

Abstract
Background
Benign vocal fold lesions, including cysts, nodules, polyps, and Reinke’s edema, are common causes of hoarseness and subsequent voice disorders. Given the prevalence of these lesions, disease etiology and pathophysiology remain unclear and their microbiota has not been studied to date secondary to the paucity of available biopsies for investigation. We sought to characterize and compare the bacterial communities in biopsies of cysts, nodules, polyps, and Reinke’s edema collected from patients in Germany and Wisconsin. These samples were then compared to the communities found in healthy saliva and throat samples from the Human Microbiome Project (HMP).
Results
454 pyrosequencing of the V3–V5 regions of the 16S rRNA gene revealed five phyla that explained most of the bacterial diversity, including Firmicutes (73.8%), Proteobacteria (12.7%), Bacteroidetes (9.2%), Actinobacteria (2.1%), and Fusobacteria (1.9%). Every lesion sample, regardless of diagnosis, had operational taxonomic units (OTUs) identified as Streptococcus, with a mean abundance of 68.7%. Most of the lesions, 31 out of 44, were indistinguishable in a principal coordinates analysis (PCoA) due to dominance by OTUs phylogenetically similar to Streptococcus pseudopneumoniae. Thirteen lesions not dominated by S. pseudopneumoniae were more similar to HMP throat and saliva samples, though 12 of them contained Pseudomonas, which was not present in any of the HMP samples. Community structure and abundance could not be correlated with lesion diagnosis or any other documented patient factor, including age, sex, or country of origin.
Conclusions
Dominance by S. pseudopneumoniae could be a factor in disease etiology, as could the presence of Pseudomonas in some samples. Likewise, decreased diversity, as compared to healthy saliva and throat samples, may be associated with disease, similar to disease models in other mucosal sites.
Electronic supplementary material
The online version of this article (doi:10.1186/2049-2618-2-43) contains supplementary material, which is available to authorized users.
Keywords: Larynx, Voice disorders, Benign lesions, Microbiota, Streptococcus
Background
At any one time, an estimated 20.7 million people in the US report problems with their voice, while 93.8 million report having problems during their lifetime [1]. Additionally, an estimated 22.6 million of the general population reports missing one or more days of work annually because of voice problems [1]. Voice problems are also associated with negative effects on quality of life including impaired communication, social isolation, and decreased occupational productivity [2–4]. Vocal folds play a key role in voice production and protection of the lungs, vibrating to produce sound and closing to protect the lungs against food and liquid aspiration during swallowing. Tissue of the vocal folds is unique and complex, composed of an epithelial layer made up of non-keratinized stratified squamous epithelium undergoing constant regeneration, which is bound to a three-layered lamina propria via basement membrane zone anchoring fibers. Vocal folds are also immunologically active serving both as a physical barrier and as a site that may detect early shifts in microbial presence [5].
While voice problems are associated with many causes, benign vocal fold lesions are one of the most frequent medical diagnoses [6]. Benign vocal fold lesions are generally classified into two broad categories: non-neoplastic lesions and neoplastic lesions. Non-neoplastic vocal fold lesions make up the majority of benign lesions and include cysts, nodules, polyps, Reinke’s edema, granulomas, ectasias, sulcus vocalis, and scar. Benign vocal fold lesions are typically distinguished by their phenotype, but there is limited knowledge of the etiology and progression of these diseases [7]. Mechanical, chemical, and thermal trauma; overuse and misuse; or combinations of these factors are thought to contribute to lesion development via remodeling of the lamina propria. Studies investigating histological differences and gene expression profiles have demonstrated differential stages of wound maturation and disordered wound healing [8–10]. While inflammation has historically been associated with polyps, Kotby et al. suggests that polyps and Reinke’s edema may represent a continuum of vocal fold injury [11]. Many comorbidities have been associated with vocal fold lesions, including smoking, laryngopharyngeal reflux, infection, and allergies [12–14]. To date, microbial contributions to the etiology of vocal fold lesions have focused mostly on microbes stereotypically considered pathogens, such as human papillomavirus and Mycobacterium tuberculosis [15–17]. However, there remains a lack of data in the literature regarding microbial community membership for normal and diseased larynx [18], secondary to a paucity of available tissue for study.
The only study published to date on laryngeal bacterial communities suggested that laryngeal squamous cell carcinoma (LSCC) could be associated with shifts in bacterial communities in the larynx [19]. Changes in the abundance of 15 genera were associated with carcinoma versus control patients with vocal fold polyps. They postulated that increases in Fusobacterium and Prevotella might be involved in initiating biofilm formation and that it could contribute to disease progression. Similar work in other mucosal sites also suggests that changes in bacterial community membership and abundance, despite the absence of classically defined pathogens, may be associated with disease. Asthmatics have been found to have altered microbial communities in their lungs [20]. While the community members responsible for this change varied between patients, a shift towards Gram negative-dominated communities was associated with elevated lipopolysaccharide levels. The authors postulated that this may be associated with increased airway cell stimulation contributing to disease. Likewise, Wang et al. found that the abundance of particular operational taxonomic units (OTUs) were shifted in feces from patients with colorectal cancer, particularly a reduction in butyrate-producing bacteria from the family Lachnospiraceae [21]. They surmised that an increase in potential pathogens coupled with a decrease in butyrate-producing bacteria could play a role in tumor formation or contribute to colorectal cancer formation.
Shifts in bacterial communities have been associated with diseases of mucosal surfaces, and we postulated that similar changes might be found in benign vocal fold lesions. In this investigation we characterized and compared the bacterial communities of benign vocal fold lesions, including biopsies of cysts, nodules, polyps, and Reinke’s edema. We hypothesized that distinct lesion types would be represented by distinct bacterial communities. This was achieved with 454 pyrosequencing of the V3-V5 region of the 16S rRNA gene using lesion biopsies in patients from the state of Wisconsin, USA, and Germany.
Results
Clinical diagnoses and patient characteristics
Forty-nine patients diagnosed with benign vocal fold lesions were enrolled in this study. Forty-four lesions in total were collected and yielded sufficient sequences, including 7 cysts, 5 nodules, 18 polyps, and 14 Reinke’s edema (Table 1). Characteristics of these patients are detailed in Table 1, including mean age, gender, and country of origin. Five of the 49 initial samples yielded less than 500 sequences; these samples were eliminated from further analyses.
