

Abstract
Purpose
Vascular endothelial growth factor receptor (VEGFR) kinases are important drug targets in oncology that affect function of systemic endothelial cells. To discover genetic markers that affect VEGFR inhibitor pharmacodynamics we performed a genome-wide association study of serum soluble vascular endothelial growth factor receptor-2 concentrations [sVEGFR2], a pharmacodynamic biomarker for VEGFR2 inhibitors.
Experimental Design
We conducted a genome-wide association study (GWAS) of [sVEGFR2] in 736 healthy Old Order Amish volunteers. Gene variants identified from the GWAS were genotyped serially in a cohort of 128 advanced solid tumor patients with baseline [sVEGR2] measurements, and in 121 renal carcinoma patients with [sVEGFR2] measured before and during pazopanib therapy.
Results
rs34231037 (C482R) in KDR, the gene encoding sVEGFR2 was found to be highly associated with [sVEGFR2], explaining 23% of the variance (p=2.7×10−37). Association of rs34231037 with [sVEGFR2] was replicated in 128 patients with cancer with comparable effect size (p = 0.025). Furthermore rs34231037 was a significant predictor of changes in [sVEGFR2] in response to pazopanib (p = 0.01).
Conclusion
Our findings suggest that genome-wide analysis of phenotypes in healthy populations can expedite identification of candidate pharmacogenetic markers. Genotyping for germ-line variants in KDR may have clinical utility in identifying cancer patients with unusual sensitivity to effects of VEGFR2 kinase inhibitors.
Keywords: Angiogenesis inhibitors, genome-wide association study, VEGFR2, biomarkers, pharmacological, carcinoma, renal cell
Introduction
Advances in our understanding of the molecular basis of cancer have led to the identification of a number of novel targets and classes of anticancer agents. However, inter-individual variability in efficacy and toxicity creates difficulties in optimizing therapy for individual patients. The discovery of germ-line and/or somatic genetic markers that predict inter-individual differences in therapeutic response promises to improve the efficacy and safety of cancer therapeutics(1–5). These markers also might accelerate the pace and increase the success rate of drug development by identifying subsets of patients who are more likely to respond to a given drug, while other markers may define which patients are more likely to experience life-threatening adverse reactions, and in whom that drug should be avoided (or dose attenuated). Unfortunately, the identification of pharmacogenetic markers for a new compound in early clinical trials is a logistical challenge, and thus the discovery of these markers has typically been deferred to late in the development of new drugs or after the drugs have become commercially available when the numbers of persons having received the drugs is sufficiently large(6). To date, most pharmacogenetic studies of anticancer agents have focused on common variants in drug-metabolizing enzymes(7–10).
Angiogenesis inhibitors are an important new class of anticancer agents, but their optimal use requires more detailed understanding of their pharmacology and the biological basis for inter-individual differences in resistance and sensitivity to treatment(11, 12). Pazopanib is an oral angiogenesis inhibitor that blocks signaling by vascular endothelial growth factor receptor 2 (VEGFR2) and other kinases, and has been approved for commercial use to treat renal cell carcinoma and soft-tissue sarcoma(13, 14). VEGFR2 is a transmembrane receptor tyrosine kinase expressed by endothelial cells, subpopulations of bone marrow-derived cells, and some tumor cells(15). It is the primary transducer of extracellular VEGF mediating endothelial cell proliferation, migration, and resistance to apoptosis(16). Alternate splicing of KDR, the gene that encodes VEGFR2, results in a 679 amino acid truncated extracellular domain product which is soluble and circulates in blood (sVEGFR2)(17). VEGFR2 kinase inhibitors decrease serum concentrations sVEGFR2 [sVEGFR2], and these concentrations return to baseline after cessation of drug administration (18–21). Additionally, the baseline concentrations among cancer patients have the same magnitude and distribution as in healthy human subjects. In rodents, the magnitude of these drug-related changes in [sVEGFR2] is dose-dependent and independent of the presence of tumors(22). In different human cancer cohorts the change in [sVEGFR2] was associated with tumor response to VEGFR2 kinase inhibitors(18, 23), suggesting that [sVEGFR2] might serve as a quantitative endophenotype with which to better understand differences among humans in response to VEGFR2 inhibitors. Furthermore, the discovery of genetic and/or other determinants of [sVEGFR2] may provide further insights into mechanisms of tumor progression and new targets for therapy.
To expedite discovery of gene variants that mark inter-individual differences in response to VEGFR2 inhibitors, we performed a genome wide association study (GWAS) of [sVEGFR2] in a non-cancer patient population, which identified a locus of linked variants on chromosome 4 associated with the phenotype. One specific missense variant, rs34231037 in KDR, was then identified as a major determinant of [sVEGFR2], which was also associated with the pharmacodynamics of pazopanib in cancer patients.
Methods
Human Subjects
This study was approved by institutional review boards (IRB) at the University of Chicago and the University of Maryland. All participants from the six different studies in the three study cohorts provided informed consent for use of their specimens in pharmacogenetic studies (see Supplementary methods for additional detail).
