

Abstract
Photoreceptor (PR) cells are highly specialized cells that convert light into electrical signals. Ten percent of their outer segment (OS) membranes (approximately 77 cm2 of membrane) are renewed every day. Therefore, PR cells must possess an extraordinary trafficking system to provide all of the needed material to build up the OS discs through a 0.3 µm diameter connecting cilium. The mechanism of trafficking of membrane proteins in the retina and corresponding degenerative diseases is still elusive. The retinal degeneration(rd3) is the gene responsible for a murine autosomal recessive hereditary retinal degeneration, which is known as Leber Congenital Amaurosis 12 (LCA12). Degeneration starts at about two weeks of age and is completed between 2–4 months. We generated the first antibody against this protein and by a protein-protein interaction analysis discovered that RD3 protein directly interacts with guanylate cyclase 1 (GC1) and partially expresses in the OS. We also detected the major binding site between these two proteins and realized that RD3 is directly involved in trafficking of this crucial protein. In a separate study, we reported that RD3 negatively regulates GC1, which is crucial for efficient trafficking of GC1 during the trafficking path, and RD3 prevents unnecessary production of cGMP. It is possible that RD3 is still involved in regulating GC1 even after targeting. Several mutations that cause visual difficulties have been reported for the mouse and human ortholog of RD3. The symptoms these mutations cause are very similar to those reported for a more severe form of blindness referred to as LCA1. Therefore, RD3 might cause a broader range of retinal diseases. Gene replacement of RD3 has shown to restore the GC1 across the retina. This makes RD3 a novel therapeutic target for retinal targeting impaired degenerative diseases.
Keywords: Retina, Protein trafficking, Retina degeneration3, Guanylate cyclase1, Leber Congenital Amaurosis 1 (LCA1), LCA12
Introduction
Malfunctioning of protein targeting in the retina has been linked to a number of retinal degenerative diseases. Nevertheless, the key elements and the pathways, which execute the targeting of membrane proteins remain elusive [1–4]. One fundamental reason for the latter, which has slowed down the progress of this field and prevents performing developmental studies, is the complexity and uniqueness of the mammalian retina. This is because mammalian photoreceptor cells tolerate drastic qualitative changes such as remodelling, inter segmental fusion, and polarity loss when they are dissociated and therefore generating an in vitro model for disc renewal and trafficking has not yet been accomplished [5]. Without a doubt, retina degeneration 3(RD3) is one of the most important findings in vision research in the past decade. A stop codon in RD3, which causes premature termination of this protein, causes Leber Congenital Amaurosis12 (LCA12) [6]. This crucial protein, which was overlooked for a long time has helped to understand the mechanism of targeting of membrane proteins, particularly guanylate cyclase 1 (GC1), one crucial element in life and death of photoreceptor cells as well as visual cycle [7]. The lack of GC1 function has already been shown to cause another severe retinal disease called LCA1. This review will summarize what is currently known about the structure and function of RD3, and we will present an overview of the published and unpublished findings. Moreover, the current understanding of the disease-related role of RD3 will be described with an emphasis on a broader role for this protein, which might be involved in other diseases directly related to the malfunction of GC1 in photoreceptor cells.
History
The rd3 mutation was originally found among four mice breeds in Switzerland in 1969. The mice were brought to the Jackson laboratory and were bred into four different lines: RBF/DnJ, RBJ/DnJ, 4Bnr, and 30Rk/J. The amount of degeneration among these four genotypes varies significantly. In the Dnj strain, degeneration is very aggressive, starting from post natal (P) 14 and by three weeks of age, most of the photoreceptors have already been eliminated. The 4Bnr strain, however, has a mild degeneration and massive cell death does not start before 4 weeks. The other two genotypes possess even slower rates of degeneration. Moreover, the degeneration in pigmented mice is slower than in the albino mice carrying this mutation. Michael Danciger et al. [8] suggested that genetic modifiers contribute to such non-uniformity. For many years RD3 had been overlooked in such a way that it was used for production of hybridoma because the spleen is slightly larger and found to be more efficient for the mentioned usage[9].