Table 1.
Demographic and characteristics of participants
| Cysts | Nodules | Polyps | Reinke’s edema | |
|---|---|---|---|---|
| Total number of participants | 7 | 5 | 18 | 14 |
| Mean age (range) | 57 (37–73) | 31 (23–38) | 44 (24–77) | 52 (27–74) |
| Female/male | 6/1 | 5/0 | 8/10 | 14/0 |
| Wisconsin/German | 6/1 | 0/5 | 6/12 | 2/12 |
The table includes information only for samples that resulted in more than 500 sequences and were included in further analyses.
Bacterial communities associated with benign vocal fold lesions
Three 454 GS Junior runs resulted in a total of 248,579 high-quality sequences with a mean length of 471 bp after quality filtering and removal of primers and adapter sequences. A total of 44 samples were successfully pyrosequenced and yielded over 500 sequences. Samples had a mean of 5,650 sequences (range 892–23,996). The lowest Good’s coverage was 0.979 suggesting that communities were well sampled. No significant differences were detected between the lesion types by Chao, inverse Simpson, or Shannon diversity indexes (Table 2).
Table 2.
Mean sequence richness, evenness, and diversity in lesion samples
| Cysts | Nodules | Polyps | Reinke’s edema | |
|---|---|---|---|---|
| Chao | 105.2 ± 25.7 | 191.0 ± 97.0 | 112.5 ± 17.4 | 111.3 ± 15.5 |
| 1/Simpson | 7.4 ± 2.9 | 6.1 ± 2.8 | 4.0 ± 1.3 | 4.1 ± 1.5 |
| Shannon | 1.9 ± 0.5 | 1.8 ± 0.6 | 1.3 ± 0.3 | 1.3 ± 0.3 |
| Good’s coverage | 0.991 ± 0.002 | 0.996 ± 0.001 | 0.994 ± 0.001 | 0.991 ± 0.001 |
Mean Good’s coverage, Chao, inverse Simpson, and Shannon are reported for each of the four lesion types, along with standard error. Values for Chao, Inverse Simpson, and Shannon were not significantly different.
After removing rare sequences, those present at less than 0.1% abundance, 334 OTUs were present at 97% similarity across all samples, representing 5 bacterial phyla, including, in order of mean abundance, Firmicutes (73.8%), Proteobacteria (12.7%), Bacteroidetes (9.2%), Actinobacteria (2.1%), and Fusobacteria (1.9%). Sequences identified as Spirochaetes, Cyanobacteria, Verrucomicrobia, Deinococcus-Thermus, Tenericutes, and unclassified were also present, but inconsistently and at low abundance (mean less than 0.2%). Individual samples had a mean of 31.6 OTUs, and only ten classes of bacteria explained most of the diversity found, including Actinobacteria, Bacilli, Bacteroidia, Clostridia, Flavobacteria, Fusobacteria, Negativicutes, and Alpha-, Beta-, and Gamma-proteobacteria (Figure 1). Most of the sequences identified as Bacilli belonged to the genus Streptococcus. This genus was the most common and abundant, found in every lesion sample. An average of 68.7% of all sequences were identified as Streptococcus (cyst, 60.3%; nodule, 58.1%; polyp, 71.5%; and Reinke’s edema, 72.9%). Many samples, 31 out of 44, were dominated (at least 50% of total sequences) by OTUs identified as Streptococcus. Sequences identified as Helicobacter were present in five samples, one from Wisconsin, and four from Germany, between 0.03%–1% relative abundance.
Figure 1.
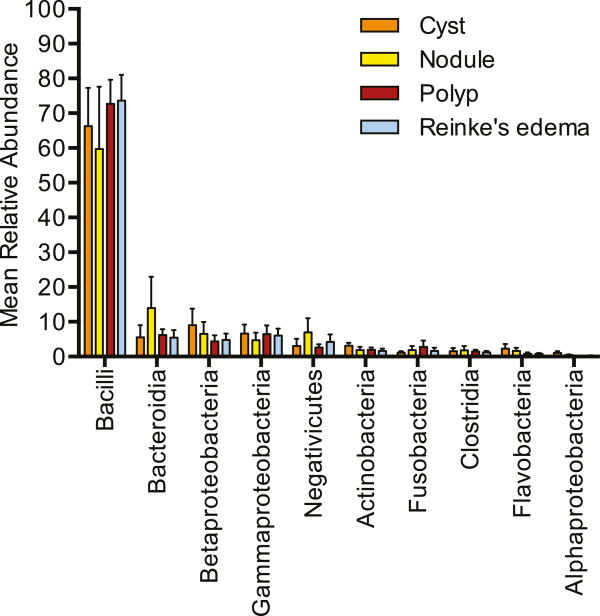
Taxonomic composition in benign vocal fold lesions. Mean relative abundance of the ten most common and abundant bacterial classes in cysts, nodules, polyps, and Reinke’s edema. Error bars represent standard error.
Bacterial community comparison of individual samples supported the above observations. Communities did not cluster in the principal coordinates analysis (PCoA) plot based on lesion type (Figure 2), gender, country of origin, or age (data not shown), nor any other documented clinical information. Dominance by Streptococcus is evident in Figure 2, where the 31 samples dominated by Streptococcus all cluster together.
Figure 2.
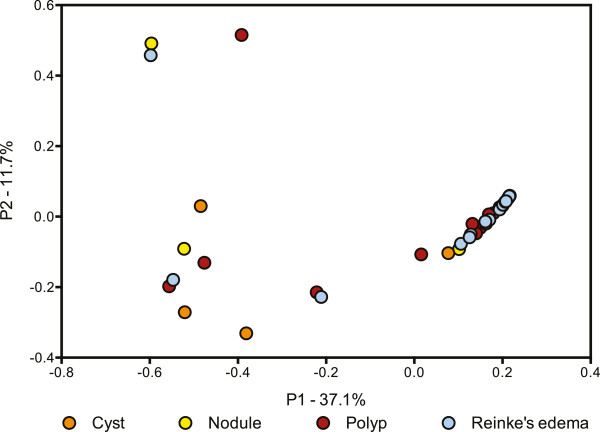
Comparison of bacterial community structure in benign vocal fold lesions. PCoA based on Theta Yue Clayton distances illustrating the clustering of most lesion samples, regardless of lesion diagnosis. Lesion samples are shown as circles, cysts as orange circles, nodules as yellow circles, polyps as red circles, and Reinke’s edema as blue circles.