Study cohort 1. The Amish Heredity and Phenotype Intervention (HAPI) Heart Study
The HAPI Heart Study began in 2003 to identify genes that interact with environmental exposures to influence risk for cardiovascular disease. Study design and phenotyping procedures have been previously described in detail(24, 25). Briefly, healthy Amish subjects, ages 20 to 80 years underwent a detailed medical examination including blood pressure, anthropometry, total fat mass by dual energy X-ray absorptiometry, fasting blood draw for lipids and other cardiovascular markers and for extraction of DNA for genetic studies, and four short-term interventions designed to challenge cardiovascular function. For this study, fresh-frozen sera, collected at 2 independent time-points either 6 hours or 1 week apart, were available for measurement of [sVEGFR2] in 736 participants.
Study cohort 2. Biomarker Validation and Cancer Patient Replication Cohorts A Pharmacokinetic, Pharmacodynamic, and Pharmacogenetic Study of Sorafenib and Blood Pressure Elevation in Patients with Advanced Malignancies
Study design and patient population have been previously described(26). Sera and DNA samples prior to administration of sorafenib were available for 41 participants with advanced cancer.
A Randomized Phase II Trial Comparing Cetuximab with Concurrent Pemetrexed/Cetuximab Therapy for Non-small Cell Lung Cancer Refractory to Primary Treatment
This study enrolled 43 patients with advanced non-small cell lung cancer whose disease progressed after initial platinum-based therapy (27). Sera and DNA samples prior to administration of the first dose of cetuximab were available for 27 participants.
A Dose Escalation Study of Sorafenib in Normotensive Patients with Advanced Malignancies (NCT00436579)
This study enrolled patients with advanced solid tumors and normal blood pressure to receive sorafenib at standard and higher doses. Sera and DNA samples prior to administration of sorafenib were available for 60 participants (28).
Study cohort 3. A Phase II Study of GW786034 (Pazopanib) Using a Randomized Discontinuation Design in Subjects with Locally Recurrent or Metastatic Clear-cell Renal Cell Carcinoma VEG 102616 (NCT00244764)
This study was used to assess the effect of genotype on pharmacodynamics of pazopanib. Details of the study design were previously reported (29). Of the 225 patients enrolled, 121 had DNA and sVEGFR2 serum measurements available from baseline and post-4-weeks of pazopanib treatment.
Serum Sample Processing
For the HAPI Heart Study, venous blood was drawn from an arm vein into serum separator tubes (SST), maintained at 4°C for 30–60 minutes prior to centrifugation, and serum separated and stored at −80°C storage until assay. For study cohorts 2–3, samples were collected from the upper extremity or a central venous port, incubated at room temperature for 30 minutes and then serum was separated by centrifugation. Samples were aliquotted into cryovials and stored at −70°C until assay.
Measurement of serum [sVEGFR2]
Serum samples were thawed on ice and processed in triplicate according to manufacturer’s specifications (R&D Systems). Samples in which the results deviated from the manufacturer’s performance specifications (CV > 7% among 3 wells) were reanalyzed. No sample required re-analysis more than once. To control for inter-plate and inter-batch variation, aliquots from a single time point blood draw from two volunteer subjects were run on each plate in triplicate (internal controls). For details of the normalization procedure based on internal control sample measurements, see Supplementary methods.
Genotyping
Genome-wide typing and quality control procedures for the Amish HAPI Heart Study have been described previously (also see Supplementary methods)(24). Of the 736 individuals with both genome-wide single nucleotide polymorphism (SNP) data (Affymetrix 500K SNP platform) and serum [sVEGFR2], 730 also had genotype data on the human cardiovascular disease (CVD) risk focused BeadChip (also known as the ITMAT-Broad-CARe (IBC) array (Illumina))(30). This array contains 48,742 markers across approximately 2,100 CVD and metabolic disease candidate genes. Twenty-seven additional SNPs at the KDR locus were present on this platform and analyzed for association with serum [VEGFR2].
Single SNP genotyping of rs7253447 and rs34231037 in samples from study cohorts 2–3 were performed by a combination of Taqman allelic discrimination assay and SNaPshot single base extension assay (Applied Biosystems).
Statistical Analysis
Analysis of variance (ANOVA) was performed to estimate the inter- and intra-individual components of variability in sVEGFR2 levels among six healthy volunteers. The basic model for the j-th serial measurement in the i-th subject is yij = μ + ai + εij, where and ε ~ N(0, σ2); reflects the between individual component and σ2 the within individual component. Estimates of and σ2 were obtained from the ANOVA breakdown and the ratio of between individual to total variability (or ICC) determined as .
Group means for [sVEGFR2] and other measures were compared by t-test. Mixed model variance components analysis was performed to identify covariates, estimate heritability, and dominance and household effects of serum [sVEGFR2] and perform GWAS in the HAPI Heart Study (31). As described in detail previously, a Bonferroni correction-based threshold for genome-wide significance was set at 1 × 10−7 (24). In the University of Chicago cancer patients (study cohort 2), multivariate regression testing for association between rs34231037 and serum [sVEGFR2] was performed with PLINK v1.07 (http://pngu.mgh.harvard.edu/purcell/plink/)(32). The q assoc command was run including age, sex, BMI, and ethnicity (encoded as European American, African American, Latino American, and Asian/Pacific Islander American) as covariates. The p-value reflects a Wald test statistic. For this replication testing, p values < 0.05 were considered significant. For the 121 renal cell cancer subjects (study cohort 3), linear regression was conducted with an additive genetic model using SAS v9.2 including age as a covariate for baseline and week 4 [sVEGFR2], and baseline [sVEGFR2] as a covariate for changes in [sVEGFR2] after pazopanib exposure.
