

Abstract
Synthesis of partially 2′/3′-O-acetylated oligoribonucleotides has been accomplished by using a 2′/3′-O-acetyl orthogonal protecting group strategy in which non-nucleophilic strong-base (DBU) labile nucleobase protecting groups and a UV-light cleavable linker were used. Strong-base stability of the photolabile linker allowed on-column nucleobase and phosphate deprotection, followed by a mild cleavage of the acetylated oligonucleotides from the solid support with UV light. Two 17nt oligonucleotides, which were synthesized possessing one specific internal 2′- or 3′-acetyl group, were used as synthetic standards in a recent report from this laboratory detailing the prebiotically plausible ligation of RNA oligonucleotides. In order to further investigate the effect of 2′/3′-O-acetyl groups on the stability of RNA duplex structure, two complementary bis-acetylated RNA oligonucleotides were also expediently obtained with the newly developed protocols. UV melting curves of 2′-O-acetylated RNA duplexes showed a consistent ∼3.1 °C decrease in Tm per 2′-O-acetyl group.
Introduction
The case for ribonucleic acid (RNA) being involved in the origin of life has been strengthened by the recent observation that pyrimidine ribonucleoside-2′,3′-cyclic phosphates can be assembled from simple building blocks under prebiotically plausible conditions.1,2 Oligomerization of these activated pyrimidine ribonucleotides to yield 3′,5′-linked RNA had been a major challenge3−6 until our recent demonstration that chemoselective acetylation of oligonucleotide-2′/3′-phosphates facilitates templated ligation with preferential formation of the native 3′,5′-phosphodiester bond.7 The first formed products of this chemistry bear a 2′/3′-acetate ester adjacent to the 3′,5′/2′,5′-phosphodiester bond formed during ligation. Standards prepared using conventional synthetic chemistry were required to probe ligation product linkage isomerism, but at the outset of this work there was no literature procedure to prepare oligoribonucleotides carrying acetyl groups at certain, predetermined 2′/3′-positions.
2′-Modification of oligonucleotides, for example, 2′-fluoro or 2′-O-alkyl, have been extensively investigated because of potential applications such as antisense technologies.8−10 However, 2′-O-acylated RNA oligonucleotides have not found widespread use perhaps due to the ease of 2′,3′-migration during monomer synthesis and lack of compatibility with other protecting groups.11−14 In a recent report, Damha and co-workers re-evaluated 2′/3′-O-acyl protection and have developed the levulinyl group as a 2′-OH protecting group in oligonucleotide synthesis with on-column deprotection of RNA being afforded by exposure to hydrazine.15 In order to avoid contamination with isomeric 3′-O-levulinyl-2′-phosphoramidites, formed by 2′–3′ migration of the acyl group, a 2′-acetal levulinyl ester (ALE) protection strategy was used by the same group in the synthesis of oligonucleotides microarrays.16 We decided to adopt a complementary approach based on an orthogonal protecting group strategy that we hoped would streamline the automated solid-phase synthesis of partially 2′/3′-O-acetylated oligoribonucleotides. It was further envisaged that once an optimized synthesis of partially acetylated-RNA was possible, the effect of this modification on secondary structure adoption could be assessed and considered in relation to an abiotic replication of RNA. In this paper, we describe the design of a 2′/3′-O-acetyl orthogonal protecting group strategy, synthesis of the required phosphoramidites and solid support, and solid-phase synthesis of several partially acetylated oligonucleotides.
Results and Discussion
Protecting group strategies employed in oligonucleotide chemistry commonly rely on acyl and formamidine protecting groups for the nucleobases. However, removal of these protecting groups requires treatment with K2CO3/MeOH or heating in concentrated aqueous methylamine and/or ammonia,17−21 conditions which would invariably cleave the target 2′- or 3′-acetate esters. Merk et al. have pioneered the use of (2-cyanoethyloxy)carbonyl (ceoc) protecting groups to protect the exocyclic amino groups of adenosine, cytidine and guanosine (A, C, and G).22 The non-nucleophilic strong base 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) was used to remove ceoc groups under nonprotic reaction conditions via a β-elimination process that was thought to be compatible with the maintenance of 2′/3′-O-acetylation.23 Moreover, another advantage of this strategy is that the deprotection of cyanoethyl-protected phosphate groups can be accomplished in the same step.23 The synthesis of partially O-acetylated-RNA also required an orthogonal protection for the 2′/3′-hydroxyl groups where acetylation was not required. tert-Butyldimethylsilyl (TBS) ethers, frequently used for 2′-hydroxyl group protection in conventional oligonucleotide synthesis, are easily cleaved under mild conditions with triethylamine trihydrofluoride (TREAT·HF), which we also believed would not result in the loss of a 2′/3′-O-acetyl group.24
Nucleobase Protection
In view of the previous results, it was decided to protect the exocyclic amino groups of A, C, and G with ceoc protecting group. In addition, the O6-position of G was protected with a (4-nitrophenyl)ethyl (npe) group to prevent nucleobase anion formation that might otherwise cause greatly reduced deprotection kinetics of the N2-ceoc group.22 The (2-cyanoethoxy)carbonylation reactions were carried out with either 2-cyanoethyl carbonochloridate (1) or 1-((2-cyanoethoxy)carbonyl)-3-methyl-1H-imidazolium chloride (2) (Scheme 1), which were synthesized by modified procedures of Merk22 and Wielser.25 The protection of the exocyclic amino groups of A and C was effected according to Pfleiderer’s strategy.22
Scheme 1. (2-Cyanoethoxy)carbonylation Reagents.
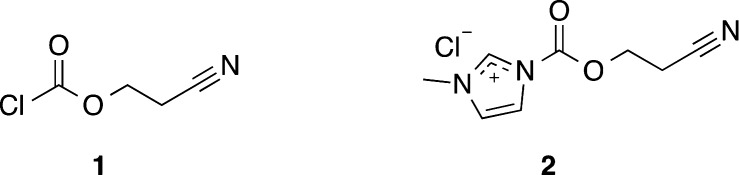
Nucleobase protection of G required several steps to install both the O6-npe and the N2-ceoc protecting groups (Scheme 2). Preliminary experiments revealed that per-acylation of guanosine was difficult because, while the hydroxyl groups underwent smooth reaction, the acetylation of the N2-position was very sluggish and did not proceed to completion. Thus, rather than relying on temporary acylation of the hydroxyl groups and the N2-position, we sought to only O-acylate, reports from Pfleiderer and co-workers indicating that a free N2-amine did not lead to extensive byproduct formation during the subsequent O6-alkylation by Mitsunobu reaction.26,27 Thus, the free hydroxyl groups of guanosine were protected with 3.6 equiv of Ac2O, Et3N and catalytic DMAP for a maximum of 30 min to give 2′,3′,5′-tri-O-acetyl-guanosine 3.28O6-Alkylation of 3 by Mitsunobu reaction gave a product that was inseparable from triphenylphosphine(oxide) byproducts, and this material was carried over to the next step. Deacetylation was accomplished with concentrated aqueous ammonia, and the limited exposure to basic conditions resulted in no loss of the O6-npe protecting group. This three-step installation of the O6-[2-(4-nitrophenyl)ethyl] protecting group gave 4 in 70% yield from guanosine. Finally, the introduction of the N2-ceoc protecting group was effected with 2-cyanoethyl carbonochloridate 1 to afford the nucleobase-protected guanosine 5 in 96% yield.22
Scheme 2. Optimized Nucleobase Protection of Guanosine.

Synthesis of the 2′/3′-O-Acetyl RNA Phosphoramidites
With the nucleobase-protected materials in hand, we envisaged two alternative ways to synthesize the 2′/3′-O-acetyl RNA phosphoramidite monomers depending on the order of the tritylation and acetylation steps. The first strategy had tritylation before selective acetylation and was initially explored starting with commercially available 5′-O-(4,4′-dimethoxytrityl)-uridine 6. This was acetylated with 1 equiv of AcCl and pyridine in THF to afford an inseparable regioisomeric mixture of 2′/3′-O-acetyl-5′-O-(4,4′-dimethoxytrityl)uridine 7a and 7b in a ratio of ca. 1:2.4 in favor of the 3′-O-acetyl regioisomer (Scheme 3). Our inability to separate the regioisomers was not seen as a problem at this stage as it was anticipated that 2′–3′-migration of the acetyl groups would occur anyway during the subsequent phosphitylation step.11
Scheme 3. Monoacetylation of 5′-O-(4,4′-Dimethoxytrityl)uridine 6.

Next, we investigated the monoacetylation of the dimethoxytritylated base protected purine and cytidine nucleosides with the same reagents and conditions as above. However, with acetyl chloride it was not possible to bring about full conversion of either starting material to monoacetylated products without also forming significant quantities of the 2′,3′-O-bisacetylated nucleosides. To overcome this problem, we considered the alternative strategy of selective acetylation before tritylation, encouraged by the prospect of being able to direct the acetylation away from the 5′-position and toward the 2′/3′-positions through the use of orthoester chemistry. Accordingly, the base-protected nucleosides 8, 9, and 5 were first quantitatively and selectively converted to the intermediate, 2′,3′-cyclic orthoesters 10 which were then hydrolyzed to regioisomeric mixtures of 2′- and 3′-acetylated nucleosides 11–13 (Scheme 4).29−31 Standard dimethoxytritylation of 11–13 then furnished the corresponding DMTr-nucleosides 14–16 in good yield, albeit with unavoidable 2′-3′-migration of the acetyl groups in favor of the 3′-O-acetyl isomer.
Scheme 4. Monoacetylation of the Base-Protected Nucleosides 8, 9, and 5.

For the subsequent phosphitylation stage, we investigated two commercially available reagents 2-cyanoethyl N,N-diisopropyl phosphoramidochloridite (17) and 2-cyanoethyl N,N,N′,N′-tetraisopropylphosphoramidite (18) (Scheme 5a). We first investigated phosphitylation of the monoacetylated purine nucleotides 14 and 16 as these had the most biased 2′:3′-OAc ratios, and we were concerned that further skewing would prevent us from using one route to make both regioisomeric monomers. Phosphitylation with 17 required Hünig’s base to neutralize in situ formed HCl; however, the base also promoted the migration of the acetyl group from the 2′-OH to the 3′-OH in line with our concerns (Table 1, entries 1 and 5).11 This result was not ideal, and so attention turned to phosphitylating agent 18, which required acid catalysis (or “activator”) to form the reactive phosphitylating species.32 Inspired by the literature,33 three different acidic activators of varying pKa values were utilized in our investigation as follows, 4,5-dicyanoimidazole (DCI, pKa = 5.2),34 1H-tetrazole (pKa = 4.9), and 5-benzylthio-1H-tetrazole (BTT, pKa = 4.1) (Scheme 5b).35 Conducting phosphitylation reactions with 18 and the three activators, it was found that the most acidic, BTT, gave the most desirable results (Table 1). In the case of 14, the product ratio was consistent with that of the starting material (Table 1, entry 4). This suggested that the increased acidity of the activator had accelerated the phosphitylation reaction and minimized the migration of the acetyl group.32,36 Beyond our expectations, the phosphitylation of 16 (Table 1, entry 6) and pyrimidine nucleosides (15 and 9) with 18 and BTT gave a more favorable 2′:3′-OAc regioisomer ratio compared to the starting materials, but the reason for this effect remains unclear.
Scheme 5. (a) Phosphitylation Reagents. (b) Phosphitylation Reactions with 18 and BTT as Activator.

Table 1. Phosphitylation Reaction Conditionsa.
| isolated
phosphoramidite ratio |
||||||
|---|---|---|---|---|---|---|
| entry | ribonucleoside mixture (1 equiv) | mixture ratio (2′-OAc:3′-OAc) | reaction conditions (equiv) | 2′-OAc (a) | 3′-OAc (b) | |
| 1 | 14a+14b | 1:3 | 17 (1.3) | DIPEA (4) | 1 | 5 |
| 2 | 14a+14b | 1:3 | 18 (2.0) | DCI (2) | 1 | 4 |
| 3 | 14a+14b | 1:3 | 18 (1.2) | 1H-Tetrazole (1) | 1 | 4 |
| 4 | 14a+14b | 1:3 | 18 (1.2) | BTT (1) | 1 | 3 |
| 5 | 16a+16b | 1:5 | 17 (1.3) | DIPEA (4) | 1 | 7 |
| 6 | 16a+16b | 1:5 | 18 (1.2) | BTT (1) | 1 | 3.7 |
Reactions were conducted in anhydrous THF (0.1 M) at room temperature (rt) and reactions times within a range of 1–3 h. 1H-Tetrazole (0.45 M) and 5-benzylthio-1H-tetrazole (BTT, 0.3 M) were added to the reaction as a solution in anhydrous MeCN. DIPEA = N,N-diisopropylethylamine (Hünig’s base), DCI = 4,5-dicyanoimidazole.
Thus, the 2′/3′-OAc regioisomeric mixtures 14, 15, 16, and 9 were all phosphitylated with 18 and BTT as the acidic activator to give eight 2′/3′-O-acetyl RNA phosphoramidites (19–22a,b, Scheme 5b), each as a pair of diastereoisomers due to the presence of a stereogenic phosphorus center. Normal-phase HPLC enabled separation of the regioisomers, and although it was on occasion possible to separate the diastereoisomers of a regioisomer, solid-phase synthesis used the regioisomerically pure phosphoramidites as pairs of diastereoisomers.
Synthesis of the 2′/3′-O-TBS RNA Phosphoramidites
For those residues in the planned oligonucleotides where a 2′/3′-O-acetate was not desired, 2′/3′-O-TBS-protected phosphoramidites were used during solid-phase synthesis. Beginning with the nucleobase-protected nucleosides 8, 9, and 5, the 5′-DMTr protecting group was introduced to give the 5′-DMTr nucleosides 23, 24, and 25 (Scheme 6). To install the TBS protecting group, we utilized the AgNO3-catalyzed method developed by Hakimelahi et al. to bring about improved selectivity for 2′-O-monosilylation of the 2′/3′-diol.37 Thus, the nucleosides 23, 24, and 25 were treated with a slight excess of TBS-Cl and AgNO3, in THF with pyridine as base, to give the monosilylated nucleosides 26, 27, and 28 each as a regioisomeric mixture with the ratio favoring the 2′-O-TBS regioisomer. The regioisomers of 26 and 27 were easily separated by silica gel flash column chromatography, but separation of 28 required normal-phase HPLC. The separated monosilylated-nucleoside derivatives were then phosphitylated using 17 under basic conditions to give the final 2′/3′-O-TBS adenosine 29a/b, 2′/3′-O-TBS cytidine 30a/b, and 2′/3′-O-TBS guanosine 31a/b phosphoramidites in high yield. The 2′-TBS uridine phosphoramidite was commercially available, and, although the regioisomeric 3′-TBS phosphoramidite was not, its precursor 32 was. Thus, 32 was also phosphitylated using the same conditions to give the 3′-TBS uridine phosphoramidite 33.
Scheme 6. Synthesis of the 2′/3′-O-TBS RNA phosphoramidites.

Synthesis of the Photolabile Linker
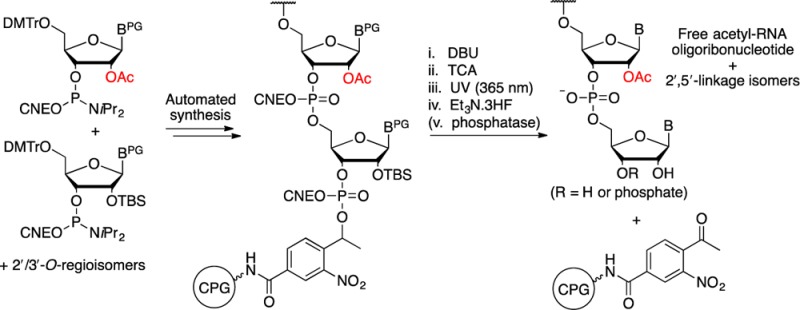
With the required phosphoramidite monomers in hand, several linkers to attach the first protected nucleoside to the solid support were considered. Typical commercial linkers such as succinate esters 34 are not compatible with the desired partially acetylated oligoribonucleotides as they require nucleophilic cleavage (Figure 1).18,38 The commercially available Q-linker 35 is cleaved by fluoride ions and was considered to be orthogonal to 2′/3′-O-acetyl groups.39 However, the Q-linker forms an ester bond with the 2′/3′-hydroxyl of the first nucleotide that upon cleavage yields a 2′,3′-diol-terminated oligoribonucleotide, and the identity of the first nucleotide is also fixed. To obtain maximum flexibility of chemistry at the 3′-oligonucleotide terminus and allow any sequences of partially acetylated RNA to be prepared, a universal linker that allowed the synthesis of 2′/3′-phosphate RNA oligonucleotides was sought. It was also deemed to be desirable, prior to removal of the product from the solid support, to be able to thoroughly remove excess DBU to prevent hydroxide formation and deprotection byproducts upon transfer to aqueous media. Hence, a strong-base-stable linker that would allow on-column deprotection with DBU was chosen as the ideal target.
Figure 1.

Structures of various solid-phase linkers.
Photolysis was deemed to offer a mild acetyl-orthogonal method for the cleavage of oligonucleotides from the solid support. Photolabile linker 36 based on the o-nitrobenzyl group has been developed by Greenberg and co-workers to allow the synthesis of oligonucleotides with 3′-hydroxyl, 3′-phosphate, and 3′-end-modified oligonucleotides.40−43 These linkers have been modified by Damha and co-workers in order to improve the photocleavage rates.44 The chain of the linker arm of 37 is extended by one carbon and is branched at the benzyl position to generate a tertiary carbon center. However, linker 37 possesses a hydrogen atom β-disposed to the oxygen atom of the leaving group, and it was suspected that exposure to DBU would cause a premature cleavage of the oligonucleotide via a β-elimination process in the case of a phosphate linkage, as suggested by analogy to the DBU cleavable o-nitrophenyl ethyl carbonate linker 38 developed by Eritja and co-workers.23,45,46 Thus, it was thought that an alternative linker 39, which has previously been utilized for the synthesis of peptides,47 and shortened by one carbon relative to 37, would meet our requirements. Thus, solid-phase synthesis would be used to routinely generate 2′/3′-phosphate-terminated oligoribonucleotides, and in the event that a diol-terminated oligoribonucleotide was required, the terminal phosphate would be removed with a phosphatase.
