

Summary
Background
Development of neutralizing anti-factor (F)VIII antibodies (‘inhibitors’) is a serious clinical problem in hemophilia A. Increased inhibitor risk has been associated with certain FVIII missense substitutions, including R593C in the A2 domain.
Objectives
The aim of the present study was to identify T-cell epitopes in FVIII and characterize T-cell responses in two unrelated hemophilia A subjects sharing F8-R593C andHLA-DRB1*1101 genotypes. We hypothesized that the hemophilic substitution site coincides with an important T-cell epitope.
Patients/methods
The binding affinities of peptides for recombinant HLA-DR proteins were measured and compared with epitope prediction results. CD4+ T cells were stimulated using peptides and stained with fluorescent, peptide-loaded tetramers.
Results
The inhibitor subjects, but not HLA-matched controls, had high-avidity HLA-DRB1* 1101-restricted T-cell responses against FVIII589–608, which contains the hemophilic missense site. Antigen-specific T cells secreted Th1 and Th2 cytokines and proliferated in response to FVIII and FVIII592–603. FVIII589–608 bound with physiologically relevant (micromolar) IC50 values to recombinant DR0101, DR1101 and DR1501 proteins.
Conclusions
Hemophilia A patients with R593C missense substitutions and these HLA haplotypes had an increased incidence of inhibitors in our cohorts, supporting a paradigm in which presentation of FVIII epitopes containing the wild-type R593 influences inhibitor risk in this hemophilia A sub-population.
Keywords: factor VIII, hemophilia A, HLA, inhibitor, T-cell clones
Introduction
FVIII-neutralizing antibodies (‘inhibitors’) develop in some hemophilia A (HA) patients who receive factor (F)VIII infusions, resulting in bleeding complications [1–3]. Inhibitors are observed in 25–35% of severe HA patients but also can occur in mild/moderately severe HA [4,5]. Inhibitors have been associated with multiple F8 missense genotypes [6], including F8-R593C [7–9]. Multiple lines of evidence, including sequences/subclasses of inhibitory antibodies [10–13], efficacy of anti-CD40L inhibition [14] and the influence of CD4+ cell counts on antibody titers [15], indicate that inhibitor induction, affinity maturation and antibody class switching involve help from CD4+ T cells. Experimental evidence [16–18] has suggested that T-cell responses in mild/moderately severe HA may be directed against epitopes that contain the wild-type FVIII sequence at the hemophilic mutation site. Several studies have also indicated that B-cell epitopes may include the missense site [9,19–21]. Although T-cell proliferation in response to FVIII protein and peptides has been investigated [22–25], further study is warranted to establish the HLA restriction of T-cell epitopes within FVIII, particularly in the context of specific F8 genotypes. This information could improve estimates of inhibitor risk in defined sub-populations, allowing individualized treatment of high-risk patients by reducing their exposure to wild-type FVIII concentrates, and would motivate the design of less immunogenic versions of FVIII.
In the present study, two unrelated HA subjects with the F8-R593C genotype and similar HLA-DR haplotypes were studied to characterize T-cell responses and to identify epitopes within FVIII. The in vitro antigenicity of synthetic, overlapping peptides spanning the FVIII-A2, FVIII-C1 and FVIII-C2 domains were evaluated. To test our hypothesis that the hemophilic substitution site coincides with an important T-cell epitope, the binding of peptides containing R593 to various recombinant HLA-DR proteins was evaluated, and the results were correlated with reported inhibitor incidences in F8-R593C patient cohorts. Our findings support a paradigm in which binding and presentation of FVIII epitopes containing the wild-type R593 by several common HLA-DR alleles may influence the relative risk of developing an inhibitor in this HA subpopulation.
Materials and methods
Subjects and blood samples
Samples from two unrelated HA subjects and from eight HLA-DRB1*1101-matched healthy controls were used. Subject 1D (HLA-DRB1*1101 and DRB1*1302), from a Dutch cohort of F8-R593C patients, had an initial inhibitor titer of 22 Bethesda units (BU) mL−1 that declined but persisted for years [26]. Before inhibitor development, his baseline FVIII clotting activity (FVIII:C) was 20%; this declined to 1% at peak inhibitor titer, indicating that the inhibitor cross-reacted to neutralize his endogenous (hemophilic) FVIII, then increased to 1.4% in subsequent years [26]. He received FVIII to support an operation, which boosted his titer to 2 BU mL−1 and elicited cross-reactive antibodies against the FVIII A2 domain [9,27]. Subject 41A (HLA-DRB1*1101 and DRB1*1303), from a cohort of American F8-R593C patients, also developed an inhibitor after receiving FVIII infusions to support surgery. His baseline FVIII:C was 26%. In the month before and after peak titer (34 BU mL−1) his FVIII:C activity ranged from approximately 1% to 4%, indicating that the initial inhibitor cross-reacted to neutralize his endogenous (hemophilic) FVIII. He was treated with Rituximab and the titer declined. His most recent titer (2007) was undetectable (< 0.5 BU mL−1). Neither patient underwent immune tolerance induction. Blood samples from both subjects were collected > 6 months after their last FVIII infusion. Peripheral blood mononuclear cells (PBMCs) were obtained by Ficoll underlay and either frozen [7% dimethylsulfoxide (DMSO) in serum] or assayed immediately. Research was performed with IRB approval from the University of Washington Human Subjects Committee or the Universiteit van Amsterdam Medical Ethics Committee, with written informed consent.
FVIII peptides and protein
About 20-mer peptides (with 12-residue overlaps) with sequences (Table S1, supporting information) spanning the FVIII A2, C1, and C2 domains were synthesized and verified by mass spectrometry (Mimotopes, Clayton Victoria, Australia; Global Peptide Inc., Ft. Collins, CO, USA; Synpep, Dublin, CA, USA; Anaspec, San Jose, CA, USA). Peptides were dissolved at 10–20 mg mL−1 in DMSO or DMSO/water. Peptide pools contained equal amounts of 3–7 peptides (10 mg mL−1 total). Recombinant FVIII was obtained from Pharmacia/Upjohn (manufactured by CSL Behring GmbH, King of Prussia, PA, USA).
Peptide-binding predictions and assays
The binding affinities of peptides spanning the FVIII-A2 sequence to theHLA-DR1101 protein were predicted using the ProPred MHC class II binding algorithm (http://www.imtech.res.in/raghava/propred/) [28]. This program predicts affinities of peptide sequences for common HLA-DR molecules that present peptides to antigen-presenting cells, by evaluating their ability to fit into the canonical 9-residue peptide binding groove that is a feature of the MHC Class II. Every possible 9-mer sequence within FVIII-A2 was analyzed with the algorithm’s threshold value set to list binding scores above 0.8. The predicted set of peptides was further narrowed by excluding sequences with valine at position 1 of the DR1101 binding motif (i.e. the fit of the peptide into the groove), as this residue has been shown to bind weakly in this pocket [29]. Peptides with sequences containing R593 or C593 were evaluated regardless of their predicted binding scores.
