

Abstract
The immunoglobulin (Ig)-like domain containing receptor 1 (ILDR1) gene encodes angulin-2/ILDR1, a recently discovered tight junction protein, which forms tricellular tight junction (tTJ) structures with tricellulin and lipolysis-stimulated lipoprotein receptor (LSR) at tricellular contacts (TCs) in the inner ear. Previously reported recessive mutations within ILDR1 have been shown to cause severe to profound nonsyndromic sensorineural hearing loss (SNHL), DFNB42. Whole-exome sequencing of a Korean multiplex family segregating partial deafness identified a novel homozygous ILDR1 variant (p.P69H) within the Ig-like domain. To address the pathogenicity of p.P69H, the angulin-2/ILDR1 p.P69H variant protein, along with the previously reported pathogenic ILDR1 mutations, was expressed in angulin-1/LSR knockdown epithelial cells. Interestingly, partial mislocalization of the p.P69H variant protein and tricellulin at TCs was observed, in contrast to a severe mislocalization and complete failure of tricellulin recruitment of the other reported ILDR1 mutations. Additionally, three-dimensional protein modeling revealed that angulin-2/ILDR1 contributed to tTJ by forming a homo-trimer structure through its Ig-like domain, and the p.P69H variant was predicted to disturb homo-trimer formation. In this study, we propose a possible role of angulin-2/ILDR1 in tTJ formation in the inner ear and a wider audiologic phenotypic spectrum of DFNB42 caused by mutations within ILDR1.
Introduction
Tight junctions (TJs) are specialized structures within the barrier of epithelial cells. TJs prevent the passage of some solutes through the paracellular pathway of adjacent epithelial cells [1,2]. TJs can be divided into bicellular TJs (bTJs) and tricellular tight junctions (tTJs), depending upon the location of the paracellular pathway. Freeze-fracture electron microscopy has shown that tTJs exist at tricellular contacts (TCs), where three epithelial cells meet [3,4]. At the TCs, the topmost apical bicellular TJ strands join and turn vertically in the basal direction. In turn, these strands recruit additional vertically oriented TJ strands to form a central tube structure surrounded by central sealing elements in the extracellular space at TC locations [4–7].
To date, tricellulin, angulin-1/lipolysis-stimulated lipoprotein receptor (LSR), angulin-2/immunoglobulin-like domain containing receptor 1 (ILDR1) and angulin-3/immunoglobulin-like domain containing receptor 2 (ILDR2) have been sequentially identified as protein components of tTJs [4,8,9]. Among the genes encoding these proteins, mutated TRIC (tricellulin) and ILDR1 (angulin-2/ILDR1) have been reported to cause autosomal recessive nonsyndromic deafness DFNB49 (MIM 610153) [10–12] and DFNB42 (MIM 609646) [13,14], respectively. Data from experiments using epithelial cell lines with overexpressed or knocked down tricellulin suggest that these structures are responsible for epithelial barrier function, thereby controlling the paracellular flux of macromolecules [4,5,15]. Recent reports on mutated tricellulin knock-in mice, which lack tricellulin at TJs, showed disrupted bicellular and tricellular TJ strands in the inner ear epithelia and rapid progression of hearing loss accompanied by loss of cochlear hair cells [16]. Interestingly, experiments have also demonstrated that the inhibition of endolymph production in mutated tricellulin knock-in mice can rescue cochlear hair cell loss, supporting the hypothesis that defective paracellular permeability due to a dysfunctional tTJ in the inner ear could result in a toxic microenvironment in the organ of Corti [16].
In the present study, we identified a novel mutant allele of ILDR1 (p.P69H) located in the immunoglobulin-like (Ig-like) domain through whole-exome sequencing (WES) that segregates as an autosomal recessive nonsyndromic sensorineural hearing loss (SNHL) allele in a Korean family. This novel ILDR1 mutant allele is associated with weakened localization of angulin-2/ILDR1 at TCs, as well as a milder auditory phenotype compared with the originally reported DFNB42 profound deafness phenotype. Using protein modeling, we predict that angulin-2/ILDR1 contributes to tTJs by forming a trimer structure through its Ig-like domain, and its variant p.P69H localizes in the interface of the trimer complex to disrupt the stability of the structure.
Materials and Methods
Subjects and audiometric analysis
This study was approved by the Institutional Review Boards (IRBs) of Seoul National University Hospital (IRBYH-0905–041–281) and Seoul National University Bundang Hospital (IRB-B-1007–105–402). Written informed consent for participation in the study was obtained from participants or from a parent/guardian in the case of child participants (SH23–52). A three-generation pedigree was established for the family (SNUH23) (Fig. 1). Among the 16 subjects, five family members were willing to participate. DNA from blood lymphocytes was isolated from all five subjects.
Figure 1. Pedigree and audiograms of SNUH23-segregating partial deafness.

All three siblings (SNUH23–98, -99 and-52) exhibited partial deafness with preservation of significant low-frequency hearing thresholds. A meaningful progression of hearing loss over 5 years was observed in SNUH23–98 in the left ear (blue), whereas the right ear experienced hearing loss directly after cochlear implantation (red).
Both pure tone audiometry (PTA) and physical examinations were performed on four members (Fig. 1). PTA, with air and bone conduction at frequencies ranging from 250 to 8,000 Hz, was performed on the four recruited subjects according to standard protocols. The auditory phenotype was inferred from medical and developmental history interviews with subject SH23–99.
WES, alignment, coverage, calculation, and variant detection
WES was performed on two siblings, SH23–98 and SH23–52, at the Samsung Genome Institute. Genomic DNA was captured using the SureSelect Human All Exon Kit V3 (Agilent, Santa Clara, CA, USA) and sequenced using a HiSeq2000 system (Illumina, San Diego, CA, USA) with 101 base-paired end reads. Sequence reads were aligned with the human genome reference sequence (hg19) using BWA-v0.6.2 with the default settings. Sam tools v0.1.18, GATK v2.4–9 and Picard v1.88 were used to process SAM/BAM files, perform local realignment, and mark duplicates. Base recalibration and variant calling were performed using the GATK unified genotyper (known single nucleotide polymorphisms [SNPs] and insertions/deletions [indels] from dbSNP137, Mills and 1000G gold standard indels b37 sites, and 1000G phase1 indels b37 sites). Variants were also recalibrated using GATK based on dbSNP137, Mills indels, Hap Map, and Omni. Perl script, offered by ANNOVAR, was used to annotate the variants. Only exonic and splicing variants including non-synonymous variants and small indels were filtered. Variants with an allele frequency greater than 1% were discarded based on NHLBI-ESP 6500, 1000 Genomes Project, and an in-house database consisting of 44 Korean individuals. Of the variants identified in both siblings, only the variants that have not been reported in dbSNP137 were included. Low quality reads (< 20) and genotyping (<30) or variant/total depth < 0.2 were ruled out subsequently.