The preparation of linker 39 on long-chain alkylamine controlled-pore glass (LCAA-CPG) began with 3-formyl-4-nitrobenzoate 40. Reaction of 40 with methylmagnesium bromide at room temperature gave the desired α-methyl alcohol 41 in admixture with the inseparable benzyl alcohol 42 as a minor byproduct (13%). The formation of 42 was suspected to proceed by a radical pathway involving single-electron transfer to the aromatic aldehyde from the Grignard reagent.48 The alcohol mixture was dimethoxytritylated under standard conditions, after which the DMTr-protected secondary alcohol 43 was separated from 44 by normal-phase HPLC with a 33% yield over two steps. The methyl ester of the photolabile linker precursor 43 was hydrolyzed using LiOH in a mixture of THF and water. The crude lithium benzoate salt was reacted with isobutylchloroformate in anhydrous pyridine to afford the mixed anhydride 45. LCAA-CPG was then derivatized with 45 at a concentration of 200–1000 μmol g–1 of CPG under anhydrous conditions to give 46 with a loading of 33.3–56.2 μmol g–1 after capping of unreacted amines (Scheme 7).
Scheme 7. Preparation of the Solid-Phase Support.

To ensure the suitability of the solid support 46 for RNA oligonucleotide synthesis, the photocleavage efficiency was tested in the synthesis of an RNA oligoribonucleotide, of sequence 5′-GCCGCCC-3′P (P = phosphate), using commercially available nucleoside phosphoramidites on a 1 μmol scale. After completion of the automated solid-phase synthesis, without any deprotection of the oligonucleotide, the CPG support was suspended in acetonitrile (1 mL) and exposed to UV irradiation at λ = 365 nm. The cleavage was followed over time by trityl assay of solubilized material (Figure 2), and this showed that photolysis was complete within 60 min with a maximal 42.3 O.D. of crude oligonucleotide released. This corresponded to approximately 683 nmol of oligonucleotide and a 68% cleavage yield, thus indicating that the photolabile linker was suitable for oligonucleotide synthesis as its cleavage was both rapid and efficient.
Figure 2.
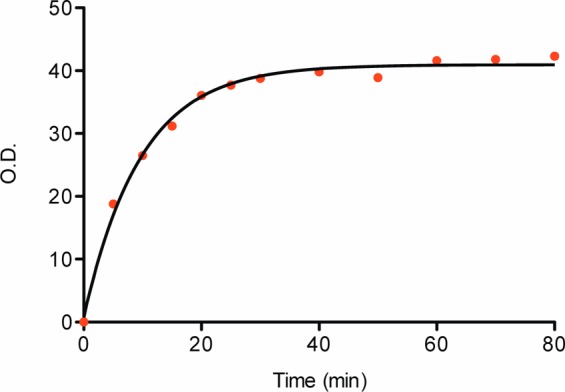
Rate of oligonucleotide release from the solid-phase support 46 when it is irradiated at λ = 365 nm.
Synthesis of Partially Acetylated-RNA Oligonucleotides
Synthesis of the acetylated oligonucleotides was conducted with a standard RNA synthesis cycle using a Bioautomation MerMade 4 automated synthesis machine. In order to improve the coupling efficiency of relatively hindered nucleoside phosphoramidites used in this work, an 8-fold excess of phosphoramidite was added to the CPG solid support during each coupling step in the automated synthesis cycle. Moreover, the coupling time was increased to 20 min, and the activator was changed to BTT from ETT (5-ethylthio-1H-tetrazole) (pKa = 4.28) to improve the coupling yield. Although the higher acidity of BTT (pKa = 4.10) helped to improve coupling yields, its poor solubility in acetonitrile (0.35 M) caused it to precipitate on ends of the reagent delivery lines used by the automated synthesis machine. This led to blockage of reagent line nozzles preventing reliable delivery of activator; this issue became a significant problem during synthesis of longer oligonucleotides as it led to the synthesis of mainly truncated oligonucleotides. Therefore, the much more soluble activator DCI (1.0 M) was employed, which did not crystallize on the nozzles. Despite DCI (pKa = 5.2) being less acidic than BTT, it has been shown to be an effective activator due to its greater nucleophilicity.34
In preliminary experiments, the original capping reagents CAP A (THF/2,6-lutidine/Ac2O) and CAP B (N-methylimidazole) were utilized to synthesize an 8nt oligonucleotide with a sequence of 5′-GCCG(2′OAc)GCCG-3′P. After deprotection and photocleavage, the MALDI-TOF analysis of the free oligonucleotide showed mass peaks corresponding to a mixture of mono-, di-, and triacetylated 8nt oligonucleotides. The extra acetyl groups were thought to result from N-acetylation on the exocyclic amine of nucleobase during the capping procedure with Ac2O, which has also been observed by Greenberg and co-workers.20,49 As suggested by the same authors, the more sterically hindered reagent, pivalic anhydride, was used in place of Ac2O in the CAP A reagent (THF/2,6-lutidine/pivalic anhydride, 4:1:1 v/v). Subsequently, the capping time was increased from 1 to 5 min to obtain efficient capping with this more sterically hindered reagent. The problem was also partly attributable to the acetyl capped solid support, and so the CPG was also capped with pivaloyl chloride in place of Ac2O to eliminate this problem completely (Figure S1, Supporting Information). At the end of the solid-phase synthesis cycle, the final DMTr-group at the 5′-terminus was not removed since it was thought that the exposed 5′-hydroxyl could act as a nucleophile under basic conditions with the possible result of acetyl migration from the 2′/3′-O-positions to the 5′-position during the deprotection step with DBU.
Next, the CPG solid support with trityl-on oligonucleotides was subjected to 0.5 M DBU in anhydrous acetonitrile at 40 °C for 4 h to bring about the complete nucleobase deprotection. Morpholine (10% v/v) was added to the deprotection solution as an acrylonitrile scavenger to prevent the alkylation of the deprotected nucleobases that is possible via a Michael-type addition under strongly basic conditions.50 After on-column deprotection, excess DBU and byproducts from the nucleobase deprotection, such as acrylonitrile, p-nitrostyrene, and their morpholinyl adducts, were washed away easily and thoroughly with anhydrous acetonitrile. After on-column removal of the 5′-terminal DMTr group with 3% TCA (trichloroacetic acid) in CH2Cl2, an accurate trityl assay was used to calculate the overall yield of full-length oligonucleotides (Table 2).
Table 2. Yields of Oligonucleotides at Various Stages of Deprotection. Characterization Using MALDI-TOF Mass Analysis Is Shown for Oligonucleotides That Were Purified.
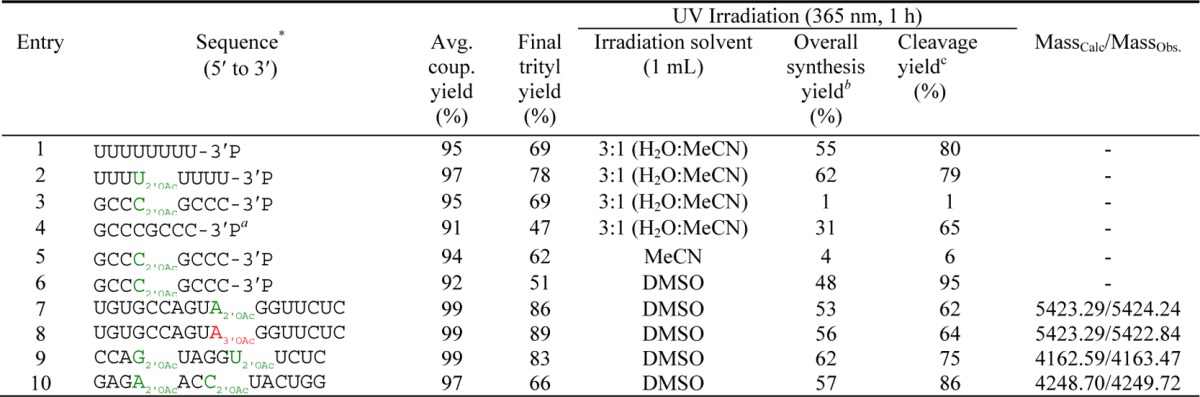
Subscript 2′-OAc indicates 3′,5′-linkage with 2′-OAc protection at the modification site while 3′-OAc indicates 2′,5′-linkage with 3′-OAc.
This oligonucleotide was synthesized using commercial amidites and was deprotected with 3:1 saturated ammonium hydroxide/EtOH.
The overall synthesis yield was obtained by comparing the total released amount of oligonucleotide from CPG to the synthesis cycle scale (1 μmol).
The cleavage yield was obtained by comparing the cleaved amount of oligonucleotide to the final trityl yield.
The next step was the UV-induced photocleavage of the oligonucleotides from CPG solid support. The solid supports were initially suspended in 3:1 mixture of H2O/MeCN and subjected to UV irradiation at λ = 365 nm for 1 h. In preliminary experiments, the sequences of 8nt polyU (Table 2, entry 1), 8nt polyU with one internal 2′-O-acetylated uridine (Table 2, entry 2), and 5′-GCCC(2′OAc)GCCC-3′P (Table 2, entry 3) were synthesized to test the photocleavage after on-column deprotection. The 8nt polyU modified with or without internal 2′-acetylated uridine gave 80% cleavage (Table 2, entries 1 and 2). However, a very poor photocleavage yield was obtained for the 5′-GCCC(2′OAc)GCCC-3′P sequence (Table 2, entry 3). The same sequence, synthesized from commercial phosphoramidites, was previously tested (Figure 2) and showed good photocleavage yield before deprotection. To investigate the effect of oligoribonucleotide deprotection status upon photocleavage, the same sequence was synthesized again with commercial phosphoramidites (Table 2, entry 4) and then on-column deprotected with 3:1 saturated ammonium hydroxide/EtOH. Finally the photolabile CPG solid support was subjected to UV light under the same conditions (3:1, H2O/MeCN). This gave a much better photocleavage yield (65% cleavage) compared to that obtained for the very close analogue with DBU deprotection (Table 2, entry 3). The different deprotection reagents of DBU or NH4OH, respectively, resulted in the DBU or ammonium salts of the oligonucleotides, and the different cleavage yields/recoveries suggested that these salts had different solubility in H2O/MeCN mixtures with the DBU-deprotected oligonucleotides having a lower solubility. Therefore, pure acetonitrile and DMSO were chosen as alternative solvents to carry out the photocleavage with the same sequence (Table 2, entries 5 and 6). It was found that DMSO was the ideal solvent and dramatically improved the cleavage yield/recovery of oligonucleotides (Table 2, entries 6–10).
The final deprotection step to remove the 2′-TBS groups of acetyl-RNA was carried out under standard conditions with TREAT·HF in anhydrous DMSO. Finally, the deprotected oligonucleotides were isolated by precipitation with sodium acetate and 1-butanol, quantitated by UV absorption, and analyzed by MALDI-TOF mass spectrometry. To obtain 2′,3′-diol-terminated oligonucleotides the terminal 2′/3′-phosphate was removed enzymatically with calf intestinal phosphatase (CIP), after which the dephosphorylated oligonucleotides were purified by strong anion exchange HPLC (SAX-HPLC). Failure sequences and small amounts of deacetylated products were efficiently separated from the desired acetylated oligonucleotides (see Figure S3, Supporting Information, for HPLC trace).
With an optimized strategy developed for the synthesis of partially acetylated oligonucleotides now available, authentic standards were synthesized that corresponded to partially acetylated products from the prebiotically plausible ligation of oligoribonucleotides as previously described by our group (Table 2, entries 7 and 8).7 Two complementary bisacetylated 13nt oligonucleotides were also made to test the effect on the duplex stability by varying the degree of acetylation (Table 2, entries 9 and 10). The first sequence was constructed from the UGUG truncation of the 17nt oligonucleotide (Table 2, entry 7), and the second 13nt oligomer was the sequence complement. Positions of acetylation were chosen so as to utilize each of the four acetylated phosphoramidite monomers and such that the resultant partially acetylated oligoribonucleotides resembled products one might expect from the templated ligation of tiled short oligoribonucleotides (3 < nt <6) under prebiotic conditions previously described by our group. Several partially acetylated oligoribonucleotides (Table 2, entries 7–10) were characterized using MALDI-TOF mass analysis, and their purity (>95%) was confirmed by analytical SAX-HPLC (see Table S1 and Figure S2, Supporting Information).
Thermal Denaturation Studies
The structural effects of partial acetylation on duplex stability were investigated using UV-melting curves from which the melting temperature (Tm) and other thermodynamic parameters could be extracted.51−53 The Tm values of duplexes that utilized a 17nt oligomer (Table 3, entries 3–6) were compared with their parent nonacetylated duplexes (Table 3, entries 1 or 2); the duplexes that contained the truncated 13nt oligonucleotides (without 5′-overhang) (Table 3, entries 8–10) were compared with the nonacetylated 13nt duplex (Table 3, entry 7). Analysis of the thermal denaturation data showed that 2′-O-acetylation, remarkably, leads to a very consistent 3.1 °C decrease in duplex Tm per acetyl group. Acetylation at the terminal positions was not explored, but it is known that other modifications that decrease duplex Tm give a less significant reduction at the strand ends.54 The destabilizing effect of 2′-O-acetylation is also evident in the thermodynamic parameters where an increasing degree of acetylation leads to a greater increase of ΔG°37 (e.g., Table 3, entry 10 vs entry 7).
Table 3. Tm and Thermodynamic Parameter Data for Partially Acetylated-RNA Duplexes.
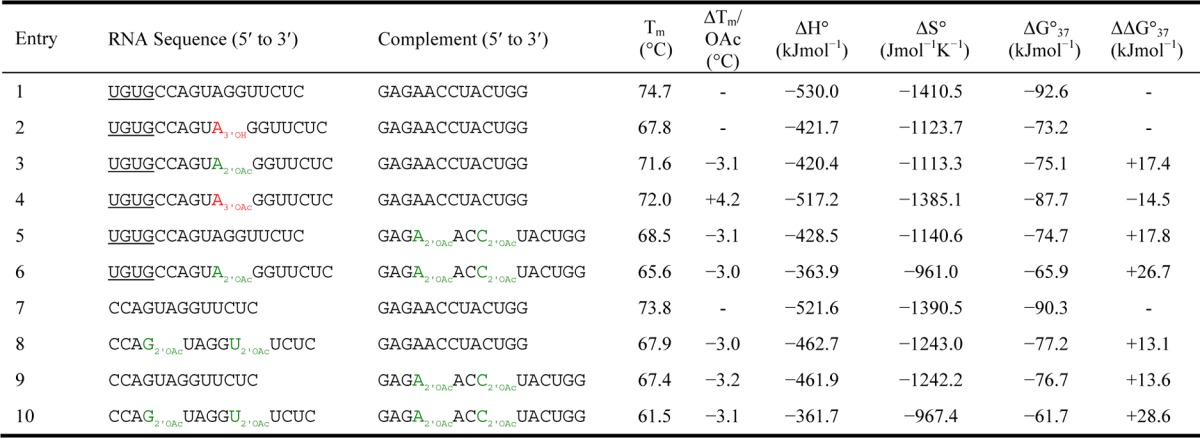
Experiments were performed in a 10 mM Na2HPO4, 0.5 mM Na2EDTA buffer (pH7) containing 1 M NaCl and using 2.5 μM of each complementary strand over a temperature range of 30–90 °C. Data is an average of three heat–cool cycles. Error for Tm values represent standard deviations of 6 values and are ±0.8 °C. Errors for thermodynamic data represent standard deviations of 6 values and are ±7.4% for ΔH°, within ±8.5% for ΔS° and within ±3.1 kJmol–1 for ΔG°37. Nonacetylated oligonucleotides were either synthesized using standard procedures or purchased in HPLC-purified Na+ form. Underlined nucleotides denote overhanging sequence, subscripts denote site of acetylation or linkage isomerism, green denotes site of 3′-5′ natural linkage and red denotes site of 2′-5′ unnatural linkage isomerism. See Figure S3 (Supporting Information) for UV melting curves.
It is known that both the major and minor grooves of A-form RNA duplexes are well hydrated with an extensive network of hydrogen-bonded water molecules.55,56 In particular, the minor groove hydration network is mediated by the 2′-hydroxyl groups that, relative to a DNA duplex, serve to provide a greater thermodynamic stabilization by acting as a scaffold to bridge both strands of the duplex. Destabilization of the 2′-O-acetylated duplexes is indicated by an increase in ΔH° and ΔS°, which correspond to an unfavorable enthalpic and a favorable entropic change. It is believed that 2′-O-acetylation blocks the hydrogen bonding ability of the 2′-hydroxyl and this reduces the degree of hydration in the minor groove.57 Reduction of the hydration network causes a loss of solvating water molecules, which accounts for the favorable entropy gain. Additionally, the number of hydrogen bonds is reduced and the formation of water bridges is hindered, which both contribute to the unfavorable enthalpy change.
