

Abstract
The arylation of sp3-hybridized C–H’s bonds is a powerful strategy to build molecular complexity and diversity. A novel and efficient palladium-catalyzed direct sp3 C–H arylation of aryl and alkyl benzyl thioether derivatives with aryl bromides is reported. The reaction involves reversible deprotonation of the benzylic C–H’s of the thioether with either LiN(SiMe3)2 or NaN(SiMe3)2 and subsequent cross-coupling to provide the functionalized products in up to 97% yield.
A screen of 24 of the most successful ligands in cross-coupling chemistry led to the identification of NiXantPhos as the only viable ligand for this challenging coupling.
Keywords: C–H functionalization, Sulfides, Palladium, Arylation, Cross-Coupling
Introduction
The direct arylation of C–H bonds[1] is an appealing alternative to classic coupling of prefunctionalized partners, because it circumvents the use of preformed organometallic reagents. In the context of a broader program to functionalize weakly acidic sp3 hybridized C–H bonds,[2–4] we became interested in the arylation of C–H bonds situated alpha to sulfur atoms, since sulfur-based compounds are commonplace in the chemistry of life and in medicinal chemistry.[5] Our initial efforts focused on the direct α-arylation of C–H bonds adjacent to sulfur(II), such as found in sulfoxides, including the parent DMSO (Scheme 1A).[6] Based on these results, we then examined α-C–H arylation of sulfur(IV) of methyl and benzyl sulfones.[7] In both cases, the reactions were promoted by a palladium catalyst employing Kwong’s indole-based phosphine[8, 9] in the presence of LiO-t-Bu at 110 °C (Scheme 1).
Scheme 1.

Direct α-arylation of sulfoxides (A) and sulfones (B) promoted by a palladium catalyst based on Kwong’s indole-based phosphine.
Our development of a successful reaction system for the arylation of sulfoxides and sulfones inspired us to conjecture about the possibility of α-arylation of less oxidized sulfur compounds such as thioethers.[10] The α-C–H’s of thioethers are more difficult to metallate than either sulfoxides or sulfones due of their higher pKa values.[11] Although thioethers can be deprotonated with organolithiums[12] and then quenched with a variety of electrophiles, the in situ metallation/arylation of thioethers has never been achieved. Given the importance of organosulfur small molecule libraries, we set out to develop conditions for the tandem reversible metallation of thioethers with subsequent arylation.
To accomplish this objective, two hurdles would need to be surmounted. First, bases must be identified that are sufficiently strong to deprotonate thioethers, yet are compatible with transition metal arylation catalysts. Second, a catalyst would need to be found that could withstand reactive organolithium,[13] -sodium, or -potassium intermediates. Furthermore, given the known propensity of palladium catalysts to cleave C–S bonds[14] the catalyst must also exhibit a high degree of chemoselectivity.
Herein we report the first examples of C–H arylation alpha to sulfur in thioethers (Scheme 2). Given the distinct chemistry of thioethers relative to sulfones and sulfoxides, and the increased reactivity of metallated sulfides compared to their sulfoxide and sulfone counterparts, it is not surprising that different bases, conditions, palladium precursor and ligand were required for the α-arylation of thioethers.
Scheme 2.

Direct α-arylation of sulfides outlined herein.
Results and Discussion
Given the high pKa’s of thioethers, it seemed wise to initiate research in this area with a class of thioethers wherein the α-lithiated substrates had been generated and studied. Benzyl phenyl sulfide satisfied our criteria: it can be deprotonated by n-BuLi and it is monomeric in solution and the solid state.[15]
Our first step toward development of the arylation of sulfides is to identify base and solvent combinations for the reversible deprotonation of the benzylic C–H’s of ArS–CH2Ar’ under conditions amenable to catalysis. To this end, we employ base and benzyl chloride to trap any metallated sulfide. By measuring the yield of the benzylation product, we can determine which bases deprotonate the benzylic C–H’s of the substrate over the course of the reaction (Table 1).[3] Thus, we screened 6 bases [LiO-tBu, NaO-tBu, KO-tBu, LiN(SiMe3)2, NaN(SiMe3)2, KN(SiMe3)2] at room temperature in CPME (cyclopentyl methyl ether). As outlined in Table 1, entries 1–3, alkoxide bases MO-tBu (M = Li, Na, K) did not generate detectable amounts of benzylation products, suggesting they would not be applicable in the arylation of thioethers. Interestingly, these results are in contrast to the arylation of aryl methyl sulfoxides[6] and sulfones,[7] where LiO-tBu was the optimal base (Scheme 1). On the other hand, benzylation products were observed in increasing conversions, with LiN(SiMe3)2 < NaN(SiMe3)2 < KN(SiMe3)2 (entries 4–6).