Affinities of FVIII peptides for HLA-DR monomers were determined experimentally by competition assays. Recombinant HLA-DR0101, DR0301, DR0401, DR1101, DR1104 or DR1501 proteins were incubated with (i) FVIII peptides at 0.05, 0.1, 0.5, 1, 5, 10, and 50 μM plus (ii) biotinylated reference peptides that bound to specific DR proteins with high affinity (Table S1, supporting information). The DR proteins were then immobilized in wells coated with anti-DR capture antibody (L243) [30]. After washing, residual bound biotinylated peptide was labeled using europium-conjugated streptavidin (Perkin Elmer, Waltham, MA, USA) and quantified using a Victor 2D fluorometer (Perkin Elmer). Sigmoidal binding curves were simulated and IC50 values (concentration displacing 50% reference peptide) calculated using SigmaPlot (Systat Software, Inc., San Jose, CA, USA).
HLA-DR tetramers
HLA-DR1101 tetramers were generated as described [31]. Briefly, biotinylated recombinant DR1101 protein was incubated with pooled or individual peptides at 37 °C for 72 h with n-octyl-β-D-glucopyranoside and Pefabloc (Sigma-Aldrich, St Louis, MO, USA) and conjugated using R-phycoerythrin (PE) streptavidin (Biosource, Camarillo, CA, USA). Tetramer quality was confirmed by staining a reference T-cell clone (not shown).
Isolation and peptide stimulation of primary CD4+ T cells
T-cell isolation was carried out as described [17,32]. Frozen PBMCs from subject 1D were thawed, washed, and CD4+ T cells were fractionated by no-touch isolation (Miltenyi Biotec, Auburn, CA, USA). For subject 41A and HLA-matched control subjects, CD4+ T cells were fractionated from freshly isolated PBMCs. Three million autologous, CD4-depleted PBMCs were plated into 48-well plates for 1 h and then washed, leaving a layer of residual adherent cells behind as antigen-presenting cells. Two million purified CD4+ responder cells were then plated into these wells. Wells were stimulated with 10 μg mL−1 pooled peptides in T-cell medium (RPMI-1640 with 10% human serum, 1 mM sodium pyruvate, 50 U mL−1 penicillin and 50 μg mL−1 streptomycin), supplemented with 40 U mL−1 interleukin-2 (IL-2) (Hemagen, Waltham, MD, USA) on day 7, and maintained with medium and IL-2.
Tetramer guided epitope mapping (TGEM)
After 2 weeks, cells were analyzed with DR1101 tetramers as described [32,33]. For subject 1D and a control subject, 0.75 × 105 cells were incubated with tetramers (labeled with PE) loaded with individual FVIII peptides predicted to bind DR1101 (Table 1) [28] at 37 °C for 1 h, then incubated with anti-CD3-PerCP (BD Biosciences, San Jose, CA, USA), anti-CD4-APC (eBioscience, San Diego, CA, USA), and anti-CD25-FITC (eBioscience) at 4 °C for 20 min, and then analyzed on a FACSCalibur (Becton Dickinson, San Jose, CA, USA). For subject 41A and a second HLA-matched control subject, 0.75 × 105 cells were stained in a similar fashion, using tetramers loaded with peptide pools spanning the A2, C1, and C2 domains of FVIII (Table S1, supporting information). Tetramer-positive responses were decoded using tetramers loaded with individual peptides. To define an objective criterion for positive tetramer staining, CD4+ T cells from six non-hemophilic DR1101 donors were ‘sham’ stimulated using DMSO for 2 weeks and subsequently stained using a panel of DR1101 tetramers. One tetramer (FVIII381–400) gave significantly higher background staining, indicating a peptide-specific effect, while all others had a statistically similar background, allowing calculation of a mean background level (Fig. S1, supporting information). Our criterion for positive staining was designated as the mean background staining plus three times the standard error of the mean: 1.53% for FVIII381–400 and 0.46% for all other specificities. The latter is consistent with the cut-off used in previous published studies [17,18,30–33].
Table 1.
FVIII-A2 domain peptides predicted to bind DR1101 with high affinity, using the ProPred algorithm [28]
| FVIII-A2 peptides | Sequence | IC50* | Predicted DR1101 binding score† | |
|---|---|---|---|---|
| 1 | 429–488 | MAYTDETFKTREAIQHESGI | 8.9 ± 8 | 1.3 |
| 2 | 453–472 | LYGEVGDTLLIIFKNQASRP | 0.2 ± 0.1 | 2.7 |
| 3 | 469–488 | ASRPYNIYPHGITDVRPLYS | > 100 | 0.8 |
| 4 | 501–520 | FPILPGEIFKYKWTVTVEDG | > 100 | 0.9 |
| 5 | 529–548 | LTRYYSSFVNMERDLASGLI | 0.2 ± 0.06 | 1.9 |
| 6 | 541–560 | RDLASGLIGPLLICYKESVD | 25 ± 24 | 1.3 |
| 7 | 581–600 | ENRSWYLTENIQRFLPNPAG | 0.5 ± 0.4 | 0.8 |
| 8 | 581–600, 593C | ENRSWYLTENIQCFLPNPAG | > 100 | 1.5 |
| 9 | 589–608 | ENIQRFLPNPAGVQLEDPEF | 0.5 ± 0.4 | 1.4 |
| 10 | 589–608, 593C | ENIQCFLPNPAGVQLEDPEF | 1.5 ± 1.7 | 1.4 |
| 11 | 605–624 | DPEFQASNIMHSINGYVFDS | 8.9 ± 20 | 3.2 |
| 12 | 610–629 | ASNIMHSINGYVFDSLQLSV | > 100 | 1.0 |
| 13 | 637–656 | WYILSIGAQTDFLSVFFSGY | 0.3 ± 0.4 | 4.3 |
| 14 | 653–672 | FSGYTFKHKMVYEDTLTLFP | 20 ± 47 | 1.9 |
| 15 | 661–680 | KMVYEDTLTLFPFSGETVFM | > 20 | 1.5 |
| 16 | 677–696 | TVFMSMENPGLWILGCHNSD | > 100 | 2.0 |
| 17 | 685–704 | PGLWILGCHNSDFRNRGMTA | > 100 | 2.0 |
Peptides subsequently pooled and used to stimulate T cells are in bold font; the three remaining peptides contained predicted MHC Class II binding motifs (the 9-residue sequences predicted to fit into the HLA-DR1101 binding groove, underlined for each peptide) that were also present in one of the other peptides. Binding scores generated by Propred for all peptides are in the far right column (higher scores indicate stronger predicted affinity). Measured IC50 values under 10 are in bold font.
IC50 values are shown in μM ± the standard error of the mean. A lower IC50 value indicates stronger binding. IC50 > 100 indicates no detectable binding in the assay.
The binding score reflects expected binding affinity. Higher scores indicate stronger binding.
Isolation of T-cell clones and a polyclonal line
For all cultures that demonstrated tetramer-positive staining, FVIII-specific T cells were stained and isolated as described [17] after staining with DR1101-PE tetramers and anti-CD4-FITC (eBioscience). CD4+ tetramer-positive cells were sorted using a FACS Vantage (Becton Dickinson) into 96-well plates containing T-cell medium at one cell per well (to produce clones) or 250 cells per well (to produce a polyclonal line) and expanded by adding 2 μg mL−1 phytohemagglutinin and 200 000 irradiated PBMCs plus IL-2. Expanded cells were stained with DR1101-PE tetramers and analyzed on a FACSCalibur (Becton Dickinson).