As expected, inclusion of an internal 2′,5′-linkage within a duplex led to a reduction in the Tm (ΔTm = −6.9 °C) and decreased the duplex stability (ΔΔG°37 = +19.4 kJmol–1, Table 3, entries 1 and 2).58 On acetylation of the 3′-hydroxyl at the internal 2′,5′-linkage the Tm increased (ΔTm = +4.2 °C) and a favorable decrease in ΔG°37 (ΔΔG°37 = −14.5 kJmol–1, Table 3, entries 2 and 4) was calculated that indicated a more stable duplex. The reason for the stabilization effect of 3′-acetylation on the RNA duplexes is not clear thus far. Szostak and co-workers have shown that partially 2′,5′-linked RNA can form an A type duplex with C2′-endo sugar puckering predominating at the 2′,5′-linkage site.59 The C2′-endo (south) sugar pucker can potentially be stabilized by acetylation of the 3′-hydroxyl group by enhancing the σC–H2′ → σ*C–O3′ stereoelectronic effect. The increased electronegativity of 3′-OAc relative to 3′-OH is thought to further stabilize the already preferred C2-endo sugar pucker (Figure 3). This could explain the relatively higher stability of the duplex with 3′-O-acetylated RNA compared to its 3′-nonacetylated counterpart.
Figure 3.

Sugar pucker at a 2′,5′-linkage site (in otherwise 3′,5′-linked RNA) can be C2′-endo or C3′-endo, although the former is preferred in A-form duplex. It is thought that the increase in Tm that is observed on acetylation of the 3′-hydroxyl of a 2′,5′-linkage site is caused by an increase in C2′-endo sugar pucker preference due to an increase in the magnitude of the σC–H2′ → σ*C–O3′ stereoelectronic effect as the antibonding orbital becomes a better acceptor due to the increased electron withdrawal.
The data obtained through these thermal denaturation experiments have interesting implications. The copying of a long single-strand of RNA would create an RNA duplex that, because of its inordinately high Tm, would be difficult to denature, and this poses a formidable challenge for the nonenzymatic replication of RNA at the origin of life.60 The stability of these “dead-end” duplexes can be reduced by incorporation of a small proportion of 2′,5′-linkages. However, 2′,5′-linkages are known to be hydrolyzed more rapidly than 3′,5′-linkages in the context of a duplex and could lead to premature chain cleavage and degradation of oligonucleotides.61 The (temporary) reduction of Tm and duplex stability by partial acetylation has the distinct potential advantage of allowing the nonenzymatic template-directed synthesis of longer oligonucleotides than is possible with native RNA, while only forming native 3′,5′-phosphodiester bonds. The more facile strand separation of partially acetylated RNA under prebiotically plausible conditions would expedite its replication relative to the replication of native RNA. After replication in partially acetylated “genotypic” form, unmodified “phenotypic” RNA could emerge simply through subsequent hydrolysis.
Conclusion
An orthogonal protecting group strategy was successfully employed for the synthesis of partially 2′/3′-O-acetylated oligoribonucleotides. Key to the synthesis was the utilization of non-nucleophilic strong base-labile nucleobase protecting groups and a photocleavable linker. This strategy allowed on-column deprotection of the nucleobases and phosphate groups under non-nucleophilic conditions and also a mild photolytic cleavage of RNA oligonucleotides from the solid support. Partial 2′/3′-O-acetylation has been incorporated into several oligonucleotides including a 17nt sequence and two complementary 13nt sequences. Their duplex-forming properties have been investigated using UV-melting (thermal denaturation). The results show that 2′-O-acetylation consistently destabilizes the RNA duplex structure (ΔT = −3.1 °C per acetylation). This effect was attributed to dehydration of the duplex minor groove and subsequent disruption of the stabilizing hydrogen-bonding network.
Experimental Section
General Experimental Methods
Chemical shifts are reported in ppm (δ). Coupling constants (J) are given in hertz, and the notations s, d, t, and br represent the multiplicities singlet, doublet, triplet, and broad signal. Assignment was based on 1H–1H COSY, HMBC, and HMQC NMR spectra.
2-Cyanoethyl Carbonochloridate (1).22
Triphosgene (5.94 g, 20.0 mmol) was dissolved in anhydrous THF (50 mL) and cooled to 0 °C. 3-Hydroxypropionitrile (2.73 mL, 40.0 mmol) was diluted with anhydrous THF (17 mL) and was added dropwise to the solution of triphosgene over a 2 h period. The resultant mixture was warmed to rt and stirred overnight. Anhydrous pyridine (4.84 mL, 60.0 mmol) was diluted with anhydrous THF (5 mL) and added dropwise to the solution at 0 °C. The resultant mixture was warmed to rt and stirred for a further 1 h. The precipitated pyridinium hydrochloride salt was removed by filtration, and the supernatant was evaporated under vacuum. The title compound was isolated as a pale yellow viscous oil in quantitative yield. The product was used immediately without further purification or stored at −30 °C under argon until required: 1H NMR (400 MHz, CDCl3) δ 4.52 (t, J = 6.3 Hz, 2H, −OCH2CH2CN), 2.84 (t, J = 6.3 Hz, 2H, −OCH2CH2CN); 13C NMR (100 MHz, CDCl3) δ 151.7 (C=O), 115.3 (CN), 65.0 (−OCH2CH2CN), 17.8 (−OCH2CH2CN).
O6-[2-(4-nitrophenyl)ethyl]guanosine (4)
2′,3′,5′-Triacetylguanosine 3 (10.8 g, 26.3 mmol) was suspended in anhydrous dioxane (100 mL). p-Nitrophenylethanol (5.27 g, 31.5 mmol) and triphenylphosphine (8.27 g, 31.5 mmol) were added, and the resultant mixture was heated at 80 °C for 45 min. Diisopropyl azodicarboxylate (6.20 mL, 31.5 mmol) was added dropwise, upon which the solution began to boil and then the solution was stirred at 60 °C for a further 1 h. The solution was cooled to rt and evaporated to dryness under vacuum to give an oil, from which the intermediate 2′,3′,5′-triacetyl-O6-[2-(4-nitrophenyl)ethyl]guanosine mixed with triphenylphosphine oxide was isolated by flash column chromatography (60:35:5, EtOAc/n-hexane/MeOH). The mixture was taken up in MeOH (200 mL) and cooled to 0 °C. To the solution was added saturated aq NH3 (200 mL) and the solution stirred in a sealed vessel at rt overnight. The solution was degassed, and then the solvent was removed under vacuum to give an orange oil. The oil was taken up in MeOH (∼80 mL). and on concentration by evaporation under vacuum a yellow solid precipitated. The mixture was cooled at 4 °C overnight and the resultant solid precipitate collected by filtration and washed with cold MeOH (3 × 20 mL). The solid contained acetamide and so was suspended in H2O (100 mL) and heated at 90 °C for 15 min. Once cooled to rt, the insoluble material was collected by filtration and washed with H2O (3 × 20 mL). The solid was air-dried and then dried under high vacuum to give the title compound over two steps as a yellow amorphous solid (8.45 g, 76%): 1H NMR (400 MHz, DMSO-d6) δ 8.18 (d, J = 8.2 Hz, 2H, aromatic), 8.09 (s, 1H, H8), 7.64 (d, J = 8.2 Hz, 2H, aromatic), 6.44 (s, 2H, NH), 5.78 (d, J = 5.9 Hz, 1H, H1′), 5.37 (d, J = 6.0 Hz, 1H, 2′-OH), 5.12–5.06 (m, 2H, 3′-OH and 5′-OH), 4.68 (t, J = 6.8 Hz, 2H, −OCH2−), 4.46 (q, J = 5.7 Hz, 1H, H2′), 4.10 (q, J = 4.3 Hz, 1H, H3′), 3.89 (q, J = 3.8 Hz, 1H, H4′), 3.65–3.50 (m, 2H, H5′), 3.25 (t, J = 6.8 Hz, 2H, −CH2ph); 13C NMR (100 MHz, DMSO-d6) δ 160.1 (C6), 159.7 (C2), 154.3 (C4), 146.7, 146.3 (aromatic), 138.1 (C8), 130.3, 123.4 (aromatic), 113.8 (C5), 86.6 (C1′), 85.3 (C4′), 73.5 (C2′), 70.4 (C3′), 65.5 (−OCH2−), 61.4 (C5′), 34.4 (−CH2ph); HRMS (ESI-TOF) m/z [M + H]+ calcd for C18H21N6O7 433.1472, found 433.1462.
N2-[(2-Cyanoethoxy)carbonyl]-O6-[2-(4-nitrophenyl)ethyl]guanosine (5)
Compound 4 (4.32 g, 10.0 mmol) was coevaporated with anhydrous pyridine (3 × 20 mL). The residue was dissolved in anhydrous pyridine (40 mL) and anhydrous CH2Cl2 (55 mL). To the solution was added dropwise Me3Si-Cl (7.61 mL, 60.0 mmol), and the resultant mixture was stirred at rt for 20 min. 2-Cyanoethyl carbonochloridate (1) (2.00 g, 15.0 mmol) diluted in anhydrous CH2Cl2 (10 mL) was added dropwise and the mixture stirred for a further 3 h. MeOH (30 mL) was added to quench the reaction and remove the Me3Si groups. The solvent was removed under vacuum, and the resultant oil was coevaporated with 1:1 MeOH/toluene (3 × 30 mL). The residue was taken up in MeOH (15 mL), and H2O was added dropwise until precipitation of a solid began. The solution was kept at 4 °C overnight to afford a slightly pink precipitate that was collected by filtration and washed with cold MeOH (3 × 20 mL). The solid contained pyridinium·HCl, and this was removed by boiling the solid as a suspension in H2O (50 mL) for 15 min. After the suspension was cooled to rt, the solid was collected, washed with H2O (3 × 20 mL), and dried under high vacuum to yield the title compound as a slightly off-white amorphous solid (5.08 g, 96%): 1H NMR (400 MHz, DMSO-d6) δ 10.54 (s, 1H, NH), 8.43 (s, 1H, H8), 8.18 (d, J = 8.7 Hz, 2H, aromatic), 7.66 (d, J = 8.7 Hz, 2H, aromatic), 5.89 (d, J = 5.9 Hz, 1H, H1′), 5.45 (d, J = 5.9 Hz, 1H, 2′-OH), 5.16 (d, J = 4.7 Hz, 1H, 3′-OH), 4.93 (t, J = 5.5 Hz, 1H, 5′-OH), 4.79 (t, J = 6.9 Hz, 2H, −OCH2−), 4.61 (q, J = 5.7 Hz, 1H, H2′), 4.31 (t, J = 6.0 Hz, 2H, −COOCH2−), 4.19 (td, J = 4.8 Hz, 3.3 Hz, 1H, H3′), 3.92 (q, J = 4.2 Hz, 1H, H4′), 3.65 (ABX, JAB = 11.8, JAX = 4.9 Hz, 1H, H-5′), 3.54 (ABX, JBA = 11.8, JBX = 4.9 Hz, 1H, H5″), 3.34–3.31 (m, 2H, −CH2ph), 2.94 (t, J = 6.0 Hz, 2H, −CH2CN); 13C NMR (100 MHz, DMSO-d6) δ 159.7 (C6), 153.1 (C4), 151.9 (C2), 151.5 (C=O), 146.4, 146.3 (aromatic), 141.4 (C8), 130.4, 123.4 (aromatic), 118.6 (CN), 117.3 (C5), 87.1 (C1′), 85.7 (C4′), 73.4 (C2′), 70.4 (C3′), 66.4 (−OCH2−), 61.4 (C5′), 59.4 (−COOCH2−), 34.2 (−CH2ph), 17.7 (−CH2CN); HRMS (ESI-TOF) m/z [M + H]+ calcd for C22H24N7O9 530.1636, found 530.1620.
2′/3′-O-Acetyl-5′-O-(4,4′-dimethoxytrityl)uridine (7a and 7b)
To a solution of commercially available 5′-O-(4,4′-dimethoxytrityl)uridine (3.00 g, 5.49 mmol) in anhydrous THF (20 mL) was added anhydrous pyridine (0.44 mL, 5.49 mmol), followed by acetyl chloride (0.39 mL, 5.49 mmol) at 0 °C. The mixture was warmed to rt, stirred for 3 h, and then quenched by addition of saturated aq NaHCO3. The organics were extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were dried over MgSO4 and finally concentrated under vacuum. The residue was purified by flash column chromatography (50:50:1:1, EtOAc/Tol/MeOH/Et3N) to give the title compounds 7a and 7b (2.20 g, 68%) as a regioisomeric mixture (note: the isomers were isolated as a mixture in a ratio of ca. 2.4:1 7b:7a calculated by integrations of both H1′ of 7a and 7b): 1H NMR (400 MHz, CDCl3) δ 9.40 (s, 0.7H, NH, b), 8.66 (d, J = 1.4 Hz, 0.3H, NH, 7a), 7.84 (d, J = 8.1 Hz, 0.7H, H6, 7b), 7.79 (d, J = 8.2 Hz, 0.3H, H6, 7a), 7.44–7.18 (m, 9H, aromatic, 7a+7b), 6.84 (d, J = 8.8 Hz, 4H, aromatic, 7a+7b), 6.14 (d, J = 4.5 Hz, 0.3H, H1′, 7a), 6.00 (d, J = 4.9 Hz, 0.7H, H1′, 7b), 5.45–5.33 (m, 1.3H, H5 and H2′), 5.26 (t, J = 4.9 Hz, 0.7H, H3′, 7b), 4.59 (q, J = 4.8 Hz, 0.3H, H3′, 7a), 4.53 (q, J = 5.4 Hz, 0.7H, H2′, 7b), 4.28 (d, J = 4.4 Hz, 0.7H, H4′, 7b), 4.14 (dt, J = 4.9, 2.3 Hz, 0.3H, H4′, 7a), 3.91 (d, J = 6.5 Hz, 0.7H, 2′-OH, 7b), 3.79 (s, 6H, OCH3, 7a+7b), 3.55 (app. dd, J = 11.0, 2.3 Hz, 1H, H5′, 7a+7b), 3.49–3.43 (m, 1H, H5″, 7a+7b), 2.48 (d, J = 3.9 Hz, 0.3H, 3′-OH, 7a) 2.18, 2.14 (2 × s, 3H, acetyl CH3, 7a+7b); 13C NMR (CDCl3, 101 MHz): δ 170.7, 170.5 (acetyl C=O), 163.7, 163.4 (C4), 158.8, 158.7 (aromatic), 151.2 (C2, 7b), 150.6 (C2, 7a), 147.4, 144.2 (aromatic), 140.2, 140.0, 139.6 (C6, 7a+7b), 135.3, 135.2, 135.1, 130.3, 130.2, 130.2, 129.2, 129.1, 128.3, 128.3, 128.2, 128.2, 127.9, 127.9, 127.3, 127.2, 113.4, 113.2 (aromatic), 102.8, 102.8 (C5, 7a+7b), 89.5 (C1′, 7b), 87.4, 87.3 (DMTr-C), 86.8 (C1′, 7a), 83.7 (C4′, 7a), 81.7 (C4′, 7b), 76.0 (C2′, 7a), 74.1 (C2′, 7b), 71.9 (C3′, 7b), 69.9 (C3′, 7a), 62.4, 62.2 (C5′, 7a+7b), 55.4 (OCH3, 7a+7b), 21.6, 20.9 (acetyl CH3, 7a+7b). HRMS (ESI-TOF) m/z [M + H]+ calcd for C32H32N2O9Na 611.2000, found 611.1979.
N6-[(2-Cyanoethoxy)carbonyl]-2′/3′-O-acetyladenosine (11a and 11b)
To a suspension of 8 (7.00 g, 19.2 mmol) in anhydrous dioxane (280 mL) were added trimethyl orthoacetate (2.07 mL, 16.4 mmol) and TFA (8.4 μL, 0.11 mmol) and the mixture stirred at 50 °C for 3 h. Water (70 mL) was added to the mixture, which was then stirred for a further 1 h at 50 °C. The solvent was removed, and the residue was purified by flash column chromatography (2–10% methanol in DCM) to give the title products as an off-white solid mixture of regioisomers (7.30 g, 94%) (note: 11b: 11a in a ratio of ca. 2.3: 1, calculated by integrations of both H-1′ of 11a and 11b from 1H NMR): 1H NMR (400 MHz, CDCl3) δ 9.92 (s, 0.70H, NH, 11b), 9.79 (s, 0.30H, NH, 11a), 8.62 (s, 0.30H, H8, 11a), 8.40 (s, 0.70H, H8, 11b), 8.27 (s, 0.30H, H2, 11a), 8.21 (s, 0.70H, H2, 11b), 6.17 (d, J = 5.7 Hz, 0.30H, H1′, 11a), 6.00 (broad, 0.40H, 5′-OH), 5.92 (d, J = 7.8 Hz, 0.70H, H1′, 11b), 5.74 (t, J = 5.5 Hz, 0.30H, H2′, 11a), 5.58 (d, J = 5.2 Hz, 0.70H, H3′, 11b), 5.14 (dd, J = 7.9, 5.3 Hz, 0.70H, H2′, 11b), 4.85 (t, J = 4.3 Hz, 0.30H, H3′, 11a), 4.64 (s, 0.60H, OH), 4.49–4.38 (m, 2H, −OCH2–, 11a+11b), 4.35 (s, 0.70H, H-4′, 11b), 4.31 (q, J = 2.4 Hz, 0.30H, H4′, 11a), 4.07–3.74 (m, 2H, H5′, 11a+11b), 2.82 (t, J = 6.1 Hz, 2H, −CH2CN, 11a+11b), 2.21 (s, 2.10H, acetyl CH3, 11b), 2.08 (s, 0.90H, acetyl CH3, 11a); 13C NMR (CDCl3, 101 MHz) δ 170.7 (acetyl C=O, 11b), 170.1 (acetyl C=O, 11a), 152.5 (C8, 11a), 151.9 (C8, 11b), 150.8 (C=O, 11b), 150.7 (C=O, 11a), 150.3 (C4, 11a+11b), 149.8 (C6, 11b), 149.6 (C6, 11a), 143.7 (C2, 11b), 143.1 (C2, 11a), 123.4 (C5, 11b), 123.0 (C5, 11a), 117.5 (CN, 11b), 117.4 (CN, 11a), 90.8 (C1′, 11b), 88.3 (C1′, 11a), 87.0 (C4′, 11a), 85.8 (C4′, 11b), 75.8 (C2′, 11a), 74.5 (C3′, 11b), 72.7 (C2′, 11b), 70.1 (C3′, 11a), 62.9 (C5′, 11b), 62.0 (C5′, 11a), 60.4 (−OCH2–, 11b), 60.4 (−OCH2–, 11a), 21.1 (acetyl CH3, 11b), 20.8 (acetyl CH3, 11a), 18.4 (−CH2CN, 11a+11b); HRMS (ESI-TOF) m/z [M + H]+ calcd for C16H19N6O7 407.1315, found 407.1310.