Table 1.
Benzylation of benzyl phenyl sulfide with benzyl chloride.

| ||
|---|---|---|
| entry | base | 1H NMR assay yield (%) |
| 1 | LiO-tBu | 0 |
| 2 | NaO-tBu | 0 |
| 3 | KO-tBu | 0 |
| 4 | LiN(SiMe3)2 | 53 |
| 5 | NaN(SiMe3)2 | 60 |
| 6 | KN(SiMe3)2 | 74 |
Having identified three potential bases for the catalytic arylation of thioethers, we next turned our attention to the hunt for a catalyst. Based on our experience with deprotonative cross-coupling processes of weakly acidic substrates,[3, 4] we chose to use van Leeuwen’s NiXantPhos ligand as a starting point (Figure 1).[16, 17] Eight different palladium sources [Pd(OAc)2, [PdCl(allyl)]2, Pd(dba)2, Pd(PPh3)4, PdCl2(cod), Pd(acac)2, Pd(tfa)2 and PdCl2], the three bases identified in the benzylation screen in Table 1 [LiN(SiMe3)2, NaN(SiMe3)2 and KN(SiMe3)2] and 4 solvents [CPME, THF, DME (dimethoxyethane) and toluene] were examined in the coupling of Ph–S–CH2Ph (1a) with bromobenzene (2a). Screens were conducted at 50°C using microscale high-throughput experimentation (HTE) techniques (see Supporting Information for details and complete results). The 7 leading hits in the HTE screen were performed on laboratory scale and the assay yields of 4a are listed in Table 2. The combination of [PdCl(allyl)]2 and LiN(SiMe3)2 in THF was the top contender and was employed in a broader screen of ligands, as outlined below. Interestingly, in contrast to the benzylation in Table 1, the results of the catalyst screening (Table 2) indicate the opposite efficacy ordering of the bases in the arylation [LiN(SiMe3)2 ~ NaN(SiMe3)2 > KN(SiMe3)2].
Figure 1.
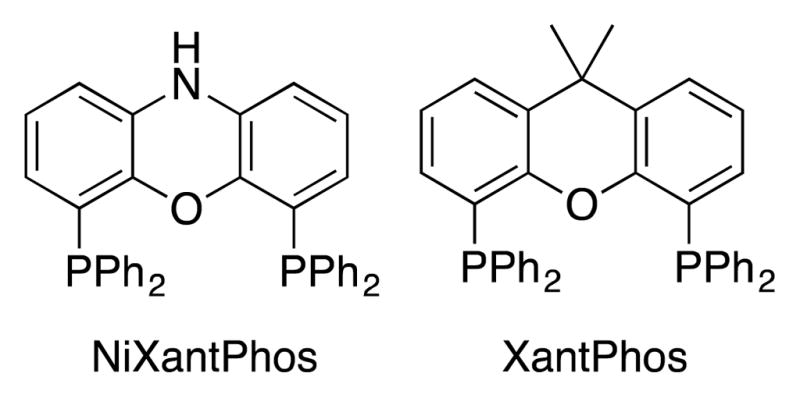
Structures of van Leeuwen’s NiXantPhos and XantPhos ligands.
Table 2.
Laboratory-scale Reactions Using the top HTE Conditions.
| ||||
|---|---|---|---|---|
| entry | base | Pd source | solvent | assay yield (%)a |
| 1 | LiN(SiMe3)2 | [PdCl(allyl)]2 | THF | 61 |
| 2 | LiN(SiMe3)2 | PdCl2(cod) | DME | 57 |
| 3 | LiN(SiMe3)2 | PdCl2(cod) | toluene | 35 |
| 4 | LiN(SiMe3)2 | PdCl2 | toluene | 0 |
| 5 | NaN(SiMe3)2 | Pd(OAc)2 | CPME | 56 |
| 6 | NaN(SiMe3)2 | PdCl2(cod) | CPME | 58 |
| 7 | NaN(SiMe3)2 | [PdCl(allyl)]2 | toluene | 45 |
Yields determined by 1H NMR analysis of crude mixture with CH2Br2 as internal standard.