Genetics
Chang et al. [9] mapped the rd3 gene to mouse chromosome 1 at 10 ± 2.5 cM distal to Akp1. This was located between markers D1Mit292 and D1Mit51. Due to closeness of rd3 to USA2A, the field originally considered rd3 as a mouse ortholog of human USH2A [10]. This gene carries mutations responsible for Usher IIa retinal degeneration/hearing loss syndrome. Crossing 4Bnr mice × C57BL/6J mice enabled Danciger et al. [8] to narrow down the location of the rd3 gene between the markers D1MIT292/D1MIT209 and D1MIT510, a far enough distance to exclude rd3 from being a USH2A ortholog. By using meiotic linkage and linkage disequilibrium mapping, Kukekova et al. [11] reduced the map of chromosome 1 to only 230 kb, which contained the three genes TRAF5, C1orf36, and SLC30A. The authors suggested that these genes could be potential positional candidates for rcd2 (rod cone dysplasia type 2). Further analysis showed that the reduced canine rcd2 interval overlapped with the murine retinal degeneration 3 (rd3). Unlike mouse and human RD3, each has a single known transcript, Kukekova et al. [11] detected three canine retinal RD3 splice variants and proposed that a sequence alteration was the cause of canine rcd2.
Structure
Rd3 mRNA contains 4287 bp of which only 588 bp contains the coding sequence, and the rest is N and C terminal un-translated regions. The rd3 gene codes a 195 amino acid protein and produces a 22–23 kDa protein [7]. It contains an N-terminal mitochondrial targeting signal, a possible coiled-coil domain, and 2 potential phosphorylation sites [12]. Our unpublished data as well as reports from other groups [12] indicate that rd3 mRNA is expressed only in the retina. In situ hybridization from our lab performed on adult mouse retina shows strong expression of rd3 in the inner segment (unpublished data) as well as outer plaxiform layer, but Lavorgna et al. [12] showed rd3 mRNA in the outer nuclear layer, the inner nuclear layer, and the ganglion cell layer. Friedman et al. [6] reported that mouse and human rd3 has three exons. Using a positional candidate approach, a homozygous transition C to T in c.319 in the third exon of mouse causes a premature stop codon at the 107th amino acid, which alters CGA to TGA, thus converting arginine to a stop codon. The same group detected this mutation in all rd3 lines mentioned above. In humans form carrying this mutation, however, the stop codon happens slightly earlier as amino acid 99th turns to be a stop codon. [6]
Localization
The first ever localization reported for RD3 was by Friedman et al. [6], who reported that this protein was associated with leukemia-gene product (PML) bodies in the nucleus when they expressed GFP-tagged RD3 in COS1 cells. By expressing rd3 cDNA, we detected a different localization than that reported by the mentioned authors. We found that RD3 in vesicular structure all across the cytoplasm [7]; however, the addition of GFP has been shown to cause mis-localization [13]. Using acid purification of one of our polyclonal antibodies, we detected RD3 protein in mouse retina mostly in outer segment. Our continuous sucrose gradient data supported the immunohistochemistry results when RD3 was in the same fractions as outer segment proteins were. The only conclusion from our localization study is that RD3 is partially localized in the OS. We believe the localization of RD3 is not conclusive. Correct localization of this protein is critical and will certainly help to identify its function. Due to the importance of this issue, we have generated several monoclonal and polyclonal antibodies and have set this as a top priority.