N4-[(2-Cyanoethyloxy)carbonyl]-2′/3′-O-acetylcytidine (12a and 12b)
Compound 12a/b (6.46 g, 82% yield, 12b:12a in a ratio of ca. 2.5:1) was obtained as a regioisomeric mixture from 9 after following the same reaction and workup procedures as for 11a/b: 1H NMR (400 MHz, DMSO-d6) δ 10.96 (s, 1H, NH), 8.39–8.34 (m, 1H, H6, 12a+12b), 7.01 (s, 1H, H5, 12a+12b), 5.95 (d, J = 3.9 Hz, 0.30H, H1′, 12a), 5.86 (d, J = 4.9 Hz, 0.70H, H1′, 12b), 5.79 (d, J = 5.7 Hz, 0.70H, 2′-OH, 12b), 5.50 (d, J = 5.6, 0.30H, 3′-OH, 12a), 5.39, 5.19 (2 × broad, 1H, 5′-OH, 12a+12b), 5.17 (t, J = 4.5 Hz, 0.30H, H2′, 12a), 5.02 (t, J = 5.0 Hz, 0.70H, H3′, 12b), 4.30 (m, 3H, H2′ and −OCH2−), 4.22 (q, J = 5.4 Hz, 0.30H, H3′, 12a), 4.13–4.10 (m, 0.70H, H4′, 12b), 3.92 (dt, J = 5.6, 2.7 Hz, 0.70H, H4′, 12a), 3.76–3.58 (m, 2H, H5′, 12a+12b), 2.93 (t, J = 6.0 Hz, 2H, −CH2CN, 12a+12b), 2.08 (s, 3H, acetyl CH3, 12a+12b); 13C NMR (101 MHz, DMSO-d6) δ 169.8 (acetyl C=O, 12b), 169.3 (acetyl C=O, 12a), 162.9, 162.8 (C4, 12a+12b), 154.5, 154.2 (C2, 12a+12b), 152.9 (C=O, 12a+12b), 145.0, 145.0 (C6, 12a+12b), 118.4 (CN, 12a+12b), 94.9, 94.8 (C5, 12a+12b), 89.6 (C1′, 12b), 87.7 (C1′, 12a), 85.0 (C4′, 12a), 82.4 (C4′, 12b), 75.8 (C2′, 12a), 72.6 (C2′, 12b), 71.9 (C3′, 12b), 67.8 (C3′, 12a), 60.3, 60.2, 60.0 (C5′ and −OCH2–, 12a+12b), 20.8, 20.7 (acetyl CH3, 12a+12b), 17.6 (−CH2CN, 12a+12b); HRMS (ESI-TOF) m/z [M + H]+ calcd for C15H19N4O8 383.1203, found 383.1213.
N2-[(2-Cyanoethoxy)carbonyl]-O6-[2-(4-nitrophenyl)ethyl]-2′/3′-O-acetyl-guanosine (13a and 13b)
Compound 13a/b (3.23 g, 99% yield, 13b: 13a in a ratio of ca. 1.5:1) was obtained as a regioisomeric mixture from 5 after following the same reaction and workup procedures as for 11a/b: 1H NMR (400 MHz, DMSO-d6) δ 10.58–10.57 (m, 1H, NH, 13a+13b), 8.46–8.45 (m, 1H, H-8, 13a+13b), 8.18, 7.66 (d, J = 8.6 Hz, 4H, aromatic, 13a+13b), 6.14 (d, J = 5.9 Hz, 0.40H, H1′, 13a), 5.89 (d, J = 7.0 Hz, 0.60H, H1′, 13b), 5.81 (d, J = 6.1 Hz, 0.60H, 2′-OH, 13b), 5.59–5.55 (m, 0.80H, H2′ and 3′-OH, 13a), 5.31 (dd, J = 5.4, 2.3 Hz, 0.60H, H3′, 13b), 5.12 (t, J = 5.6 Hz, 0.60H, 5′-OH, 13b), 5.02 (t, J = 5.4 Hz, 0.40H, 5′-OH, 13a), 4.91 (q, J = 6.2 Hz, 0.6H, H-2′, 13b), 4.82–4.77 (m, 2H, −OCH2–, 13a+13b), 4.53 (td, J = 5.3, 3.7 Hz, 0.40H, H3′, 13a), 4.31 (t, J = 6.0 Hz, 2H, −COOCH2–, 13a+13b), 4.10 (q, J = 4.1 Hz, 0.60H, H4′, 13b), 3.97 (q, J = 4.1 Hz, 0.40H, H4′, 13a), 3.73–3.56 (m, 2H, H5′, 13a+13b), 3.34–3.31 (m, 2H, −CH2ph, 13a+13b), 2.95 (t, J = 6.0 Hz, 2H, −CH2CN, 13a+13b), 2.12 (s, 1.80H, acetyl CH3, 13b), 2.03 (s, 1.20H, acetyl CH3, 13a); 13C NMR (101 MHz, DMSO-d6) δ 169.6 (acetyl CO, 13a+13b), 159.8 (C6, 13a+13b), 153.2 (C4, 13b), 152.9 (C4, 13a), 152.0 (C2, 13a+13b), 151.5 (C=O, 13a+13b), 146.5, 146.4, 146.3 (aromatic), 141.4 (C8, 13a), 141.2 (C8, 13b), 130.4, 123.4 (aromatic), 118.6 (CN, 13a+13b), 117.3 (C5, 13a+13b), 86.9 (C1′, 13b), 86.1 (C4′, 13a), 84.8 (C1′, 13a), 83.5 (C4′, 13b), 75.3 (C2′, 13a), 73.2 (C3′, 13b), 71.8 (C2′, 13b), 68.8 (C3′, 13a), 66.5 (−OCH2–, 13a+13b), 61.3, 61.3 (C5′, 13a+13b), 59.5, 59.3 (−COOCH2–, 13a+13b), 34.3, 34.2 (−CH2ph, 13a+13b), 20.8 (acetyl CH3, 13b), 20.6 (acetyl CH3, 13a), 17.8, 17.7 (−CH2CN, 13a+13b); HRMS (ESI-TOF) m/z [M + H]+ calcd for C24H26N7O10 572.1741, found 572.1757.
N6-[(2-Cyanoethoxy)carbonyl]-2′/3′-O-acetyl-5′-O-(4,4′-dimethoxytrityl)adenosine (14a and 14b)
Compound 13a/b (1.50 g, 3.69 mmol) was coevaporated with anhydrous pyridine (3 × 20 mL). The residue was taken up in anhydrous pyridine (35 mL). DMTr-Cl (2.50 g, 7.38 mmol) was added and the mixture stirred for 3 h. MeOH (20 mL) was added and the mixture stirred for 10 min. The solvent was removed under vacuum, and the residue was taken up in CH2Cl2 (30 mL). The organics were washed with saturated aq NaHCO3 (3 × 50 mL). The organics were separated, dried over Na2SO4, and evaporated to dryness. The residue was coevaporated with toluene (3 × 20 mL), followed by CH2Cl2 (3 × 20 mL), after which point the residue was purified by flash column chromatography (0–5% MeOH in 50% toluene in EtOAc with 1% Et3N) to give a regioisomeric mixture (1.64 g, 63%) (note: 14b:14a in a ratio of ca. 3.2:1, calculated by integrations of both H-1′ in 1H NMR): 1H NMR (400 MHz, CDCl3) δ 9.75, 9.63 (2 × s, 1H, NH, 14a+14b), 8.69, 8.67 (2 × s, 1H, H8, 14a+14b), 8.26, 8.24 (2 × s, 1H, H2, 14a+14b), 7.39–6.74 (m, 13H, aromatic, 14a+14b), 6.27 (d, J = 4.5 Hz, 0.24H, H1′, 14a), 6.10 (d, J = 6.5 Hz, 0.76H, H1′, 14b), 5.87 (t, J = 4.9 Hz, 0.24H, H2′, 14a), 5.47 (dd, J = 5.4, 2.4 Hz, 0.76H, H3′, 14b), 5.14 (t, J = 6.0 Hz, 0.76H, H2′, 14b), 4.88 (t, J = 5.2 Hz, 0.24H, H3′, 14a), 4.40–4.33 (m, 2.76H, −OCH2– and H-4′), 4.27 (q, J = 3.9 Hz, 0.24H, H4′, 14a), 3.74–3.73 (m, 6H,-OCH3, 14a+14b), 3.56–3.35 (m, 2H, H5′, 14a+14b), 2.72–2.68 (m, 2H, −CH2CN, 14a+14b), 2.15 (s, 2.28H, acetyl CH3, 14b), 2.08 (s, 0.72H, acetyl CH3, 14a); 13C NMR (CDCl3, 101 MHz) δ 170.5 (acetyl CH3, 14b), 170.2 (acetyl CH3, 14a), 158.6 (aromatic, 14a+14b), 152.9 (C8, 14a), 152.6 (C8, 14b), 151.3 (C4, 14a+14b), 150.6 (−OCH2–, 14a+14b), 149.4 (C6, 14b), 149.3 (C6, 14a), 144.4, 144.3 (aromatic, 14a+14b), 142.1 (C2, 14a), 141.8 (C2, 14b), 135.6, 135.5, 135.4, 135.4, 130.1, 130.1, 128.2, 128.1, 128.0, 127.1 (aromatic, 14a+14b), 122.5 (C5, 14a+14b), 117.0 (CN, 14a+14b), 113.3 (aromatic, 14a+14b), 89.2 (C1′, 14b), 86.9 (DMTr-C, 14b), 86.7 (DMTr-C, 14a), 86.6 (C1′, 14a), 83.9 (C4′, 14a), 83.4 (C4′, 14b), 75.8 (C2′, 14a), 73.8, 73.7 (C2′, C3′, 14b), 70.2 (C3′, 14a), 63.3 (C5′, 14a), 63.0 (C5′, 14b), 60.1 (−OCH2–, 14a+14b), 55.3 (−OCH3, 14a+14b), 21.0 (acetyl CH3, 14b), 20.8 (acetyl CH3, 14a), 18.2 (−CH2CN, 14a+14b); HRMS (ESI-TOF) m/z [M + H]+ calcd for C37H37N6O9 709.2610, found 709.2617.
N4-[(2-Cyanoethyloxy)carbonyl]-2′/3′-O-acetyl-5′-O-(4,4′-dimethoxytrityl)cytidine (15a and 15b)
Compound 15a/b (11 g, 96% yield, 15b:15a in a ratio of ca. 3:1) was obtained as a regioisomeric mixture from 12a/b after following the same reaction and workup procedures as for 14a/b: 1H NMR (400 MHz, CDCl3) δ 8.34 (d, J = 7.5 Hz, 1H, H6, 15a+15b), 7.43–7.21 (m, 9H, aromatic, 15a+15b), 7.03 (s, 1H, H5, 15a+15b), 6.86–6.83 (m, 4H, aromatic, 15a+15b), 6.05 (d, J = 1.7 Hz, 0.25H, H1′, 15a), 5.92 (d, J = 2.7 Hz, 0.75H, H1′, 15b), 5.47 (dd, J = 4.8, 1.9 Hz, 0.25H, H2′, 15a), 5.21 (t, J = 5.7 Hz, 0.75H, H3′, 15b), 4.67–4.63 (m, 1H, H3′, 15a and H2′, 15b), 4.33 (m, 2.75H, H4′, 15b and −OCH2–, 15a+15b), 4.20–4.16 (m, 0.25H, H4′, 15a), 3.79–3.78 (m, 6H,-OCH3, 15a+15b), 3.61–3.49 (m, 1.25H, H5′, 15a+15b), 3.39 (dd, J = 11.2, 2.7 Hz, 0.75H, H5″, 15b), 2.72 (m, 2H, −CH2CN, 15a+15b), 2.14 (s, 0.75H, acetyl CH3, 15a), 2.11 (s, 2.25H, acetyl CH3, 15b); 13C NMR (101 MHz, CDCl3) δ 170.6 (acetyl C=O, 15a), 170.4 (acetyl C=O, 15b), 162.9 (C4, 15a+15b), 158.8 (aromatic, 15a+15b), 155.9 (C2, 15a+15b), 152.1 (C=O, 15a+15b), 144.4, 144.2, 144.1 (C6 and aromatic, 15a+15b), 135.6, 135.4, 135.2 (aromatic, 15a+15b), 130.1, 130.1, 128.3, 128.1, 127.2 (aromatic, 15a+15b), 117.0, 116.9 (CN, 15a+15b), 113.4 (aromatic, 15a+15b), 95.6 (C5, 15a+15b), 92.7 (C1′, 15b), 89.0 (C1′, 15a), 87.3, 87.2 (DMTr-C, 15a+15b), 82.8 (C4′, 15a), 81.8 (C1′, 15b), 76.7 (C2′, 15a), 74.5 (C2′, 15b), 71.5 (C3′, 15b), 68.5 (C3′, 15a), 61.6 (C5′, 15b), 61.4 (C5′, 15a), 60.2 (−OCH2–, 15a+15b), 55.3 (−OCH3, 15a+15b), 20.8 (acetyl CH3, 15a+15b), 18.1 (−CH2CN, 15a+15b); HRMS (ESI-TOF) m/z [M + H]+ calcd for C36H37N4O10 685.2510, found 685.2528.
N2-[(2-Cyanoethoxy)carbonyl]-O6-[2-(4-nitrophenyl)ethyl]-2′/3′-O-acetyl-5′-O-(4,4′-dimethoxytrityl)guanosine (16a and 16b)
Compound 16a/b (3.63 g, 74% yield, 16b:16a in a ratio of ca. 5:1) was obtained as a regioisomeric mixture from 12a/b after following the same reaction and workup procedures as for 14a/b: 1H NMR (400 MHz, CDCl3) δ 8.17–8.13 (m, 2H, aromatic, 16a+16b), 8.08 (s, 0.83H, H8, 16b), 7.94 (s, 0.17H, H8, 16a), 7.64 (s, 1H, NH), 7.51–7.46 (m, 2H, aromatic, 16a+16b), 7.39–7.09 (m, 9H, aromatic, 16a+16b), 6.77–6.68 (m, 4H, aromatic, 16a+16b), 6.42 (s, 0.75H, 2′-OH, 16b), 6.09 (d, J = 4.0 Hz, 0.17H, H1′, 16a), 5.92–5.89 (m, 1H, H1′ and H2′), 5.48 (d, J = 5.5 Hz, 0.83H, H3′, 16b), 5.18 (t, J = 5.3 Hz, 0.83H, H2′, 16b), 5.11 (t, J = 5.6 Hz, 0.17H, H3′, 16a), 4.78 (t, J = 6.8 Hz, 2H, −OCH2–, 16a+16b), 4.47–4.32 (m, 2.83H, H4′ and −COOCH2−), 4.19 (q, J = 4.5 Hz, 0.17H, H4′, 16a), 3.75, 3.74 (2 × s, 6H, OCH3, 16a+16b), 3.48–3.23 (m, 4H, H5′ and −CH2ph, 16a+16b), 2.78 (t, J = 6.1 Hz, 1.67H, −CH2CN, 16b), 2.69–2.66 (m, 0.33H, −CH2CN, 16a), 2.18 (s, 2.50H, acetyl CH3, 16b), 2.15 (s, 0.50H, acetyl CH3, 16a); 13C NMR (CDCl3, 101 MHz) δ 170.5 (acetyl C=O, 16b), 170.2 (acetyl C=O, 16a), 161.0, 160.8 (C6, 16a+16b), 158.6 (aromatic), 152.7 (C4, 16a), 151.8 (C4, 16b), (C2, 16b), 151.4 (C2, 16a) 150.9, 150.4 (C=O, 16a+16b), 147.0, 145.8, 145.6, 144.6, 144.2 (aromatic), 141.0 (C8, 16a), 140.5 (C8, 16b), 135.8, 135.4, 135.3, 130.2, 130.1, 130.0, 128.3, 128.0, 127.9, 126.9, 123.9 (aromatic), 118.5 (C5, 16a+16b), 116.8 (CN, 16a+16b), 113.2 (aromatic), 91.6 (C1′, 16b), 86.9 (DMTr-C), 86.6 (C1′, 16a), 85.4 (C4′, 16b), 83.7 (C4′, 16a), 75.8 (C2′, 16a), 75.2, 75.1 (C2′, C3′, 16b), 70.1 (C3′, 16a), 67.2 (−OCH2–, 16a+16b), 63.7 (C5′, 16b), 63.3 (C5′, 16a), 60.1, 59.7 (−COOCH2–, 16a+16b), 55.3 (OCH3), 35.1 (−CH2ph, 16a+16b), 21.2, 20.8 (acetyl CH3, 16a+16b), 18.4, 18.3 (−CH2CN, 16a+16b); HRMS (ESI-TOF) m/z [M + H]+ calcd for C45H44N7O12 874.3048; Found 874.3030.