Results
Intra- and Inter-individual Variation in sVEGFR2 Levels in Normal Human Serum
In serial serum samples from healthy volunteers 87% of the total measurement variance of [sVEGFR2] was inter-individual, with the remaining small fraction due to within individual measurement differences over time(33). The high ratio of inter-individual to intra-individual variance suggested that the basis for the inter-individual variance could be studied further in larger populations without the need for serial measures. To further characterize the sources of inter-individual variance, serum [sVEGFR2] was measured in 736 Old Order Amish subjects who had participated in the HAPI Heart Study(24, 25, 34)[402 men, 334 women; mean age ± SD = 42.8 ± 13.7 years; mean body mass index (BMI) ± SD = 26.5 ± 4.4 kg/m2]. Median serum [sVEGFR2] was 9.05 ng/mL (interquartile range: 8.03 to 10.11) with an approximate normal distribution (Fig. 1). Men had higher concentrations (mean ± SD = 9.33 ± 1.49 ng/mL) than women (mean ± SD = 8.74 ± 1.67 ng/mL; p < 0.0001). Variance component analysis revealed [sVEGFR2] to be associated with age, BMI, and diastolic blood pressure (Table 1). Sex and age accounted for 4.4% of the variation, with age having the greater impact on [sVEGFR2]. Systolic blood pressure, fat mass, and pulse wave velocity (PWV), a measure of large vessel arterial stiffness, were also associated with variation in [sVEGFR2] but explained a trivial proportion of the variance.
Figure 1.

Distribution of serum [sVEGFR2] in 736 Amish subjects. Histogram plot of serum concentrations. Horizontal axis reflects 0.5 ng/ml quantiles among the 3-fold range of measurements from less than 4.5ng/ml to 15.5ng/ml. Vertical axis represents number of subjects with measurements in the interval.
Table 1.
Correlation of serum [sVEGFR2] with selected variables.
| Covariate | β ± SE | p-value | Age and sex adjusted β ± SE | p-value |
|---|---|---|---|---|
| Age (y) | −17 ± 4 | <0.0001 | --- | --- |
| BMI (kg/m2) | 34 ± 13 | 0.008 | 74 ± 13 | <0.0001 |
| Fat Mass (kg) | 0.004 ± 0.009 | 0.64 | 0.03 ± 0.01 | 0.002 |
| SBP (mmHg) | 4 ± 4 | 0.27 | 13 ± 4 | 0.002 |
| DBP (mmHg) | 24 ± 6 | 0.0002 | 30 ± 7 | <0.0001 |
| Carotid/Femoral PWV (m/s) | −6 ± 60 | 0.63 | 99 ± 58 | 0.087 |
BMI, body mass index; SBP, systolic blood pressure; DBP, diastolic blood pressure; PWV, pulse-wave velocity; β, effect size (pg/mL); SE, standard error, Total fat mass was measured by dual energy x-ray absorptiometry.
Heritability of [sVEGFR2]
The 736 Amish subjects of the HAPI Heart Study constituted 458 sibling pairs, 213 parent-offspring pairs, 334 avuncular pairs, and 153 first cousin pairs. A polygenic model of [sVEGFR2], adjusting for sex and age, demonstrated [sVEGFR2] to be a strongly heritable trait (h2 = 0.69). This estimate was virtually unchanged with inclusion of a random household and dominance effect into the model.
Genome-wide Association Analysis of [sVEGFR2]
The initial agnostic genome-wide association analysis of 736 HAPI Heart Study subjects with the Affymetrix 500K SNP genotype data identified a cluster of SNPs on chromosome 4 significantly associated with [sVEGFR2]. The top 687 serum [sVEGFR2]-associated SNPs were all on chromosome 4 and spanned a 3.5 Mb region that included several candidate genes including FIP1L1, PDGFRA, KIT, and KDR (Fig. 2 and Table 2). For the most significant SNP, rs725344, the effect size of each copy of the minor T allele (allele frequency = 0.12) was a decrease of 1.32 ng/mL [sVEGFR2] (p = 8×10−26).
Figure 2.

Genomewide association of SNPs with serum [sVEGFR2]. Manhattan plot of each SNP on the genotyping platform by chromosome (horizontal axis) vs. the −log P value for association with serum [sVEGFR2] (vertical axis). Chr = chromosome.
Table 2.
Top 20 SNPs on genome-wide association scan for [sVEGFR2].