Localization of ILDR1 at TCs
The rat anti-tricellulin antibody (Ab) used in this study has been described previously [4]. Mouse anti-FLAG monoclonal antibody (mAb), rabbit anti-claudin-3 polyclonal antibody (pAb) and Cy3-conjugated donkey anti-mouse IgG were purchased from WAKO Chemicals (Osaka, Japan), Zymed (San Francisco, CA, USA) and Jackson ImmunoResearch Laboratories (Bar Harbor, ME, USA), respectively. Alexa 488-conjugated donkey anti-rat IgG and Alexa 647-conjugated donkey anti-rabbit IgG Molecular Probes were obtained from Life Technologies (Eugene, OR, USA).
Mouse mammary epithelial EpH4 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal calf serum (FCS). The knockdown of angulin-1/LSR in EpH4 cells and the cDNA sequence encoding human angulin-2/ILDR1 have been described previously [8]. The p.P69H mutation was introduced by site-directed mutagenesis. DNA fragments encoding angulin-2/ILDR1 were subcloned into the pCAG-cFLAG2 vector. To evaluate the tricellulin-recruiting property of human angulin-2/ILDR1, angulin-1/LSR knockdown EpH4 cells were transfected with plasmids encoding either wild-type human angulin-2/ILDR1, p.P69H, p.R97Q, p.Q195X, or a mock plasmid, and stable clones were selected using G418 added to the medium.
For immunofluorescent staining, each clone was cultured on coverslips, fixed with 1% formaldehyde in phosphate-buffered saline (PBS), blocked with 2% bovine serum albumin (BSA) in PBS, and incubated with the above-described primary and secondary antibodies. After washing with PBS, samples were embedded in FluorSave reagents (Calbiochem, San Diego, CA, USA) and observed under a fluorescence microscope (IX71; Olympus, Tokyo, Japan) equipped with a cooled charge-coupled device camera (ORCA-ER; Hamamatsu Photonics K. K., Hamamatsu City, Japan) controlled by Power Macintosh G5 and IPLab V3.9.5 software (BD Biosciences, San Jose, CA, USA) at room temperature.
Three-dimensional protein modeling of the Ig-like domain
To obtain structural insight regarding the effects of the p.P69H mutation on tricellular localization of the angulin-2/ILDR1 protein, a structural model of the Ig-like domain of angulin-2/ILDR1 was constructed. Since the Ig-like domain is located within the extracellular domain at the site where the mutation occurs, only the Ig-like domain was modeled rather than the entire protein. First, we identified feasible template structures for the Ig-like domain of angulin-2/ILDR1 from its homologs using BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and HHsearch (http://toolkit.tuebingen.mpg.de/hhpred). Among these structures, 1XT5 (crystal structure of VCBP3, domain 1, from Branchiostomafloridae), which demonstrated the highest alignment score, was chosen as the template structure. The three-dimensional (3D) structural model for a single Ig-like domain of angulin-2/ILDR1was then constructed using Modeller (http://salilab.org/modeller/) (S1 Fig.). In addition, loop structures within the model were refined using Rosetta loop modeling (https://www.rosettacommons.org/). To assess the physiochemical impact of the p.P69H mutation, we built a complex structural model of the Ig-like domains of angulin-2/ILDR1, assuming that the complex of Ig-like domains of angulin-2/ILDR1 formed a homo-trimeric structure based on their colocalization at the extracellular region of three neighboring epithelial cells [8]. First, 50,000 symmetric trimer models of angulin-2/ILDR1 Ig-like domains were built using Rosetta Dock (https://www.rosettacommons.org/). These structures were then ranked by their interface energy score. After choosing the upper 500 (1%) structures, the most plausible trimeric structure was then selected as the final trimer model. The criteria used were as follows: (i) the putative membrane location for each chain was plausible (i.e., the C-terminus of each domain was not buried inside the complex), (ii) the interface energy score, and (iii) the number of polar interactions between interface residues capable of forming hydrogen bonds. The mutant (p.P69H) trimer model was then constructed from the wild-type trimer model using Fold X (http://foldx.crg.es/) (Fig. 2). A molecular dynamics (MD) simulation was also performed using Gromacs (http://www.gromacs.org/) to compare the dynamic stability of wild-type and mutant trimer models to ultimately assess the impact of the p.P69H mutation.
Figure 2. Trimer structure model of ig-like domains of ILDR1.

The red and orange residues are the 69th and 97th residue, respectively. The cell membrane of each epithelial cell is located after the C-terminus (red color). The grey bars schematically represent the cell membranes surrounding the tricellular junction (dashed circle). Both 69th residues and 97th residues are gathered near interface region, which explains why p.P69H and p.R97Q mutations weakening the localization. (A) wild type trimer (B) p.P69H mutant trimer.
Results
Clinical features of the NSHL family
PTA was performed on five family members, three of whom (SH23–98, SH23–99 and SH23–52) exhibited a post-lingual onset and partial deafness (Fig. 1). SH23–98 manifested significant progression of hearing loss over 5 years, and parents recalled that SH23–99 and SH23–54 had near-normal hearing and speech development until 10 years of age. Three members (SH23–98, SH23–99 and SH23–52) demonstrated normal cognitive function in the absence of any anomalous-looking features indicative of syndromic hearing loss. Detailed imaging studies, including brain magnetic resonance imaging and temporal bone computed tomography scans, did not reveal any inner ear abnormality. Both parents (SH23–97 and SH23–54) showed normal hearing, indicating autosomal recessive hearing loss in this family (Fig. 1). Since TMPRSS3, the gene encoding transmembrane protease serine 3, is the most frequently detected gene in subjects with ski-slope type hearing loss in the Korean population [17], we examined the TMPRSS3 sequence present in SH23–98 and SH23–52 to identify any mutation(s) located within the exons and/or splice junctions; however, we failed to identify any mutations within this gene.