Comparison of healthy HMP samples to lesion communities
Due to the unethical nature of taking biopsies from healthy human vocal folds, for further analyses we included 15 randomly selected saliva and throat samples from the Human Microbiome Project (HMP) as proxies for healthy vocal fold bacterial communities. Saliva was the most diverse sample type and was significantly different from all other samples by both inverse Simpson and Shannon (p < 0.0001) (Table 3). Throat samples were intermediary, and the differences in diversity compared to lesion samples were not always statistically supported.
Table 3.
Mean sequence richness, evenness, and diversity in lesion and HMP samples
| Cysts | Nodules | Polyps | Reinke’s edema | Saliva | Throat | |
|---|---|---|---|---|---|---|
| Chao | 96.9 ± 23.4b | 121.7 ± 43.5 | 98.1 ± 15.6c | 90.2 ± 11.6a | 190.1 ± 12.2abc | 140.6 ± 11.5 |
| 1/Simpson | 7.4 ± 2.9 | 6.0 ± 2.8 | 4.0 ± 1.3e | 4.1 ± 1.5d | 17.7 ± 1.6* | 11.0 ± 1.4de |
| Shannon | 1.9 ± 0.5 | 1.8 ± 0.6 | 1.3 ± 0.3g | 1.3 ± 0.3f | 3.4 ± 0.1** | 2.8 ± 0.1fg |
| Good’s coverage | 0.991 ± 0.002 | 0.997 ± 0.001 | 0.994 ± 0.001 | 0.993 ± 0.001 | 0.988 ± 0.001 | 0.992 ± 0.001 |
Mean Good’s coverage, Chao, inverse Simpson, and Shannon are reported for each of the four lesion types, and saliva and throat samples from the HMP, along with standard error.
*Saliva was significantly different from all other groups for inverse Simpson (p < 0.0001).
**Saliva was significantly different from all lesion samples for Shannon, but not throat (p < 0.0001).
a–gPairwise comparisons that were significantly different (p < 0.0001).
Tight clustering of lesion samples was not altered in the PCoA plot with the inclusion of the HMP samples (Figure 3). However, some lesions had communities more similar to saliva and throat samples than to other lesions. OTUs identified as Pseudomonas were present in 12 of the 13 lesions that did not cluster based on dominance by Streptococcus, though Pseudomonas was found in only one throat sample.
Figure 3.
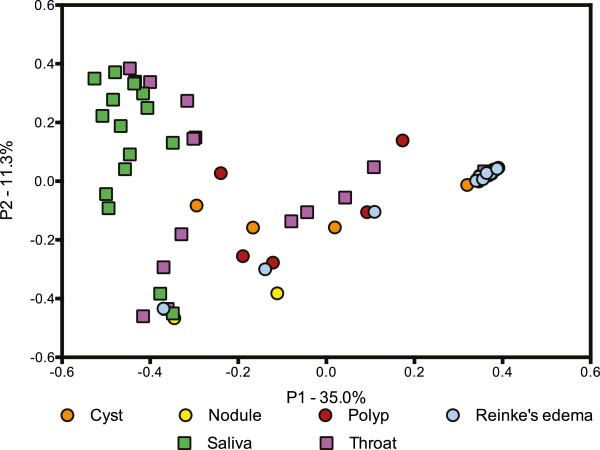
Comparison of bacterial community structure in benign vocal fold lesions and throat and saliva samples from the HMP. PCoA based on Theta Yue Clayton distances illustrating the clustering of most lesion samples, regardless of lesion diagnosis with the inclusion of 15 randomly selected throat and saliva HMP samples. Lesion samples are shown as circles, cysts as orange circles, nodules as yellow circles, polyps as red circles, and Reinke’s edema as blue circles. HMP samples are shown as squares, saliva as green squares, and throat as purple squares.
The abundance of Streptococcusin lesion samples
At 97% sequence identity, one OTU identified as Streptococcus was present in every lesion sample at a mean abundance of 66.21% (range 0.76%–99.76%, 5.01 SE). The sequence selected by mothur to represent this OTU was identified as S. pseudopneumoniae. However, 16S sequences in the mitis group, of which S. pseudopneumoniae is a member, often have greater than 99% sequence identity [22, 23]. We used the 100 most abundant unique OTUs identified as Streptococcus to further explore the diversity and phylogenetic relationship of this genus present in our samples and the included HMP samples (Figure 4). Forty of the top 100 Streptococcus OTUs claded with S. pneumoniae and S. pseudopneumoniae, labeled as clade I, which differ by only one base pair in the region sequenced. Thirty-six of the 44 lesion samples contained OTUs in clade I, while this clade was not present in saliva or throat samples (Figure 4, panel inset). Manual inspection of the alignment file indicated that OTUs within clade I differed in only short homopolymer sequences, 2–6 bp in length. Clades II and III were less common and abundant (Figure 4, panel inset). OTUs in clade II grouped with Streptococcus dentisani, a recently described Streptococcus species [24]. Clade III OTUs could not be identified to the species level, as some species in the mitis group are indistinguishable in this region of the 16S rRNA gene [23]. OTUs that grouped outside these clades were also present, but at very low abundance (data not shown).
Figure 4.