Disease
Friedman et al. [6] performed a mutation analysis on 461 probands with retinopathies and reported an alteration of G to A in exon 2 of rd3 in one patient, which caused a stop codon at amino acid 100 and resulted in a premature truncation. In the course of screening they found several other alterations: W6R, E23D, K130M. G57V, W6R/ E23D, R68W, R167K and D195V. They generated p.W6R, p.E23D, p.K130M, and p.W6R; E23D mutations in the RD3-GFP expression construct. The localization of the mutants did not differ from WT counterpart. We constructed the 6 former mutants and performed a binding analysis with the full-length GC1 when we co-expressed them in HEK293 cells. The binding of GC1 and above mentioned mutants were the same as WT [17], which confirmed the results from Friedman et al. [6]. It seems that the mentioned mutation can hardly be the cause for the vision difficulties of the probands carrying these mutaions. Perrault, et al. [19] performed a comprehensive survey of RD3 mutations and of their clinical expression in 852 patients affected with LCA or early-onset and severe retinal degeneration. They identified three RD3 mutations: c.112C.T, which caused p.R38*; c.136G.T, which caused p.E46*; and a 2 bp deletion in c.137–138, which caused a stop codon in amino acid 84. They suggested that the LCA12 phenotype in studied probands were highly similar to those of LCA1. Preising et al. [20] studied retinal degeneration in a consanguineous Kurdish family with LCA/early onset retinal dystrophy. They observed a severe form of retinal dystrophy that showed up at a very early age. Taken together, the mutants carrying defected RD3 resemble LCA1 and due to overlapping in the mechanism of action, distinguishing these two diseases is not feasible.
Conclusion
Results from our laboratory as well as the data from others have revealed an understanding that RD3 is a very crucial protein for trafficking, regulation as well as execution of the diseases, which involves GC1. The overlapping symptoms in patients carrying rd3 mutations (LCA12) with LCA1 would raise a possibility that the same mechanism of disease may be the cause of both diseases. In both cases, we believe that the impaired targeting of GC1 is the causal factor of the diseases. We are currently investigating the molecular mechanisms behind the targeting of GC1. The outcome will identify pathways and the key molecules, which execute such a sophisticated mechanism of action. The knowledge from the latter could be adapted for unraveling the trafficking mechanisms of other important photoreceptor membrane proteins. RD3 is a target for development of new therapeutic treatments for the debilitating LCA1.
Figure 1.
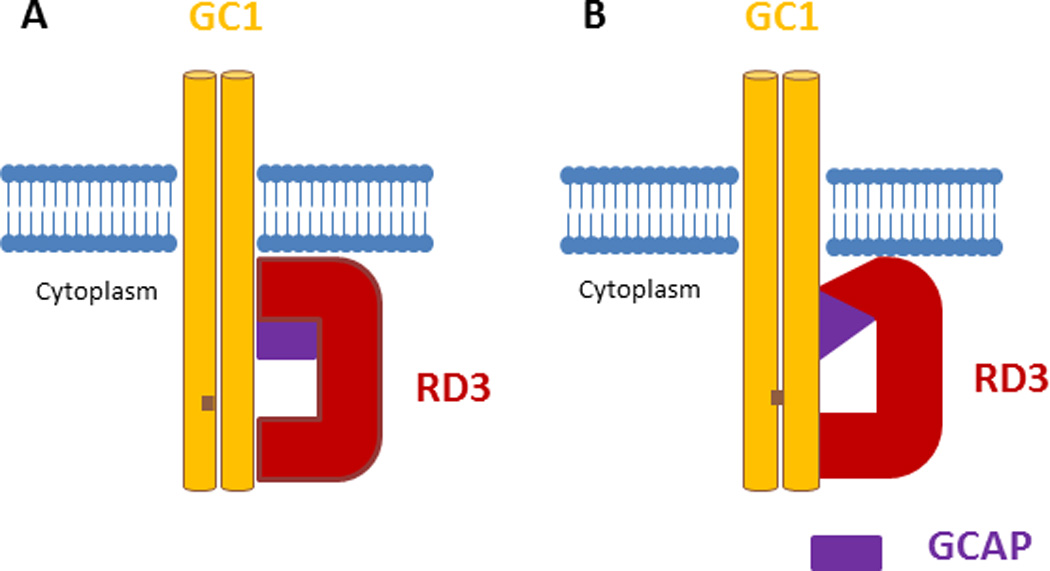
RD3 regulates GC1 in either of the following possible mechanisms: (A) the RD3-GC1 second binding site (see the text) competes with one or a combination of the GC1-GCAP1 binding site [26], and consequently negatively regulates the GC1 by preventing binding of GCAP1 to GC1. The major RD3-GC1 binding sites (amino acid 1055–1068) [13] regulate the targeting of GC1 to photoreceptor outer segment; or (B) RD3 interacts with GCAP1 and causes conformational changes in this protein; therefore, GCAP1 cannot activate GC1. In this model, the RD3-GC1 main binding site (amino acid 1055–1068) regulates the targeting of GC1 to outer.