N6-[(2-Cyanoethoxy)carbonyl]-5′-O-(4,4′-dimethoxytrityl)adenosine (23)
Compound 8 (0.92 g, 2.52 mmol) was coevaporated with anhydrous pyridine (3 × 20 mL). The residue was taken up in anhydrous pyridine (20 mL). DMTr-Cl (1.03 g, 3.4 mmol) was added, and the mixture was stirred for 3 h. The solvent was removed under vacuum, and the residue was taken up in CH2Cl2 (20 mL). The organic layer was washed with saturated aq NaHCO3 (3 × 30 mL) and dried over Na2SO4, and the solvent was removed under vacuum. The crude residue was coevaporated with toluene (3 × 20 mL), followed by CH2Cl2 (2 × 20 mL) to remove residual pyridine, and finally purified by flash column chromatography (75:25:2, EtOAc/Tol/Et3N → 40:2:2:2, EtOAc/Tol/MeOH/Et3N) to give the title compound as a slightly yellow foam (1.41 g, 83%): 1H NMR (400 MHz, CDCl3) δ 8.67 (s, 1H, H8), 8.28 (s, 1H, H2), 7.31–7.14 (m, 9H, aromatic), 6.74 (d, J = 8.8 Hz, 4H, aromatic), 6.11 (d, J = 5.5 Hz, 1H, H1′), 4.90 (t, J = 5.4 Hz, 1H, H-2′), 4.51 (dd, J = 5.0, 2.8 Hz, 1H, H3′), 4.42–4.36 (m, 3H, H4′ and −OCH2−), 3.74 (s, 6H, OCH3), 3.46 (ABX, JAB = 10.6, JAX = 3.3 Hz, 1H, H5′), 3.34 (ABX, JBA = 10.6, JBX = 3.8 Hz, 1H, H5″), 2.71 (t, J = 6.2 Hz, 2H, −CH2CN); 13C NMR (CDCl3, 101 MHz) δ 158.5 (aromatic), 152.3 (C8), 150.9 (C4), 150.3 (C=O), 149.1 (C6), 144.3 (aromatic), 141.8 (C2), 135.4, 135.4, 129.9, 127.9, 127.8, 126.9 (aromatic), 122.3 (C5), 116.8 (CN), 113.1 (aromatic), 90.0 (C1′), 86.5 (DMTr-C), 85.5 (C4′), 75.3 (C2′), 72.1 (C3′), 63.4 (C5′), 60.0 (−OCH2−), 55.2 (OCH3), 18.1 (−CH2CN); HRMS (ESI-TOF) m/z [M + H]+ calcd for C35H35N6O8 667.2511, found 667.2533.
N4-[(2-Cyanoethyloxy)carbonyl]-5′-O-(4,4′-dimethoxytrityl)cytidine (24)
Compound 24 (15 g, 89% yield) was obtained from 9 g of 9 after following the same reaction and workup procedure as for 23: 1H NMR (400 MHz, CDCl3) δ 8.32 (d, J = 7.5 Hz, 1H, H6), 7.39–7.16 (m, 9H, aromatic), 6.97 (s, 1H, H5), 6.82 (dd, J = 9.0, 2.3 Hz, 4H, aromatic), 5.90 (d, J = 1.8 Hz, 1H, H1′), 4.48–4.24 (m, 5H, H2′, H3′, H4′, and −OCH2−), 3.77 (s, 6H, OCH3), 3.52–3.39 (m, 2H, H5′), 2.74 (t, J = 6.3 Hz, 2H, −CH2CN); 13C NMR (CDCl3, 100 MHz) δ 162.8 (C4), 158.7, 158.7 (aromatic), 156.3 (C2), 152.0 (C=O), 144.8 (C6), 144.2, 135.6, 135.3, 130.2, 130.1, 128.2, 128.1, 127.2 (aromatic), 116.8 (CN), 113.4 (aromatic), 95.6 (C5), 92.9 (C1′), 87.1 (DMTr-C), 84.9, 76.5, 70.7 (C2′, C3′, C4′), 62.3 (C5′), 60.3 (−OCH2−), 55.3 (OCH3), 18.1 (−CH2CN); HRMS (ESI-TOF) m/z [M + H]+ calcd for C34H34N4O9Na 665.2218, found 665.2233.
N2-[(2-Cyanoethoxy)carbonyl]-O6-[2-(4-nitrophenyl)ethyl]-5′-O-(4,4′-dimethoxytrityl)guanosine (25)
Compound 25 (4.27 g, 91% yield) was obtained from 2.97 g of 9 after following the same reaction and workup procedure as for 23: 1H NMR (400 MHz, CDCl3) δ 8.15–8.12 (m, 3H, H8 and aromatic), 7.81 (s, 1H, NH), 7.47 (d, J = 8.7 Hz, 2H, aromatic), 7.18–7.08 (m, 9H, aromatic), 6.99 (br s, 1H, 2′-OH), 6.69 (2 × d, J = 8.8 Hz, 4H, aromatic), 5.90 (d, J = 6.2 Hz, 1H, H1′), 4.93 (t, J = 5.7 Hz, 1H, H2′), 4.79 (t, J = 6.7 Hz, 2H, −OCH2−), 4.49–4.38 (m, 4H, H3′, H4′, −COOCH2−), 3.74 (s, 6H, OCH3), 3.38 (ABX, JAB = 10.6, JAX = 3.2 Hz, 1H, H5′), 3.29 (t, J = 6.8 Hz, 2H, −CH2ph), 3.17 (ABX, JBA = 10.6, JBX = 3.2 Hz, 1H, H5″), 2.77 (t, J = 6.1 Hz, 2H, −CH2CN); 1H NMR (100 MHz, CDCl3) δ 161.0 (C6), 158.6 (aromatic), 151.8 (C4), 151.1 (C2), 150.7 (C=O), 147.0, 145.6, 144.3 (aromatic), 140.3 (C8), 135.5, 135.3, 130.0, 128.0, 127.8, 126.9, 123.9 (aromatic), 118.6 (C5), 116.7 (CN), 113.2 (aromatic), 92.1 (C1′), 87.1 (C4′), 86.7 (DMTr-C), 76.7 (C2′) 74.0 (C3′), 67.2 (−OCH2−), 63.9 (C5′), 60.1 (−COOCH2−), 55.3 (OCH3), 35.1 (−CH2ph), 18.4 (−CH2CN); HRMS (ESI-TOF) m/z [M + H]+ calcd for C43H42N7O11 832.2942, found 832.2941.
N6-[(2-Cyanoethoxy)carbonyl]-2′-O-(tert-butyldimethylsilyl)-5′-O-(4,4′-dimethoxytrityl)adenosine (26a) and N6-[(2-Cyanoethoxy)carbonyl]-3′-O-(tert-butyldimethylsilyl)-5′-O-(4,4′-dimethoxytrityl)adenosine (26b)
Compound 23 (4.00 g, 6.00 mmol) was dissolved in anhydrous THF (50 mL). Anhydrous pyridine (1.80 mL, 22.2 mmol) and AgNO3 (1.22 g, 7.20 mmol) were added. The mixture was warmed until the AgNO3 was fully dissolved. While the mixture was still warm, TBDMS-Cl (1.18 g, 7.80 mmol) was added, resulting in a colorless precipitate. The mixture was stirred in the dark for 5 h. The solid was removed by filtration and the supernatant immediately filtered into saturated aq NaHCO3 (50 mL). The aqueous phase was extracted with EtOAc (3 × 50 mL). The combined organic phases were dried over MgSO4. Finally, the solvent was removed under vacuum. The crude residue was purified, and the regioisomers were separated by flash column chromatography (3:1 → 2:1 → 1:1, Et2O/EtOAc) to give 26a (2.50 g, 53%) and 26b (0.91 g, 20%) both as colorless foams. 26a: 1H NMR (400 MHz, DMSO) δ 10.80 (s, 1H, NH), 8.58, 8.57 (2 × s, 2H, H2 and H8), 7.43–7.35 (m, 2H, aromatic), 7.31–7.16 (m, 7H, aromatic), 6.85 (dd, J = 9.0, 3.1 Hz, 4H, aromatic), 6.05 (d, J = 4.8 Hz, 1H, H-1′), 5.18 (d, J = 5.9 Hz, 1H, 3′-OH), 4.86 (t, J = 4.9 Hz, 1H, H2′), 4.29 (m, 3H, −OCH2– and H3′), 4.13 (q, J = 4.5 Hz, 1H, H4′), 3.73 (s, 6H, OCH3), 3.29 (m, 2H, H5′), 2.93 (t, J = 6.0 Hz, 2H, −CH2CN), 0.75 (s, 9H, SiC(CH3)3), −0.04 (s, 3H, Si(CH3)2), −0.14 (s, 3H, Si(CH3)2); 13C NMR (DMSO, 101 MHz) δ 158.0 (aromatic), 151.6, 151.6 (C8, C=O), 149.6 (C4, C6), 144.8 (aromatic), 142.9 (C2), 135.5, 135.4, 129.7, 127.8, 127.6, 126.7 (aromatic), 124.0 (C5), 118.5 (CN), 113.1 (aromatic), 88.2 (C1′), 85.5 (DMTr-C), 83.5 (C4′), 74.8 (C2′), 70.1 (C3′), 63.4 (C5′), 59.9 (−OCH2−), 55.0, 54.9 (OCH3), 25.5 (SiC(CH3)3), 17.8, 17.6 (−CH2CN, SiC(CH3)3), −4.8 (Si(CH3)2), −5.3 (Si(CH3)2); HRMS (ESI-TOF) m/z [M + H]+ calcd for C41H49N6O8Si 781.3381, found 781.3354. 26b: 1H NMR (400 MHz, DMSO) δ 10.81 (s, 1H, NH), 8.63 (s, 1H, H2), 8.56 (s, 1H, H8), 7.39–7.17 (m, 9H, aromatic), 6.88–6.79 (m, 4H, aromatic), 6.00 (d, J = 5.1 Hz, 1H, H1′), 5.47 (d, J = 6.0 Hz, 1H, 2′-OH), 4.88 (q, J = 5.4 Hz, 1H, H2′), 4.49 (t, J = 4.6 Hz, 1H, H3′), 4.32 (t, J = 6.0 Hz, 2H, −OCH2−), 4.06 (q, J = 4.5 Hz, 1H, H4′), 3.72 (s, 6H, OCH3), 3.36 (m, 1H, H5′), 3.15 (ABX, JBA = 10.5, JBX = 4.9 Hz, 1H, H5″), 2.93 (t, J = 6.0 Hz, 2H, −CH2CN), 0.84 (s, 9H, Si(C(CH3)3), 0.08 (s, 3H, Si(CH3)2), 0.05 (s, 3H, Si(CH3)2); 13C NMR (DMSO, 101 MHz) δ = 158.1 (aromatic), 151.7, 151.6, 151.5 (C=O, C8, C4), 149.6 (C6), 144.7 (aromatic), 143.7 (C2), 135.4, 129.6, 129.6, 127.7, 127.6, 126.7 (aromatic), 124.2 (C5), 118.5 (CN), 113.1 (aromatic), 88.3 (C1′), 85.6 (DMTr-C), 83.6 (C4′), 72.2 (C3′), 72.0 (C2′), 63.0 (C5′), 59.9 (−OCH2−), 55.0 (OCH3), 25.8 (SiC(CH3)3), 18.0 (−CH2CN), 17.6 (SiC(CH3)3), −4.5 (Si(CH3)2), −5.1 (Si(CH3)2); HRMS (ESI-TOF) m/z [M + H]+ calcd for C41H49N6O8Si 781.3381, found 781.3380.
N4-[(2-Cyanoethyloxy)carbonyl]-2′-O-(tert-butyldimethylsilyl)-5′-O-(4,4′-dimethoxytrityl)cytidine (27a) and N4-[(2-Cyanoethyloxy)carbonyl]-3′-O-(tert-butyldimethylsilyl)-5′-O-(4,4′-dimethoxytrityl)cytidine (27b)
Compounds 27a (8.12 g, 46%) and 27b (4.26 g, 24%) were obtained as separated isomers from 15 g of 24 after following the same reaction and workup procedures as for 26a/b. 27a: 1H NMR (400 MHz, CDCl3) δ 9.10 (s, 1H, NH), 8.52 (s, 1H, H6), 7.49–7.21 (m, 9H, aromatic), 6.86 (m, 5H, aromatic and H5), 5.89 (s, 1H, H1′), 4.45–4.25 (m, 4H, H2′, H3′ and −OCH2−), 4.10 (d, J = 7.9 Hz, 1H, H–C4′), 3.80 (s, 6H, OCH3), 3.65–3.50 (m, 2H, H5′), 2.83–2.67 (m, 2H, −CH2CN), 2.43 (d, J = 8.1 Hz, 1H, 3′-OH), 0.93 (s, 9H, SiC(CH3)3), 0.31 (s, 3H, Si(CH3)2), 0.19 (s, 3H, Si(CH3)2); 13C NMR (CDCl3, 100 MHz) δ 162.6 (C4), 158.8 (aromatic), 154.7 (C2), 151.9 (C=O), 145.1 (C6), 144.2, 135.6, 135.3, 130.2, 130.2, 128.3, 128.1, 127.2 (aromatic), 116.6 (CN), 113.4 (aromaic), 94.8 (C5), 90.8 (C1′), 87.2 (aromatic), 83.1 (C4′), 76.6, 69.1 (C2′ and C3′), 61.4 (C5′), 60.1 (−OCH2−), 55.3 (OCH3), 25.9 (SiC(CH3)3), 18.1 (SiC(CH3)3 and -CH2CN), −4.2 (Si(CH3)2), −5.3 (Si(CH3)2); HRMS (ESI-TOF) m/z [M + H]+ calcd for C40H49N4O9Si 757.3269, found 757.3281.
N2-[(2-Cyanoethoxy)carbonyl]-O6-[2-(4-nitrophenyl)ethyl]-2′-O-(tert-butyldimethylsilyl)-5′-O-(4,4′-dimethoxytrityl)guanosine (28a) and N2-[(2-Cyanoethoxy)carbonyl]-O6-[2-(4-nitrophenyl)ethyl]-3′-O-(tert-butyldimethylsilyl)-5′-O-(4,4′-dimethoxytrityl)guanosine (28b)
The separated regioisomers 28a (1.64 g, 44%) and 28b (1.29 g, 35%) were obtained as colorless foams from 3.2 g of 25 after following the same reaction and workup procedures as for 26a/b. 28a: 1H NMR (400 MHz, CDCl3) δ 8.16 (d, J = 8.6 Hz, 2H, aromatic), 7.99 (s, 1H, H8), 7.53 (d, J = 8.6 Hz, 2H, aromatic), 7.44 (d, J = 7.0 Hz, 2H, aromatic), 7.33 (2 × d, J = 8.9 Hz, 4H, aromatic), 7.25–7.17 (m, 3H, aromatic), 6.78 (2 × d, J = 8.7 Hz, 4H, aromatic), 5.93 (d, J = 5.7 Hz, 1H, H1′), 5.03 (t, J = 5.4 Hz, 1H, H2′), 4.84 (t, J = 6.6 Hz, 2H, −OCH2−), 4.42 (q, J = 3.5 Hz, 1H, H3′), 4.30 (t, J = 6.2 Hz, 2H, −COOCH2−), 4.23 (q, J = 3.1 Hz, 1H, H4′), 3.77 (m, 6H, OCH3), 3.50 (ABX, JAB= 10.6, JAX= 2.6 Hz, 1H, H5′), 3.40 - 3.32 (m, 3H, H5″ and −CH2ph), 2.72 (d, J = 3.7 Hz, 1H, 3′-OH), 2.66 (t, J = 6.2 Hz, 2H, −CH2CN), 0.84 (s, 9H, SiC(CH3)3), 0.00 (s, 3H, Si(CH3)2), −0.18 (s, 3H, Si(CH3)2); 13C NMR (100 MHz, CDCl3) δ 160.9 (C6), 158.7 (aromatic), 153.0 (C4), 151.5 (C2), 150.4 (C=O), 147.0, 145.8, 144.8 (aromatic), 141.0 (C8), 135.9, 135.8, 130.2, 128.2, 128.0, 127.1, 123.9 (aromatic), 118.8 (C5), 116.8 (CN), 113.3 (aromatic), 88.5 (C1′), 86.7 (DMTr-C), 84.5 (C4′), 75.4 (C2′), 71.6 (C3′), 67.1 (−OCH2−), 63.8 (C5′), 59.6 (−COOCH2−), 55.4 (2 × OCH3), 35.2 (−CH2ph), 25.7 (SiC(CH3)3), 18.3 (−CH2CN), 18.0 (SiC(CH3)3), −4.9, −5.0 (Si(CH3)2); HRMS (ESI-TOF) m/z [M + H]+ calcd for C49H56N7O11Si 946.3807, found 946.3785. 28b: 1H NMR (400 MHz, CDCl3) δ 8.15 (d, J = 8.7 Hz, 2H, aromatic), 8.05 (s, 1H, H8), 7.50 (d, J = 8.7 Hz, 2H, aromatic), 7.46 (s, 1H, NH), 7.34–7.32 (m, 2H, aromatic), 7.24–7.14 (m, 7H, aromatic), 6.74 (d, J = 8.6 Hz, 4H, aromatic), 5.93 (d, J = 5.2 Hz, 1H, H1′), 4.80 (t, J = 6.8 Hz, 2H, −OCH2−), 4.69 (q, J = 5.3 Hz, 1H, H2′), 4.56 (dd, J = 5.2, 3.3 Hz, 1H, H3′), 4.42–4.33 (m, 2H, −COOCH2−), 4.23–4.15 (m, 2H, H4′, 2′-OH), 3.76 (s, 6H, OCH3), 3.43 - 3.21 (m, 4H, H5′ and −CH2ph), 2.75 (t, J = 6.2 Hz, 2H, −CH2CN), 0.89 (s, 9H, SiC(CH3)3), 0.11 (s, 3H, Si(CH3)2), 0.04 (s, 3H, Si(CH3)2); 13C NMR (101 MHz, CDCl3) δ 160.8 (C6), 158.7 (aromatic), 152.5 (C4), 151.1 (C2), 150.5 (C=O), 147.0, 145.8, 144.5 (aromatic), 140.9 (C8), 135.8, 135.7, 130.1, 128.2, 127.9, 127.0, 123.9 (aromatic), 118.8 (C5), 116.8 (CN), 113.2 (aromatic), 90.3 (C1′), 86.7 (DMTr-C), 85.9 (C4′), 75.2 (C2′), 73.1 (C3′), 67.1 (−OCH2−), 63.5 (C5′), 59.8 (−COOCH2−), 55.3 (OCH3), 35.2 (−CH2Ar), 25.9 (SiC(CH3)3), 18.37 (−CH2CN), 18.27 (SiC(CH3)3), −4.56, −4.71 (Si(CH3)2); HRMS (ESI-TOF) m/z [M + H]+ calcd for C49H56N7O11Si 946.3807, found 946.3787.