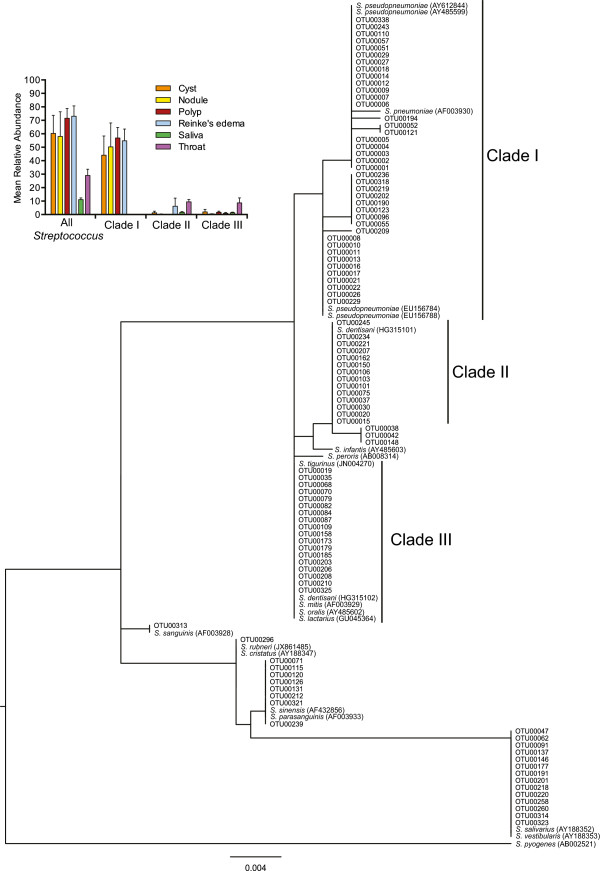
Phylogenetic relationship and abundance of the top 100 unique Streptococcus OTUs. The top 100 unique Streptococcus OTUs along with 21 reference Streptococcus sequences were phylogenetically analyzed. Three major clades were found. Sequences from clade I dominated the lesion samples while being completely absent from HMP samples. Panel inset represents the mean relative abundance of all Streptococcus OTUs in cysts, nodules, polyps, Reinke’s edema, saliva, and throat, in addition to the portion represented by clades I, II, and III. Error bars represent standard error.
Discussion
Maintenance of vocal fold function is essential for human health, where the consequences of impaired voice production hold profound implications for individual health and wellness, social and occupational function, and societal productivity [3, 4]. Benign vocal fold lesions are one of the most common diagnoses when problems with voice arise, but a full pathophysiologic understanding of how and why these lesions form is still lacking. We sought to describe the bacterial communities in a large set of vocal fold biopsies that included cysts, nodules, polyps, and Reinke’s edema. Surprisingly, the majority of our samples, 31 out of 44, had highly similar bacterial communities dominated by S. pseudopneumoniae, regardless of lesion type, or any other documented patient characteristic. To date, only one other study has looked at whole bacterial communities in the larynx, comparing LSCC to nearby “healthy” tissue, and a control group of patients with polyps [19]. Broad differences are apparent between their control polyp patients and ours, from the phyla level to the genus level, where we found considerably more Streptococcus (71% vs. 56%), but less of all other genera they deemed dominant, including Fusobacterium (0.6% vs. 8%), Prevotella (5% vs. 7%), Neisseria (2% vs. 5%), and Gemella (0.5% vs. 2%). It is widely accepted that even minor differences in sample preparation can lead to discrepancies in community composition, and the differences found between our polyp samples and those in Gong et al. could be due to any one of these factors, including the use of different primer pairs or DNA extraction protocols, and modifications to PCR protocols [19, 25–32].
The presence of Streptococcus is well noted in healthy oral and respiratory sites [33–35]. However, due to the high similarity of the 16S gene in many Streptococcus species, finer resolution based on non-full length sequences is difficult. A phylogeny of the OTUs identified as Streptococcus in our study demonstrated that many of them were either S. pseudopneumoniae or S. pneumoniae. These two Streptococcus species vary by a single base pair in the region sequenced here, and a manual check of the alignment showed that all sequences in clade I contained a cytosine at that position, like S. pseudopneumoniae, and no OTU out of the top 100 could be identified as S. pneumoniae. Dominance by S. pseudopneumoniae in the majority of our samples may be playing a role in disease progression. This recently described member of the mitis group has many genes associated with host cell interaction, including some of which are thought to be virulence factors [36, 37]. S. pseudopneumoniae, previously thought to be atypical S. pneumoniae, has been isolated from patients with a number of respiratory diseases including chronic obstructive pulmonary disease (COPD), cystic fibrosis, pneumonia, bronchitis, and chronic sinusitis [38–40]. However, its status as a pathogen, or mutualist, is still not clear. All 20 human subjects in a tonsillar crypt study were found to have S. pseudopneumoniae, regardless of tonsil health status [41].
Helicobacter pylori is perhaps one of the most prevalent “infections” in the world, estimated to colonize the stomach of over 50% of the world’s human population [42]. Work in the last few decades has shown a direct relation between H. pylori and stomach cancer [42], and more recently, correlations have been made with other aerodigestive tract diseases, such as otitis media with effusion [43] and oral aphthous ulcers [44]. H. pylori has also been associated with LSCC [45–47], though there is debate about whether it is associated with benign vocal fold lesions [48] or not [47, 49]. Only a few of the samples presented here contained Helicobacter and at very low abundances. Many studies seeking to tie vocal fold lesions to H. pylori infections have looked for the specific presence of the bacterium in the larynx [47], but not necessarily at its abundance. It could be that H. pylori is common in vocal folds, but at a low enough abundance that it was mostly missed in our patient population and with our sampling technique, even in those with previously diagnosed infections.
One limitation in our study design was the inability to compare these lesions to the microbial community that may be found in biopsied healthy human vocal folds. The lamina propria of the vocal folds is very thin, only 3 mm thick, and the possibility of creating vocal scar and impairing voice production is present with every surgical procedure including biopsy. As such, it is considered unethical to biopsy vocal folds of healthy human subjects. As a proxy, we included in our analyses 15 randomly selected saliva and throat samples on the basis that many of the habitats above the stomach have similar microbial communities due to the buffering nature of saliva, regular nutrient availability in the form of mucin, and a common epithelial lining (non-keratinized, stratified, squamous epithelium) [33]. Of the 15–18 body sites sampled by the HMP, non-tooth oral sites, including saliva and throat, were deemed to be highly similar [50]. Samples from these body sites might have the most in common with our samples, considering that they all face similar environmental exposures such as inhaled air, ingested substances (food and beverage), and constant contact via saliva and a continuous mucus layer. Indeed, 13 of our 44 samples were highly similar to these throat and saliva samples. However, the majority harbored a distinct community, one dominated by S. pseudopneumoniae, as described above. Interestingly, of the 13 samples more similar to saliva and throat, 12 of them contained Pseudomonas, a genus found in only 1 throat sample. However, many of the common and dominant genera found in the saliva and throat samples were also found in these 13 samples, including Actinomyces, Prevotella, Fusobacterium, Leptotrichia, Neisseria, Haemophilus, Gemella, Granulicatella, Oribacterium, Veillonella, unclassified Prevotellaceae, and unclassified Lachnospiraceae [33]. Pseudomonas has been associated with diseases of the lower respiratory system including bronchiolitis obliterans syndrome [51] and cystic fibrosis [52] and could be playing a role in benign vocal fold lesion etiology.