Antigen-specific T-cell proliferation assay
T-cell proliferation was assessed as described [17,18]. Briefly, irradiated PBMCs from an HLA-matched (DRB1*1101) non-HA donor were plated at 105 cells per well in 100 μL T-cell medium. Peptides (final concentrations 10, 1, 0.1 and 0 μM) and T cells (104 cells per well) were added in 100 μL T-cell medium and plates were incubated at 37 °C. Wells were pulsed with [3H]thymidine (1 μCi per well) after 48 h and cells were harvested 18 h later. [3H]thymidine uptake was measured with a scintillation counter, and stimulation indices (SIs) were calculated as the counts per minute (cpm) of peptide-stimulated cultures divided by the cpm with no peptide added.
Cytokine sandwich ELISAs
Interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), IL-4, IL-10 and IL-17A were measured in supernatants by ELISA. Plates were coated with 100 μL of 2–4 μg mL−1 cytokine-specific antibody (anti-IFN-γ MD-1, anti-TNF-α MAb1, anti-IL-4 8D4-8, anti-IL-10 JES3-9D7 and anti-IL-17A eBio64-CAP17; eBioscience) in coating buffer (eBioscience) overnight at 4 °C, washed in phosphate-buffered saline (PBS) with 0.05% Tween 20, blocked with diluent solution (eBioscience) for 1 h at room temperature and washed again. Cytokine standard (100 μL; Cell Sciences or eBioscience) or 20–50 μL cell supernatant (plus diluent) was added to each well, and plates were incubated overnight at 4 °C and washed. Biotin-labeled antibody (100 μL at 2 μg mL−1) (anti-IFN-γ clone 4S.B3, anti-TNF-α MAb11, anti-IL-4 MP4-25D2, anti-IL-10 JES3-12G8 and anti-IL-17 eBio64DEC17; eBioscience) was added and incubated at room temperature for 1 h. Avidin horseradish peroxidase (eBioscience) was added (1:1000 dilution), incubated at room temperature for 30 min and washed. Super Aquablue substrate (100 μL; eBioscience) was then added and A405measured using a Bio-Rad 550 reader (Bio-Rad, Hercules, CA, USA). Cytokine concentrations were calculated from linear standard curves for each cytokine. Th1/Th2 ratios were calculated as: .
Results
Binding of FVIII peptides to DR1101
The two R593C subjects had the DRB1*1101 allele in common. An MHC class II binding computer prediction algorithm [28] was used to predict which FVIII-A2 peptides might bind to DR1101. For these predictions a higher score (see Table 1) indicates a greater likelihood that the corresponding peptide is capable of binding. Seventeen synthetic peptides corresponding to sequences with the highest predicted binding scores were then tested to empirically determine their in vitro affinities for recombinant DR1101 protein. Observed IC50 values ranged from 0.2 to > 100 μM, the detection limit. As summarized in Table 1, 8 of the 17 peptides with predicted binding scores above 0.8 bound to DR1101 with an IC50 under 10 μM. Notably, FVIII581–600, FVIII589–608 and FVIII589–608,593C, all of which contain the missense site, bound to DR1101 with reasonable affinity as compared with the influenza HA306–318 control peptide (Table 2), whereas FVIII581–600,593C did not.
Table 2.
Binding of peptides to DRB1 proteins
| Class II protein | Reference peptide* (IC50 in μM) | IC50†(μM) FVIII581–600 |
IC50† (μM) FVIII581–600,593C |
IC50† (μM) FVIII589–608 |
IC50† (μM) FVIII589–608,593C |
|---|---|---|---|---|---|
| DR0101 | HA306–318 (0.26) | 38 ± 30 | 50 ± 3 | 4.2 ± 0.3 | 8.3 ± 0.7 |
| DR0301 | Myo137–148 (0.82) | 44 ± 7 | NB | 50 ± 4 | NB |
| DR0401 | HA306–318 (3.1) | 48 ± 7 | NB | 38 ± 3 | NB |
| DR1101 | HA306–318 (5.0) | 1.1 ± 0.1 | NB | 1.1 ± 0.1 | 6.3 ± 0.6 |
| DR1104 | VP1634–44 (3.1) | 9.8 ± 0.8 | NB | 59 ± 3 | NB |
| DR1501 | MBP84–102 (0.05) | 3.7 ± 0.4 | 56 ± 4 | 4.6 ± 0.4 | 9.8 ± 0.6 |
NB, no binding.
IC50 indicates the strength of interaction between the class II protein and FVIII peptide compared to a reference peptide (sequences shown in Table S1, supporting information). IC50 values for reference peptides are listed in parentheses. Lower numbers indicate stronger interactions.
Values shown ± standard error of the mean.
T-cell responses to selected peptides
For inhibitor subject 1D, the number of cryo-preserved cells available for study was only sufficient to test responses to a limited number of peptides. Therefore, peptides that contained predicted FVIII-A2 domain epitopes (Table 1) were utilized to query his T-cell responses. These were divided into two 7-peptide pools, which were then used to stimulate CD4+ T cells from the subject and a control subject. T cells from the inhibitor and control subjects were cultured for 14 days and then stained using DR1101 tetramers loaded with individual peptides. A clear population of CD4+ T cells was stained by tetramers loaded with FVIII589–608 (Fig. 1), which bound to DR1101 with high affinity (IC50 = 0.5 ± 0.4 μM). Weaker positive staining was observed for FVIII429–448, FVIII469–488 and FVIII581–600, which bound to DR1101 with IC50 values of 0.5 ± 0.4, 8.9 ± 8 and approximately 100 μM. Notably, tetramer staining was negative for CD4+ T cells stimulated by the hemophilic peptide FVIII589–608,593C. Attempts to stain T cells from the control subject using tetramers loaded with each of the 14 peptides containing predicted epitopes (Table 1) yielded negative results (not shown).
Fig. 1.
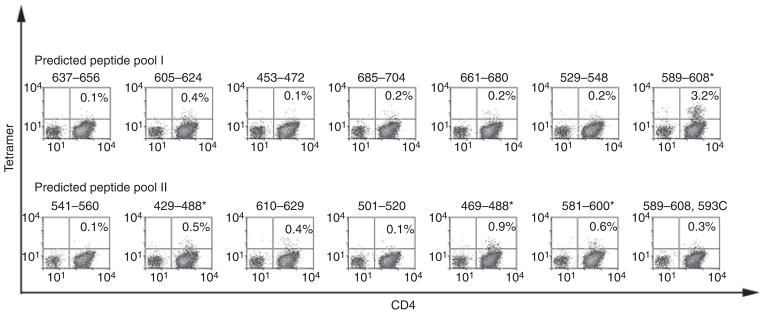
T-cell epitopes recognized by subject 1D. CD4+ cells were stimulated using two pools of seven factor (F)VIII peptides each with predicted HLA-DRB1*1101-restricted epitopes. Peptides that elicited a tetramer-positive CD4+ population (greater than three times the standard error of the mean above background) are indicated by asterisks. These included FVIII429–448, FVIII469–488, FVIII581–600 and FVIII581–600.