We have also performed a rigorous systemic physical examination to detect, if any, subtle abnormalities relevant to the recently proposed role of angulin-2/ILDR1 [18], however, we failed to identify any other abnormality than partial deafness.
WES and data analysis
The whole exomes of two siblings (SH23–98 and SH23–52) with hearing loss were sequenced; the mean depth of the target regions was greater than 80× coverage, with nearly 90% with at least 10× coverage (S1 Table). To identify causal variants, 27,648 common single nucleotide variants and indels were filtered and prioritized in two family members (Fig. 3). Of the 141 variants, eight homozygous and six compound heterozygous variants located in 11 genes were selected under the recessive model (Table 1). The 14 variants were filtered further by Sanger sequencing and segregation analysis within the family members. Three variants from two genes (MUC12 and CHD3) were excluded by Sanger sequencing. An additional four variants from two genes (BRCA1 and ABCA10) did not co-segregate with the auditory phenotype among the remaining family members. Four homozygous variants from ZSCAN26, NR1H2, VSIG10L, and PLK1S1 were also excluded, since they were recently reported as polymorphisms (rs61622742, rs34296657, rs11402251, and rs11087346, respectively) in dbSNP138. Finally, MICA and ATXN3 variants were detected in three and four unrelated Korean normal hearing controls, respectively, of 276 chromosomes, suggesting that these variants are Korean-specific polymorphisms. Finally, only one novel homozygous missense variant, c.C206A in ILDR1, resulting in the substitution of p.P69H, remained and was validated by Sanger sequencing. This variant was found in a partially deafened SH23–99 (Fig. 4). The novel variant p.P69H is described as ‘damaging’ by SIFT and PolyPhen2 (0.03 and 1, respectively) and is well-conserved in various species based on the GERP++ score (5.64) (Table 2). This variant was not detected among 476 chromosomes from unrelated Korean normal hearing controls.
Figure 3. Filtering and prioritization steps for novel variant identification.

Of the 27,648 variants identified in two siblings, all synonymous SNPs were excluded, and only rare variants (< 1%) based on NIHBL-ESP6500, the 1000 Genomes Project and our in-house database (44 Koreans) were included for subsequent analysis. Only variants that have not been reported in dbSNP137 were selected. Low-quality read depth (DP) and genotype (GP) under 20 and 30, respectively, or variant/total depth less than 0.2 were also discarded. Under the recessive model, eight variants of homozygotes and six variants of compound heterozygotes (11 genes in total) were identified. Further filtering of these variants was performed by Sanger sequencing and by confirming co-segregation with the phenotype. The remaining variants were also cross-checked against dbSNP138 to further exclude the irrelevant variants to the phenotype.
Table 1. List of final candidate SNVs in two siblings with hearing loss.
| Inheritance | Gene | Func | GenbankID | Exon | Nucleotide | AA | Chr | Start | End | Ref | Alt | SH23_52 | SH23_98 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DP | GQ | DP | GQ | ||||||||||||
| Homo | ILDR1 | NS | NM_001199799 | Exon2 | c.C206A | p.P69H | chr3 | 121725861 | 121725861 | G | T | 25 | 75 | 40 | 99 |
| Homo | ZSCAN26 | - | - | - | - | - | chr6 | 28239932 | 28239932 | - | G | 28 | 84 | 40 | 78 |
| Homo | MICA | FI | NM_000247 | exon6 | c.949–1->CTGCTGCTGCT | NA | chr6 | 31380161 | 31380161 | - | CTGCTGCTGCT | 23 | 66 | 24 | 72 |
| Homo | MUC12 | NS | NM_001164462 | exon2 | c.C7682T | p.T2561M | chr7 | 100641526 | 100641526 | C | T | 203 | 99 | 238 | 99 |
| Homo | ATXN3 | NI | NM_001164782 | exon2 | c.68_69insAGCAGCAGCAGC | p.G23delinsGAAAA | chr14 | 92537353 | 92537353 | - | GCTGCTGCTGCT | 50 | 99 | 66 | 99 |
| Homo | NR1H2 | NI | NM_001256647 | exon5 | c.225_226insCAG | p.K75delinsKQ | chr19 | 50881822 | 50881822 | - | CAG | 64 | 99 | 73 | 99 |
| Homo | VSIG10L | FI | NM_001163922 | exon10 | c.2577dupC | p.A859fs | chr19 | 51835892 | 51835892 | - | G | 47 | 99 | 72 | 99 |
| Homo | PLK1S1 | - | - | - | - | - | chr20 | 21186161 | 21186161 | - | G | 67 | 99 | 96 | 99 |
| Com het | CHD3 | NS | NM_001005271 | exon5 | c.G898C | p.A300P | chr17 | 7796815 | 7796815 | G | C | 60 | 99 | 84 | 99 |
| NS | NM_001005271 | exon5 | c.T902C | p.L301P | chr17 | 7796819 | 7796819 | T | C | 70 | 87 | 102 | 99 | ||
| Com het | BRCA1 | NS | NM_007297 | exon9 | c.T1789A | p.C597S | chr17 | 41245618 | 41245618 | A | T | 138 | 99 | 206 | 99 |
| NS | NM_007297 | exon3 | c.C13T | p.L5F | chr17 | 41258531 | 41258531 | G | A | 52 | 99 | 61 | 99 | ||
| Com het | ABCA10 | NS | NM_080282 | exon32 | c.C3779T | p.A1260V | chr17 | 67150383 | 67150383 | G | A | 107 | 99 | 158 | 99 |
| NS | NM_080282 | exon32 | c.G3778C | p.A1260P | chr17 | 67150384 | 67150384 | C | G | 109 | 99 | 162 | 99 | ||
Candidate variants were identified under the recessive model (either homozygotes or compound heterozygotes). AA: Amino Acid; Ref: Reference; Alt: Alternative; NS: Nonsynonymous SNV; FD: Frameshift Deletion; FI: Frameshft Insertion; NI: Nonframeshift Insertion; DP: Depth; GQ: Genotype Quality; Homo: Homozygotes; Com het: Compound heterozygotes.