| SNP | CHR | POS | FREQ | β SNP | P-value |
|---|---|---|---|---|---|
| rs725344 | 4 | 54371725 | 0.88 | 1.32 | 4.76E-25 |
| rs7674066 | 4 | 54417731 | 0.80 | 0.88 | 1.51E-17 |
| rs12650282 | 4 | 57428361 | 0.67 | 0.70 | 4.64E-16 |
| rs2898989 | 4 | 54375636 | 0.21 | −0.79 | 8.53E-16 |
| rs10018606 | 4 | 57833169 | 0.78 | 0.81 | 1.03E-15 |
| rs4865033 | 4 | 56324164 | 0.55 | −0.65 | 7.69E-15 |
| rs12641893 | 4 | 56323282 | 0.45 | 0.65 | 7.74E-15 |
| rs11133292 | 4 | 54402456 | 0.71 | 0.71 | 1.23E-14 |
| rs10780091 | 4 | 54400288 | 0.28 | −0.68 | 4.08E-13 |
| rs4272068 | 4 | 56304297 | 0.33 | 0.63 | 2.03E-12 |
| rs4864562 | 4 | 56283606 | 0.31 | 0.67 | 4.79E-12 |
| rs2703469 | 4 | 55142857 | 0.78 | 0.69 | 7.41E-12 |
| rs17087713 | 4 | 57794798 | 0.79 | 0.71 | 8.58E-12 |
| rs17183291 | 4 | 57628147 | 0.83 | 0.73 | 8.66E-12 |
| rs4865181 | 4 | 57658226 | 0.29 | −0.61 | 9.76E-12 |
| rs10016091 | 4 | 53686267 | 0.63 | 0.59 | 1.01E-11 |
| rs17463708 | 4 | 47580771 | 0.15 | −0.80 | 1.62E-11 |
| rs4865165 | 4 | 57460303 | 0.45 | −0.56 | 5.36E-11 |
| rs17698672 | 4 | 53406391 | 0.90 | 0.88 | 5.46E-11 |
| rs9999844 | 4 | 57797310 | 0.21 | −0.69 | 5.64E-11 |
CHR- chromosome, POS- position, FREQ- frequency of the coded allele, β SNP- effect size (ng/mL) for the coded allele on serum sVEGFR2 concentrations.
The strong association of [sVEGFR2] with multiple SNPs on chromosome 4q11-q12 and inferences from the known founder population structure of the Lancaster County Amish guided further analysis. The causative SNP was expected to be in strong linkage disequilibrium with rs725344 and might have been introduced into the Lancaster County Amish population through a single or small number of founders. The rs725344 mutation therefore marks a broader founder haplotype such that all SNPs on this haplotype show association with [sVEGFR2]. Because this haplotype would have been passed down from the founder(s) to the present day Lancaster County Amish for only 12–14 generations, opportunity for recombination would be limited and the causative variants could be up to several Mb away. Consistent with this assumption, we performed multivariate regression association analysis with single time-point serum [sVEGFR2] in 128 unrelated advanced solid tumor patients enrolled in clinical trials at the University of Chicago. We detected no association between rs725344 (allele frequency = 0.20) and serum [sVEGFR2] (β= 0.23 ng/mL, p = 0.51) in this heterogeneous cancer patient population.
Based on the assumption that rs725344 was in linkage disequilibrium (LD) with a functional variant within a broad linkage peak in the Amish, but not in LD in the heterogeneous cancer patient population, we pursued two approaches to identify variants with detectable effects on [sVEGFR2]. First, we identified candidate variants in the larger population within the 3.5 Mb linkage peak and tested their association with [sVEGFR2] directly in the 128 cancer patients. Subsequently, we evaluated candidate loci in the KDR gene in the Amish using the Illumina cardiovascular disease (IBC) chip, which had more extensive coverage of KDR, including rare coding region alleles(30).
Evaluation of Candidate Gene Variants for Association with [sVEGFR2]
We used the bioinformatics tool SCAN(35) to identify all genes within the locus and then accessed the 1000 Genomes Project(36) to identify all missense polymorphisms within those genes. This approach identified four coding variants (Supplementary Table 1) within FIP1L, PDGFRA, and KDR with minor allele frequencies > 0.02 in populations of European ancestry. Additionally, the common polymorphism in the 5′-untranslated region of KDR, rs7667298, had been demonstrated to affect VEGFR2 expression in vitro (37). None of these candidate variants demonstrated association with [sVEGFR2] in the heterogeneous cancer patient population (study 2 cohort, see Materials and Methods) (Supplementary Table 1).
As KDR was within 1.3 Mb of the top signal, we considered this gene the prime candidate for more in depth analysis. During the course of our investigation, data from the IBC chip became available from 730 of the 736 original Amish subjects. In the Amish there were 27 non-monomorphic SNPs in or near KDR. Single SNP association analysis with [sVEGFR2] revealed a missense mutation in exon 11, C482R caused by an A to G change (rs34231037) to have the lowest p value, 2.7×10−37 (Supplementary Table 2). Among the Amish, in an additive genetic model, the minor G allele lowered [sVEGFR2] approximately 1.80 ng/mL per allele (Fig. 3a). This finding was consistent with our original hypothesis, that 1) rs725344 would be, in the Amish, a proxy on our initial genotyping array in linkage disequilibrium with a SNP not on the array that has a stronger association with the [sVEGFR2] phenotype, and 2) the SNP would not be in linkage disequilibrium with rs725344 among a more heterogeneous population of European ancestry (such as the 128 University of Chicago cancer patient cohort) (see Supplementary Results for a description of the linkage disquilibrium assessment).
Figure 3.


Association of rs34231037 with serum [sVEGFR2]. (a) in 730 Amish subjects. Cohorts defined by genotype status (AA= homozygous major allele, AG= heterozygous, GG= homozygous minor allele) with boxplots of distribution of serum [sVEGFR2]. (b) in 128 unrelated cancer patients with dotplots of distribution of serum [sVEGFR2]. Given the rarity of GG homozygotes in the population, none were detected among the cancer patients.
