pharmacol 134: 127–131, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Soares AC, Leite R, Tatsuo MAKF, and Duarte IDG. Activation of ATP-sensitive K+ channels: mechanism of peripheral antinociceptive action of the nitric oxide donor, sodium nitroprusside. Eur J Pharmacol 400: 67–71, 2000 [DOI] [PubMed] [Google Scholar]
- 172.Stadtman ER. Ascorbic acid and oxidative inactivation of proteins. Am J Clin Nutr 54: 1125S–1128S, 1991 [DOI] [PubMed] [Google Scholar]
- 173.Stansfeld CE, Marsh SJ, Parcej DN, Dolly JO, and Brown DA. Mast cell degranulating peptide and dendrotoxin selectively inhibit a fast-activating potassium current and bind to common neuronal proteins. Neuroscience 23: 893–902, 1987 [DOI] [PubMed] [Google Scholar]
- 174.Starkov AA. and Fiskum G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J Neurochem 86: 1101–1107, 2003 [DOI] [PubMed] [Google Scholar]
- 175.Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, and Beal MF. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci 24: 7779–7788, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Staruschenko A, Jeske NA, and Akopian AN. Contribution of TRPV1-TRPA1 interaction to the single channel properties of the TRPA1 channel. J Biol Chem 285: 15167–15177, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Steel JH, Terenghi G, Chung JM, Na HS, Carlton SM, and Polak JM. Increased nitric oxide synthase immunoreactivity in rat dorsal root ganglia in a neuropathic pain model. Neurosci Lett 169: 81–84, 1994 [DOI] [PubMed] [Google Scholar]
- 178.Steiner AA, Branco LG, Cunha FQ, and Ferreira SH. Role of the haeme oxygenase/carbon monoxide pathway in mechanical nociceptor hypersensitivity. Br J Pharmacol 132: 1673–1682, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Steinert JR, Chernova T, and Forsythe ID. Nitric oxide signaling in brain function, dysfunction, and dementia. Neuroscientist 16: 435–452, 2010 [DOI] [PubMed] [Google Scholar]
- 180.This reference has been deleted
- 181.Stone JR. An assessment of proposed mechanisms for sensing hydrogen peroxide in mammalian systems. Arch Biochem Biophys 422: 119–124, 2004 [DOI] [PubMed] [Google Scholar]
- 182.Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, Hricik TR, Earley TJ, Hergarden AC, Andersson DA, Hwang SW, McIntyre P, Jegla T, Bevan S, and Patapoutian A. ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 112: 819–829, 2003 [DOI] [PubMed] [Google Scholar]
- 183.Stoyanova II. and Lazarov NE. Localization of nitric oxide synthase in rat trigeminal primary afferent neurons using NADPH-diaphorase histochemistry. J Mol Histol 36: 187–193, 2005 [DOI] [PubMed] [Google Scholar]
- 184.Sullivan JM, Traynelis SF, Chen HS, Escobar W, Heinemann SF, and Lipton SA. Identification of two cysteine residues that are required for redox modulation of the NMDA subtype of glutamate receptor. Neuron 13: 929–936, 1994 [DOI] [PubMed] [Google Scholar]
- 185.Sultana R, Poon HF, Cai J, Pierce WM, Merchant M, Klein JB, Markesbery WR, and Butterfield DA. Identification of nitrated proteins in Alzheimer's disease brain using a redox proteomics approach. Neurobiol Dis 22: 76–87, 2006 [DOI] [PubMed] [Google Scholar]
- 186.Susankova K, Tousova K, Vyklicky L, Teisinger J, and Vlachova V. Reducing and oxidizing agents sensitize heat-activated vanilloid receptor (TRPV1) current. Mol Pharmacol 70: 383–394, 2006 [DOI] [PubMed] [Google Scholar]
- 187.Takahashi N, Mizuno Y, Kozai D, Yamamoto S, Kiyonaka S, Shibata T, Uchida K, and Mori Y. Molecular characterization of TRPA1 channel activation by cysteine-reactive inflammatory mediators. Channels (Austin) 2: 287–298, 2008 [DOI] [PubMed] [Google Scholar]
- 188.Takahashi N. and Mori Y. TRP channels as sensors and signal integrators of redox status changes. Front Pharmacol 2: 58, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Talley EM, Solorzano G, Lei Q, Kim D, and Bayliss DA. Cns distribution of members of the two-pore-domain (KCNK) potassium channel family. J Neurosci 21: 7491–7505, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Tandon RK. and Garg PK. Oxidative stress in chronic pancreatitis: pathophysiological relevance and management. Antioxid Redox Signal 15: 2757–2766, 2011 [DOI] [PubMed] [Google Scholar]