Mapping epitopes in the FVIII A2, C1 and C2 domains
CD4+ T cells freshly isolated from subject 41A were stimulated with peptides spanning the FVIII A2, C1 and C2 domains, including two peptides with the R593C substitution (Table S1, supporting information). Cells were cultured and evaluated for responses by staining with fluorescent, peptide-loaded DR1101 tetramers. Representative results are shown in Fig. 2A. Tetramer staining was above background for CD4+ cells stimulated with FVIII-A2 peptide pools 1, 2 and 6 and with FVIII-C2 pool 1. Therefore, T cells stimulated with these pools were selected for further analysis (decoding) using tetramers loaded with single peptides that comprised these pools (Fig. 2B). T cells stimulated using peptide pool 6 showed positive staining by tetramers loaded with FVIII589–608 and FVIII581–600, both of which bound with IC50 values of 0.5 ± 0.4 μM. FVIII-A2 peptide pool 2 and FVIII-C2 peptide pool 1 showed weaker positive staining by tetramers loaded with FVIII421–440 and FVIII2187–2205, respectively. The IC50 values for these peptides were 5.0 ± 18 and 12 ± 26 μM. The apparent positive staining of A2 peptide pool 1 was because of FVIII381–400, which caused high peptide-specific background staining. Tetramer-stained cells were generally CD25+, suggesting they were activated (not shown). Notably, staining with tetramers loaded with FVIII-A2 peptide pool 11, which contains two peptides with the hemophilic R593C substitution, was negative, indicating that neither peptide containing C593 elicited a high-avidity T-cell response. The same peptide-loaded tetramers were used to evaluate T-cell responses for an HLA-DRB1*1101 control subject. All staining results using T cells from this subject were negative (not shown).
Fig. 2.

T-cell epitopes recognized by subject 41A. (A) CD4+ cells were stimulated for 2 weeks with pooled, overlapping peptides spanning the factor FVIII A2, C1, and C2 domains. Positive and representative negative tetramer staining results are shown (fluorescent labeling greater than three times the standard error of the mean above background was considered positive). (B) Decoding by staining the same cells with HLA-DR1101 tetramers loaded with individual peptides. Peptides that elicited a tetramer-positive CD4+ population are indicated by asterisks. These included FVIII421–440, FVIII581–600, FVIII581–600 and FVIII2187–2205 (note that the tetramer loaded with FVIII381–400 had an uncharacteristically high background, indicating nonspecific binding to CD4+ cells).
Isolating T-cell clones and evaluating additional control subjects
To facilitate further study of FVIII-specific T-cell responses, cells from each positive well were stained again and single-cell sorted to obtain FVIII-specific T-cell clones and lines (as described in Materials and Methods). Multiple high-affinity FVIII589–608-specific T-cell clones and lines were isolated. Sorted cells with other specificities did not expand. To evaluate the disease specificity of the DR1101-restricted T-cell responses observed in these two-inhibitor subjects, T cells from six additional non-HA subjects were stimulated with FVIII peptides and stained with tetramers after 2 weeks of in vitro culture. In all cases, tetramer staining was below the positivity threshold (not shown). In spite of the limited number of subjects, the magnitude of FVIII589–608-specific tetramer staining observed for hemophilic subjects with inhibitors was significantly higher than for healthy subjects (P = 0.045). No other tetramer-positive signals were statistically different for patients and controls.
Binding of truncated peptides to DR1101
To determine the minimal T-cell epitope within FVIII589–608, binding of truncated peptides to recombinant DR1101 was measured in a competition assay (Fig. 3A). While FVIII592–603 bound with affinity comparable to FVIII589–608, the FVIII593–603 and FVIII594–603 peptides bound with 10- and 25-fold lower affinity, respectively. This suggests that residue F594 occupies position 1 of the canonical, nine-residue peptide-binding groove in HLA-DR1101 (Fig. 3B), consistent with an epitope predicted by the computer program Propred [28].
Fig. 3.

Defining the minimal DR1101-restricted epitope within FVIII589–608. (A) In vitro binding of truncated peptides FVIII592–603, FVIII593–603 and FVIII594–603 and the influenza HA306–318 control to HLA-DR1101 protein (arrow indicates increasing affinity). (B) Schematic of the core HLA-DR1101 binding region within FVIII592–603, based on experimental results and the published DR1101 binding motif [29]. Arrows indicate DR1101 contact residues (pointing downward) and possible T-cell receptor contact residues (pointing upward).
T-cell clone proliferation and cytokine secretion
Three antigen-specific T-cell clones and one polyclonal T-cell line were isolated from the same peptide-stimulated cultures used for epitope mapping. Clone 1D-1 was stained by tetramers loaded with FVIII589–608 but not with FVIII581–600 or an unrelated influenza control peptide, HA306–318 (Fig. 4A). T cells isolated from subject 41A gave similar results (not shown), indicating that these cells recognize FVIII589–608. Proliferation assays were conducted for these T cells using FVIII589–608 and truncated versions of this peptide to determine the functional epitope. In all cases, residue R593 was essential for maximal proliferation (Fig. 4B–E). Interestingly, peptides containing either R593 (wild-type sequence) or C593 (hemophilic sequence) elicited similar proliferation. These T cells proliferated well above background in response to wild-type FVIII protein (Fig. 5).
Fig. 4.

Tetramer staining and proliferation of T-cell clones and a polyclonal T-cell line. (A) Staining of clone 1D-1 using tetramers loaded with FVIII581–600, FVIII589–608 or the control influenzaHA306–318 peptide. (B–E) Clones from subject 1D(clone 1D-1, B), subject 41A (clones 41A-1 and 41A-2; C,D) and a polyclonal T-cell line from subject 41A (41A Line, E) were stimulated with FVIII589–608, FVIII592–603, FVIII593–603, FVIII594–603 and the hemophilic FVIII589–608,593C peptide at 0, 0.1, 1.0 and 10 μM. [3H]thymidine uptake was measured in triplicate wells. Data are expressed as stimulation index values ± standard deviation (SI ± SD), where SI = measured counts/baseline counts.
Fig. 5.

Proliferation of T-cell clones and polyclonal line in response to factor FVIII. Clones 1D-1, 41A-1 and 41A-2 and a polyclonal T-cell line from subject 41A were stimulated with 0, 0.1 or 0.2 μg mL−1 of FVIII protein. [3H]thymidine uptake was measured in triplicate wells (data expressed as SI ± SD).
Supernatants harvested 48 h after incubation with FVIII589–608 were assayed to determine the cytokines secreted in response to FVIII peptide stimulation. Both the T-cell clones and the polyclonal line secreted robust levels of IFN-γ, significant amounts of TNF-α, IL-4 and IL-10, but no IL-17 (Fig. 6). Th1/Th2 ratios ranged from 1.8 to 31.6. In the absence of peptide stimulation, cytokine secretion was negligible.
Fig. 6.
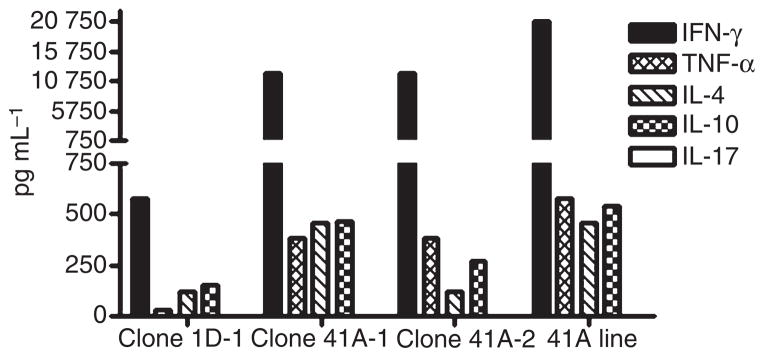
Cytokine secretion by T-cell clones and polyclonal line. Clones from subject 1D and 41A and a polyclonal T-cell line from subject 41A were stimulated with various concentrations of FVIII589–608 peptide for 48 h. Supernatants were collected and analyzed by ELISA to quantify interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), interleukin-4 (IL-4), IL-10 and IL-17 secretion. Cytokines elicited at peptide concentrations of 10 μg mL−1 are shown, representing averages from triplicate wells.