Rs34231037 is a Determinant of [sVEGFR2] in Cancer Patients
The candidate SNP rs34231037 met our expectations for a SNP that would be associated with serum [sVEGFR2] in the heterogeneous population of cancer patients of European ancestry in the University of Chicago cohort. Mechanistically, we considered that rs34231037 encodes a nonsynonymous variant, C482R, located in the fifth of 7he immunoglobulin-like (Ig-like) domains of the extracellular region, formed by amino acids 421–548;. This cysteine is one of four in the fifth Ig-like domain conserved throughout homologues of KDR in the chordates [from T. nigroviridis (pufferfish), to G. gallus (domestic chicken), to M. musculus (mouse), through H. sapiens] (see Supplementary Figure 2). We genotyped rs34231037 in 128 advanced cancer patients enrolled in University of Chicago trials (study 2 cohort, see Materials and Methods). Using a regression model for additive effects of the allele and incorporating sex, age, body mass index, and self-reported race categories, the rs34231037 minor allele was associated with serum [sVEGFR2] with the same direction and a similar effect size as in the Amish (1.85 ng/mL lower in G allele carriers, p = 0.025) (Fig. 3b).
Rs34231037 is Associated with sVEGFR2 Response to Pazopanib
VEGFR2 kinase inhibitors routinely cause circulating [sVEGFR2] measures to decline within the first 4 weeks of exposure(18–23) and the magnitude of change in [sVEGFR2] has been associated with increased response to therapy in thyroid and lung cancer(18, 23). With the biological relevance of rs34231037 established as a significant predictor of [sVEGFR2] in the Amish and unrelated cancer patients, we hypothesized it might also play a role in differential responses to inhibitors of the VEGF signaling pathway. In an independent group of 121 renal cell cancer patients treated on clinical trials with pazopanib (study 3 cohort, see Materials and Methods), serial [sVEGFR2] measures were available prior to, and 4 weeks after initiation of continuous oral pazopanib therapy 800 mg daily. Patients with the G allele not only had lower baseline [sVEGFR2] measures (Fig. 4a), but also experienced greater decline over 4 weeks in [sVEGFR2] with pazopanib exposure compared to non-carriers (mean decrease −3.5 ng/mL vs −2.3 ng/mL, respectively; p = 0.01) (Fig. 4b and 4c).
Figure 4.
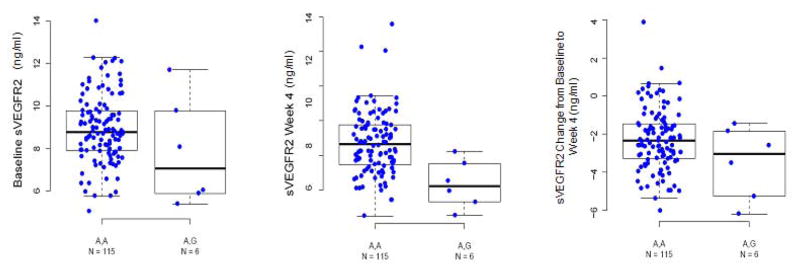
Association of rs34231037 with changes in serum [sVEGFR2] after pazopanib therapy. Dot and boxplots demonstrate: (a) replication of the association between the SNP and baseline serum [sVEGFR2] among an independent cohort of 121 renal cancer patients, (b) the serum [sVEGFR2] measurements by genotype in the same 121 subjects after 4 weeks pazopanib therapy, and (c) the fractional change in serum [sVEGFR2] among these subjects by genotype.
Other Variants Associated with [sVEGFR2]
In the Amish, conditional analyses of both the GWAS and IBC chips were run including rs34231037 as a covariate in the model to determine the impact on other variants in the chromosome 4 region. Several SNPs on chromosome 4 in the 56–57Mb region remained highly significant from the imputed data, but had lower effect size than rs34231037. The most significant of these other SNPs was rs10050046 located 56kb from rs34231037. The minor allele C has frequency 0.37 and was associated with decreased levels of [sVEGFR2] (0.53 ng/mL per allele; p = 3.0 x10−11), less than 1/3 the magnitude of effect of rs34231037. In the primary GWAS there was marginal association between rs10050046 and [sVEGFR2] (p = 10−3). The LD between rs10050046 and rs34231037 is r2=0.04 and |D′|=0.8 suggesting that the two [sVEGFR2]-lowering alleles act independently in the Amish. However, rs10050046 was not associated with serum [sVEGFR2] in either of the cancer cohorts (−0.04 ng/mL, p = 0.88 in the study 2 cohort, and 0.09 ng/mL, p= 0.67 in the study 3 cohort).
Discussion
We performed a genome-wide association study of a serum peptide in the Amish to discover a SNP with reproducible effects on that peptide in cancer patients. This phenotype [sVEGFR2] is now recognized as a pharmacodynamic marker of VEGFR2 inhibition and this SNP affects the pharmacodynamic response to pazopanib. As in genetic studies, for example of grasshopper body size(38) and human schizophreniform behavior(39), we employed the approach of endophenotyping to the discovery of a novel cancer pharmacogenetic biomarker. In a step-wise approach, we first characterized the repeatability, interindividual variance, and heritability of [sVEGFR2] as an endophenotype for response to VEGFR2 kinase inhibitors. Second, we performed an agnostic GWAS in a healthy population and identified rs34231037/C482R in KDR, the gene encoding the VEGFR2 protein, to be associated with [sVEGFR2]. Third, we replicated these findings in two cancer populations. Fourth, we extended these findings by demonstrating that this variant is a marker for pharmacodynamics response to the kinase inhibitor, pazopanib.