Conclusions
Benign vocal fold lesions are usually diagnosed based on phenotypic differences present upon videostroboscopic examination. While histological and gene expression differences have been noted, the data presented here adds to the idea that despite phenotypic differences, benign vocal fold lesions share many similarities, including highly similar bacterial communities that are distinct from healthy throat and saliva samples [8, 9]. The possibility remains that these shifts in the bacterial community could inhibit wound healing, or that the presence of inflammation creates a niche for a community dominated by S. pseudopneumoniae.
Methods
Subjects and collection of benign lesions
Benign vocal fold lesions were collected at University of Wisconsin-Madison, USA, and the University of Hamburg, Germany. Laryngeal microsurgical techniques were used to remove lesions. For each lesion, the clinical diagnosis was made based on initial videostroboscopic exam and confirmed under direct visualization by the surgeon at the time of surgical removal. Samples were immediately placed in RNAlater (Ambion Inc., Austin, Texas) and stored at -80°C until use. The University of Wisconsin Madison Health Sciences IRB and the University of Hamburg Ethics Committee approved the protocol for attainment of all tissue samples. All subjects provided written consent to participate in this study. Meta data for each sample is detailed in Additional file 1: Table S1.
DNA extraction and PCR for 454 pyrosequencing
DNA was extracted from tissue with the EpiCenter MasterPure Complete DNA and RNA Purification Kit (Illumina, Madison, WI) with modifications to the manufacturer’s protocol. Samples were gently thawed at room temperature and briefly centrifuged to collect tissue. RNAlater was removed by pipetting and 300 μl of tissue and cell lysis solution was added to the tube with the tissue. Lysis solution and tissue were then transferred to a sterile screw top tube containing 150–200 mg of 400 μM silica beads. Proteinase K, 100 μg, was added; the tubes were vortexed and incubated at 55°C for 1 h, with vortexing every 15 min. Bead tubes were then shaken in a horizontal adapter on the vortex for 10 min. Rnase A, 5 μg, was added; the tubes were vortexed and incubated at 37°C for 30 min. The remainder of the manufacturer’s protocol was followed as written. DNA was resuspended in TE buffer and stored at 4°C until use.
PCRs were performed in triplicates containing 50–100 ng of template DNA, 0.2 μl AccuPrime Taq DNA Polymerase (Life Technologies, Grand Island, NY), 2.5 μl Buffer II, 400 nM both forward and reverse primers, and water to 25 μl total. Thermocycling conditions were as follows: 95°C 2 min, followed by 30 cycles of 95°C 20 s, 56°C 30 s, 72°C 1 min, and a final extension of 72°C 8 min. The primers included 357 F and 926R, as suggested by the HMP [53], where 357 F contained the B adapter for 454 pyrosequencing, and 926R contained both the A adapter and a 10-base pair multiplex identifier. Triplicate PCRs were pooled and cleaned using Purelink PCR purification kit (Invitrogen, Grand Island, NY) as per manufacturer’s directions for removal of primer dimers and short PCR products <300 bp. Cleaned products were eluted in 30 μl of elution buffer. Samples were then gel extracted from a low-melt agarose gel using Zymoclean Gel DNA Recovery Kit (Zymo Research, Irvine, CA) by visualizing on a blue light transilluminator (Clare Chemical Research, Dolores, CO). Cleaned PCR products were quantified using a Qubit fluorometer (Invitrogen, Grand Island, NY). Products were diluted and pooled at equal concentrations for 454 pyrosequencing.
454 pyrosequencing, data analysis, and statistics
454 pyrosequencing was conducted on a Roche GS Junior (Roche, Indianapolis, IN) using titanium chemistry and long read modifications found in Hanshew et al. [26]. Samples were sequenced across three picotiter plates. Raw data were processed using mothur (v. 1.33.1) [54], with most of the defaults put forth in the Schloss 454 SOP (http://www.mothur.org/wiki/Schloss_SOP; accessed Jan 27, 2014) [55], but with minflows = 350 and maxflows = 720 [56]. Sequences were aligned to a Silva-derived reference data base (v. 102 as implemented for mothur) [57]. Chimeras were detected using UCHIME and removed [58]. Sequences were assigned to taxonomic groups using the Ribosomal Database Project (RDP)-derived reference database [59]. All eukaryotic and unclassifiable reads were removed after classify.seqs. Sequences were assigned to OTUs at 97% sequence identity, used to construct a distance matrix using theta Yue and Clayton values, and analyzed with PCoA plots in Prism. Good’s coverage, Chao, inverse Simpson, and Shannon were calculated in mothur. One-way ANOVA with TukeyHSD p value correction for pairwise comparison was used to assess differences in Chao, inverse Simpson, and Shannon. In addition to lesion diagnosis, bacterial communities were also compared based on age, gender, date of surgery, and geographic location. The data sets supporting the results of this article are available in the NCBI sequence read archive, SRP047304.
Thirty HMP samples, including 15 haphazardly selected saliva and 15 throat (oropharynx) samples, were included in further analyses as proxies for “healthy” (Additional file 1: Table S1). As described above, Good’s coverage, Chao, inverse Simpson, and Shannon diversity indexes were calculated along with theta Yue and Clayton values for PCoA at 97% sequence identity [53, 60].