Binding of FVIII peptides to additional HLA-DR proteins
To determine which common HLA-DR proteins [34] can effectively present FVIII peptides containing the wild-type R593, the binding of FVIII589–608, FVIII589–608,593C, FVIII581–600, and FVIII581–600,593C to DR0101, DR0301, DR0401, DR1101, DR1104 and DR1501 proteins, which represent prevalent HLA-DR haplotypes in the Dutch and American study population, was measured. As summarized in Table 2, FVIII589–608 and FVIII589–608,593C bound to DR0101, DR1101 and DR1501. FVIII581–600 bound to DR1101, DR1104 and DR1501. These alleles are found in 33% of individuals in European and non-indigenous North American populations [34]. This suggests that a substantial fraction of HA patients with F8-R593C, those with DRB1*01, DRB1*11, or DRB1*15 haplotypes, may be at increased risk of inhibitor formation. Of course, additional alleles that were not tested in the present study may also be associated with increased inhibitor risk as well.
Discussion
Inhibitory antibodies are the most severe complication affecting HA patients with access to FVIII replacement therapy. However, predicting inhibitor development for individuals remains challenging because risk factors include genetic and environmental components [35–43]. Clinical and experimental evidence suggests that responses to FVIII in mild/moderately severe HA can be triggered by differences between endogenous and infused FVIII and can be potentiated by immune challenges [17,26]. The present study of two unrelated HA subjects with established inhibitors (sharing the F8-R593C genotype and HLA-DRB1*1101 allele) demonstrated robust T-cell responses directed against an epitope that contains the wild-type FVIII sequence at the hemophilic mutation site. Mild HA patients would only be exposed to this epitope upon treatment or prevention of bleeding episodes by infusions with wild-type FVIII concentrates. Our experiments also showed that the invitro binding affinity of the wild-type FVIII peptide containing R593 for DR1101 was stronger than that of several other peptides containing predicted high-affinity epitopes. In fact, there was only a weak correlation (R2 = 0.14) between the observed IC50 value and the predicted binding score. These results indicate the importance of complementing epitope prediction methods with physical peptide-binding measurements and T-cell assays in order to obtain an accurate assessment of immunogenicity. Many FVIII peptides bound to DR1101 with high affinity but did not elicit T-cell responses, suggesting that both the mild HA subjects and non-hemophilic individuals have central tolerance to these sequences. Some of these sequences may, however, elicit immune responses in severe HA subjects with no circulating FVIII protein.
In agreement with previous studies of mild HA subjects [16,17,44], the experimental results indicate robust T-cell responses directed against an epitope that contains the wild-type sequence at the hemophilic mutation site. For subject 1D (Fig. 1), analysis with a limited set of peptides revealed a high-affinity T-cell response directed against FVIII589–608 and weaker responses directed against an overlapping peptide (FVIII581–600) and two distinct sequences (FVIII429–448 and FVIII469–488) which appeared to be of lower affinity. T-cell responses of subject 41A were queried using a much larger panel of overlapping FVIII peptides that spanned the FVIII A2, C1 and C2 domains (Fig. 2), and FVIII589–608 again elicited a high-affinity response. Weaker, apparently low-affinity responses were directed against FVIII421–440, FVIII581–600 and FVIII2187–2205. Expanded FVIII589–608-specific T cells from both HA subjects proliferated in response to FVIII protein, indicating that this peptide mimics a naturally processed epitope. Although it is still possible that additional T-cell responses to regions of FVIII not tested here, for example, the A1, A3 or B domains, may also contribute to FVIII immunogenicity, our results suggest that high-affinity HLA-DRB1*1101-restricted T-cell responses to an epitope within FVIII589–608 contributed to inhibitor formation in both of these HA subjects. Among the peptides that elicited positive responses, only FVIII589–608 had significantly higher staining for HA subjects (P = 0.045) than for healthy control subjects. However, it should be noted that because of the limited number of HA subjects analyzed, there were insufficient data to conclude that responses to FVIII589–608 occur only in hemophilic subjects with inhibitors. In fact, in a previous study of brothers who shared the DR0101 haplotype and had mild HA as a result of the A2201P missense genotype, both subjects had T-cell responses to the same peptide (which included the mutation site) even although they were discordant for inhibitor development [18]. However, T-cell clones isolated from their blood had distinctly different phenotypes, and IgG concentrated from plasma donated by the ‘non-inhibitor’ brother had a measurable Bethesda titer, indicating he in fact had a circulating but sub-clinical inhibitor [18,44]. Therefore, there is accumulating evidence that T-cell responses such as those characterized here indicate the presence of anti-FVIII antibodies, although actual titers may vary significantly.
T-cell help can drive development and maturation of antibody responses. T cells can also exhibit regulatory phenotypes, including FoxP3 expression, anergy and IL-10 secretion [45]. Therefore, analysis of tetramer-stained, FVIII-specific T-cell clones and the polyclonal T-cell line included quantification of representative Th1 and Th2 cytokines, IL-10 and IL-17. FVIII-specific T cells from both inhibitor subjects secreted robust levels of IFN-γ and detectable TNF-α, IL-4 and IL-10, with Th1/Th2 ratios suggesting varying degrees of Th1 polarization. This is consistent with previous observations that IFN-γ and IL-4 are both secreted by FVIII-stimulated CD4+ T cells from inhibitor subjects [46]. A recent study using a HA mouse model suggested that Th1-polarization was associated with tolerance [47]. A study of a mild HA subject [44] showed that HLA-DRB1*0101-restricted T-cell clones isolated 2 years after inhibitor formation were strongly Th2 polarized, whereas clones isolated at earlier time points secreted IFN-γ and IL-17. Another study of human inhibitor responses concluded that Th2-driven inhibitors occur when the anti-FVIII antibody response is intense, whereas Th1 cells may be involved in the long-term maintenance of anti-FVIII antibody synthesis [48]. Additional studies evaluating changes in T-cell phenotypes and responses over time, particularly in subjects matched by disease severity, genetic characteristics including F8 genotype and HLA haplotype, and treatment regime, are needed to determine mechanisms leading to tolerance vs. high-titer anti-FVIII antibodies.
Initial T-cell proliferation experiments revealed the existence of an epitope within the FVIII589–608 peptide. Although responses of the single clone obtained from subject 1D were not as vigorous as those of the cells isolated from subject 41A, proliferation assays indicated robust responses to FVIII592–603 for all three clones and for the polyclonal line. Their proliferation was less pronounced in response to FVIII594–603, highlighting the importance of the R593 residue. The experimental results and prediction algorithms both indicated that F594 occupies position 1 in the DR1101 peptide-binding groove, while N597, A599 and Q602 fit into the pockets at positions 4, 6 and 9, and adjacent and intervening side chains project outward to interact with T-cell receptors [49].