Figure 4. Validation of variants within ILDR1 using Sanger sequencing.

(A) Sanger sequencing trace of p.P69H within ILDR1 from SNUH23. (B) Conservation of the p.P69 residue among orthologs and paralogs of ILDR1.
Table 2. Causal variant ILDR1.
| Gene | Patient | Inheritance | DP | GQ | Type | Genomic location | Genbank No. | Exon | Nucleotide | Protein | R | A | ESP6500 | 1000G | dbSNP137 | SIFT | PolyPhen2 (HVAR) | GERP++ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ILDR1 | SH23_52 | homo | 25 | 75 | nonsynonymous SNV | chr3:121725861 | NM_001199799 | exon2 | c.C206A | p.P69H | G | T | - | - | - | 0.03 | 1 | 5.64 |
| SH23_98 | homo | 40 | 99 |
R: Reference; A: Alternative; DP: Depth; GQ: Genotype Quality.
Localization of angulin-2/ILDR1 at TCs
Most, if not all, angulin-2/ILDR1 protein-containing mutations reported to date in DFNB42 patients exhibit failure to localize at TCs and defectiveness in tricellulin recruitment in angulin-1/LSR knockdown EpH4 cells [8]. Therefore, we determined whether the p.P69H mutation affects the localization of angulin-2/ILDR1 or tricellulin recruitment. As reported previously, tricellulin was mislocalized to the lateral membrane in angulin-1/LSR knockdown EpH4 cells, in contrast to its normal localization at TCs in parental EpH4 cells (Fig. 5A and 5B). The exogenously expressed wild-type angulin-2/ILDR1 was nearly always localized at TCs and able to recruit tricellulin to TCs (Fig. 5D). On the other hand, the p.P69H mutant was concentrated at approximately half of the TCs of angulin-1/LSR knockdown cells. These data indicate that the p.P69H mutation affects the ability of angulin-2/ILDR1 to localize at TCs (411/722 for p.P69H vs. 593/618 for wild-type; p = 1.14 x 10–60) (Fig. 5E and 5H). Tricellulin was also concentrated at approximately half of all TCs in p.P69H-expressing angulin-1/LSR knockdown cells (360/722 for P69H vs. 573/618 for wild-type; p = 7.34 x 10–65) (Fig. 5D and 5H). p.R97Q and p.Q195X mutants, which served as negative controls, were not concentrated at TCs and showed no involvement in tricellulin recruitment (Fig. 5F, 5G, and 5H). These results indicate that the p.P69H mutation significantly affects the localization of angulin-2/ILDR1, although the extent of mislocalization was less severe than that of the p.R97Q and p.Q195X mutations. Therefore, the difference in the deafness phenotype may reflect the severity of mislocalization.
Figure 5. Effect of the p.P69H mutation on the localization and tricellulin recruitment function of ILDR1.
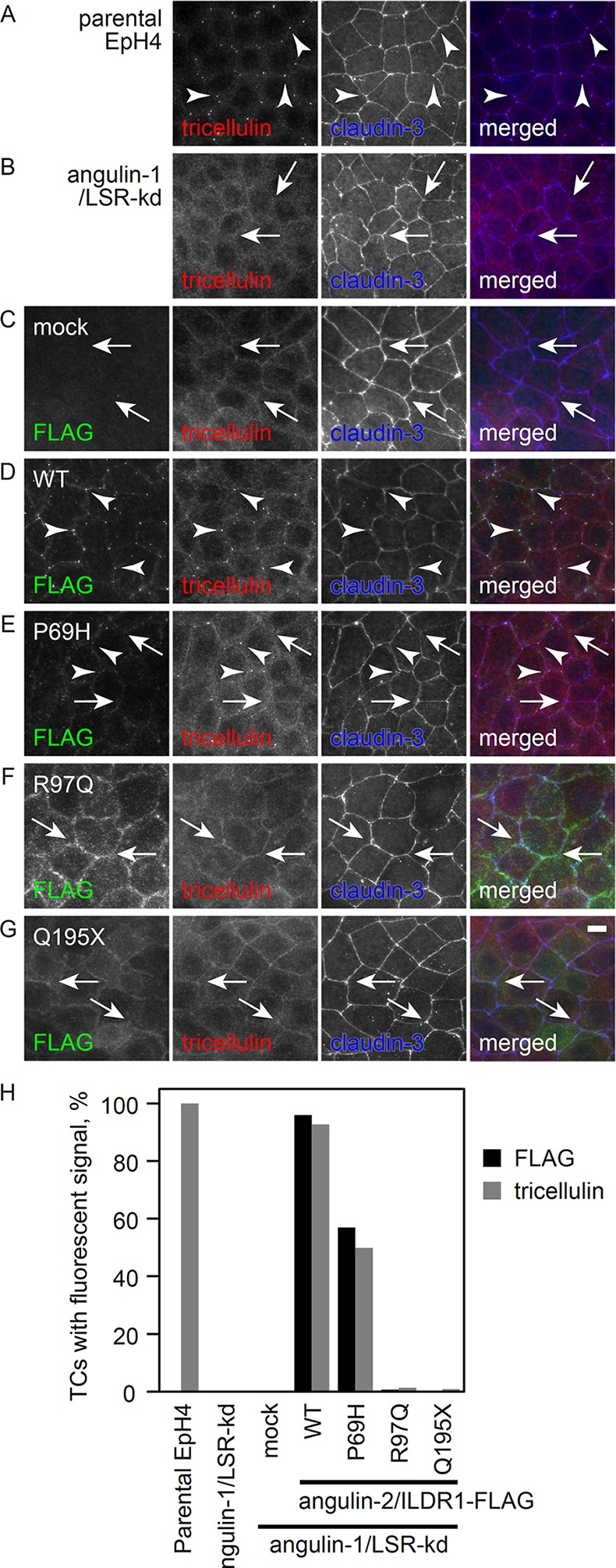
(A-G) Parental EpH4 cells, (A) LSR knockdown cells. (B) LSR knockdown cells stably expressing the mock FLAG vector (C) or wild-type (WT) human ILDR1. Immunostaining of (D) p.P69H, (E) p.R97Q, (F) or p.Q195X (G) was performed using anti-FLAG mAb (green), anti-tricellulin mAb (red) and anti-claudin-3 pAb (blue). Arrowheads and arrows indicate tricellular contacts with and without tricellulin localization, respectively. Scale bar: 10 μm. (H) Statistical analysis of FLAG and tricellulin localization at TCs. More than 600 TCs for each cell clone were examined for FLAG and tricellulin accumulation.