Based on the results in Table 2, we subsequently examined 24 sterically and electronically diverse ligands in the arylation of PhSCH2Ph with bromobenzene using HTE techniques with [PdCl(allyl)]2 and LiN(SiMe3)2 in THF (see the Supporting Information for details and a full list of ligands tested). Interestingly, of the 24 ligands examined, only NiXantPhos generated the coupling product in meaningful amounts. Structurally similar XantPhos[18] (Figure 1) generated only trace product. Well known dppf derivatives and bulky monodentate phosphines (members of the Buchwald family,[19] Kwong’s indole-based phosphine,[8] Q-Phos,[20] etc.) failed to generate product under these conditions, attesting to the challenging nature of this cross-coupling reaction. These results further indicate that NiXantPhos, with an N–H that can be deprotonated under the basic reaction conditions, is a uniquely effective ligand.[17]
In order to optimize the reaction conditions with the NiXantPhos/[PdCl(allyl)]2 system, we tested different ratios of thioether (1a), bromobenzene (2a), LiN(SiMe3)2 (3a) and various reaction times (Table 3). At this point, we did not yet understand the sensitivity of the yield to reaction time (due to product decomposition). The yield of the reaction was higher when 2 equiv each of LiN(SiMe3)2 and bromobenzene were used (entries 1–3, up to 52% yield).
Table 3.
Optimization of palladium-catalyzed DCCP of benzyl phenyl sulfide.
| ||||||
|---|---|---|---|---|---|---|
| entry | 1a:2a:3a | solvent | T (°C) | time (h) | Pd: NiXantPhos (mol %) | 4aa (%) |
| 1 | 2:1:2 | THF | 80 | 12 | 10:20 | 45 |
| 2 | 2:1:3 | THF | 80 | 12 | 10:20 | 0 |
| 3 | 1:2:2 | THF | 80 | 12 | 10:20 | 52 |
| 4 | 1:2:2 | THF | 50 | 12 | 10:20 | 53 |
| 5 | 1:2:2 | THF | R.T. | 12 | 10:20 | 60 |
| 6 | 1:2:2 | THF | R.T. | 6 | 10:20 | 67 |
| 7 | 1:2:2 | THF | R.T. | 1 | 10:20 | 80 |
| 8 | 1:2:2 | THF | R.T. | 0.5 | 10:20 | 86 |
| 9 | 1:2:2 | THF | R.T. | 0.25 | 10:20 | 76 |
| 10 | 1:2:2 | THF | R.T. | 0.5 | 5:10 | 67 |
| 11 | 1:2:2 | THF | R.T. | 0.5 | 5:10 | 64b |
| 12 | 1:2:2 | CPME | R.T. | 0.5 | 10:20 | 0 |
| 13 | 1:2:2 | DME | R.T. | 0.5 | 10:20 | 58 |
| 14 | 1:2:2 | dioxane | R.T. | 0.5 | 10:20 | 0 |
| 15 | 1:2:2 | hexanes | R.T. | 0.5 | 10:20 | 0 |
| 16 | 1:2:2 | toluene | R.T. | 0.5 | 10:20 | 0 |
Yields determined by 1H NMR analysis of crude mixture with CH2Br2 as internal standard.
concentration 0.2 M.
To minimize byproduct formation, we reduced the reaction temperature from 80 °C to 50 °C and to room temperature with 12 h reaction time. The yield of 4a increased to 60% at rt (entries 3–5). The effect of the reaction time was next evaluated. The yield of 4a in entries 6–9 reached a maximum when the reaction was quenched after 30 min (entry 8, 86% yield). We next decreased the palladium and ligand loading from 10 mol % Pd to 5 mol %. Unfortunately, the yield decreased from 86 to 67% (entries 8 vs. 10). Furthermore, increasing the reaction concentration at the lower catalyst loading did not improve the yield of 4a (entry 11). Substituting other solvents for THF did not lead to the desired product, except in the case of DME where the yield was not improved (entries 12–16).
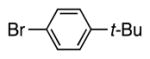
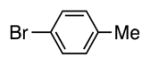
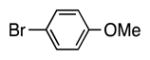
With our optimized conditions (entry 8, Table 3), we examined the substrate scope of the arylation of benzyl phenyl sulfide (4a) with aryl bromides (2a–k, Table 4). The DCCP showed good to excellent yields for alkyl substituted aryl bromides (82–84%, 4b–d, entries 2–4) and those with methoxy substituents in the para or meta positions (73–93%, 4e–f, entries 5–6). Electron-withdrawing groups in the para position led to lower yields (50–63%, 4g–h, entries 7–8). Both 1- and 2-bromonaphthalene were good substrates, forming product in 70 and 72% yield (4i–j, entries 9 and 10, respectively). Nitrogen-protected 5-bromo indole 2k was also a good coupling partner, producing the heterocyclic product 4k in 68% yield (entry 11). Reactions with heteroaryl bromides bearing 2- or 3-pyridyl, 2- or 3-furyl, or 2- or 3-thiophenyl did not yield coupling products. This is not totally unexpected given the sensitivity of these heterocycles and the strongly basic conditions in these coupling reactions.