General Procedure for the Synthesis of 2′/3′-O-Acetylated RNA Phosphoramidites
To a solution of substrate (1 equiv) and 2-cyanoethyl N,N,N′,N′-tetraisopropylphosphoramidite 18 (2 equiv) in dry THF was added a solution of 5-benzylthio-1H-tetrazole (BTT) in anhydrous MeCN (0.35 M, 1 equiv) dropwise. The reaction mixture was stirred at rt for 3 h, after which saturated aq NaHCO3 was added to quench the reaction. The organics were extracted with EtOAc (3 × 10 mL), combined, and dried over MgSO4. The solvent was removed, and the residue was applied to a short flash chromatography column. The regioisomeric mixture thus obtained was dissolved in DCM (∼200 mg/mL), purified, and separated by normal-phase HPLC.
General Procedure for the Synthesis of 2′/3′-O-TBS RNA Phosphoramidites
To a solution of substrate (1 equiv) in anhydrous THF was added N,N-diisopropylethylamine (3.5 equiv) and 2-cyanoethyl N,N-diisopropyl phosphoamidochloridite 17 (1.4 equiv) at 0 °C. The mixture was warmed to rt and stirred for 5 h. Anhydrous methanol was added to quench the reaction, and the mixture was stirred for a further 30 min. The reaction was diluted with EtOAc and washed with saturated aq NaHCO3 (3 × 20 mL). The combined organic layers were dried over MgSO4, and the solvent was evaporated under vacuum. The crude product was purified by flash column chromatography to give a mixture of two diastereoisomers.
NMR and MS Data for Phosphoramidites
N6-[(2-Cyanoethoxy)carbonyl]-2′-O-acetyl-5′-O-(4,4′-dimethoxyltrityl)adenosine-3′-O-(2-cyanoethyl-N,N-diisopropyl)phosphoramidite (19a):
1H NMR (400 MHz, CDCl3) δ 8.71, 8.69 (2 × s, 2H, H8, NH), 8.19 (s, 1H, H2), 7.49–7.12 (m, 9H, aromatic), 6.79 (dd, J = 8.6, 4.8 Hz, 4H, aromatic), 6.29 (2 × d, J = 5.9 Hz, 1H, H1′), 6.00 (t, J = 5.4 Hz, 0.5H, H2′), 5.90 (t, J = 5.7 Hz, 0.5H, H2′), 5.01–4.78 (m, 1H, H3′), 4.52–4.38 (m, 2.5H, −OCH2–, H4′), 4.35 (q, J = 3.6 Hz, 0.5H, H4′), 3.98–3.46 (m, 11H, −OCH2–, OCH3, iPr CH, H5′), 3.38 (ABX, JBA = 10.7 Hz, JBX = 4.0 Hz, 1H, H5″), 2.79 (t, J = 6.3 Hz, 2H, −CH2CN), 2.64 (t, J = 6.4 Hz, 1H, −CH2CN), 2.36 (m, 1H, −CH2CN), 2.12, 2.08 (2 × s, 3H, acetyl CH3), 1.23–1.00 (m, 12H, iPr CH3); 13C NMR (CDCl3, 101 MHz) δ 169.9 (acetyl C=O), 158.7 (aromatic), 153.0 (C8), 151.6 (C4), 150.3 (C=O), 149.1 (C6), 144.4, 144.3 (aromatic), 141.9 (C2), 135.6, 135.5, 135.4, 130.3, 130.2, 128.4, 128.2, 128.0, 127.2, 127.1 (aromatic), 122.6 (C5), 117.7, 117.4 (CN), 116.8 (CN), 113.3 (aromatic), 86.9 (DMTr-C), 86.1 (C1′), 84.7, 84.4 (C4′), 74.8, 74.7 (C2′), 71.5, 71.3, 71.0, 70.8 (C3′), 63.0 (C5′), 60.3 (−OCH2−), 59.0, 58.8, 58.3, 58.1 (−OCH2−), 55.4 (OCH3), 43.5, 43.4, 43.3 (iPr CH), 24.9, 24.8, 24.7, 24.6 (iPr CH3), 21.0, 20.9 (acetyl CH3), 20.3, 20.2 (−CH2CN), 18.3 (−CH2CN); 31P NMR (162 MHz, CDCl3) δ 151.17 (s), 150.22 (s); HRMS (ESI-TOF) m/z [M + H]+ calcd for C46H54N8O10P 909.3701, found 909.3665.
N6-[(2-cyanoethoxy)carbonyl]-3′-O-acetyl-5′-O-(4,4′-dimethoxyltrityl)adenosine-2′-O-(2-cyanoethyl-N,N-diisopropyl)phosphoramidite (19b):
1H NMR (400 MHz, CDCl3) δ 8.74, 8.71, 8.69 (3 × s, 2H, NH, H8), 8.26, 8.23 (2 × s, 1H, H2), 7.47–7.13 (m, 9H, aromatic), 6.88–6.73 (m, 4H, aromatic), 6.26 (d, J = 5.3 Hz, 0.5H, H1′), 6.22 (d, J = 5.7 Hz, 0.5H, H1′), 5.57, 5.52 (t, J = 4.4 Hz, 1H, H3′), 5.21, 5.15 (2 × dt, J = 10.4, 5.4 Hz, 1H, H2′), 4.46 (t, J = 6.2 Hz, 2H, −OCH2−), 4.33 (q, J = 3.7 Hz, 1H, H4′), 3.88–3.64 (m, 7H, OCH3, −OCH2−), 3.63–3.36 (m, 5H, H5′, −OCH2–, iPr CH), 2.80 (t, J = 6.2 Hz, 2H, −CH2CN), 2.55 (td, J = 6.4, 2.9 Hz, 1H, −CH2CN), 2.33 (t, J = 6.4 Hz, 1H, −CH2CN), 2.14, 2.10 (2 × s, 3H, acetyl CH3), 1.20–1.00 (m, 9H, iPr CH3), 0.88 (d, J = 6.7 Hz, 3H, iPr CH3); 13C NMR (CDCl3, 101 MHz) δ 169.9, 169.8 (acetyl C=O), 158.7 (aromatic), 153.0, 152.8 (C8), 151.7, 151.6 (C4), 150.4, 150.3 (C=O), 149.1, 149.0 (C6), 144.5 (aromatic), 142.0, 141.8 (C2), 135.6, 135.5, 130.2, 128.3, 128.2, 128.1, 128.0, 127.2 (aromatic), 122.6 (C5), 117.5, 117.4 (CN), 116.8 (CN), 113.4, 113.3 (aromatic), 87.7, 87.6, 87.5 (C1′), 87.0, 87.0 (DMTr-C), 82.5, 82.1 (C4′), 74.7, 74.5, 74.2, 74.0 (C2′), 72.2, 72.1 (C3′), 63.0 (C5′), 60.3 (−OCH2−), 58.8, 58.6, 58.1, 57.9 (−OCH2−), 55.4 (OCH3), 43.5, 43.4 (iPr CH), 24.8, 24.7, 24.6, 24.5, 24.4, 24.3 (iPr CH3), 21.1, 21.0 (acetyl CH3), 20.3, 20.2, 20.1, 20.0 (CH2CN), 18.3 (CH2CN); 31P NMR (162 MHz, CDCl3) δ 151.56 (s), 151.18 (s); HRMS (ESI-TOF) m/z [M + H]+ calcd for C46H54N8O10P 909.3701, found 909.3699.
N4-[(2-Cyanoethyloxy)carbonyl]-2′-O-acetyl-5′-O-(4,4′-dimethoxytrityl)cytidine-3′-O-(2-cyanoethyl-N,N-diisopropyl)phosphoramidite (20a):
1H NMR (400 MHz, CDCl3) δ 8.28 (s, 1H, H6), 7.51–7.21 (m, 9H, aromatic), 6.96–6.72 (m, 5H, aromatic and H5), 6.16 (2 × d, J = 2.9 Hz, 1H, H1′), 5.56 (dd, J = 4.9, 3.5 Hz, 0.44H, H2′), 5.52–5.45 (m, 0.56H, H2′), 4.72–4.58 (m, 1H, H3′), 4.37 (t, J = 6.4 Hz, 2H, −OCH2−), 4.32–4.22 (m, 1H, H4′), 3.92–3.35 (m, 12H, −OCH2–, OCH3, H5′ and iPr CH), 2.77 (m, 2H, −CH2CN), 2.64 (q, J = 6.4 Hz, 0.86H, −CH2CN), 2.36 (q, J = 6.0 Hz, 1.14H, −CH2CN), 2.14, 2.11 (2 × s, 3H, acetyl CH3), 1.34–1.00 (m, 12H, iPr CH3); 13C NMR (CDCl3, 101 MHz) δ 169.2, 169.1 (acetyl C=O), 162.9 (C4), 158.8 (aromatic), 154.8 (C2), 152.1 (C=O), 144.7 (C6), 144.1, 144.0, 135.5, 135.3, 135.1, 130.3, 128.5, 128.4, 128.1, 127.3 (aromatic), 117.9, 117.5, 117.1, 116.9, 116.8 (CN), 113.4 (aromatic), 95.4 (C5), 88.8 (C1′), 87.2 (DMTr-C), 83.2, 82.9 (C4′), 75.3, 74.8 (C2′), 69.8 (C3′), 61.4 (C5′), 60.1 (−OCH2−), 58.5, 58.3, 58.2, 58.0 (−OCH2−), 55.3 (OCH3), 45.7, 45.6, 45.4, 43.4, 43.3, 43.2 (iPr CH), 24.7, 24.6, 24.5, 23.2, 23.1, 23.1, 23.0 (iPr CH3), 21.1, 20.9 (acetyl CH3), 20.4, 20.3, 20.2, 20.1 (−CH2CN), 18.1 (−CH2CN); 31P NMR (162 MHz, CDCl3) δ 150.62 (s), 150.07 (s); HRMS (ESI-TOF) m/z [M + H]+ calcd for C45H54N6O11P 885.3588, found 885.3617.
N4-[(2-Cyanoethyloxy)carbonyl]-3′-O-acetyl-5′-O-(4,4′-dimethoxytrityl)cytidine-2′-O-(2-cyanoethyl-N,N-diisopropyl)phosphoramidite (20b):
1H NMR (400 MHz, CDCl3) δ 8.52 (s, 1H, H6), 8.13 (br s, 1H, NH), 7.42–7.21 (m, 9H, aromatic), 6.85 (m, 5H, H5 and aromatic), 6.14 (d, J = 1.8 Hz, 0.54H, H1′), 6.06 (s, 0.46H, H1′), 5.25–5.06 (m, 1H, H3′), 4.74–4.66 (m, 0.54H, H2′), 4.65–4.56 (m, 0.46H, H2′), 4.42–4.29 (m, 3H, −OCH2–, H4′), 4.09–3.50 (m, 11H, −OCH2–, OCH3, iPr CH and H5′), 3.41 (d, J = 11.4 Hz, 1H, H5″), 2.77 (t, J = 6.5 Hz, 2.50H, −CH2CN), 2.64 (dt, J = 16.6, 5.9 Hz, 1.50H, −CH2CN), 2.06 (m, 3H, acetyl CH3), 1.23–1.11 (m, 12H, iPr CH3); 13C NMR (CDCl3, 101 MHz) δ 169.9, 169.8 (acetyl C=O), 162.2 (C4), 158.8 (aromatic), 154.7 (C2), 151.6 (C=O), 144.9 (C6), 144.1, 135.4, 135.3, 130.2, 128.3, 128.2, 127.3 (aromatic), 118.2, 117.8 (−CH2CN), 116.5 (−CH2CN), 113.5 (aromatic), 94.9 (C5), 90.7, 90.3 (C1′), 87.5 (DMTr-C), 80.8, 80.7, 80.5 (C4′), 75.7, 75.5, 74.5, 74.4 (C2′), 69.9, 69.5, 69.4 (C3′), 61.0, 60.7 (C5′), 60.3 (−OCH2−), 59.0, 58.8, 58.6 (−OCH2−), 55.3 (OCH3), 43.6, 43.5 (iPr-CH), 24.8, 24.7, 24.6 (iPr-CH3), 20.9, 20.4, 20.3 (acetyl CH3), 18.2 (−CH2CN); 31P NMR (162 MHz, CDCl3) δ 152.32 (s), 150.29 (s); HRMS (ESI-TOF) m/z [M + H]+ calcd for C45H53N6O11NaP 907.3402, found 907.3374.
N2-[(2-Cyanoethoxy)carbonyl]-O6-[2-(4-nitrophenyl)ethyl]-2′-O-acetyl-5′-O-(4,4′-dimethoxytrityl)guanosine-3′-O-(2-cyanoethyl-N,N-diisopropyl)phosphoramidite (21a):
1H NMR (400 MHz, CDCl3) δ 8.16 (d, J = 8.6 Hz, 2H, aromatic), 7.96, 7.95 (2 × s, 1H, H8), 7.52 (2 × d, J = 8.6 Hz, 2H, aromatic), 7.47–7.15 (m, 9H, aromatic), 6.77 (d, J = 8.8 Hz, 4H, aromatic), 6.16 (d, J = 6.5 Hz, 0.40H, H1′), 6.09 (d, J = 6.2 Hz, 0.60H, H1′), 6.04 (t, J = 5.7 Hz, 0.45H, H2′), 5.95 (t, J = 5.9 Hz, 0.55H, H2′), 4.90–4.78 (m, 3H, H3′ and −OCH2−), 4.41–4.26 (m, 3H, H4′, −COOCH2−), 3.97–3.25 (m, 14H, −OCH2–, OCH3, iPr CH, H5′ and −CH2ph), 2.76–2.62 (m, 3H, −CH2CN), 2.31 (t, J = 6.4 Hz, 1H, −CH2CN), 2.09 (m, 3H, acetyl CH3), 1.21–1.02 (m, 12H, iPr CH3); 13C NMR (CDCl3, 101 MHz) δ 169.8 (acetyl CO), 160.8 (C6), 158.7 (aromartic) 153.1, 152.9 (C4), 151.6 (C2), 150.5, 150.4 (C=O), 147.0, 145.9, 144.5, 144.3 (aromatic), 141.0, 140.7 (C8), 135.8, 135.7, 135.6, 135.6, 130.3, 130.2, 128.5, 128.3, 128.0, 127.1, 123.9 (aromatic), 118.8, 118.7 (C5), 117.7, 117.4, 116.9 (CN), 113.3, 113.2 (aromatic), 86.8, 86.7 (DMTr-C), 86.3, 85.8 (C1′), 84.8, 84.5 (C4′), 74.3, 74.1, 74.0 (C2′), 71.4, 71.3, 70.9, 70.7 (C3′), 67.1 (−OCH2−), 63.4, 63.3 (C5′), 59.6, 59.5 (−COOCH2−), 59.0, 58.8, 58.1, 57.8 (−OCH2−), 55.4 (OCH3), 43.5, 43.4, 43.2 (iPr CH), 35.2 (−CH2ph), 24.8, 24.7, 24.6 (iPr CH3), 21.1, 20.9 (acetyl CH3), 20.5, 20.4, 20.2, 20.1, 18.3 (−CH2CN); 31P NMR (162 MHz, CDCl3) δ 150.90 (s), 150.28 (s); HRMS (ESI-TOF) m/z [M + H]+ calcd for C54H61N9O13P 1074.4126, found 1074.4131.
N2-[(2-Cyanoethoxy)carbonyl]-O6-[2-(4-nitrophenyl)ethyl]-3′-O-acetyl-5′-O-(4,4′-dimethoxytrityl)guanosine-2′-O-(2-cyanoethyl-N,N-diisopropyl)phosphoramidite (21b):
1H NMR (400 MHz, CDCl3) δ 8.17 (2 × d, J = 8.7 Hz, 2H, aromatic), 8.02 (s, 0.5H, H8), 7.97 (s, 0.5H, H8), 7.53 (d, J = 8.7 Hz, 2H, aromatic), 7.40–7.16 (m, 9H, aromatic), 6.79–6.74 (m, 4H, aromatic), 6.10 (d, J = 5.5 Hz, 0.5H, H1′), 6.02 (d, J = 5.9 Hz, 0.5H, H1′), 5.63–5.60 (m, 1H, H3′), 5.37 (dt, J = 10.9, 5.6 Hz, 0.5H, H2′), 5.19 (dt, J = 10.6, 5.5 Hz, 0.5H, H2′), 4.87–4.82 (m, 2H, −OCH2−), 4.34 (t, J = 6.2 Hz, 2H, −COOCH2−), 4.29–4.25 (m, 1H, H4′), 3.84–3.66 (m, 7H, OCH3, and −OCH2−), 3.58–3.41 (m, 5H, H5′, −OCH2–, and iPr CH), 3.34 (t, J = 6.9 Hz, 2H, −CH2CN), 2.75–2.70 (m, 2H, −CH2CN), 2.58 (t, J = 6.3 Hz, 1H, −CH2CN), 2.38–2.25 (m, 1H, −CH2CN), 2.14–2.14, 2.11 (2 × s, 3H, acetyl CH3), 1.14–1.08 (m, 9H, iPr CH3), 0.90 (d, J = 6.8 Hz, 3H, iPr CH3); 13C NMR (101 MHz, CDCl3) δ 169.9 (acetyl CO), 160.8 (C6), 158.7 (aromatic), 153.1, 152.9 (C4), 151.5 (C2), 150.4, 150.3 (C=O), 147.0, 145.8, 144.7, 144.6 (aromatic), 141.0, 140.9 (C8), 135.8, 135.7, 135.6, 130.2, 130.1, 128.2, 128.0, 127.9, 127.1, 127.0, 123.8 (aromatic), 118.8, 118.6 (C5), 117.6, 117.3 (CN), 116.9 (CN), 113.3, 113.2 (aromatic), 87.9, 87.8 (C1′), 86.8, 86.7 (DMTr-C), 82.6, 82.1 (C4′), 74.4, 74.2, 73.4, 73.2 (C2′), 72.4, 72.3 (C3′), 67.1 (−OCH2−), 63.4, 63.3 (C5′), 59.6 (−COOCH2−), 58.6, 58.4, 58.1, 58.0 (−OCH2−), 55.3 (OCH3), 43.5, 43.4, 43.3 (iPr CH), 35.2 (−CH2ph), 24.8, 24.7, 24.6, 24.4 (iPr CH3), 21.1, 21.0 (acetyl CH3), 20.3, 20.0, 19.9, 18.3 (−CH2CN); 31P NMR (162 MHz, CDCl3) δ 151.62, 151.01; HRMS (ESI-TOF) m/z [M + H]+ calcd for C54H61N9O13P 1074.4126, found 1074.4169.