The sVEGFR2 protein is an alternative spliced product of KDR and functions as a physiologic inhibitor of developmental and reparative lymphangiogenesis(17). In situ hybridization demonstrated mammalian expression of the alternative splice product in epithelium such as in the cornea and skin. The product of this transcript appears to be monomeric and to have higher relative avidity for VEGF-C than VEGF-A. In the mouse cornea, sVEGFR2 preferentially regulates lymphanigogenesis. The extent to which this alternative splice product contributes to the sVEGFR2 protein detected in human serum and whether the circulating protein plays additional roles in regulating human angiogenesis and lymphangiogenesis is not known.
The function of the rs34231037/C482R variant has been previously characterized in studies of hemangiomata(40). The amino acid is located in the extracellular Ig-like domain V, distant from the VEGF ligand binding site (41). Recombinant cell transfection studies revealed no differences in VEGF-induced phosphorylation or expression of VEGFR2. However, the substitution of arginine for cysteine diminished formation of complexes by VEGFR2 with β1 integrin and the integrin receptor-like protein TEM8. This complex was associated with VEGFR2-mediated activation of VEGFR1 transcription and protein expression. With less VEGFR1 expression, endothelial cells demonstrate greater sensitivity to activation of VEGFR2 signaling by VEGF binding. This amplified sensitivity to VEGF/VEGFR2 ligation might explain the potential for greater sensitivity of G allele carriers to pazopanib treatment and potentially greater anti-cancer activity of VEGFR2 inhibitors in patients with this variant.
The rs34231037 polymorphism is a common variant in the Amish (minor allele frequency = 0.1) but an uncommon variant in the larger out-bred Caucasian population (minor allele frequency = 0.02). We expect this SNP might have a clinically significant effect on outcomes of therapy, since the magnitude of change in sVEGFR2 has been associated with increased response to therapy in thyroid and lung cancer (18, 23). Since allele carriers comprise only a small subset (i.e., approximately 3%) of all patients, almost all individual trials will be underpowered to demonstrate clinically significant effects. We have not excluded additional genetic variants contributing to inter-individual differences in baseline [sVEGFR2] and important interactive covariates for explaining differences in response to VEGFR2 kinase inhibitors is the subject of ongoing investigation.
Despite the commercial availability of 9 drugs in the class since 2006, a clinically useful biomarker to guide therapy with VEGFR2 inhibitors remains elusive. Our finding of a germline variant within KDR with reproducible effects on the response of the pharmacodynamic marker sVEGFR2 warrants further investigation. This might provide important insights into mechanisms underlying inter-individual variation in response to kinase inhibitors, and approaches to deliver anti-cancer therapy more effectively.
Supplementary Material
Translational Relevance.
Germ-line genetic variation can play an important role in the course of cancer and treatment responses. Conventional strategies for discovering germ-line factors in disease and therapeutic responses in other health conditions can be challenging to apply to cancer pharmacogenomics research. We employed a step-wise approach of studying a reproducibly measurable serum protein, sVEGFR2, in a population of volunteers who did not have cancer, to identify a genetic variant (rs34231037/C482R) as a marker for response to VEGFR2 kinase inhibitors. The association of rs34231037 with serum concentrations of sVEGFR2 [sVEGFR2] was replicated first in unrelated cancer patients. In a separate cohort of renal cancer patients, the uncommon gene variant was associated with lower baseline [sVEGFR2] measures and carriers experienced greater decline in [sVEGFR2] over 4 weeks of pazopanib exposure compared to non-carriers. We expect this genetic marker might have a clinically significant effect on treatment outcomes with VEGFR2 kinase inhibitors.
Acknowledgments
We thank Carole Ober for her initial contributions to the study concept. This project was supported by U.S. National Institutes of Health grants: K23CA124802 NCI Career Development Award (MLM), U01CA69852 including study 15002 supported by the Cancer Therapy Evaluation Program of the NCI (MJR), NIGMS U01GM61393 Pharmacogenetics of Anticancer Agents Research Group (MJR, NJC, EG, SD, and MLM), U01HL105918 The Amish Pharmacogenomics of Anti-Platelet Intervention Study and U01 HL072515 The Amish Heredity and Phenotype Intervention Study (ARS, BDM, JRO, KAR, Y-CC), a Preclinical Pilot Translational Science Award from the University of Chicago CTSA UL1RR024999, and the University of Chicago Comprehensive Cancer Center. Additional support was provided by a CALGB Foundation Faculty Fellowship (MLM), the Conquer Cancer Foundation of the American Society of Clinical Oncology (MLM and MJR), research funding from the O’Connor Foundation, and contributions from the friends and families of Joseph S. Berger Jr., and Robert Wesselhoff. Glaxo SmithKline supported the contributions of its employees and provided data on pazopanib.
Footnotes
Conflict of Interest: C-F. Xu, N. Bing, P.A. Wilson, Y. Liu, L.R. Cardon and L.N. Pandite are employees of GlaxoSmithKline and own company stock.
Author contributions: MLM, C-FX, Y-CC, EOK-G, TK, JRO, NJC, BDM, MJR, and ARS designed experiments and analyzed and interpreted results. KAR, SD, DT, EG, MRL, PW, NB, and YL conducted analyses and interpreted results. LC and LP provided data and with C-FX participated in planning of analyses. NJC, BDM, MJR, ARS, and MLM structured and modified the overall project design. MLM conceived of and directed the project and wrote the manuscript with VT, JRO, BDM, MJR and ARS. All authors reviewed and had the opportunity to comment on the manuscript.