Acknowledgment
I sincerely thank the following persons: 1) Dr. Robert Eugene Anderson, for his support and for assistance in revising the manuscript, 2) Dr. Robert S. Molday for all his intellectual input and his support, and 3) Dr. Muna I. Naash for all her scientific input and for her support.
Funding Acknowledgment:This study was supported by NIH/NEI P30 core grant (EY021725 to REA)and Knight Templar Eye Foundation (KTEF).
References
- Deretic D. Post-Golgi trafficking of rhodopsin in retinal photoreceptors. Eye (Lond) 1998;12(Pt 3b):526–530. doi: 10.1038/eye.1998.141. [DOI] [PubMed] [Google Scholar]
- 2.Li L, et al. Ablation of the x-linked retinitis pigmentosa 2 (rp2) gene in mice results in opsin mislocalization and photoreceptor degeneration. Invest Ophthalmol Vis Sci. 2013;54(7):4503–4511. doi: 10.1167/iovs.13-12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nojima S, et al. A point mutation in Semaphorin 4A associates with defective endosomal sorting and causes retinal degeneration. Nat Commun. 2013;4:1406. doi: 10.1038/ncomms2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hollingsworth TJ, Gross AK. Defective trafficking of rhodopsin and its role in retinal degenerations. Int Rev Cell Mol Biol. 2012;293:1–44. doi: 10.1016/B978-0-12-394304-0.00006-3. [DOI] [PubMed] [Google Scholar]
- 5.Sung CH, Chuang JZ. The cell biology of vision. J Cell Biol. 2010;190(6):953–963. doi: 10.1083/jcb.201006020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedman JS, et al. Premature truncation of a novel protein RD3 exhibiting subnuclear localization is associated with retinal degeneration. Am J Hum Genet. 2006;79(6):1059–1070. doi: 10.1086/510021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Azadi S, Molday LL, Molday RS. RD3, the protein associated with Leber congenital amaurosis type 12, is required for guanylate cyclase trafficking in photoreceptor cells. Proc Natl Acad Sci U S A. 2010;107(49):21158–21163. doi: 10.1073/pnas.1010460107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Danciger JS, et al. Genetic and physical maps of the mouse rd3 locus; exclusion of the ortholog of USH2A. Mamm Genome. 1999;10(7):657–661. doi: 10.1007/s003359901067. [DOI] [PubMed] [Google Scholar]
- 9.Chang B, et al. New mouse primary retinal degeneration (rd-3) Genomics. 1993;16(1):45–49. doi: 10.1006/geno.1993.1138. [DOI] [PubMed] [Google Scholar]
- 10.Pieke-Dahl S, et al. Hearing loss in the RBF/DnJ mouse, a proposed animal model of Usher syndrome type IIa. Hear Res. 1997;112(1–2):1–12. doi: 10.1016/s0378-5955(97)00087-7. [DOI] [PubMed] [Google Scholar]
- 11.Kukekova AV, et al. Canine RD3 mutation establishes rod-cone dysplasia type 2 (rcd2) as ortholog of human and murine rd3. Mamm Genome. 2009;20(2):109–123. doi: 10.1007/s00335-008-9163-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lavorgna G, et al. Identification and characterization of C1orf36, a transcript highly expressed in photoreceptor cells, and mutation analysis in retinitis pigmentosa. Biochem Biophys Res Commun. 2003;308(3):414–421. doi: 10.1016/s0006-291x(03)01410-4. [DOI] [PubMed] [Google Scholar]
- 13.Lisenbee CS, Karnik SK, Trelease RN. Overexpression and mislocalization of a tail-anchored GFP redefines the identity of peroxisomal ER. Traffic. 2003;4(7):491–501. doi: 10.1034/j.1600-0854.2003.00107.x. [DOI] [PubMed] [Google Scholar]