Similar to Jensen et al. and Scholz et al., the 100 most abundant unique OTUs identified as Streptococcus were chosen for phylogenetic analysis [41, 61]. Sequences were aligned in MEGA6 using ClustalW and trimmed at the ends [62]. A phylogenetic tree was created in MEGA6 using maximum likelihood with 500 bootstraps. Sequences were searched in the Ribosomal Database Project, and matching type strain sequences were included in the phylogenetic analysis, including S. cristatus (AY188347), S. infantis (AY485603), S. lactarius (GU045364), S. mitis (AF003929), S. oralis (AY485602), S. parasanguinis (AF003933), S. peroris (AB008314), S. pneumoniae (AF003930), S. pseudopneumoniae (AY612844, AY485599, EU156784, and EU156788), S. salivarius (AY188352), S. sanguinis (AF003928), S. sinensis (AF432856), S. vestibularis (AY188353) [59, 63]. Non-type strains S. dentisani (HG315101 and HG315102), S. rubneri (JX861485), and S. tigurinus (JN004270) were included due to the high match to some OTUs. S. pyogenes (AB002521) was included as an outgroup.
Electronic supplementary material
Additional file 1: Table S1: Meta data for each lesion sample along with a list of the haphazardly chosen HMP samples. (XLSX 16 KB)
Acknowledgements
The National Institutes of Health NIDCD R01 9600 and T32 9401 funded this work. We thank Nicole Li, Charles N. Ford, and Seth H. Dailey at the University of Wisconsin Madison, and Markus Hess, Susanne Fleischer, and Frank Muller at the University of Hamburg, Germany, for lesion collection. We thank Garret Suen for the use of server space in his lab and Glen Leverson for assistance with statistical analyses.
Abbreviations
- OTU
operational taxonomic units
- PCoA
principal coordinates analysis
- HMP
Human Microbiome Project
- LSCC
laryngeal squamous cell carcinoma
- COPD
chronic obstructive pulmonary disease
- RDP
Ribosomal Database Project.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MEJ and SLT conceived of the study. ASH, MEJ, and SLT designed the experiment. ASH carried out the experiment. All authors wrote, read, and approved the final manuscript.
Contributor Information
Alissa S Hanshew, Email: hanshew@surgery.wisc.edu.
Marie E Jetté, Email: jette@surgery.wisc.edu.
Susan L Thibeault, Email: THIBEAUL@surgery.wisc.edu.
References
- 1.Roy N, Merrill RM, Gray SD, Smith EM. Voice disorders in the general population: prevalence, risk factors, and occupational impact. Laryngoscope. 2005;115:1988–1995. doi: 10.1097/01.mlg.0000179174.32345.41. [DOI] [PubMed] [Google Scholar]
- 2.Cohen SM, Dupont WD, Courey MS. Quality-of-life impact of non-neoplastic voice disorders: a meta-analysis. Ann Otol Rhinol Laryngol. 2006;115:128–134. doi: 10.1177/000348940611500209. [DOI] [PubMed] [Google Scholar]
- 3.Jacobson BH, Johnson A, Grywalski C, Silbergleit A, Jacobson G, Benninger MS, Newman CW. The voice handicap index (VHI): development and validation. Am J Speech Lang Pathol. 1997;6:66–70. doi: 10.1044/1058-0360.0603.66. [DOI] [Google Scholar]
- 4.Ma EP-M, Yiu EM-L. Voice activity and participation profile: assessing the impact of voice disorders on daily activities. J Speech Lang Hear Res. 2001;44:511–524. doi: 10.1044/1092-4388(2001/040). [DOI] [PubMed] [Google Scholar]
- 5.Thibeault SL, Rees L, Pazmany L, Birchall MA. At the crossroads: mucosal immunology of the larynx. Mucosal Immunol. 2009;2:122–128. doi: 10.1038/mi.2008.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen SM, Kim J, Roy N, Asche C, Courey M. Prevalence and causes of dysphonia in a large treatment-seeking population. Laryngoscope. 2012;122:343–348. doi: 10.1002/lary.22426. [DOI] [PubMed] [Google Scholar]
- 7.Cipriani NA, Martin DE, Corey JP, Portugal L, Caballero N, Lester R, Anthony B, Taxy JB. The clinicopathologic spectrum of benign mass lesions of the vocal fold due to vocal abuse. Int J Surg Pathol. 2011;19:583–587. doi: 10.1177/1066896911411480. [DOI] [PubMed] [Google Scholar]
- 8.Duflo SM, Thibeault SL, Li W, Smith ME, Schade G, Hess MM. Differential gene expression profiling of vocal fold polyps and Reinke’s edema by complementary DNA microarray. Ann Otol Rhinol Laryngol. 2006;115:703–714. doi: 10.1177/000348940611500910. [DOI] [PubMed] [Google Scholar]
- 9.Thibeault SL, Gray SD, Li W, Ford C, Smith ME, Davis RK. Genotypic and phenotypic expression of vocal fold polyps and Reinke’s edema: a preliminary study. Ann Otol Rhinol Laryngol. 2002;111:302–309. doi: 10.1177/000348940211100404. [DOI] [PubMed] [Google Scholar]
- 10.Marcotullio D, Magliulo G, Pietrunti S, Suriano M. Exudative laryngeal diseases of Reinke’s space: a clinicohistopathalogical framing. J Otolaryngol. 2002;31:376–380. doi: 10.2310/7070.2002.34517. [DOI] [PubMed] [Google Scholar]
- 11.Kotby M, Nassar A, Seif E, Helal E, Saleh M. Ultrastructural features of vocal fold nodules and polyps. Acta Otolaryngol. 1988;105:477–482. doi: 10.3109/00016488809119505. [DOI] [PubMed] [Google Scholar]
- 12.Kuhn J, Toohill RJ, Ulualp SO, Kulpa J, Hofmann C, Arndorfer R, Shaker R. Pharyngeal acid reflux events in patients with vocal cord nodules. Laryngoscope. 1998;108:1146–1149. doi: 10.1097/00005537-199808000-00008. [DOI] [PubMed] [Google Scholar]
- 13.Ulualp SO, Toohill RJ, Shaker R. Pharyngeal acid reflux in patients with single and multiple otolaryngologic disorders. Otolaryngol Head Neck Surg. 1999;121:725–730. doi: 10.1053/hn.1999.v121.a98010. [DOI] [PubMed] [Google Scholar]
- 14.Hocevar-Boltezar I, Radsel Z, Zargi M. The role of allergy in the etiopathogenesis of laryngeal mucosal lesions. Acta Otolaryngol Suppl. 1997;527:134–137. doi: 10.3109/00016489709124056. [DOI] [PubMed] [Google Scholar]