Interestingly, all three expanded T-cell clones and the polyclonal line proliferated in response to the hemophilic FVIII589–608,593C peptide, in spite of the fact that neither primary nor cloned T cells were stained by tetramers loaded with this peptide, suggesting a lower-avidity interaction of T cells with tetramers or antigen-presenting cells when the hemophilic peptide was presented on the DR1101 surface. Peptide affinities for DR1101 are determined by the fit of peptide ‘anchor’ residues into specific pockets in the class II binding groove, whereas tetramer staining of cells has the additional requirement that the DR1101-peptide complex be recognized by the T-cell receptor on the surface of the responding T cell. Residue 593 is adjacent to the classic 9-residue class II binding motif, but it clearly contributes to binding affinities. The results imply that although the tetramer loaded with the hemophilic peptide was less effective in staining the T cells (so that labeled cells were below the threshold for a ‘tetramer-positive’ response) this lower-avidity interaction was nevertheless strong enough to stimulate T-cell proliferation. This raises the possibility that T cells initially activated by wild-type FVIII can cross-react with wild-type and hemophilic FVIII. This cross-reactivity at the T-cell level may be analogous with cross-reactivity seen at the B-cell level for both subjects, whose inhibitors neutralized their endogenous FVIII. Cross-maintenance of FVIII589–608-specific T cells by the endogenous peptide/protein containing the substitution R593C may also contribute to the persistence of immune responses to FVIII; indeed, inhibitors and epitope-specific T-cell responses to FVIII have been observed in mild HA subjects even years after their last infusion [17,44].
Peptide affinities for a series of HLA-DR proteins indicated that DR0101, DR1104 and DR1501, but not DR0301 and DR0401, can present FVIII peptides containing R593. This reinforces previous suggestions that while HLA haplotypes are not a general risk factor for inhibitor development, certain combinations of FVIII genotype and HLA haplotype may confer an increased risk [7,50]. In the American and Dutch cohorts of F8-R593C hemophilia subjects (69 total subjects) 9 of the 10 (90%) inhibitor subjects had DRB1*01, DRB1*11 or DRB1*15 haplotypes, whereas 26 of the 59 (44%) subjects without inhibitors had these haplotypes [7 and unpublished data]. These alleles are found in 33% of individuals in European and non-indigenous North American populations [34]. Fisher’s exact probability test indicates that this is a significant increase (P-value = 0.0076) in inhibitor risk for subjects with these alleles, as compared with all other class II HLA types. However, these results should be replicated using larger populations and accounting for confounding factors such as intensity of treatment [9] and genetic determinants such as IL-10 [36] and TNF-α [38] polymorphisms, before drawing firm conclusions about HLA-associated inhibitor risks.
T-cell responses to FVIII were characterized for two unrelated individuals in the present study. Both demonstrated Th1-polarized responses (with accompanying low-level IL-4 secretion) directed against a common HLA-DRB1*1101-restricted epitope, supporting the notion that T-cell responses to epitopes that contain the hemophilic substitution site contribute to inhibitor formation in mild/moderately severe HA. These T-cell responses may occur whenever epitopes containing the wild-type sequence at a missense site are bound to and presented by particular DR proteins at the surface of an antigen-presenting cell. Knowledge of HLA-restricted T-cell epitopes in FVIII and their binding affinities for HLA-DR and possibly other MHC class II proteins should improve predictions of inhibitor risk. Only certain MHC class II proteins on the surface of antigen-presenting cells will be capable of effectively presenting particular FVIII peptides. Additionally, the characterization of immunodominant FVIII T-cell epitopes and T-cell responses will facilitate rational modifications of FVIII, for example, amino acid substitutions at residues shown to be critical for the interaction with particular MHC Class II molecules. Such FVIII sequence modification could prevent effective epitope presentation and subsequent T-cell activation. This approach should be particularly promising to develop less immunogenic proteins to treat bleeding in individuals with HA genotypes that are associated with increased inhibitor risk.
Supplementary Material
Acknowledgments
Arthur R. Thompson, MD, PhD, Charles Cooper, RN and Susan J. Geraghty, RN managed clinical aspects of the American cohort studies. Aru Arumuganathan provided technical assistance and Douglas C. Bolgiano, MS assisted with statistical analysis. We are grateful to all subjects for their generous blood donations. Supported by a grant from CSL Behring, Inc. (KPP, 2007–2009), a Bayer Hemophilia Award (KPP, 2007–2009), NIH HL R01 07109 (ART) and NIH 1RC2HL101851-01 (KPP).
Footnotes
Disclosure of Conflict of Interest
The authors state that they have no conflict of interest.
Additional Supporting Information may be found in the online version of this article:
Figure S1. Background staining threshold for tetramer reagents.
Table S1. Peptide sequences.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Hoyer LW. Factor VIII Inhibitors. Curr Opin Hematol. 1995;2:365–71. doi: 10.1097/00062752-199502050-00007. [DOI] [PubMed] [Google Scholar]
- 2.Bray GL, Gomperts ED, Courter S, Gruppo R, Gordon EM, Manco-Johnson M, Shapiro A, Scheibel E, White G, 3rd, Lee M. A multicenter study of recombinant factor VIII (recombinate): safety, efficacy, and inhibitor risk in previously untreated patients with hemophilia A. The Recombinate Study Group. Blood. 1994;83:2428–35. [PubMed] [Google Scholar]
- 3.Kreuz W, Ettingshausen CE, Zyschka A, Oldenburg J, Saguer IM, Ehrenforth S, Klingebiel T. Inhibitor development in previously untreated patients with hemophilia A: a prospective long-term follow-up comparing plasma-derived and recombinant products. Semin Thromb Hemost. 2002;28:285–90. doi: 10.1055/s-2002-32664. [DOI] [PubMed] [Google Scholar]
- 4.Hay CR. Factor VIII inhibitors in mild and moderate-severity haemophilia A. Haemophilia. 1998;4:558–63. doi: 10.1046/j.1365-2516.1998.440558.x. [DOI] [PubMed] [Google Scholar]