2′-O-Acetyl-5′-O-(4,4′-dimethoxytrityl)uridine-3′-O-(2-cyanoethyl-N,N-diisopropyl)phosphoramidite (22a):
1H NMR (400 MHz, CDCl3) δ 8.28 (s, 1H, NH), 7.81–7.69 (m, 1H, H6), 7.45–7.20 (m, 9H, aromatic), 6.90–6.78 (m, 4H, aromatic), 6.24–6.12 (m, 1H, H–C1′), 5.53 (t, J = 5.3 Hz, 0.4H, H2′), 5.38 (t, J = 5.7 Hz, 0.6H, H2′), 5.36–5.27 (m, 1H, H5), 4.76–4.61 (m, 1H, H3′), 4.30 (d, J = 2.6 Hz, 0.5H, H4′), 4.20 (d, J = 3.4 Hz, 0.5H, H4′), 3.98–3.85 (m, 0.5H, −OCH2−), 3.84–3.74 (m, 6H, OCH3), 3.73–3.39 (m, 5.5H, −OCH2–, H5′ and iPr CH), 2.66 (td, J = 6.2, 1.9 Hz, 0.8H, −CH2CN), 2.46–2.31 (m, 1.2H, CH2CN), 2.20–2.07 (m, 3H, acetyl CH3), 1.32–1.01 (m, 12H, iPr CH3); 13C NMR (101 MHz, CDCl3) δ 169.9, 169.7 (acetyl CO), 162.7 (C4), 158.9 (aromatic), 150.3 (C2), 144.2, 144.1 (aromatic), 140.1 (C6), 135.3, 135.2, 135.1, 135.0, 134.4, 130.4, 130.3, 128.5, 128.4, 128.2, 127.4 (aromatic), 117.8, 117.4 (CN), 113.5, 113.4 (aromatic), 102.9 (C5), 87.5, 87.4 (DMTr-C), 86.5, 86.3 (C1′), 84.2, 83.8, 83.7 (C4′), 74.8, 74.5 (C2′), 71.5, 71.3, 70.7, 70.5 (C3′), 62.7, 62.5 (C5′), 58.9, 58.7, 58.3, 58.1, 57.9 (−OCH2−), 55.4 (OCH3), 43.5, 43.4, 43.3 (iPr CH), 24.8, 24.7 (iPr CH3), 21.1, 20.9 (acetyl CH3), 20.5, 20.3 (−CH2CN); 31P NMR (162 MHz, CDCl3) δ 151.04 (s), 150.25 (s); HRMS (ESI-TOF) m/z [M + H]+ calcd for C41H49N4O10PNa 811.3084, found 811.3083.
3′-O-Acetyl-5′-O-(4,4′-dimethoxytrityl)uridine-2′-O-(2-cyanoethyl-N,N-diisopropyl)phosphoramidite (22b):
1H NMR (400 MHz, CDCl3) δ 8.57 (br s, 1H, NH), 7.86 (dd, J = 10.1, 8.1 Hz, 1H, H6), 7.38–7.22 (m, 9H, aromatic), 6.85–6.83 (m, 4H, aromatic), 6.13, 6.09 (2 × d, J = 4.4 Hz, 1H, H1′), 5.39–5.27 (m, 2H, H5 and H3′), 4.63–4.56 (m, 1H, H2′) 4.23–4.22 (m, 1H, H4′), 3.89–3.55 (m, 11H, −OCH2–, OCH3, iPr CH and H5′), 3.46–3.41 (m, 1H, H5″), 2.67–2.53 (m, 2H, −CH2CN), 2.10 (2 × s, 3H, acetyl CH3), 1.18–1.14 (m, 12H, iPr CH3); 13C NMR (CDCl3, 101 MHz) δ 170.0, 169.9 (acetyl CO), 162.9 (C4), 158.9 (aromatic), 150.3, 150.2 (C2), 144.3 (aromatic), 140.3, 140.1 (C6), 135.2, 135.1, 130.3, 130.2, 128.2, 128.2, 127.4 (aromatic), 117.8, 117.6 (CN), 113.5 (aromatic), 102.6 (C5), 88.3, 88.2 (C1′), 87.6, 87.5 (DMTr-C), 81.5 (C4′), 74.9, 74.8, 74.6, 74.5 (C2′), 71.3, 71.1 (C3′), 62.2, 62.0 (C5′), 58.8, 58.6, 58.4 (−OCH2−), 55.4 (OCH3), 43.7, 43.6, 43.5 (iPr CH), 24.9, 24.8, 24.7, 24.6 (iPr CH3), 21.1, 21.0 (acetyl CH3), 20.4, 20.3 (−CH2CN); 31P NMR (162 MHz, CDCl3) δ 151.80 (s), 151.11 (s); HRMS (ESI-TOF) m/z [M + H]+ calcd for C41H49N4O10PNa 811.3084, found 811.3072.
N6-[(2-Cyanoethoxy)carbonyl]-2′-O-(tert-butyldimethylsilyl)-5′-O-(4,4′-dimethoxytrityl)adenosine-3′-O-(2-cyanoethyl-N,N-diisopropyl)phosphoramidite (29a):
1H NMR (400 MHz, CDCl3) δ 8.67, 8.65 (s, 1H, H8), 8.49 (s, 1H, NH), 8.24, 8.21 (s, 1H, H2), 7.51–7.19 (m, 9H, aromatic), 6.92–6.73 (m, 4H, aromatic), 6.08 (d, J = 6.3 Hz, 0.55H, H1′), 6.03 (d, J = 6.1 Hz, 0.45H, H1′), 5.12–4.98 (m, 1H, H2′), 4.52–4.32 (m, 4H, −OCH2–, H3′ and H4′), 4.03–3.52 (m, 11H, −OCH2–, OCH3, iPr CH and H5′), 3.41–3.27 (m, 1H, H5″), 2.81 (m, 2H, −CH2CN), 2.73–2.57 (m, 1H, −CH2CN), 2.44–2.22 (m, 1H, −CH2CN), 1.23–1.11 (m, 9H, iPr CH3), 1.05 (d, J = 6.8 Hz, 3H, iPr CH3), 0.75 (s, 9H, SiC(CH3)3), −0.03, −0.06 (2 × s, 3H, Si(CH3)2), −0.22, −0.23 (2 × s, 3H, Si(CH3)2); 13C NMR (CDCl3, 101 MHz) δ 158.7 (aromatic), 152.9 (C8), 151.5 (C4), 150.2 (C=O), 148.9 (C6), 144.7, 144.6 (aromatic), 142.3 (C2), 135.8, 135.8, 135.6, 135.6, 130.3, 130.3, 130.2, 128.4, 128.3, 128.1, 128.0, 127.1 (aromatic), 122.7, 122.6 (C5), 117.7, 117.4 (CN), 116.8 (CN), 113.3, 113.3 (aromatic), 88.6, 88.4 (C1′), 86.9, 86.8 (aromatic), 84.4, 84.1, 84.1 (C4′), 75.4, 74.8, 74.8 (C2′), 73.5, 73.4, 72.9, 72.7 (C3′), 63.4, 63.2 (C5′), 60.2 (−OCH2−), 59.0, 58.8, 57.8, 57.6 (−OCH2−), 55.4 (OCH3), 43.6, 43.5, 43.2, 43.0 (iPr CH), 25.7 (SiC(CH3)3), 24.9, 24.8, 24.7 (iPr CH3), 20.6, 20.3, 20.2, 18.3 (−CH2CN), 18.1, 18.0 (SiC(CH3)3), −4.5, −4.6, −5.0 (Si(CH3)2); 31P NMR (162 MHz, CDCl3) δ 151.04 (s), 149.06 (s); HRMS (ESI-TOF) m/z [M + H]+ calcd for C50H66N8O9SiP 981.4460, found 981.4433
N6-[(2-Cyanoethoxy)carbonyl]-3′-O-(tert-butyldimethylsilyl)-5′-O-(4,4′-dimethoxytrityl)adenosine-2′-O-(2-cyanoethyl-N,N-diisopropyl)phosphoramidite (29b):
1H NMR (400 MHz, CDCl3) δ 8.70, 8.68 (s, 1H, H8), 8.56 (s, 1H, NH), 8.30, 8.27 (s, 1H, H-2), 7.44–7.14 (m, 9H, aromatic), 6.79 (m, 4H, aromatic), 6.27 (d, J = 4.3 Hz, 0.62H, H1′), 6.18 (d, J = 4.7 Hz, 0.38H, H1′), 5.01 (dt, J = 11.3, 4.6 Hz, 0.38H, H2′), 4.82 (dt, J = 9.5, 4.5 Hz, 0.62H, H2′), 4.57–4.49 (m, 1H, H3′), 4.45 (t, J = 6.2 Hz, 2H, −OCH2−), 4.29–4.13 (m, 1H, H4′), 3.87–3.42 (m, 11H, −OCH2–, OCH3, iPr CH and H5′), 3.36–3.20 (m, 1H, H5″), 2.83–2.76 (m, 2H, −CH2CN), 2.50 (t, J = 6.4 Hz, 1H, −CH2CN), 2.38 (t, J = 6.3, 1H, −CH2CN), 1.15–1.02 (m, 9H, iPr CH3), 0.95–0.79 (m, 12H, iPr CH3 and SiC(CH3)3), 0.09, 0.06 (2 × s, 3H, Si(CH3)2), 0.01, −0.00 (2 × s, 3H, Si(CH3)2); 13C NMR (CDCl3, 101 MHz) δ 158.7, 158.7 (aromatic), 152.7, 152.6 (C8), 151.4, 151.4 (C4), 150.3, 150.3 (C=O), 149.0, 149.0 (C6), 144.6 (aromatic), 142.6, 142.4 (C2), 135.7, 135.7, 130.2, 130.2, 130.2, 128.3, 128.3, 128.0, 128.0, 127.1, 127.1 (aromatic), 122.7 (C5), 117.6 (CN), 116.8 (CN), 113.3, 113.3 (aromatic), 88.1, 88.0, 87.9 (C1′), 86.9, 86.7 (DMTr-C), 84.8, 84.4 (C4′), 76.0, 75.9, 75.6, 75.4 (C2′), 71.9, 71.6, 71.5 (C3′), 63.1, 62.8 (C5′), 60.3, 60.2 (−OCH2−), 58.6, 58.4, 57.9, 57.7 (−OCH2−), 55.4 (OCH3), 43.5, 43.3, 43.2 (iPr CH), 25.9 (SiC(CH3)3), 24.8, 24.7, 24.4 (iPr CH3), 20.4, 20.3, 20.2, 20.1, 18.3 (−CH2CN), 18.2 (SiC(CH3)3), −4.2, −4.3, −4.7, −4.8 (Si(CH3)2); 31P NMR (162 MHz, CDCl3) δ 150.44 (s), 150.15 (s); HRMS (ESI-TOF) m/z [M + H]+ calcd for C50H66N8O9SiP 981.4460, found 981.4424.
N4-[(2-Cyanoethyloxy)carbonyl]-2′-O-(tert-butyldimethylsilyl)-5′-O-(4,4′-dimethoxytrityl)cytidine-3′-O-(2-cyanoethyl-N,N-diisopropyl)phosphoramidite (30a):
1H NMR (400 MHz, CDCl3) δ 8.70–8.11 (m, 2H, H6, NH), 7.50–7.21 (m, 9H, aromatic), 6.92–6.59 (m, 5H, aromatic and H5), 5.92 (d, J = 2.3 Hz, 0.32H, H1′), 5.82 (s, 0.68H, H1′), 4.45–4.21 (m, 5H, H2′, H3′, H4′ and −OCH2−), 3.92–3.38 (m, 12H, OCH3, H5′, iPr CH and −OCH2−), 2.76 (t, J = 6.5 Hz, 2H, −CH2CN), 2.59 (t, J = 6.3 Hz, 0.68H, −CH2CN), 2.41 (t, J = 6.4 Hz, 1.32H, −CH2CN), 1.31–0.97 (m, 12H, iPr CH3), 0.94–0.86 (m, 9H, SiC(CH3)3), 0.25 (s, 3H, Si(CH3)2), 0.17–0.09 (m, 3H, Si(CH3)2); 13C NMR (CDCl3, 101 MHz) δ 162.5, 162.3 (C4), 158.8 (aromatic), 154.7 (C2), 151.9 (C=O), 145.2 (C6), 144.2, 144.1, 135.5, 135.4, 135.2, 130.4, 130.3, 128.5, 128.0, 127.3 (aromatic), 117.6, 117.5, 116.6 (CN), 113.4, 113.3 (aromatic), 94.7 (C5), 91.5 (C1′), 87.3, 87.2 (DMTr-C), 81.7, 81.5, 75.9, 75.3, 71.5, 69.6 (C2′/C3′/C4′), 61.6, 61.0 (C5′/–OCH2−), 60.1 (−OCH2−), 58.4, 58.3, 58.2 (C(5′)/–OCH2−), 55.3, 55.3 (OCH3), 45.5, 45.4, 43.4, 43.2, 43.1 (iPr CH), 26.0, 25.9 (SiC(CH3)3), 25.0, 24.9, 24.9, 24.8, 24.7, 24.6, 23.1, 23.0 (iPr CH3), 20.6, 20.5, 20.3, 20.3, 20.2 (−CH2CN), 18.2, 18.1 (−CH2CN/SiC(CH3)3), −4.2, −4.3, −4.9, −5.0 (Si(CH3)2); 31P NMR (162 MHz, CDCl3) δ 150.30 (s), 149.10 (s); HRMS (ESI-TOF) m/z [M + H]+ calcd for C49H66N6O10PSi 957.4347, found 957.4380.
N4-[(2-Cyanoethyloxy)carbonyl]-3′-O-(tert-butyldimethylsilyl)-5′-O-(4,4′-dimethoxytrityl)cytidine-2′-O-(2-cyanoethyl-N,N-diisopropyl)phosphoramidite (30b):
1H NMR (400 MHz, CDCl3) δ 8.68 (br s, 1H, H6), 7.46–7.17 (m, 9H, aromatic), 6.86–6.65 (m, 5H, aromatic and H5), 6.17–6.13 (m, 1H, H1′), 4.36–4.24 (m, 4H, H2′, H3′ and −OCH2−), 4.21–4.09 (m, 1H, H4′), 4.08–3.93 (m, 1H, −OCH2−), 3.87–3.70 (m, 8H, OCH3, H5′ and −OCH2−), 3.69–3.57 (m, 2H, iPr CH), 3.35–3.29 (m, 1H, H5″), 2.83–2.54 (m, 4H, −CH2CN), 1.21–1.01 (m, 12H, iPr CH3), 0.73, 0.72 (2 × s, 9H, SiC(CH3)3), 0.03, −0.02 (2 × s, 3H, Si(CH3)2), −0.09, −0.10 (2 × s, 3H, Si(CH3)2); 13C NMR (CDCl3, 101 MHz) δ 162.4, 162.3 (C4), 158.7, 158.7 (aromatic), 154.4 (C2), 151.9 (C=O), 144.9 (C6), 143.6, 143.6, 135.0, 135.0, 130.3, 130.2, 130.2, 130.2, 128.5, 128.5, 127.9, 127.3, 127.2 (aromatic), 118.3, 117.9, 117.1, 116.6 (CN), 113.2 (aromatic), 94.9 (C5), 90.0 (C1′), 87.1, 87.1 (DMTr-C), 82.3, 82.1 (C4′), 76.0, 75.9 (C2′), 69.0, 68.9 (C3′), 60.6, 60.5 (C5′), 60.0 (−OCH2−), 58.6, 58.4, 58.3, 58.2, 58.2, 57.9 (−OCH2−), 55.2 (OCH3), 43.5, 43.3, 43.2, 43.0 (iPr CH), 25.7, 25.6, 25.5 (SiC(CH3)3), 24.7, 24.7, 24.6, 24.6, 24.6, 24.5, 24.4 (iPr CH3), 20.1, 20.1, 20.0, 18.0 (−CH2CN), 17.9 (SiC(CH3)3), −4.3, −5.2 (Si(CH3)2); 31P NMR (162 MHz, CDCl3) δ 151.23 (s), 147.97 (s); HRMS (ESI-TOF) m/z [M + H]+ calcd for C49H65N6O10PSiNa 979.4161, found 979.4132.