- 15.Altman KW. Vocal fold masses. Otolaryngol Clin North Am. 2007;40:1091–1108. doi: 10.1016/j.otc.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 16.Shin J-E, Nam SY, Yoo SJ, Kim SY. Changing trends in clinical manifestations of laryngeal tuberculosis. Laryngoscope. 2000;110:1950–1953. doi: 10.1097/00005537-200011000-00034. [DOI] [PubMed] [Google Scholar]
- 17.Wang C-C, Lin C-C, Wang C-P, Liu S-A. Laryngeal tuberculosis: a review of 26 cases. Otolaryngol Head Neck Surg. 2007;137:582–588. doi: 10.1016/j.otohns.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 18.Hong P, Liu CM, Nordstrom L, Lalwani AK. The role of the human microbiome in otolaryngology-head and neck surgery: a contemporary review. Laryngoscope. 2014;124:1352–1357. doi: 10.1002/lary.24490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gong H-L, Shi Y, Zhou L, Wu C-P, Cao P-Y, Tao L, Xu C, Hou D-S, Wang Y-Z. The composition of microbiome in larynx and the throat biodiversity between laryngeal squamous cell carcinoma patients and control population. PLoS One. 2013;8:e66476. doi: 10.1371/journal.pone.0066476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goleva E, Jackson LP, Harris JK, Robertson CE, Sutherland ER, Hall CF, Good JT, Gelfand EW, Martin RJ, Leung DYM. The effects of airway microbiome on corticosteroid responsiveness in asthma. Am J Respir Crit Care Med. 2013;188:1193–1201. doi: 10.1164/rccm.201304-0775OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang T, Cai G, Qiu Y, Fei N, Zhang M, Pang X, Jia W, Cai S, Zhao L. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2012;6:320–329. doi: 10.1038/ismej.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thompson CC, Emmel VE, Fonseca EL, Marin MA, Vicente ACP. Streptococcal taxonomy based on genome sequence analyses. F1000Res. 2013;2:67. doi: 10.12688/f1000research.2-67.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawamura Y, Hou X-G, Sultana F, Miura H, Ezaki T. Determination of 16S rRNA sequences of Streptococcus mitis and Streptococcus gordonii and phylogenetic relationships among members of the genus Streptococcus. Int J Syst Bacteriol. 1995;45:406–408. doi: 10.1099/00207713-45-2-406. [DOI] [PubMed] [Google Scholar]
- 24.Camelo-Castillo A, Benítez-Páez A, Belda-Ferre P, Cabrera-Rubio R, Mira A. Streptococcus dentisani sp. nov., a novel member of the mitis group. Int J Syst Evol Microbiol. 2014;64:60–65. doi: 10.1099/ijs.0.054098-0. [DOI] [PubMed] [Google Scholar]
- 25.Sergeant MJ, Constantinidou C, Cogan T, Penn CW, Pallen MJ. High-throughput sequencing of 16S rRNA gene amplicons: effects of extraction procedure, primer length and annealing temperature. PLoS One. 2012;7:e38094. doi: 10.1371/journal.pone.0038094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanshew AS, Mason CJ, Raffa KF, Currie CR. Minimization of chloroplast contamination in 16S rRNA gene pyrosequencing of insect herbivore bacterial communities. J Microbiol Methods. 2013;95:149–155. doi: 10.1016/j.mimet.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jumpstart Consortium Human Microbiome Project Data Generation Working Group Evaluation of 16S rDNA-based community profiling for human microbiome research. PLoS One. 2012;7:e39315. doi: 10.1371/journal.pone.0039315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar PS, Brooker MR, Dowd SE, Camerlengo T. Target region selection is a critical determinant of community fingerprints generated by 16S pyrosequencing. PLoS One. 2011;6:e20956. doi: 10.1371/journal.pone.0020956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Engelbrektson A, Kunin V, Wrighton KC, Zvenigorodsky N, Chen F, Ochman H, Hugenholtz P. Experimental factors affecting PCR-based estimates of microbial species richness and evenness. ISME J. 2010;4:642–647. doi: 10.1038/ismej.2009.153. [DOI] [PubMed] [Google Scholar]
- 30.Ahn J-H, Kim B-Y, Song J, Weon H-Y. Effects of PCR cycle number and DNA polymerase type on the 16S rRNA gene pyrosequencing analysis of bacterial communities. J Microbiol. 2012;50:1071–1074. doi: 10.1007/s12275-012-2642-z. [DOI] [PubMed] [Google Scholar]
- 31.Sipos R, Székely AJ, Palatinszky M, Révész S, Márialigeti K, Nikolausz M. Effect of primer mismatch, annealing temperature and PCR cycle number on 16S rRNA gene-targetting bacterial community analysis. FEMS Microbiol Ecol. 2007;60:341–350. doi: 10.1111/j.1574-6941.2007.00283.x. [DOI] [PubMed] [Google Scholar]
- 32.Jewell KA. Characterization of the Ruminal Bacterial Microbiota in Relation to Host Parameters. University of Wisconsin-Madison, Microbiology Doctoral Training Program: PhD thesis; 2014. [Google Scholar]
- 33.Segata N, Haake SK, Mannon P, Lemon KP, Waldron L, Gevers D, Huttenhower C, Izard J. Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome Biol. 2012;13:R42. doi: 10.1186/gb-2012-13-6-r42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huse SM, Ye Y, Zhou Y, Fodor AA. A core human microbiome as viewed through 16S rRNA sequence clusters. PLoS One. 2012;7:e34242. doi: 10.1371/journal.pone.0034242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edlund A, Yang Y, Hall AP, Guo L, Lux R, He X, Nelson KE, Nealson KH, Yooseph S, Shi W, Mclean JS. An in vitro biofilm model system maintaining a highly reproducible species and metabolic diversity approaching that of the human oral microbiome. Microbiome. 2013;1:25. doi: 10.1186/2049-2618-1-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shahinas D, Thornton CS, Tamber GS, Arya G, Wong A, Jamieson FB, Ma JH, Alexander DC, Low DE, Pillai DR. Comparative genomic analyses of Streptococcus pseudopneumoniae provide insight into virulence and commensalism dynamics. PLoS One. 2013;8:e65670. doi: 10.1371/journal.pone.0065670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arbique JC, Poyart C, Trieu-Cuot P, Quesne G, Carvalho Mda GS, Steigerwalt AG, Morey RE, Jackson D, Davidson RJ, Facklam RR. Accuracy of phenotypic and genotypic testing for identification of Streptococcus pneumoniae and description of Streptococcus pseudopneumoniae sp nov. J Clin Microbiol. 