3D protein modeling of the Ig-like domain
The localization of angulin-2/ILDR1 at TCs was reported recently [8]. In the study described herein, the p.P69H mutation was observed in our patients with partial deafness. These data suggest that the Ig-like domain of angulin-2/ILDR1, where p.P69H resides, may be involved in the localization of angulin-2/ILDR1 at TCs. However, the mechanism of localization is not clear. It has yet to be determined whether angulin-2/ILDR1 is able to localize at TCs without assistance from other protein(s). Most Ig-like domains, such as that in angulin-2/ILDR1, are important in cell adhesion [9,19]. Therefore, we hypothesized that angulin-2/ILDR1 may function by forming direct interactions with tTJ proteins. Thus, we plan to study the interaction between angulin-2/ILDR1 and other components that drive the localization of angulin-2/ILDR1 at TCs by modeling their complex structure, although any additional components have yet to be elucidated. Correct localization of angulin-2/ILDR1 at TCs was also observed in TricR497X/R497X mice lacking tricellulin at TCs, indicating that tricellulin does not drive angulin-2/ILDR1 localization [16]. We further believe that angulin-2/ILDR1 may be the main driver of localization, and three TCs from each cell could form a trimer to initiate the tTJ. In order to test this hypothesis, we constructed a 3D model of the angulin-2/ILDR1 trimer structure (Fig. 2). In this model, the p.P69H mutation and the previously reported deleterious p.R97Q mutation [13] were both located at the interface of the complex, explaining why these mutations weaken its localization. The pathogenicity of the p.P69H mutation can be predicted by the trimer model. Since residue 69 was mutated from proline to histidine, the loop conformation may have been shifted due to differences in flexibility. Additionally, the space for placement of residue 69 is narrow. Since histidine has a larger side chain than proline, repulsive forces may be increased in the mutant. Moreover, unlike proline, histidine can exist in a positively charged form (~10% at physiological pH), which can further amplify the repulsion.
To further evaluate the stability of the model and to evaluate the mutational effect in greater detail, we performed molecular dynamics (MD) simulations. MD simulations were executed a total of eight times (four wild-type, four mutant). We found that the root-mean-square deviation (RMSD) of residue 69 converged to ~5 Å in the wild-type trimer model, but the RMSD in the p.P69H mutant trimer model did not converge, varying between 3~7 Å (S2 Fig.). This result indicates that the space around residue 69 can become unstable as a result of the p.P69H mutation.
Discussion
Systematic exclusion of the variants either in the homozygous or compound heterozygous state from 10 autosomal genes based upon a segregation study among family members of SNUH23 indicated that the p.P69H allele of ILDR1 most likely accounts for the partial deafness in this family. Less severe, but significant mislocalization of the p.P69H substitution of angulin-2/ILDR1 was noted in angulin-1/LSR knockdown EpH4 cells, further supporting its pathogenic potential. Recently, WES contributed significantly to the detection of a series of new deafness genes [20–27]. Although the SNUH23 family was not a consanguineous family and did not have a significant linkage interval prior to this study, unlike those in other studies, the presence of three affected siblings enabled detection of a causative gene through WES, coupled with segregation analysis.
A ski-slope type sensorineural hearing loss, now referred to as ‘partial deafness’, characterized by an abrupt downsloping from mild to severe/profound hearing loss across high frequencies, is a challenge for conventional auditory rehabilitation methods, such as hearing aids or cochlear implants. Based upon the progression of hearing loss and the degree of residual low-frequency hearing, appropriate auditory rehabilitation varies from patient to patient. However, the molecular etiology underlying this special configuration of hearing loss has not been elucidated, with the exception of the previously identified recessive mutations within the TMPRSS3 gene [28,29]. Depending upon the proteolytic activity of the mutant TMPRSS3 protein [17,30], two discrete types of phenotypes can manifest. In this study, we showed that mutations within ILDR1 can also manifest as discrete, audiologic phenotypes depending upon the degree of localization of angulin-2/ILDR1 at the TC.
Angulin-2/ILDR1 protein, a component of tTJs, provides a strong barrier property to the epithelium in cultured epithelial cells. The majority of DFNB42-associated mutations within the angulin-2/ILDR1 protein did not localize at TCs in the angulin-1/LSR knockdown epithelial cells [8]. Higashi et al. showed that angulin-2/ILDR1 interacts with tricellulin and recruits tricellulin at TCs in LSR knockdown cells, proposing the failure of angulin-2/ILDR1-mediated tricellulin recruitment as a possible pathogenesis of DFNB42 deafness. Consistent with our previous results [8], partial mislocalization of the p.P69H ILDR1 mutant in this study led to the incomplete recruitment of tricellulin to TCs in LSR knockdown epithelial cells. This incomplete recruitment of tricellulin due to partial mislocalization of angulin-2/ILDR1 could be a direct cause of progressive partial deafness in the patients described in this study since mislocalization of tricellulin causes deafness in humans and mice regardless of angulin-2/ILDR1 [11,16]. The correlation between the degree of localization of angulin-2/ILDR1 and tricellulin at TCs in angulin-1/LSR knockdown EpH4 cells strongly supports the hypothesis that angulin-2/ILDR1 interacts directly with tricellulin.