- 191.Tang XD, Daggett H, Hanner M, Garcia ML, McManus OB, Brot N, Weissbach H, Heinemann SH, and Hoshi T. Oxidative regulation of large conductance calcium-activated potassium channels. J Gen Physiol 117: 253–274, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Telezhkin V, Reilly JM, Thomas AM, Tinker A, and Brown DA. Structural requirements of membrane phospholipids for M-type potassium channel activation and binding. J Biol Chem 287: 10001–10012, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Terenghi G, Riveros-Moreno V, Hudson LD, Ibrahim NB, and Polak JM. Immunohistochemistry of nitric oxide synthase demonstrates immunoreactive neurons in spinal cord and dorsal root ganglia of man and rat. J Neurol Sci 118: 34–37, 1993 [DOI] [PubMed] [Google Scholar]
- 194.Thippeswamy T, McKay JS, Quinn J, and Morris R. Either nitric oxide or nerve growth factor is required for dorsal root ganglion neurons to survive during embryonic and neonatal development. Brain Res Dev brain Res 154: 153–164, 2005 [DOI] [PubMed] [Google Scholar]
- 195.Thomas DD, Espey MG, Vitek MP, Miranda KM, and Wink DA. Protein nitration is mediated by heme and free metals through Fenton-type chemistry: an alternative to the NO/O2-reaction. Proc Natl Acad Sci U S A 99: 12691–12696, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Thompson JW, Narayanan SV, and Perez-Pinzon MA. Redox signaling pathways involved in neuronal ischemic preconditioning. Curr Neuropharmacol 10: 354–369, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Todorovic SM, Jevtovic-Todorovic V, Meyenburg A, Mennerick S, Perez-Reyes E, Romano C, Olney JW, and Zorumski CF. Redox modulation of T-type calcium channels in rat peripheral nociceptors. Neuron 31: 75–85, 2001 [DOI] [PubMed] [Google Scholar]
- 198.Trauner D. and Kramer RH. Metabolic modulation of potassium channels. Sci STKE 2004: pe22, 2004 [DOI] [PubMed] [Google Scholar]
- 199.Tretter L. and Adam-Vizi V. Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. J Neurosci 24: 7771–7778, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Tretter L, Takacs K, Hegedus V, and Adam-Vizi V. Characteristics of alpha-glycerophosphate-evoked H2O2 generation in brain mitochondria. J Neurochem 100: 650–663, 2007 [DOI] [PubMed] [Google Scholar]
- 201.van Dam PS. Oxidative stress and diabetic neuropathy: pathophysiological mechanisms and treatment perspectives. Diabetes Metab Res Rev 18: 176–184, 2002 [DOI] [PubMed] [Google Scholar]
- 202.Vay L, Gu C, and McNaughton PA. The thermo-TRP ion channel family: properties and therapeutic implications. Br J Pharmacol 165: 787–801, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Verge VM, Xu Z, Xu XJ, Wiesenfeld-Hallin Z, and Hokfelt T. Marked increase in nitric oxide synthase mRNA in rat dorsal root ganglia after peripheral axotomy: in situ hybridization and functional studies. Proc Natl Acad Sci U S A 89: 11617–11621, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Vydyanathan A, Wu ZZ, Chen SR, and Pan HL. A-type voltage-gated K+ currents influence firing properties of isolectin B4-positive but not isolectin B4-negative primary sensory neurons. J Neurophysiol 93: 3401–3409, 2005 [DOI] [PubMed] [Google Scholar]
- 205.Vyklicky L, Lyfenko A, Susankova K, Teisinger J, and Vlachova V. Reducing agent dithiothreitol facilitates activity of the capsaicin receptor VR-1. Neuroscience 111: 435–441, 2002 [DOI] [PubMed] [Google Scholar]
- 206.Wagner BA, Buettner GR, and Burns CP. Free radical-mediated lipid peroxidation in cells: oxidizability is a function of cell lipid bis-allylic hydrogen content. Biochemistry 33: 4449–4453, 1994 [DOI] [PubMed] [Google Scholar]
- 207.Wang HJ, Li YL, Zhang LB, Zucker IH, Gao L, Zimmerman MC, and Wang W. Endogenous reactive oxygen species modulates voltage-gated sodium channels in dorsal root ganglia of rats. J Appl Physiol 110: 1439–1447, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, and McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282: 1890–1893, 1998 [DOI] [PubMed] [Google Scholar]
- 209.Wang R. Shared signaling pathways among gasotransmitters. Proc Natl Acad Sci U S A 109: 8801–8802, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Wei EP, Moskowitz MA, Boccalini P, and Kontos HA. Calcitonin gene-related peptide mediates nitroglycerin and sodium nitroprusside-induced vasodilation in feline cerebral arterioles. Circ Res 70: 1313–1319, 1992 [DOI] [PubMed] [Google Scholar]