- 5.d’Oiron R, Pipe SW, Jacquemin M. Mild/moderate haemophilia A: new insights into molecular mechanisms and inhibitor development. Haemophilia. 2008;14(S3):138–46. doi: 10.1111/j.1365-2516.2008.01730.x. [DOI] [PubMed] [Google Scholar]
- 6.Oldenburg J, Pavlova A. Genetic risk factors for inhibitors to factors VIII and IX. Haemophilia. 2006;12(S6):15–22. doi: 10.1111/j.1365-2516.2006.01361.x. [DOI] [PubMed] [Google Scholar]
- 7.Bril WS, MacLean PE, Kaijen PH, van den Brink EN, Lardy NM, Fijnvandraat K, Peters M, Voorberg J. HLA class II genotype and factor VIII inhibitors in mild haemophilia A patients with an Arg593 to Cys mutation. Haemophilia. 2004;10:509–14. doi: 10.1111/j.1365-2516.2004.01011.x. [DOI] [PubMed] [Google Scholar]
- 8.Thompson AR, Murphy ME, Liu M, Saenko EL, Healey JF, Lollar P, Scandella D. Loss of tolerance to exogenous and endogenous factor VIII in a mild hemophilia A patient with an Arg593 to Cys mutation. Blood. 1997;90:1902–10. [PubMed] [Google Scholar]
- 9.Eckhardt CL, Menke LA, van Ommen CH, van der Lee JH, Geskus RB, Kamphuisen PW, Peters M, Fijnvandraat K. Intensive peri-operative use of factor VIII and the Arg593 → Cys mutation are risk factors for inhibitor development in mild/moderate hemophilia A. J Thromb Haemost. 2009;7:930–7. doi: 10.1111/j.1538-7836.2009.03357.x. [DOI] [PubMed] [Google Scholar]
- 10.van den Brink EN, Turenhout EA, Davies J, Bovenschen N, Fijnvandraat K, Ouwehand WH, Peters M, Voorberg J. Human antibodies with specificity for the C2 domain of factor VIII are derived from VH1 germline genes. Blood. 2000;95:558–63. [PubMed] [Google Scholar]
- 11.Fulcher CA, de Graaf Mahoney S, Zimmerman TS. FVIII inhibitor IgG subclass and FVIII polypeptide specificity determined by immunoblotting. Blood. 1987;69:1475–80. [PubMed] [Google Scholar]
- 12.Shima M. Characterization of factor VIII inhibitors. Int J Hematol. 2006;83:109–18. doi: 10.1532/IJH97.05160. [DOI] [PubMed] [Google Scholar]
- 13.Lacroix-Desmazes S, Misra N, Bayry J, Mohanty D, Kaveri SV, Kazatchkine MD. Autoantibodies to factor VIII. Autoimmun Rev. 2002;1:105–10. doi: 10.1016/s1568-9972(01)00017-9. [DOI] [PubMed] [Google Scholar]
- 14.Ewenstein BM, Hoots WK, Lusher JM, DiMichele D, White GC, 2nd, Adelman B, Nadeau K. Inhibition of CD40 ligand (CD154) in the treatment of factor VIII inhibitors. Haematologica. 2000;85:35–9. [PubMed] [Google Scholar]
- 15.Bray GL, Kroner BL, Arkin S, Aledort LW, Hilgartner MW, Eyster ME, Ragni MV, Goedert JJ. Loss of high-responder inhibitors in patients with severe hemophilia A and human immunodeficiency virus type 1 infection: a report from the Multi-Center Hemophilia Cohort Study. Am J Hematol. 1993;42:375–9. doi: 10.1002/ajh.2830420408. [DOI] [PubMed] [Google Scholar]
- 16.Jacquemin M, Vantomme V, Buhot C, Lavend’homme R, Burny W, Demotte N, Chaux P, Peerlinck K, Vermylen J, Maillere B, van der Bruggen P, Saint-Remy JM. CD4+ T-cell clones specific for wild-type factor VIII: a molecular mechanism responsible for a higher incidence of inhibitor formation in mild/moderate hemophilia A. Blood. 2003;101:1351–8. doi: 10.1182/blood-2002-05-1369. [DOI] [PubMed] [Google Scholar]
- 17.James EA, Kwok WW, Ettinger RA, Thompson AR, Pratt KP. T-cell Responses over time in a mild hemophilia A inhibitor subject: epitope identification and transient immunogenicity of the corresponding self-peptide. Thromb Haemost. 2007;5:2399–407. doi: 10.1111/j.1538-7836.2007.02762.x. [DOI] [PubMed] [Google Scholar]
- 18.Ettinger RA, James EA, Kwok WW, Thompson AR, Pratt KP. HLA-DR-restricted T-cell responses to Factor VIII epitopes in a mild haemophilia A family with missense substitution A2201P. Haemophilia. 2010;16:44–55. doi: 10.1111/j.1365-2516.2008.01905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peerlinck K, Jacquemin MG, Arnout J, Hoylaerts MF, Gilles JG, Lavend’homme R, Johnson KM, Freson K, Scandella D, Saint-Remy JM, Vermylen J. Antifactor VIII antibody inhibiting allogeneic but not autologous factor VIII in patients with mild hemophilia A. Blood. 1999;93:2267–73. [PubMed] [Google Scholar]
- 20.d’Oiron R, Lavergne JM, Lavend’homme R, Benhida A, Bordet JC, Negrier C, Peerlinck K, Vermylen J, Saint-Remy JM, Jacquemin M. Deletion of alanine 2201 in the FVIII C2 domain results in mild hemophilia A by impairing FVIII binding to VWF and phospholipids and destroys a major FVIII antigenic determinant involved in inhibitor development. Blood. 2004;103:155–7. doi: 10.1182/blood-2003-04-1321. [DOI] [PubMed] [Google Scholar]
- 21.Jacquemin M, Benhida A, Peerlinck K, Desqueper B, Vander Elst L, Lavend’homme R, d’Oiron R, Schwaab R, Bakkus M, Thielemans K, Gilles JG, Vermylen J, Saint-Remy JM. A human antibody directed to the factor VIII C1 domain inhibits factor VIII cofactor activity and binding to von Willebrand factor. Blood. 2000;95:156–63. [PubMed] [Google Scholar]
- 22.Hu GL, Okita DK, Conti-Fine BM. T cell recognition of the A2 domain of coagulation factor VIII in hemophilia patients and healthy subjects. J Thromb Haemost. 2004;2:1908–17. doi: 10.1111/j.1538-7836.2004.00918.x. [DOI] [PubMed] [Google Scholar]
- 23.Reding MT, Wu H, Krampf M, Okita DK, Diethelm-Okita BM, Key NS, Conti-Fine BM. CD4+ T cell response to factor VIII in hemophilia A, acquired hemophilia and healthy subjects. Thromb Haemost. 1999;82:509–15. [PubMed] [Google Scholar]
- 24.Jones TD, Phillips WJ, Smith BJ, Bamford CA, Nayee PD, Baglin TP, Gaston JS, Baker MP. Identification and removal of a promiscuous CD4+ T cell epitope from the C1 domain of factor VIII. J Thromb Haemost. 2005;3:991–1000. doi: 10.1111/j.1538-7836.2005.01309.x. [DOI] [PubMed] [Google Scholar]
- 25.Reding MT, Wu H, Krampf M, Okita DK, Diethelm-Okita BM, Christie BA, Key NS, Conti-Fine BM. Sensitization of CD4+ T cells to coagulation factor VIII: response in congenital and acquired hemophilia patients and in healthy subjects. ThrombHaemost. 2000;84:643–52. [PubMed] [Google Scholar]
- 26.Fijnvandraat K, Turenhout EA, van den Brink EN, ten Cate JW, van Mourik JA, Peters M, Voorberg J. The missense mutation Arg593 → Cys is related to antibody formation in a patient with mild hemophilia A. Blood. 1997;89:4371–7. [PubMed] [Google Scholar]
- 27.Bril WS, Turenhout EA, Kaijen PH, van den Brink EN, Koopman MM, Peters M, Voorberg J. Analysis of factor VIII inhibitors in a haemophilia A patient with an Arg593 → Cys mutation using phage display. Br J Haematol. 2002;119:393–6. doi: 10.1046/j.1365-2141.2002.03856.x. [DOI] [PubMed] [Google Scholar]
- 28.Singh H, Raghava GP. ProPred: prediction of HLA-DR binding sites. Bioinformatics. 2001;17:1236–7. doi: 10.1093/bioinformatics/17.12.1236. [DOI] [PubMed] [Google Scholar]