However, there may be another mechanism for hearing loss due to alteration of angulin-2/ILDR1 in vivo. Recently, Morozko et al. proposed that angulin-2/ILDR1 was not required for initial recruitment of tricellulin to TCs in vivo. Their data indicated that failure in recruitment of tricellulin itself does not account for the early- onset severe hearing loss in DFNB49 and in angulin-2/ILDR1 deficient in mice. They observed gradual mislocalization of tricellulin at TCs after the first postnatal week in angulin-2/ILDR1 deficient mice [31]. These in vivo results contradict our in vitro findings showing that tricellulin localization was tightly correlated with that of angulin-2/ILDR1 in angulin-1/LSR knockdown EpH4 cells. The presence of other tight junction proteins in vivo, such as angulin-1/LSR and occludin could contribute to this discrepancy; LSR is co-expressed with angulin-2/ILDR1 in the inner ear [8], and occludin is involved in tricellulin localization [9,32]. Therefore, angulin-1/LSR or others that were lacked by our in vitro system may be able to recruit tricellulin to the TCs in the inner ear in the absence of angulin-2/ILDR1 expression. Given these in vivo findings, it is also possible that the mutation within ILDR1 may lead to deafness via a different mechanism unrelated to failed tricellulin recruitment at TCs, or through a combination of these mechanisms. Rather, we proposed a direct effect of ILDR1 alterations upon the tTJ structure irrespective of the status of tricellulin recruitment, based upon our protein modeling of the Ig-like domain. In fact, the position of the p.P69H mutation within the Ig-like domain and the instability of the predicted homo-trimer complex of angulin-2/ILDR1 caused by this mutation suggest that p.P69H may exert pathogenicity irrespective of the failure to recruit tricellulin at TCs. Specifically, the Ig-like domain of angulin-2/ILDR1 shares the same motif as other cell adhesion molecules (CAM) [9,19], indicating that the Ig-like domain of angulin-2/ILDR1 may attach to other proteins or the extracellular matrix (ECM) to localize proteins at TCs. In this study, we assumed that the Ig-like domain of angulin-2/ILDR1 was bound to the same type of molecule (homophilic CAM) for the following reasons. First, when a tTJ is formed, the tricellular corner would become highly narrow due to the tight junctions between epithelial cells (3~12 nm) [33]. It is difficult to expect any ECM binding motifs in this narrow space. Secondly, if CAM binding occurs via an alternative mechanism for angulin-2/ILDR1 localization, those CAMs must also be localized at the TCs. Such localized CAMs have not been found at the tricellular corner as of yet, and moreover such a mechanism seems inefficient considering that angulin-2/ILDR1 is the key protein that recruits tricellulin to the TC [8]. If we assume that the Ig-like domain of angulin-2/ILDR1 is a homophilic CAM, then a trimer would be the most plausible minimum-sized complex. Therefore, the homo-trimeric ILDR1 complex is the most practical model to currently explain angulin-2/ILDR1 localization. The lateral size of the trimer model (Fig. 2A) is 4.21 nm, which does not contradict the known size of tight junctions. Evaluation of ILDR1 knockout or knock-in mice of a specific ILDR1 mutation, as well as further biostructural studies, will be needed to address this issue. Indeed, freeze-fracture electron microscopy has revealed alteration of the ultrastructure of inner ear tTJ from angulin-2/ILDR1 deficient mice [31].
Results from our study are intriguing, in that a milder auditory phenotype characterized by a ‘partial deafness’ was observed in all three siblings from this family. A higher degree of hearing loss with a flat or slightly downsloping configuration was the main audiological feature of the DFNB42 family [13]. What accounts for this partial deafness? To address this question, it is important to note that partial mislocalization of angulin-2/ILDR1 at TCs was reported previously for the three ILDR1 variants (p.Glu379X, p.Thr345ProfsX20 and p.Glu394SerfsX15) [8], albeit the degree of mislocalization of the three mutants appeared to be more severe than that of p.P69H. All ILDR1 variants reported thus far, including the three variants described herein, are associated with severe or a more severe degree of flat-type hearing loss [8,13,14]. In this sense, the p.P69H mutant protein was the first and only variant that showed both weakened localization at TCs of angulin-1/LSR knockdown cells and a milder auditory phenotype ‘partial deafness’. It is possible that the degree of weakened localization of angulin-2/ILDR1 protein at TCs is correlated with the severity of the initial auditory phenotype. Examination of TricR497X/R497X mice, which lack tricellulin at TCs at P16, revealed that cochlear hair cell degeneration was more pronounced in the mid and basal turns compared with the apical turn [16], suggesting that the reticular lamina of the organ of Corti in the mid and basal turn is more sensitive to various types of continually varying mechanical stressors in the disrupted tTJ, thereby giving rise to a ski-slope type of partial deafness.
Alternatively, mutations within the Ig-like domain of ILDR1 could result in the auditory phenotype that is more pronounced in mid and high frequencies. Disruption of the tTJ, due to abnormal angulin-2/ILDR1, may damage the organ of Corti preferentially in the mid and basal turn of the cochlea. To date, only three ILDR1 variants, including p.P69H, have been reported to reside in this domain. The p.Asn109_Pro111dup mutation, reported in a consanguineous Saudi family, was associated with a non-progressive downsloping audiogram from severe to profound across higher frequencies. The residual low-frequency hearing in this Saudi family was not as significant as that in our study [14]. With respect to p.R97Q, another variant in this domain [13], the detailed audiogram of a homozygous carrier of p.R97Q has not yet been reported, precluding any genotype-phenotype correlation.
It should also be noted that significant progression of hearing loss was observed in homozygous carriers of p.P69H in this study. This is in contrast to a previous description in which DFNB42 deafness was characterized by bilateral non-progressive moderate to profound sensorineural hearing impairment [13]. This worsened hearing threshold in homozygous carriers of p.P69H within ILDR1 in this study was compatible with aggravated hearing thresholds in a mouse model lacking tricellulin (Tricp.R497X/p.R497X mice) [16]. It has been suggested that hearing loss due to impaired TJs is caused by defective permeability via the paracellular pathway, which would expose the sensory epithelium to a toxic environment [16]. Significant residual hearing at low frequencies and progressiveness never reported before with other ILDR1 mutations suggest that the pathogenic mechanism of p.P69H may differ from those of other variants. It may be that the p.P69H mutation in the Ig-like domain affects TJ structure in the inner ear to the extent that the inner ear sensory epithelium develops normally and can be maintained for a while, but subsequently begins to degenerate due to a toxic microenvironment in the extracellular space of the hair cells especially from the basal turns. In addition, this progression should be considered for appropriate auditory rehabilitation of partial deafness.