Table 4.
Substrate scope of aryl bromides in the α-arylation of benzyl phenyl sulfide.
| |||
|---|---|---|---|
| entry | Br–Ar | product | isolated yield (%) |
| 1 | Br–Ph | 4a | 86 |
| 2 |
|
4b | 84a |
| 3 |
|
4c | 82a |
| 4 |
|
4d | 83 |
| 5 |
|
4e | 73b |
| 6 |
|
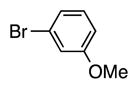
4f | 93 |
| 7 |
|
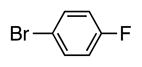
4g | 50 |
| 8 |
|
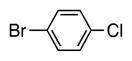
4h | 63 |
| 9 |
|
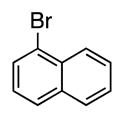
4i | 72a |
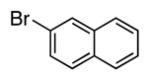
| 10 |
|
4j | 70a |
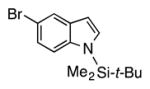
| 11 |
|
4k | 68c |
Slow addition of base for 40 min, 30 min reaction time.
Slow addition of base for 40 min at 10 °C, 15 min reaction time.
4 h reaction time.
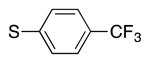
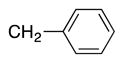
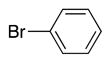
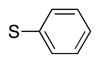
We next turned our attention to the scope of sulfide substrates (Table 5). Sulfides with electron-donating substituents in the S–Ar group resulted in good to excellent yields (4l–n, 80–97%, entries 1–3) while one with a para-CF3 group gave lower yield (4o, 60%, entry 4).
Table 5.
Substrate scope of aryl bromides in the α-arylation of aryl benzyl thioethers.
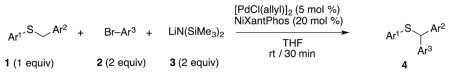
| |||||
|---|---|---|---|---|---|
| entry | S–Ar | –CH2–Ar | Br–Ar | product | yield (%) |
| 1 |
|
|
|
4l | 80a |
| 2 |
|
|
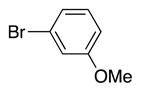
|
4m | 97a |
| 3 |
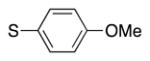
|
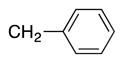
|
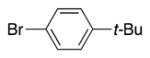
|
4n | 83b |
| 4 |
|
|
|
4o | 60 |
| 5 |
|
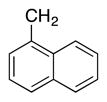
|
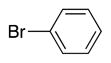
|
4i | 77b |
| 6 |
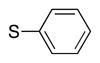
|
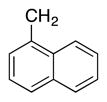
|
|
4p | 70 |
| 7 |
|
|
|
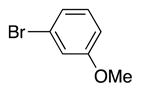
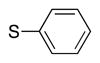
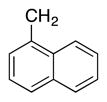
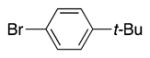
4q | 65b |
| 8 |
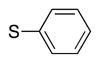
|
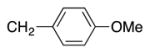
|
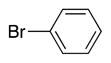
|
4e | 60a |
| 9 |
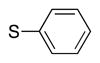
|
|
|
4r | 74a |
| 10 |
|
|
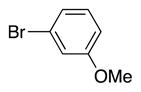
|
4s | 74 |
| 11 |
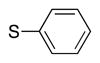
|
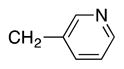
|
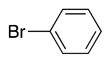
|
4t | 56c |
| 12 |
|
|
|
4u | 70c |
| 13 |
|
|
|
4v | 71c |
| 14 |
|
|
|
4x | 57c |
1 h.
Slow addition of base for 40 min, 15 min.
KO-tBu as base, 50°C for 30 min reaction time.
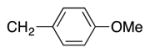
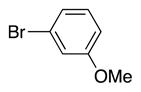
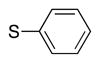
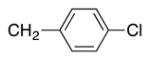
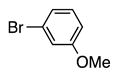
Variation of the benzylic group was also examined. Substitution of 1-naphthyl for phenyl (Ar2) and coupling with Ph–Br, 3-MeO-C6H4–Br, 4-t-Bu-C6H4–Br resulted in formation of 4i, 4p, and 4q in 77, 70, and 65% yield, respectively (entries 5–7). An electron donating 4-MeO-C6H4–CH2 group is expected to decrease the acidity of the benzylic hydrogens. Nonetheless, coupling with Ph–Br or 3-MeO-C6H4–Br resulted in formation of 4e and 4r in 60 and 74%, respectively (entries 8 and 9). Electron poor 4-Cl-C6H4–CH2SPh coupled with 3-MeO-C6H4–Br to furnish product 4s in 74% yield (entry 10), illustrating the chemoselectivity of the catalyst for the C–Br bond over the C–Cl bond.