2004;42:4686–4696. doi: 10.1128/JCM.42.10.4686-4696.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laurens C, Michon A-L, Marchandin H, Bayette J, Didelot M-N, Jean-Pierre H. Clinical and antimicrobial susceptibility data of 140 Streptococcus pseudopneumoniae isolates in France. Antimicrob Agents Chemother. 2012;56:4504–4507. doi: 10.1128/AAC.06374-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keith ER, Podmore RG, Anderson TP, Murdoch DR. Characteristics of Streptococcus pseudopneumoniae isolated from purulent sputum samples. J Clin Microbiol. 2006;44:923–927. doi: 10.1128/JCM.44.3.923-927.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harf-Monteil C, Granello C, Le Brun C, Monteil H, Riegel P. Incidence and pathogenic effect of Streptococcus pseudopneumoniae. J Clin Microbiol. 2006;44:2240–2241. doi: 10.1128/JCM.02643-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jensen A, Fagö-Olsen H, Sørensen CH, Kilian M. Molecular mapping to species level of the tonsillar crypt microbiota associated with health and recurrent tonsillitis. PLoS One. 2013;8:e56418. doi: 10.1371/journal.pone.0056418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Conteduca V, Sansonno D, Lauletta G, Russi S, Ingravallo G, Dammacco F. H. pylori infection and gastric cancer: state of the art (review) Int J Oncol. 2013;42:5–18. doi: 10.3892/ijo.2012.1701. [DOI] [PubMed] [Google Scholar]
- 43.Yilmaz MD, Aktepe O, Cetinkol Y, Altuntas A. Does Helicobacter pylori have role in development of otitis media with effusion? Int J Pediatr Otorhinolaryngol. 2005;69:745–749. doi: 10.1016/j.ijporl.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 44.Birek C, Grandhi R, McNeill K, Singer D, Ficarra G, Bowden G. Detection of Helicobacter pylori in oral aphthous ulcers. J Oral Pathol Med. 1999;28:197–203. doi: 10.1111/j.1600-0714.1999.tb02024.x. [DOI] [PubMed] [Google Scholar]
- 45.Burduk PK. Association between infection of virulence cagA gene Helicobacter pylori and laryngeal squamous cell carcinoma. Med Sci Monit. 2013;19:584–591. doi: 10.12659/MSM.889011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhuo X-L, Wang Y, Zhuo W-L, Zhang X-Y. Possible association of Helicobacter pylori infection with laryngeal cancer risk: an evidence-based meta-analysis. Arch Med Res. 2008;39:625–628. doi: 10.1016/j.arcmed.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 47.Titiz A, Ozcakir O, Ceyhan S, Yilmaz YF, Unal A, Akyon Y. The presence of Helicobacter pylori in the larynx pathologies. Auris Nasus Larynx. 2008;35:534–538. doi: 10.1016/j.anl.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 48.Tiba M, Fawaz S, Osman H. Helicobacter pylori and its role in vocal folds’ minimal lesions. Clin Respir J. 2009;4:237–240. doi: 10.1111/j.1752-699X.2009.00182.x. [DOI] [PubMed] [Google Scholar]
- 49.Masoud N, Manouchehr K, Najmeh D, Monireh H. Lack of association between Helicobacter pylori and laryngeal carcinoma. Asian Pac J Cancer Prev. 2008;9:81–82. [PubMed] [Google Scholar]
- 50.Aagaard K, Riehle K, Ma J, Segata N, Mistretta T-A, Coarfa C, Raza S, Rosenbaum S, Van den Veyver I, Milosavljevic A, Gevers D, Huttenhower C, Petrosino J, Versalovic J. A metagenomic approach to characterization of the vaginal microbiome signature in pregnancy. PLoS One. 2012;7:e36466. doi: 10.1371/journal.pone.0036466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dickson RP, Erb-Downward JR, Freeman CM, Walker N, Scales BS, Beck JM, Martinez FJ, Curtis JL, Lama VN, Huffnagle GB. Changes in the lung microbiome following lung transplantation include the emergence of two distinct Pseudomonas species with distinct clinical associations. PLoS One. 2014;9:e97214. doi: 10.1371/journal.pone.0097214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guss AM, Roeselers G, Newton ILG, Young CR, Klepac-Ceraj V, Lory S, Cavanaugh CM. Phylogenetic and metabolic diversity of bacteria associated with cystic fibrosis. ISME J. 2011;5:20–29. doi: 10.1038/ismej.2010.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.The Human Microbiome Project Consortium A framework for human microbiome research. Nature. 2012;486:215–221. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schloss PD, Gevers D, Westcott SL. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One. 2011;6:e27310. doi: 10.1371/journal.pone.0027310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Quince C, Lanzen A, Davenport RJ, Turnbaugh PJ. Removing noise from pyrosequenced amplicons. BMC Bioinformatics. 2011;12:38. doi: 10.1186/1471-2105-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glöckner FO. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cole JR, Wang Q, Fish JA, Chai B, Mcgarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, Tiedje JM. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014;42:633–642. doi: 10.1093/nar/gkt1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.The Human Microbiome Project Consortium Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scholz CFP, Poulsen K, Kilian M. Novel molecular method for identification of Streptococcus pneumoniae applicable to clinical microbiology and 16S rRNA sequence-based microbiome studies. J Clin Microbiol. 2012;50:1968–1973. doi: 10.1128/JCM.00365-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol Biol Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1: Meta data for each lesion sample along with a list of the haphazardly chosen HMP samples. (XLSX 16 KB)