In this study, we reported a novel missense mutation within the ILDR1 gene associated with partial deafness, and identified partial mislocalization of the mutant angulin-2/ILDR1 protein at TCs. More importantly, we have proposed a new pathogenic mechanism of ILDR1 mutations residing in the Ig-like domain, based upon the calculated effect of our novel ILDR1 mutation on the stability of a predicted trimer model.
Supporting Information
The red and orange residues are the 69th and 97th residue, respectively. Those two residues are the sites for the mutation p.P69H and p.R97Q, respectively.
(TIF)
The MD simulations executed 8 times; 4 times for a wild type model (wt1~wt4) and 4 times for a mutant model (m1~m4). The RMSD values tend to converge on around 5 Å for the wild type model, while they tend to widely vary between 3~7 Å for the mutant model.
(TIF)
Statistical analysis was performed using Picard with the module ‘CalculateHsMetrics’. ‘UNIQUE READS’ represent the number of reads not marked as duplicates. ‘UQ READS ALIGNED’ represent the number of unique reads that align with a mapping score > 0 to the reference. ‘ON+NEAR BAIT’ represents the percentage of on + near bait bases against bases aligned, and ‘OFF BAIT’ represents the percentage of aligned bases that mapped neither on nor near a bait. ‘ZERO CVG TARGETS PCT’ represents the number of targets that did not reach coverage = 2 over any base. PCT: percent, UQ: unique, CVG: coverage.
(DOCX)
Acknowledgments
We thank Dr. Thomas Friedman (National Institute on Deafness and Other Communication Disorders/National Institutes of Health) for critically reading this manuscript.
This work was supported by the Korea Healthcare Technology R&D Project, Ministry for Health, Welfare and Family Affairs, Republic of Korea, Grant HI11C13310000 (previously A111377 to B. Y. Choi), the Seoul National University Bundang Hospital Research Fund 14–2014–033 (to B.Y. Choi), the Korean Health Technology R&D Project, Ministry of health and Welfare, Republic of Korea (Hl12C0014) and the Funding Program for Next Generation World Leading Researchers (NEXT Program) from the Japan Society for the Promotion of Science initiated by the Council for Science and Technology Policy [Grand number LS084] (to M.Furuse). T.Higashi is a research fellow of the Japan Society for the Promotion of Science.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the Korea Healthcare Technology R&D Project, Ministry for Health, Welfare and Family Affairs, Republic of Korea, Grant HI11C13310000 (previously A111377 to B. Y. Choi), the Seoul National University Bundang Hospital Research Fund 14-2014-033 (to B.Y. Choi), the Korean Health Technology R&D Project, Ministry of health and Welfare, Republic of Korea (Hl12C0014) and the Funding Program for Next Generation World Leading Researchers (NEXT Program) from the Japan Society for the Promotion of Science initiated by the Council for Science and Technology Policy [Grand number LS084] (to M. Furuse). T. Higashi is a research fellow of the Japan Society for the Promotion of Science. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Schneeberger EE, Lynch RD (2004) The tight junction: a multifunctional complex. Am J Physiol Cell Physiol 286: C1213–1228. [DOI] [PubMed] [Google Scholar]
- 2. Tsukita S, Furuse M, Itoh M (2001) Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol 2: 285–293. [DOI] [PubMed] [Google Scholar]
- 3. Friend DS, Gilula NB (1972) Variations in tight and gap junctions in mammalian tissues. J Cell Biol 53: 758–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ikenouchi J, Furuse M, Furuse K, Sasaki H, Tsukita S, et al. (2005) Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J Cell Biol 171: 939–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Krug SM, Amasheh S, Richter JF, Milatz S, Gunzel D, et al. (2009) Tricellulin forms a barrier to macromolecules in tricellular tight junctions without affecting ion permeability. Mol Biol Cell 20: 3713–3724. 10.1091/mbc.E09-01-0080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mariano C, Sasaki H, Brites D, Brito MA (2011) A look at tricellulin and its role in tight junction formation and maintenance. Eur J Cell Biol 90: 787–796. 10.1016/j.ejcb.2011.06.005 [DOI] [PubMed] [Google Scholar]
- 7. Staehelin LA (1973) Further observations on the fine structure of freeze-cleaved tight junctions. J Cell Sci 13: 763–786. [DOI] [PubMed] [Google Scholar]
- 8. Higashi T, Tokuda S, Kitajiri S, Masuda S, Nakamura H, et al. (2013) Analysis of the ’angulin' proteins LSR, ILDR1 and ILDR2—tricellulin recruitment, epithelial barrier function and implication in deafness pathogenesis. J Cell Sci 126: 966–977. 10.1242/jcs.116442 [DOI] [PubMed] [Google Scholar]
- 9. Masuda S, Oda Y, Sasaki H, Ikenouchi J, Higashi T, et al. (2011) LSR defines cell corners for tricellular tight junction formation in epithelial cells. J Cell Sci 124: 548–555. 10.1242/jcs.072058 [DOI] [PubMed] [Google Scholar]
- 10. Chishti MS, Bhatti A, Tamim S, Lee K, McDonald ML, et al. (2008) Splice-site mutations in the TRIC gene underlie autosomal recessive nonsyndromic hearing impairment in Pakistani families. J Hum Genet 53: 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Riazuddin S, Ahmed ZM, Fanning AS, Lagziel A, Kitajiri S, et al. (2006) Tricellulin is a tight-junction protein necessary for hearing. Am J Hum Genet 79: 1040–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Safka Brozkova D, Lastuvkova J, Stepankova H, Krutova M, Trkova M, et al. (2012) DFNB49 is an important cause of non-syndromic deafness in Czech Roma patients but not in the general Czech population. Clin Genet 82: 579–582. 10.1111/j.1399-0004.2011.01817.x [DOI] [PubMed] [Google Scholar]
- 13. Borck G, Ur Rehman A, Lee K, Pogoda HM, Kakar N, et al. (2011) Loss-of-function mutations of ILDR1 cause autosomal-recessive hearing impairment DFNB42. Am J Hum Genet 88: 127–137. 10.1016/j.ajhg.2010.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramzan K, Taibah K, Tahir AI, Al-Tassan N, Berhan A, et al. (2014) ILDR1: Novel mutation and a rare cause of congenital deafness in the Saudi Arabian population. Eur J Med Genet. [DOI] [PubMed]