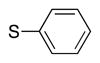
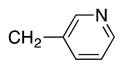
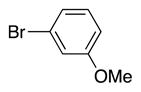
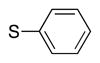
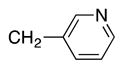
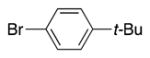
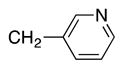
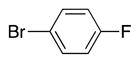
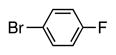
Heterocycle-containing compounds are important in medicinal chemistry.[21] We, therefore, examined heterocycles in the benzylic position of the thioether. Initially, coupling reactions with bromobenzene and Ph–S–CH2(3-pyridyl) failed due to decomposition of the pyridyl-containing substrate in the presence of LiN(SiMe3)2. We, therefore, screened alkoxide bases LiO-tBu, NaO-tBu and KO-tBu at room temperature, which led to trace products with NaO-tBu and KO-tBu. Heating reaction mixtures with these bases to 50 °C resulted in generation of the coupling product 4t in 56% yield with KO-tBu (entry 11). Coupling of Ph–S–CH2(3-pyridyl) with 3-MeO-C6H4–Br, 4-t-Bu-C6H4–Br, and 4-F-C6H4–Br resulted in moderate to good yields of the coupled products 4u–4x (57–71%, entries 12–14).
To examine the scalability of the reaction, we preformed the arylation of benzyl phenyl sulfide (1a) with bromobenzene (2a) on a 5 mmol (1.00 g) scale (Scheme 3). The desired product 4a was afforded in 78% isolated yield (1.076 g).
Scheme 3.
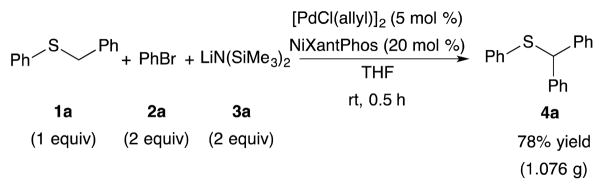
Arylation of benzyl phenyl sulfide with bromobenzene on a 5 mmol scale.
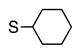
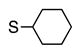
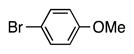
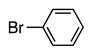
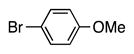
Finally, we wanted to expand our method to arylation of alkyl benzyl sulfides, R–S–CH2Ar. Under our standard conditions with LiN(SiMe3)2 and c-Hex–S–CH2Ph the reaction failed and we observed only recovered starting materials. We hypothesized that this was due to the decreased acidity of the alkyl thioether substrate. Based on the difference between estimated pKa’s of dimethyl sulfide (pKa~45)[22] and Me–S–Ph (pKa~42),[11] it is likely that c-Hex–S–CH2Ph is about 3 pKa units less acidic than Ph–S–CH2Ph (pKa 30.8 in DMSO).[11] Following this line of reasoning, we tested the more reactive bases NaN(SiMe3)2 and KN(SiMe3)2.[4] At rt in THF, we observed trace formation of products after 0.5 h with both NaN(SiMe3)2 and KN(SiMe3)2. Heating reaction mixtures with NaN(SiMe3)2 or KN(SiMe3)2 to 50°C for 1 h resulted in coupled product 6a in 84% yield for the benzyl cyclohexyl sulfide with NaN(SiMe3)2 (entry 1, Table 6). Coupling of c-Hex–S–CH2Ph with 4-MeO-C6H4–Br, 3-MeO-C6H4–Br and 4-F-C6H4–Br resulted in generation 6b–d in 60–95% yields of the coupled products (entries 2–4). These conditions also worked well with t-Bu–S–CH2Ph, which underwent DCCP with Ph–Br, 4-MeO-C6H4–Br, 3-MeO-C6H4–Br and 4-F-C6H4–Br to provide 6e–h in 75–87% yield (entries 5–8).
Table 6.
Substrate scope of aryl bromides in the α-arylation of alkyl phenyl sulfides.