- 29.Verreck FA, van de Poel A, Drijfhout JW, Amons R, Coligan JE, Konig F. Natural peptides isolated from Gly86/Val86-containing variants of HLA-DR1, -DR11, -DR13, and -DR52. Immunogenetics. 1996;43:392–7. doi: 10.1007/BF02199809. [DOI] [PubMed] [Google Scholar]
- 30.James EA, Moustakas AK, Berger D, Huston L, Papadopoulos GK, Kwok WW. Definition of the peptide binding motif within DRB1*1401 restricted epitopes by peptide competition and structural modeling. Mol Immunol. 2008;45:2651–9. doi: 10.1016/j.molimm.2007.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Novak EJ, Liu AW, Nepom GT, Kwok WW. MHC class II tetramers identify peptide-specific human CD4(+) T cells proliferating in response to influenza A antigen. J Clin Invest. 1999;104:R63–7. doi: 10.1172/JCI8476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.James EA, Bui J, Berger D, Huston L, Roti M, Kwok WW. Tetramer-guided epitope mapping reveals broad, individualized repertoires of tetanus toxin-specific CD4+ T cells and suggests HLA-based differences in epitope recognition. Int Immunol. 2007;19:1291–301. doi: 10.1093/intimm/dxm099. [DOI] [PubMed] [Google Scholar]
- 33.Novak EJ, Liu AW, Gebe JA, Falk BA, Nepom GT, Koelle DM, Kwok WW. Tetramer-guided epitope mapping: rapid identification and characterization of immunodominant CD4+ T cell epitopes from complex antigens. J Immunol. 2001;166:6665–70. doi: 10.4049/jimmunol.166.11.6665. [DOI] [PubMed] [Google Scholar]
- 34.Meyer D, Singe RM, Mack SJ, Lancaster A, Nelson MP, Erlich H, Fernandez-Vina M, Thomson G. Single Locus Polymorphism of ClassicalHLA Genes. In: Hansen JA, editor. Immunobiology of theHuman MHC: Proceedings of the 13th International Histocompatibility Workshop and Conference. Vol. 1. Seattle, WA: IHWG Press; 2007. pp. 653–704. [Google Scholar]
- 35.Oldenburg J, Schröder J, Brackmann HH, Müller-Reible C, Schwaab R, Tuddenham E. Environmental and genetic factors influencing inhibitor development. Semin Hematol. 2004;41(S1):82–8. doi: 10.1053/j.seminhematol.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 36.Astermark J, Oldenburg J, Pavlova A, Berntorp E, Lefvert AK MIBS Study Group. Polymorphisms in the IL10 but not in the IL1beta and IL4 genes are associated with inhibitor development in patients with hemophilia A. Blood. 2006;107:3167–72. doi: 10.1182/blood-2005-09-3918. [DOI] [PubMed] [Google Scholar]
- 37.Lee CA, Lillicrap D, Astermark J. Inhibitor development in hemophiliacs: the roles of genetic versus environmental factors. Semin Thromb Hemost. 2006;32(S2):10–4. doi: 10.1055/s-2006-946909. [DOI] [PubMed] [Google Scholar]
- 38.Astermark J, Oldenburg J, Carlson J, Pavlova A, Kavakli K, Berntorp E, Lefvert AK. Polymorphisms in the TNFA gene and the risk of inhibitor development in patients with hemophilia A. Blood. 2006;108:3739–45. doi: 10.1182/blood-2006-05-024711. [DOI] [PubMed] [Google Scholar]
- 39.Repessé Y, Slaoui M, Ferrandiz D, Gautier P, Costa C, Costa JM, Lavergne JM, Borel-Derlon A. Factor VIII (FVIII) gene mutations in 120 patients with hemophilia A: detection of 26 novel mutations and correlation with FVIII inhibitor development. J Thromb Haemost. 2007;5:1469–76. doi: 10.1111/j.1538-7836.2007.02591.x. [DOI] [PubMed] [Google Scholar]
- 40.Pavlova A, Delev D, LaCrois-Desmazes S, Schwaab R, Mende M, Fimmers R, Astermark J, Oldenburg J. Impact of polymorphisms of the major histocompatibility complex class II, interleukin-10, tumor necrosis factor-α and cytotoxic T-lymphocyte antigen-4 genes on inhibitor development in severe hemophilia A. J Thromb Haemost. 2009;7:2006–15. doi: 10.1111/j.1538-7836.2009.03636.x. [DOI] [PubMed] [Google Scholar]
- 41.Gouw SC, van den Berg M. The multifactorial etiology of inhibitor development in hemophilia: genetics and environment. Semin Thromb Hemost. 2009;35:723–34. doi: 10.1055/s-0029-1245105. [DOI] [PubMed] [Google Scholar]
- 42.Astermark J, Altisent C, Batarova A, Diniz MJ, Gringeri A, Holme PA, Karafoulidou A, Lopez-Fernández MF, Reipert BM, Rocino A, Schiavoni M, von Depka M, Windyga J, Fijnvandraat K. Non-genetic risk factors and the development of inhibitors in haemophilia: a comprehensive review and consensus report. Haemophilia. 2010;16:747–66. doi: 10.1111/j.1365-2516.2010.02231.x. [DOI] [PubMed] [Google Scholar]
- 43.Bafunno V, Santacroce R, CHetta M, D’Andrea G, Pisanelli D, Sessa F, Trota T, Tagariello G, Peyvandi F, Margaglione M. Polymorphisms in genes involved in autoimmune disease and the risk of FVIII inhibitor development in Italian patients with haemophilia A. Haemophilia. 2010;16:469–73. doi: 10.1111/j.1365-2516.2009.02150.x. [DOI] [PubMed] [Google Scholar]
- 44.Ettinger RA, James EA, Kwok WW, Thompson AR, Pratt KP. Lineages of human T-cell clones, including T helper 17/T helper 1 cells, isolated at different stages of anti-factor VIII immune responses. Blood. 2009;114:1423–8. doi: 10.1182/blood-2009-01-200725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fehérvari Z, Sakaguchi S. CD4+ Tregs and immune control. J Clin Invest. 2004;114:1209–17. doi: 10.1172/JCI23395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu G, Guo D, Key NS, Conti-Fine BM. Cytokine production by CD4+ T cells specific for coagulation factor VIII in healthy subjects and haemophilia A patients. Thromb Haemost. 2007;97:788–94. [PubMed] [Google Scholar]
- 47.Waters B, Qadura M, Burnett E, Chegeni R, Labelle A, Thompson P, Hough C, Lillicrap D. Anti-CD3 prevents factor VIII inhibitor development in hemophilia A mice by a regulatory CD4+ CD25+ dependent mechanism and by shifting cytokine production to favor a Th1 response. Blood. 2009;113:193–203. doi: 10.1182/blood-2008-04-151597. [DOI] [PubMed] [Google Scholar]
- 48.Reding MT, Lei S, Lei H, Green D, Gill J, Conti-Fine BM. Distribution of Th1- and Th2-induced anti-factor VIII IgG subclasses in congenital and acquired hemophilia patients. Thromb Haemost. 2002;88:568–75. [PubMed] [Google Scholar]
- 49.Hammer J, Valsasnini P, Tolba K, Bolin D, Higelin J, Takacs B, Sinigaglia F. Promiscuous and allele-specific anchors in HLA-DR-binding peptides. Cell. 1993;74:197–203. doi: 10.1016/0092-8674(93)90306-b. [DOI] [PubMed] [Google Scholar]
- 50.White GC, 2nd, Kempton CL, Grimsley A, Nielsen B, Roberts HR. Cellular immune responses in hemophilia: why do inhibitors develop in some, but not all hemophiliacs? J Thromb Haemost. 2005;3:1676–81. doi: 10.1111/j.1538-7836.2005.01375.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.