- 15. Raleigh DR, Marchiando AM, Zhang Y, Shen L, Sasaki H, et al. (2010) Tight junction-associated MARVEL proteins marveld3, tricellulin, and occludin have distinct but overlapping functions. Mol Biol Cell 21: 1200–1213. 10.1091/mbc.E09-08-0734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nayak G, Lee SI, Yousaf R, Edelmann SE, Trincot C, et al. (2013) Tricellulin deficiency affects tight junction architecture and cochlear hair cells. J Clin Invest 123: 4036–4049. 10.1172/JCI69031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chung J, Park SM, Chang SO, Chung T, Lee KY, et al. (2014) A novel mutation of TMPRSS3 related to milder auditory phenotype in Korean postlingual deafness: a possible future implication for a personalized auditory rehabilitation. J Mol Med (Berl). [DOI] [PubMed]
- 18. Chandra R, Wang Y, Shahid RA, Vigna SR, Freedman NJ, et al. (2013) Immunoglobulin-like domain containing receptor 1 mediates fat-stimulated cholecystokinin secretion. J Clin Invest 123: 3343–3352. 10.1172/JCI68587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Martin-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, et al. (1998) Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol 142: 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Diaz-Horta O, Duman D, Foster J 2nd, Sirmaci A, Gonzalez M, et al. (2012) Whole-exome sequencing efficiently detects rare mutations in autosomal recessive nonsyndromic hearing loss. PLoS One 7: e50628 10.1371/journal.pone.0050628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huebner AK, Gandia M, Frommolt P, Maak A, Wicklein EM, et al. (2011) Nonsense mutations in SMPX, encoding a protein responsive to physical force, result in X-chromosomal hearing loss. Am J Hum Genet 88: 621–627. 10.1016/j.ajhg.2011.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rehman AU, Morell RJ, Belyantseva IA, Khan SY, Boger ET, et al. (2010) Targeted capture and next-generation sequencing identifies C9orf75, encoding taperin, as the mutated gene in nonsyndromic deafness DFNB79. Am J Hum Genet 86: 378–388. 10.1016/j.ajhg.2010.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Santos-Cortez RL, Lee K, Azeem Z, Antonellis PJ, Pollock LM, et al. (2013) Mutations in KARS, encoding lysyl-tRNA synthetase, cause autosomal-recessive nonsyndromic hearing impairment DFNB89. Am J Hum Genet 93: 132–140. 10.1016/j.ajhg.2013.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schraders M, Haas SA, Weegerink NJ, Oostrik J, Hu H, et al. (2011) Next-generation sequencing identifies mutations of SMPX, which encodes the small muscle protein, X-linked, as a cause of progressive hearing impairment. Am J Hum Genet 88: 628–634. 10.1016/j.ajhg.2011.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sirmaci A, Edwards YJ, Akay H, Tekin M (2012) Challenges in whole exome sequencing: an example from hereditary deafness. PLoS One 7: e32000 10.1371/journal.pone.0032000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tekin M, Chioza BA, Matsumoto Y, Diaz-Horta O, Cross HE, et al. (2013) SLITRK6 mutations cause myopia and deafness in humans and mice. J Clin Invest 123: 2094–2102. 10.1172/JCI65853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yariz KO, Duman D, Seco CZ, Dallman J, Huang M, et al. (2012) Mutations in OTOGL, encoding the inner ear protein otogelin-like, cause moderate sensorineural hearing loss. Am J Hum Genet 91: 872–882. 10.1016/j.ajhg.2012.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bonne-Tamir B, DeStefano AL, Briggs CE, Adair R, Franklyn B, et al. (1996) Linkage of congenital recessive deafness (gene DFNB10) to chromosome 21q22.3. Am J Hum Genet 58: 1254–1259. [PMC free article] [PubMed] [Google Scholar]
- 29. Scott HS, Kudoh J, Wattenhofer M, Shibuya K, Berry A, et al. (2001) Insertion of beta-satellite repeats identifies a transmembrane protease causing both congenital and childhood onset autosomal recessive deafness. Nat Genet 27: 59–63. [DOI] [PubMed] [Google Scholar]
- 30. Lee YJ, Park D, Kim SY, Park WJ (2003) Pathogenic mutations but not polymorphisms in congenital and childhood onset autosomal recessive deafness disrupt the proteolytic activity of TMPRSS3. J Med Genet 40: 629–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morozko EL, Nishio A, Ingham NJ, Chandra R, Fitzgerald T, et al. (2014) ILDR1 null mice, a model of human deafness DFNB42, show structural aberrations of tricellular tight junctions and degeneration of auditory hair cells. Hum Mol Genet. [DOI] [PMC free article] [PubMed]
- 32. Ikenouchi J, Sasaki H, Tsukita S, Furuse M, Tsukita S (2008) Loss of occludin affects tricellular localization of tricellulin. Mol Biol Cell 19: 4687–4693. 10.1091/mbc.E08-05-0530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martinez-Palomo A, Erlij D (1975) Structure of tight junctions in epithelia with different permeability. Proc Natl Acad Sci U S A 72: 4487–4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The red and orange residues are the 69th and 97th residue, respectively. Those two residues are the sites for the mutation p.P69H and p.R97Q, respectively.
(TIF)
The MD simulations executed 8 times; 4 times for a wild type model (wt1~wt4) and 4 times for a mutant model (m1~m4). The RMSD values tend to converge on around 5 Å for the wild type model, while they tend to widely vary between 3~7 Å for the mutant model.
(TIF)
Statistical analysis was performed using Picard with the module ‘CalculateHsMetrics’. ‘UNIQUE READS’ represent the number of reads not marked as duplicates. ‘UQ READS ALIGNED’ represent the number of unique reads that align with a mapping score > 0 to the reference. ‘ON+NEAR BAIT’ represents the percentage of on + near bait bases against bases aligned, and ‘OFF BAIT’ represents the percentage of aligned bases that mapped neither on nor near a bait. ‘ZERO CVG TARGETS PCT’ represents the number of targets that did not reach coverage = 2 over any base. PCT: percent, UQ: unique, CVG: coverage.
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.