References
- 1.Evans WE, Relling MV. Moving towards individualized medicine with pharmacogenomics. Nature. 2004;429:464–8. doi: 10.1038/nature02626. [DOI] [PubMed] [Google Scholar]
- 2.Roses AD. Pharmacogenetics and the practice of medicine. Nature. 2000;405:857–65. doi: 10.1038/35015728. [DOI] [PubMed] [Google Scholar]
- 3.Gillis NK, Patel JN, Innocenti F. Clinical implementation of germ line cancer pharmacogenetic variants during the next-generation sequencing era. Clinical pharmacology and therapeutics. 2014;95:269–80. doi: 10.1038/clpt.2013.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wheeler HE, Maitland ML, Dolan ME, Cox NJ, Ratain MJ. Cancer pharmacogenomics: strategies and challenges. Nature reviews Genetics. 2013;14:23–34. doi: 10.1038/nrg3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCarthy JJ, McLeod HL, Ginsburg GS. Genomic medicine: a decade of successes, challenges, and opportunities. Science translational medicine. 2013;5:189sr4. doi: 10.1126/scitranslmed.3005785. [DOI] [PubMed] [Google Scholar]
- 6.Roses AD. Pharmacogenetics in drug discovery and development: a translational perspective. Nat Rev Drug Discov. 2008;7:807–17. doi: 10.1038/nrd2593. [DOI] [PubMed] [Google Scholar]
- 7.Higgins MJ, Stearns V. Pharmacogenetics of endocrine therapy for breast cancer. Annu Rev Med. 2011;62:281–93. doi: 10.1146/annurev-med-070909-182545. [DOI] [PubMed] [Google Scholar]
- 8.Huang RS, Ratain MJ. Pharmacogenetics and pharmacogenomics of anticancer agents. CA Cancer J Clin. 2009;59:42–55. doi: 10.3322/caac.20002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maitland ML, Vasisht K, Ratain MJ. TPMT, UGT1A1 and DPYD: genotyping to ensure safer cancer therapy? Trends Pharmacol Sci. 2006;27:432–7. doi: 10.1016/j.tips.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 10.Swen JJ, Nijenhuis M, de Boer A, Grandia L, Maitland-van der Zee AH, Mulder H, et al. Pharmacogenetics: from bench to byte--an update of guidelines. Clinical pharmacology and therapeutics. 2011;89:662–73. doi: 10.1038/clpt.2011.34. [DOI] [PubMed] [Google Scholar]
- 11.Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. 2008;8:579–91. doi: 10.1038/nrc2403. [DOI] [PubMed] [Google Scholar]
- 12.Jubb AM, Harris AL. Biomarkers to predict the clinical efficacy of bevacizumab in cancer. Lancet Oncol. 2010;11:1172–83. doi: 10.1016/S1470-2045(10)70232-1. [DOI] [PubMed] [Google Scholar]
- 13.Sternberg CN, Davis ID, Mardiak J, Szczylik C, Lee E, Wagstaff J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061–8. doi: 10.1200/JCO.2009.23.9764. [DOI] [PubMed] [Google Scholar]
- 14.van der Graaf WT, Blay JY, Chawla SP, Kim DW, Bui-Nguyen B, Casali PG, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379:1879–86. doi: 10.1016/S0140-6736(12)60651-5. [DOI] [PubMed] [Google Scholar]
- 15.Quinn TP, Peters KG, De Vries C, Ferrara N, Williams LT. Fetal liver kinase 1 is a receptor for vascular endothelial growth factor and is selectively expressed in vascular endothelium. Proc Natl Acad Sci U S A. 1993;90:7533–7. doi: 10.1073/pnas.90.16.7533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–71. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 17.Albuquerque RJ, Hayashi T, Cho WG, Kleinman ME, Dridi S, Takeda A, et al. Alternatively spliced vascular endothelial growth factor receptor-2 is an essential endogenous inhibitor of lymphatic vessel growth. Nat Med. 2009;15:1023–30. doi: 10.1038/nm.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bass MB, Sherman SI, Schlumberger MJ, Davis MT, Kivman L, Khoo HM, et al. Biomarkers as predictors of response to treatment with motesanib in patients with progressive advanced thyroid cancer. J Clin Endocrinol Metab. 2010;95:5018–27. doi: 10.1210/jc.2010-0947. [DOI] [PubMed] [Google Scholar]
- 19.Cohen EE, Rosen LS, Vokes EE, Kies MS, Forastiere AA, Worden FP, et al. Axitinib is an active treatment for all histologic subtypes of advanced thyroid cancer: results from a phase II study. J Clin Oncol. 2008;26:4708–13. doi: 10.1200/JCO.2007.15.9566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Motzer RJ, Michaelson MD, Redman BG, Hudes GR, Wilding G, Figlin RA, et al. Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J Clin Oncol. 2006;24:16–24. doi: 10.1200/JCO.2005.02.2574. [DOI] [PubMed] [Google Scholar]
- 21.Pena C, Lathia C, Shan M, Escudier B, Bukowski RM. Biomarkers predicting outcome in patients with advanced renal cell carcinoma: Results from sorafenib phase III Treatment Approaches in Renal Cancer Global Evaluation Trial. Clin Cancer Res. 2011;16:4853–63. doi: 10.1158/1078-0432.CCR-09-3343. [DOI] [PubMed] [Google Scholar]