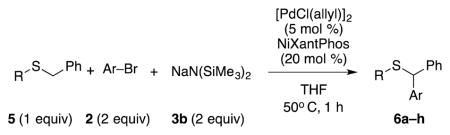
| ||||
|---|---|---|---|---|
| entry | S–R | Ar–Br | product | yield (%) |
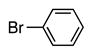
| 1 |
|
|
6a | 84 |
| 2 |
|
|
6b | 95 |
| 3 |
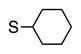
|
|
6c | 68 |
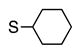
| 4 |
|
|
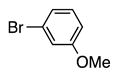
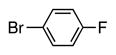
6d | 60 |
| 5 |
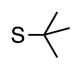
|
|
6e | 87 |
| 6 |
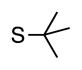
|
|
6f | 89 |
| 7 |
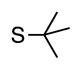
|
|
6g | 75 |
| 8 |
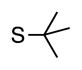
|
|
6h | 76 |
These results suggest that a variety of alkyl benzyl thioethers can be selectively coupled with aryl bromides at the benzylic position.
Conclusion
Direct and selective functionalization of C–H bonds by transition metal catalysts is a powerful method to facilitate the synthesis of diverse products from simple and readily available precursors. An expanding repertoire of substrates bearing various functional groups is now amenable to such reactions. Herein we have advanced the first method for the arylation adjacent to sulfur of thioethers. This method enables the synthesis of various aryl and alkyl (diarylmethyl) sulfides, which have demonstrated bioactivity.[23]
Our approach involves deprotonative cross-coupling of aryl benzyl sulfides, wherein the weakly acidic sulfide is reversibly metallated by LiN(SiMe3)2 or NaN(SiMe3)2 in the presence of a palladium-NiXantPhos-based catalyst. The reaction proved challenging due to the difficult deprotonation of the weakly acidic benzyl thioether. Nonetheless, moderate to excellent yields were obtained (50–97%). This study highlights the compatibility of the NiXantPhos-based palladium catalyst to highly reactive organolithium, -sodium, and -potassium intermediates that are structurally quite different than those employed in prior previously as coupling partners.[3, 18]
Experimental Section
General Procedures for α-arylation of thioethers
General Procedure A
An oven-dried 10 mL reaction vial equipped with a stir bar was charged with LiN(SiMe3)2 2 equiv) and the benzyl thioether (1 equiv) under a nitrogen atmosphere. A 1 mL solution (from a stock solution) of [PdCl(allyl)]2 (5 mol %) and NiXantPhos (20 mol %) in dry THF was added to the vial via a syringe. After stirring for 5 min at 24 °C, aryl bromide (2 equiv) was added to the reaction mixture. The reaction mixture was stirred for 30 min at 24 °C, quenched with three drops of H2O, diluted with 1 mL of dichloromethane, and filtered over a pad of silica. The pad was rinsed with additional dichloromethane, and the solution was concentrated under reduced pressure. The crude material was loaded onto a silica gel column and purified by flash chromatography.
General Procedure B
An oven-dried 10 mL reaction vial equipped with a stir bar was charged with the benzyl thioether (1 equiv) under a nitrogen atmosphere. A 1 mL solution (from a stock solution) of [PdCl(allyl)]2 (5 mol %) and NiXantPhos (20 mol %) in dry THF was added to the vial via a syringe. After stirring for 5 min at 24 °C, aryl bromide (2 equiv) was added to the reaction mixture followed by slow addition of a solution of LiN(SiMe3)2 (2 equiv) in 0.5 mL of THF for 40 min. The reaction mixture was stirred for 15 min at 24 °C, quenched with three drops of H2O, diluted with 1 mL of dichloromethane, and filtered over a pad of silica. The pad was rinsed with additional dichloromethane, and the solution was concentrated under reduced pressure. The crude material was loaded onto a silica gel column and purified by flash chromatography.
General Procedure C
An oven-dried 10 mL reaction vial equipped with a stir bar was charged with KO-tBu (2 equiv) and the benzyl thioether (1 equiv) under a nitrogen atmosphere. A 1 mL solution (from a stock solution) of [PdCl(allyl)]2 (5 mol %) and NiXantPhos (20 mol %) in dry THF was added to the vial via a syringe. After stirring for 5 min at 24 °C, aryl bromide (2 equiv) was added to the reaction mixture. The reaction mixture was stirred for 30 min at 50 °C, cooled to room temperature, quenched with three drops of H2O, diluted with 1 mL of dichloromethane, and filtered over a pad of silica. The pad was rinsed with additional dichloromethane and the solution was concentrated under reduced pressure. The crude material was loaded onto a silica gel column and purified by flash chromatography.
General Procedure D
An oven-dried 10 mL reaction vial equipped with a stir bar was charged with NaN(SiMe3)2 (2 equiv) and the benzyl thioether (1 equiv) under a nitrogen atmosphere. A 1 mL solution (from a stock solution) of [PdCl(allyl)]2 (5 mol %) and NiXantPhos (20 mol %) in dry THF was added to the vial via a syringe. After stirring for 5 min at 24 °C, aryl bromide (2 equiv) was added to the reaction mixture. The reaction mixture was stirred for 1 hour at 50 °C, cooled to rt, quenched with three drops of H2O, diluted with 1 mL of dichloromethane, and filtered over a pad of silica. The pad was rinsed with additional dichloromethane, and the solution was concentrated under reduced pressure. The crude material was loaded onto a silica gel column and purified by flash chromatography.