- 211.White G, Lovinger DM, and Weight FF. Transient low-threshold Ca2+current triggers burst firing through an afterdepolarizing potential in an adult mammalian neuron. Proc Natl Acad Sci U S A 86: 6802–6806, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Wilson SR, Nelson AM, Batia L, Morita T, Estandian D, Owens DM, Lumpkin EA, and Bautista DM. The ion channel TRPA1 is required for chronic itch. J Neurosci 33: 9283–9294, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Wolff SP. and Dean RT. Glucose autoxidation and protein modification. The potential role of ‘autoxidative glycosylation’ in diabetes. Biochem J 245: 243–250, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Wood KC, Batchelor AM, Bartus K, Harris KL, Garthwaite G, Vernon J, and Garthwaite J. Picomolar nitric oxide signals from central neurons recorded using ultrasensitive detector cells. J Biol Chem 286: 43172–43181, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Xia Y, Dawson VL, Dawson TM, Snyder SH, and Zweier JL. Nitric oxide synthase generates superoxide and nitric oxide in arginine-depleted cells leading to peroxynitrite-mediated cellular injury. Proc Natl Acad Sci U S A 93: 6770–6774, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Yamada K. and Inagaki N. Neuroprotection by KATP channels. J Mol Cell Cardiol 38: 945–949, 2005 [DOI] [PubMed] [Google Scholar]
- 217.Yamakage M, Hirshman CA, and Croxton TL. Sodium nitroprusside stimulates Ca2+-activated K+ channels in porcine tracheal smooth muscle cells. Am J Physiol 270: L338–L345, 1996 [DOI] [PubMed] [Google Scholar]
- 218.Yamamoto Y, Katsumata O, Furuyama S, and Sugiya H. Ca2+, calmodulin and phospholipids regulate nitricoxide synthase activity in the rabbit submandibular gland. J Comp Physiol B 174: 593–599, 2004 [DOI] [PubMed] [Google Scholar]
- 219.Yang EK, Takimoto K, Hayashi Y, de Groat WC, and Yoshimura N. Altered expression of potassium channel subunit mRNA and alpha-dendrotoxin sensitivity of potassium currents in rat dorsal root ganglion neurons after axotomy. Neuroscience 123: 867–874, 2004 [DOI] [PubMed] [Google Scholar]
- 220.Yang HC, Farooqui AA, and Horrocks LA. Plasmalogen-selective phospholipase A2 and its role in signal transduction. J Lipid Mediat Cell Signal 14: 9–13, 1996 [DOI] [PubMed] [Google Scholar]
- 221.Yang WP, Levesque PC, Little WA, Conder ML, Ramakrishnan P, Neubauer MG, and Blanar MA. Functional expression of two KvLQT1-related potassium channels responsible for an inherited idiopathic epilepsy. J Biol Chem 273: 19419–19423, 1998 [DOI] [PubMed] [Google Scholar]
- 222.Yoshida T, Inoue R, Morii T, Takahashi N, Yamamoto S, Hara Y, Tominaga M, Shimizu S, Sato Y, and Mori Y. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat Chem Biol 2: 596–607, 2006 [DOI] [PubMed] [Google Scholar]
- 223.Yoshimura N, White G, Weight FF, and de Groat WC. Different types of Na+ and A-type K+ currents in dorsal root ganglion neurones innervating the rat urinary bladder. J Physiol 494: 1–16, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Zaika O, Lara LS, Gamper N, Hilgemann DW, Jaffe DB, and Shapiro MS. Angiotensin II regulates neuronal excitability via phosphatidylinositol 4,5-bisphosphate-dependent modulation of Kv7 (M-type) K+ channels. J Physiol 575: 49–67, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Zaika O, Tolstykh GP, Jaffe DB, and Shapiro MS. Inositol triphosphate-mediated Ca2+ signals direct purinergic P2Y receptor regulation of neuronal ion channels. J Neurosci 27: 8914–8926, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Zhang G. and Horrigan FT. Cysteine modification alters voltage- and Ca2+-dependent gating of large conductance (BK) potassium channels. J Gen Physiol 125: 213–236, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Zhang X, Verge V, Wiesenfeld-Hallin Z, Ju G, Bredt D, Synder SH, and Hokfelt T. Nitric oxide synthase-like immunoreactivity in lumbar dorsal root ganglia and spinal cord of rat and monkey and effect of peripheral axotomy. J Comp Neurol 335: 563–575, 1993 [DOI] [PubMed] [Google Scholar]
- 228.Zheng Q, Fang D, Liu M, Cai J, Wan Y, Han JS, and Xing GG. Suppression of KCNQ/M (Kv7) potassium channels in dorsal root ganglion neurons contributes to the development of bone cancer pain in a rat model. Pain 154: 434–448, 2013 [DOI] [PubMed] [Google Scholar]
- 229.Zoga V, Kawano T, Liang MY, Bienengraeber M, Weihrauch D, McCallum B, Gemes G, Hogan Q, and Sarantopoulos C. KATP channel subunits in rat dorsal root ganglia: alterations by painful axotomy. Mol Pain 6: 6, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]