- 22.Ebos JM, Lee CR, Christensen JG, Mutsaers AJ, Kerbel RS. Multiple circulating proangiogenic factors induced by sunitinib malate are tumor-independent and correlate with antitumor efficacy. Proc Natl Acad Sci U S A. 2007;104:17069–74. doi: 10.1073/pnas.0708148104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nikolinakos PG, Altorki N, Yankelevitz D, Tran HT, Yan S, Rajagopalan D, et al. Plasma cytokine and angiogenic factor profiling identifies markers associated with tumor shrinkage in early-stage non-small cell lung cancer patients treated with pazopanib. Cancer Res. 2010;70:2171–9. doi: 10.1158/0008-5472.CAN-09-2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng YC, Kao WH, Mitchell BD, O’Connell JR, Shen H, McArdle PF, et al. Genome-wide association scan identifies variants near Matrix Metalloproteinase (MMP) genes on chromosome 11q21-22 strongly associated with serum MMP-1 levels. Circ Cardiovasc Genet. 2009;2:329–37. doi: 10.1161/CIRCGENETICS.108.834986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mitchell BD, McArdle PF, Shen H, Rampersaud E, Pollin TI, Bielak LF, et al. The genetic response to short-term interventions affecting cardiovascular function: rationale and design of the Heredity and Phenotype Intervention (HAPI) Heart Study. Am Heart J. 2008;155:823–8. doi: 10.1016/j.ahj.2008.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maitland ML, Kasza KE, Karrison T, Moshier K, Sit L, Black HR, et al. Ambulatory monitoring detects sorafenib-induced blood pressure elevations on the first day of treatment. Clin Cancer Res. 2009;15:6250–7. doi: 10.1158/1078-0432.CCR-09-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maitland ML, Levine MR, Lacouture ME, Wroblewski KE, Chung CH, Gordon IO, et al. Evaluation of a novel rash scale and a serum proteomic predictor in a randomized phase II trial of sequential or concurrent cetuximab and pemetrexed in previously treated non-small cell lung cancer. BMC cancer. 2014;14:5. doi: 10.1186/1471-2407-14-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karovic S, Wen Y, Karrison TG, Bakris GL, Levine MR, House LK, et al. Sorafenib dose escalation is not uniformly associated with blood pressure elevations in normotensive patients with advanced malignancies. Clinical pharmacology and therapeutics. 2014;96:27–35. doi: 10.1038/clpt.2014.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hutson TE, Davis ID, Machiels JH, de Souza PL, Baker K, Bordogna W, et al. Biomarker analysis and final efficacy and safety results of a phase II renal cell carcinoma trial with pazopanib (GW786034), a multi-kinase angiogenesis inhibitor. ASCO Meeting Abstracts. 2008;26:5046. [Google Scholar]
- 30.Keating BJ, Tischfield S, Murray SS, Bhangale T, Price TS, Glessner JT, et al. Concept, design and implementation of a cardiovascular gene-centric 50 k SNP array for large-scale genomic association studies. PLoS One. 2008;3:e3583. doi: 10.1371/journal.pone.0003583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Connell JR. Optimizing Measured Genotype Genome-wide Association Analysis for Quantitative Traits in Pedigrees Abstract 2409 American Society of Human Genetics Annual Meeting; 2008; Philadelphia. 2008. software available at http://edn.som.umaryland.edu/mmap/index.php. [Google Scholar]
- 32.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thomeas V, Chow S, Gutierrez JO, Karovic S, Wroblewski K, Kistner-Griffin E, et al. Technical considerations in the development of circulating peptides as pharmacodynamic biomarkers for angiogenesis inhibitors. Journal of clinical pharmacology. 2014;54:682–7. doi: 10.1002/jcph.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pollin TI, Damcott CM, Shen H, Ott SH, Shelton J, Horenstein RB, et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science. 2008;322:1702–5. doi: 10.1126/science.1161524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gamazon ER, Zhang W, Konkashbaev A, Duan S, Kistner EO, Nicolae DL, et al. SCAN: SNP and copy number annotation. Bioinformatics. 2010;26:259–62. doi: 10.1093/bioinformatics/btp644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clarke L, Zheng-Bradley X, Smith R, Kulesha E, Xiao C, Toneva I, et al. The 1000 Genomes Project: data management and community access. Nat Methods. 2012;9:459–62. doi: 10.1038/nmeth.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Glubb DM, Cerri E, Giese A, Zhang W, Mirza O, Thompson EE, et al. Novel functional germline variants in the VEGF receptor 2 gene and their effect on gene expression and microvessel density in lung cancer. Clin Cancer Res. 2011;17:5257–67. doi: 10.1158/1078-0432.CCR-11-0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.John B, Lewis KR. Chromosome variability and geographic distribution in insects. Science. 1966;152:711–21. doi: 10.1126/science.152.3723.711. [DOI] [PubMed] [Google Scholar]
- 39.Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry. 2003;160:636–45. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- 40.Jinnin M, Medici D, Park L, Limaye N, Liu Y, Boscolo E, et al. Suppressed NFAT-dependent VEGFR1 expression and constitutive VEGFR2 signaling in infantile hemangioma. Nat Med. 2008;14:1236–46. doi: 10.1038/nm.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shinkai A, Ito M, Anazawa H, Yamaguchi S, Shitara K, Shibuya M. Mapping of the sites involved in ligand association and dissociation at the extracellular domain of the kinase insert domain-containing receptor for vascular endothelial growth factor. J Biol Chem. 1998;273:31283–8. doi: 10.1074/jbc.273.47.31283. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.