Supplementary Material
Acknowledgments
We thank the NSF (CHE-1152488) and NIH (NIGMS 104349) for partial support of this work. G. F. thanks the Brazilian Science Without Borders program (237849/2012-7) for financial support.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/adsc.201######.
References
- 1.a) Baudoin O. Chem Soc Rev. 2011;40:4902–4911. doi: 10.1039/c1cs15058h. [DOI] [PubMed] [Google Scholar]; Bellina F, Rossi R. Chem Rev. 2010;110:1082–1146. doi: 10.1021/cr9000836. [DOI] [PubMed] [Google Scholar]; b) Daugulis O, Do HQ, Shabashov D. Acc Chem Res. 2009;42:1074–1086. doi: 10.1021/ar9000058. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Engle KM, Mei TS, Wasa M, Yu JQ. Acc Chem Res. 2011;45:788–802. doi: 10.1021/ar200185g. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lyons TW, Sanford MS. Chem Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Colby DA, Bergman RG, Ellman JA. Chem Rev. 2010;110:624–655. doi: 10.1021/cr900005n. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Kakiuchi F, Kochi T. Synthesis. 2008;2008:3013–3039. [Google Scholar]; g) Alberico D, Scott ME, Lautens M. Chem Rev. 2007;107:174–238. doi: 10.1021/cr0509760. [DOI] [PubMed] [Google Scholar]; h) Ackermann L. Chem Rev. 2011;111:1315–1345. doi: 10.1021/cr100412j. [DOI] [PubMed] [Google Scholar]; i) Rubin M, Rubina M, Gevorgyan V. Chem Rev. 2007;107:3117–3179. doi: 10.1021/cr050988l. [DOI] [PubMed] [Google Scholar]; j) Wencel-Delord J, Droge T, Liu F, Glorius F. Chem Soc Rev. 2011;40:4740–4761. doi: 10.1039/c1cs15083a. [DOI] [PubMed] [Google Scholar]; k) Wencel-Delord J, Glorius F. Nat Chem. 2013;5:369–375. doi: 10.1038/nchem.1607. [DOI] [PubMed] [Google Scholar]; l) Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem Rev. 2010;110:890–931. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]; m) Campeau LC, Fagnou K. Chem Soc Rev. 2007;36:1058–1068. doi: 10.1039/b616082d. [DOI] [PubMed] [Google Scholar]
- 2.a) McGrew GI, Temaismithi J, Carroll PJ, Walsh PJ. Angew Chem, Int Ed. 2010;49:5541–5544. doi: 10.1002/anie.201000957. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang J, Stanciu C, Wang B, Hussain MM, Da CS, Carroll PJ, Dreher SD, Walsh PJ. J Am Chem Soc. 2011;133:20552–20560. doi: 10.1021/ja208935u. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) McGrew GI, Stanciu C, Zhang J, Carroll PJ, Dreher SD, Walsh PJ. Angew Chem, Int Ed. 2012;51:11510–11513. doi: 10.1002/anie.201201874. [DOI] [PubMed] [Google Scholar]; d) Sha SC, Zhang J, Carroll PJ, Walsh PJ. J Am Chem Soc. 2013;135:17602–17609. doi: 10.1021/ja409511n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang J, Bellomo A, Creamer AD, Dreher SD, Walsh PJ. J Am Chem Soc. 2012;134:13765–13772. doi: 10.1021/ja3047816. [DOI] [PubMed] [Google Scholar]
- 4.Bellomo A, Zhang J, Trongsiriwat N, Walsh PJ. Chem Sci. 2013;4:849–857. [Google Scholar]
- 5.a) Cremlyn RJ. An Introduction to Organosulfur Chemistry. Wiley; 1996. [Google Scholar]; b) Top 200 Prescription Drugs of 2009. 2010 avaiable at http://www.pharmacytimes.com/publications/issue/2010/may2010/rxfocustopdrugs-0510.
- 6.Jia T, Bellomo A, Baina KE, Dreher SD, Walsh PJ. J Am Chem Soc. 2013;135:3740–3743. doi: 10.1021/ja4009776. [DOI] [PubMed] [Google Scholar]
- 7.Zheng B, Jia T, Walsh PJ. Org Lett. 2013;15:1690–1693. doi: 10.1021/ol400472v. [DOI] [PubMed] [Google Scholar]
- 8.So CM, Lau CP, Kwong FY. Org Lett. 2007;9:2795–2798. doi: 10.1021/ol070898y. [DOI] [PubMed] [Google Scholar]
- 9.Lee HW, Lam FL, So CM, Lau CP, Chan ASC, Kwong FY. Angew Chem, Int Ed. 2009;48:7436–7439. doi: 10.1002/anie.200904033. [DOI] [PubMed] [Google Scholar]
- 10.For representitive methods to prepare sulfides see: Schaffner AP, Montermini F, Pozzi D, Darmency V, Scanlan EM, Renaud P. Adv Synth Catal. 2008;350:1163–1167.Shirakawa S, Kobayashi S. Org Lett. 2007;9:311–314. doi: 10.1021/ol062813j.Muramatsu W, Nakano K, Li CJ. Org Lett. 2013;15:3650–3653. doi: 10.1021/ol401534g.
- 11.Bordwell FG. Acc Chem Res. 1988;21:456–463. [Google Scholar]
- 12.a) Gilman H, Webb FJ. J Am Chem Soc. 1949;71:4062–4066. [Google Scholar]; b) Corey EJ, Seebach D. J Org Chem. 1966;31:4097–4099. [Google Scholar]; c) Peterson DJ. J Org Chem. 1967;32:1717–1720. [Google Scholar]; d) Krief A. Tetrahedron. 1980;36:2531–2640. [Google Scholar]
- 13.a) Murahashi S, Yamamura M, Yanagisawa K, Mita N, Kondo K. J Org Chem. 1979;44:2408–2417. [Google Scholar]; b) Nagaki A, Kenmoku A, Moriwaki Y, Hayashi A, Yoshida J-i. Angew Chem, Int Ed. 2010;49:7543–7547. doi: 10.1002/anie.201002763. [DOI] [PubMed] [Google Scholar]; c) Vila C, Giannerini M, Hornillos V, Fananas-Mastral M, Feringa BL. Chem Sci. 2014;5:1361–1367. doi: 10.1039/c4sc03117b. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Giannerini M, Fañanás-Mastral M, Feringa BL. Nat Chem. 2013;5:667–672. doi: 10.1038/nchem.1678. [DOI] [PubMed] [Google Scholar]
- 14.a) Umakoshi K, Yamasaki T, Fukuoka A, Kawano H, Ichikawa M, Onish M. Inorg Chem. 2002;41:4093–4095. doi: 10.1021/ic020282z. [DOI] [PubMed] [Google Scholar]; b) Jia T, Bellomo A, Montel S, Zhang M, Baina KEL, Zheng B, Walsh PJ. Angew Chem, Int Ed. 2014;53:260–264. doi: 10.1002/anie.201307172. [DOI] [PubMed] [Google Scholar]
- 15.Schade S, Boche G. J Organomet Chem. 1998;550:359–379. [Google Scholar]
- 16.Van der Veen L, Keeven PH, Schoemaker GC, Reek JNH, Kamer PCJ, van Leeuwen PWNM, Lutz M, Spek AL. Organometallics. 2000;19:872–883. [Google Scholar]
- 17.Zhang J, Bellomo A, Trongsiriwat N, Jia T, Carroll PJ, Dreher SD, Tudge MT, Yin H, Robinson JR, Schelter EJ, Walsh PJ. J Am Chem Soc. 2014;136:6276–6287. doi: 10.1021/ja411855d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Leeuwen PWNM, Kamer PCJ, Reek JNH, Dierkes P. Chem Rev. 2000;100:2741–2770. doi: 10.1021/cr9902704. [DOI] [PubMed] [Google Scholar]
- 19.Martin R, Buchwald SL. Acc Chem Res. 2008;41:1461–1473. doi: 10.1021/ar800036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shelby Q, Kataoka N, Mann G, Hartwig J. J Am Chem Soc. 2000;122:10718–10719. [Google Scholar]
- 21.Dua R, Shrivastava S, Sonwane SK, Srivastava SK. Advan Biol Res. 2011;5:120–144. [Google Scholar]
- 22.Bordwell FG, Bares JE, Bartmess JE, Drucker GE, Gerholg J, McCollum GJ, Van der Puy M, Vanier NR, Matthews WS. J Org Chem. 1977;42:326–332. [Google Scholar]
- 23.Goyal M, Singh P, Alam A, Das SK, Iqbal MS, Dey S, Bindu S, Pal C, Das SK, Panda G, Bandyopadhyay U. Free Radical Biology and Medicine. 2012;53:129–142. doi: 10.1016/j.freeradbiomed.2012.04.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.