N2-[(2-Cyanoethoxy)carbonyl]-O6-[2-(4-nitrophenyl)ethyl]-2′-O-(tert-butyldimethylsilyl)-5′-O-(4,4′-dimethoxytrityl)guanosine-3′-O-(2-cyanoethyl-N,N-diisopropyl)phosphoramidite (31a):
1H NMR (400 MHz, CDCl3) δ 8.16 (d, J = 8.7 Hz, 2H, aromatic), 8.04, 8.02 (2 × s, 1H, H8), 7.58–7.30 (m, 9H, aromatic), 7.30–7.17 (m, 2H, aromatic), 7.09 (s, 1H, NH), 6.80 (m, 4H, aromatic), 6.01 (d, J = 7.2 Hz, 0.40H, H1′), 5.86 (d, J = 6.9 Hz, 0.60H, H1′), 5.12 (dd, J = 6.9, 5.2 Hz, 0.60H, H2′), 5.00 (dd, J = 7.2, 4.5 Hz, 0.40H, H2′), 4.86 (t, J = 6.8 Hz, 2H, −OCH2−), 4.43–4.19 (m, 4H, H3′, H4′ and −COOCH2−), 3.98–3.87 (m, 0.47H, −OCH2−), 3.98–3.87 (m, 0.53H, −OCH2−), 3.81–3.75 (m, 6H, OCH3), 3.68–3.46 (m, 4H, iPr CH, H5′ and −OCH2−), 3.39–3.22 (m, 3H, −CH2ph and H5″), 2.34–2.16 (m, 3H, −CH2CN), 2.34–2.16 (m, 1H, −CH2CN), 1.23–1.13 (m, 9H, iPr CH3), 1.01 (d, J = 6.7 Hz, 3H, iPr CH3), 0.75 (s, 9H, SiC(CH3)3), −0.01, −0.06 (2 × s, 3H, Si(CH3)2), −0.25, −0.26 (2 × s, 3H, Si(CH3)2); 13C NMR (100 MHz, CDCl3) δ 160.8 (C6), 158.8, 158.5 (aromatic), 153.3, 153.1 (C4), 151.6, 151.4 (C2), 150.5, 150.4 (C=O), 147.0, 146.8, 145.9, 145.7, 144.8, 144.6 (aromatic), 141.4, 140.6 (C8), 136.0, 135.8, 135.7, 135.6 (aromatic), 130.2, 130.2, 130.1, 130.0, 129.9, 128.3, 128.2, 128.1, 128.0, 127.2, 126.9, 123.8, 123.6 (aromatic), 118.9, 118.6 (C5), 118.0, 117.4, 116.9, 116.8 (CN), 113.4, 113.3, 113.2, 113.1 (aromatic), 88.4, 87.5 (C1′), 86.9, 86.6 (DMTr-C), 84.7, 84.3 (C4′), 75.8, 74.1, 74.0 (C2′), 73.6, 73.5, 72.7, 72.6 (C3′), 67.1, 67.0 (−OCH2−), 63.6, 63.5 (C5′), 59.5, 59.7 (−OCH2–, −COOCH2−), 59.1, 57.8, 57.6 (−OCH2−), 55.4, 55.1 (OCH3), 43.6, 43.5, 43.1, 43.0 (iPr CH), 35.2, 35.0 (−CH2ph), 25.7, 25.5, 25.4 (SiC(CH3)3), 24.9, 24.8, 24.7, 24.5 (iPr CH3), 20.6, 20.5, 20.2, 20.1 (−CH2CN), 18.3, 18.2, 18.1, 18.0 (−CH2CN and SiC(CH3)3), −4.5, −4.6, −5.1, −5.3 (Si(CH3)2); 31P NMR (162 MHz, CDCl3) δ 151.07 (s), 149.03 (s); HRMS (ESI-TOF) m/z [M + H]+ calcd for C58H73N9O12SiP 1146.4886, found 1146.4889.
N2-[(2-Cyanoethoxy)carbonyl]-O6-[2-(4-nitrophenyl)ethyl]-3′-O-(tert-butyldimethylsilyl)-5′-O-(4,4′-dimethoxytrityl)guanosine-2′-O-(2-cyanoethyl-N,N-diisopropyl)phosphoramidite (31b):
1H NMR (400 MHz, CDCl3) δ 8.15 (d, J = 7.6 Hz, 2H, aromatic), 8.07, 8.05 (2 × s, 1H, H8), 7.52 (d, J = 8.6 Hz, 2H, aromatic), 7.45–7.17 (m, 9H, aromatic), 6.78 (d, J = 8.6 Hz, 4H, aromatic), 6.14 (d, J = 5.2 Hz, 0.51H, H1′), 6.04 (d, J = 5.6 Hz, 0.49H, H1′), 5.08 (dt, J = 10.8, 5.1 Hz, 0.50H, H2′), 4.86–4.76 (m, 2.50H, −OCH2–, H2′), 4.47 (t, J = 4.1 Hz, 1H, H3′), 4.36 (q, J = 6.0 Hz, 2H, −COOCH2−), 4.17–4.13 (m, 1H, H4′), 3.84–3.45 (m, 11H, −OCH2–, OCH3, iPr CH and H5′), 3.35–3.27 (m, 3H, −CH2ph and H5″), 2.74 (q, J = 6.2 Hz, 2H, −CH2CN), 2.51 (t, J = 6.3 Hz, 1H, −CH2CN), 2.40–2.32 (m, 1H, −CH2CN), 1.14–1.03 (m, 9H, iPr CH3), 0.91–0.85 (m, 12H, iPr CH3 and SiC(CH3)3), 0.11–0.07 (2 × s, 3H, Si(CH3)2), 0.00––0.01 (2 × s, 3H, Si(CH3)2); 13C NMR (CDCl3, 101 MHz) δ 160.7 (C6), 158.6 (aromatic), 153.1, 153.0 (C4), 151.4 (C2), 150.5, 150.4 (C=O), 147.0, 145.9, 144.7, 144.7 (aromatic), 141.2, 141.1 (C8), 135.9, 135.8, 130.2, 128.3, 128.0, 127.0, 123.8 (aromatic), 118.8, 118.7 (C5), 117.6, 117.6 (CN), 116.9 (CN), 113.3 (aromatic), 87.7, 87.6 (C1′), 86.8, 86.7 (DMTr-C), 85.3, 84.8 (C4′), 75.8, 75.7, 75.4, 75.2 (C2′), 72.2, 71.9 (C3′), 67.1 (−OCH2−), 63.5, 63.3 (C5′), 59.6, 59.6 (−COOCH2−), 58.3, 58.1, 58.0, 57.8 (−OCH2−), 55.3 (OCH3), 43.4, 43.3, 43.1 (iPr CH), 35.2 (−CH2ph), 25.9 (SiC(CH3)3), 24.8, 24.8, 24.7, 24.6, 24.4, 24.3 (iPr CH3), 20.4, 20.4, 20.1, 20.0, 18.4 (−CH2CN), 18.2 (SiC(CH3)3), −4.3, −4.7, −4.8 (Si(CH3)2); 31P NMR (162 MHz, CDCl3) δ 150.24 (s), 149.97 (s); HRMS (ESI-TOF) m/z [M + H]+ calcd for C58H72N 9O12NaSiP 1168.4700, found 1168.4669.
3′-O-(tert-Butyldimethylsilyl)-5′-O-(4,4′-dimethoxytrityl)uridine-2′-O-(2-cyanoethyl-N,N-diisopropyl)phosphoramidite (33):
1H NMR (400 MHz, CDCl3) δ 8.72, 8.53 (2 × s, 1H, NH), 8.14, 8.06 (2 × d, J = 8.2 Hz, 1H, H6), 7.38–7.23 (m, 9H, aromatic), 6.87–6.81 (m, 4H, aromatic), 6.15 (d, J = 3.2 Hz, 0.4H, H1′), 6.06 (d, J = 2.2 Hz, 0.6H, H1′), 5.35, 5.27 (2 × d, J = 8.1 Hz, 1H, H5), 4.37–4.19 (m, 2H, H2′, H3′), 4.14–4.06 (m, 1H, H4′), 3.99–3.85 (m, 1H, −OCH2−), 3.84–3.54 (m, 10H, OCH3, −OCH2–, iPr CH, H5′), 3.37–3.29 (m, 1H, H5″), 2.72–2.47 (m, 2H, −CH2CN), 1.18–1.15 (m, 12H, iPr CH3), 0.80–0.78 (m, 9H, SiC(CH3)3), 0.09, 0.04 (2 × s, 3H, Si(CH3)2), −0.01, −0.03 (2 × s, 3H, Si(CH3)2); 13C NMR (CDCl3, 101 MHz) δ 163.2, 163.1 (C4), 158.9, 158.9 (aomatic), 150.3, 150.1 (C2), 144.2, 144.2 (aromatic), 140.6, 140.4 (C6), 135.2, 135.1, 135.0, 130.4, 130.4, 128.5, 128.4, 128.1, 127.4, 127.4 (aromatic), 118.1, 117.8 (CN), 113.4, 113.4 (aromatic), 102.2, 102.1 (C5), 88.7, 88.6, 88.5 (C1′), 87.4, 87.3 (DMTr-C), 83.6, 83.5 (C4′), 76.6, 76.5 (C2′), 70.5, 70.5, 70.4 (C3′), 61.7, 61.6 (C5′), 58.5, 58.3, 58.3, 58.1 (−OCH2−),), 55.4, 55.4 (OCH3), 43.5, 43.4, 43.3 (iPr CH), 25.9 (SiC(CH3)3), 24.9, 24.9, 24.8, 24.8, 24.6, 24.5 (iPr CH3), 20.5, 20.4, 20.4, 20.3 (−CH2CN), 18.2, 18.1 (SiC(CH3)3), −4.1, −4.2, −4.8, −4.9 (Si(CH3)2); 31P NMR (162 MHz, CDCl3) δ 150.42 (s), 149.37 (s); HRMS (ESI-TOF) m/z [M + H]+ calcd for C45H61O9N4PSiNa 883.3838, found 883.3816.
Procedure for the Synthesis of Photolabile Linker
Methyl 4-(1-(Bis(4-methoxyphenyl)(phenyl)methoxy)ethyl)-3-nitrobenzoate (43)
Methyl 3-formyl-4-nitrobenzoate 40 (purchased from Sigma) (5.00 g, 23.9 mmmol) was dissolved in anhydrous diethyl ether (150 mL), and methylmagnesium bromide (1 M in dibutyl ether, 59.7 mL, 59.7 mmol) was added dropwise slowly, maintaining the reaction vessel at rt. The reaction mixture was stirred for 4 h and quenched by the addition of saturated ammonium chloride (50 mL). The organics were extracted with Et2O (3 × 50 mL). The combined organic layers were washed with water (50 mL) and brine (50 mL) and dried over MgSO4. The solvent was removed under vacuum, and the residue was purified by flash column chromatography. The product was found to also contain a benzyl alcohol side product 42 that could not be separated by flash column chromatography. The material was used in the next step without further purification. Alcohol 41 containing 42 (2.60 g, 11.5 mmol) was coevaporated with anhydrous pyridine (3 × 10 mL). The residue and DMTr-Cl (3.90 g, 11.5 mmol) were dissolved in anhydrous pyridine (20 mL) and anhydrous CH2Cl2 (10 mL), and the reaction mixture was stirred at rt overnight. MeOH (20 mL) was added, and the mixture was stirred for a further 10 min before the solvent was removed under vacuum. The residue was dissolved in CHCl3 (50 mL) and washed with saturated aq NaHCO3 (2 × 40 mL). The organic layer was dried over MgSO4 and evaporated under vacuum. The residue was purified by flash column chromatography (99:1, toluene/Et3N) to afford a yellow oil, which was further purified by normal-phase HLPC give the title product 43 (4.10 g, 33% for two steps) as a colorless solid: 1H NMR (400 MHz, CDCl3) δ 8.21 (d, J = 1.7 Hz, 1H, H2), 7.97 (dd, J = 8.2, 1.7 Hz, 1H, H6), 7.82 (d, J = 8.2 Hz, 1H, H5), 7.48–7.43 (m, 2H, DMTr), 7.29–7.10 (m, 7H, DMTr), 6.68–6.59 (m, 4H, DMTr), 5.23 (q, J = 6.2 Hz, 1H, H7), 3.92 (s, 3H, COOCH3), 3.74, 3.73 (2 × s, 6H, DMTr OCH3), 1.65 (d, J = 6.2 Hz, 3H, H8); 13C NMR (CDCl3, 101 MHz) δ 165.0, 158.5, 158.4, 147.6, 146.0, 145.3, 136.2, 135.7, 132.0, 130.1, 130.02, 130.0, 128.7, 127.8, 127.8, 126.8, 124.7, 113.1, 113.0, 68.0, 55.2, 55.2, 52.5, 24.8; HRMS (ESI-TOF) m/z [M + H]+ calcd for C31H29O7N1Na 550.1836, found 550.1827.
Photolabile-CPG (45)
Compound 43 (500 mg, 0.94 mmol) was dissolved in THF (1.50 mL) and water (0.50 mL). LiOH (24.9 mg, 1.04 mmol) was added, and the mixture was stirred at rt for 24 h, the reaction being monitored by TLC until no starting material was observable. The solvent was removed to give a white solid, and this material was used in the next step without further purification. The residue was coevaporated with anhydrous pyridine (3 × 4 mL) and redissolved in anhydrous pyridine (4 mL). Isobutyl chloroformate (72.0 mg, 0.53 mmol) was added, and the formation of a white precipitate was observed. The reaction mixture was stirred for 30 min before the precipitate was removed by filtration under argon, and the supernatant was filtered directly into oven-dried glassware. The solvent was removed under vacuum and the resultant crude mixed anhydride was dissolved in anhydrous CH2Cl2 (4 mL), followed by the addition of N,N-diisopropylethylamine (68 mg, 0.53 mmol) and long-chain alkylamine controlled pore glass (250 mg, 120–200 mesh, nominal diameter 500 Å) (Sigma). The suspension was rotated gently under argon at rt for 24 h. The CPG was filtered and washed with CH2Cl2, MeCN, water, MeCN, and finally CH2Cl2 (15 mL each). The modified CPG was dried overnight under vacuum and then treated with CAP A (80:10:10, THF/2,6-lutidine/pivaloyl chloride) (2 mL) and commercially available CAP B (Link Technologies) (90:10, THF/N-methylimidazole) (2 mL) solution, and the mixture was rotated gently for 1.5 h. The CPG was filtered, washed with CH2Cl2 (15 mL), and dried under vacuum. The loading was determined by Trityl assay, and loading values of 33.3–56.2 μmol/g were obtained. The prepared CPG was stored in the dark at 4 °C.
Automated Synthesis of Acetylated RNA Oligonucleotides
RNA oligonucleotide synthesis was performed on 1 μmol scale with modified monomers (0.1 M in 1:1 CH2Cl2/MeCN) and utilized the photolabile-CPG 46. The synthesis cycle began with detritylation using 3% dichloroacetic acid (DCA) in CH2Cl2. The coupling step utilized 80 μL of each amidite (0.1 M, 1:1, MeCN/CH2Cl2) and 1 M 4,5-dicyanoimidazole (DCI) as the activator with a single 20 min coupling, with the exception that the acetylated amidites were given a 15 min double coupling step. A 5 min capping step was carried out with CAP A (80:10:10, THF/2,6-lutidine/pivalic anhydride), and CAP B (90:10, THF/N-methylimidazole) solutions. The oxidation of the phosphites was carried out with 0.02 M iodine (THF/pyridine/water, 7:2:1). The automation program was set as DMTr-ON such that the final DMTr group was not removed.
Deprotection, Cleavage, and Purification of the Acetylated-RNA Oligonucleotides
Without removal of the CPG from the synthesis column, the CPG was dried under vacuum for 15 min. A solution of 0.5 M DBU in anhydrous MeCN (3.5 mL, 10% morpholine) was initially passed through the column for 5 min, and then the column and CPG were immersed in the DBU solution under an atmosphere of argon for 6 h at 40 °C with sonication every hour. The column was washed with anhydrous acetonitrile (10 mL) and CH2Cl2 (10 mL). The final DMTr group was removed by passing 3% TCA in CH2Cl2 through the column until the washings became colorless. The collected orange solution was diluted to 50 mL with CH2Cl2, and the yield of full-length product was calculated by trityl assay. At this point, the CPG was placed into a 4 mL UV transparent vessel and suspended in DMSO (0.5 mL). A single wavelength LED light (λ = 365) was placed above the UV transparent vessel, focusing on the region containing CPG, in a black box to prevent extra UV light escaping into the environment. The CPG was irradiated at 365 nm (max = 34.5 mW/cm2, Prizmatix Mic-LED-365) for 1 h. The CPG was removed by filtration and washed with DMSO (2 × 0.5 mL), and the fractions were combined. The DMSO was removed by lyophilization, and the residue was redissolved in anhydrous DMSO (200 μL). To the solution was added Et3N·3HF (125 μL), the mixture was thoroughly mixed and then heated at 65 °C for 3 h. Sodium acetate (3 M, 25 μL, pH 7) was added, followed by thorough mixing. After the addition of n-butanol (1 mL) the mixture was cooled at −80 °C for 30 min and then centrifuged for 10 min at 13200 rpm. The n-butanol was decanted, followed by washing of the pellet with ethanol (2 × 0.75 mL) and finally drying the pellet in a SpeedVac at 65 °C for 1 h. The dried pellet was dissolved in RNAase-free water (0.5–1 mL), and the RNA oligomer was quantified by UV absorbance at λ 260 nm and analyzed by MALDI-TOF MS to assess the synthesis. Dephosphorylation (if required) of the acetylated RNA oligonucleotide began by dilution to a concentration of 1 μg/10 μL with PBS buffer (0.01 M phosphate, 0.138 M NaCl, 0.0027 M KCl, 1 mM MgCl2, pH 7.4). To the dissolved oligoribonucleotide calf intestinal alkaline phosphatase (0.5u/μg) was added, and the mixture was heated at 37 °C for 2 h. To the solution was added one volume of 1 M TEAA buffer (pH 7.0), and the oligonucleotide was desalted using a Waters C18 Sep-Pak column prior to HPLC purification of the oligonucleotide. Acetylated RNA oligomers were purified by preparative SAX-HPLC. The target fractions were combined and desalted by dialysis against 10 mM TEAA buffer (pH 7.0) at 4 °C. Finally, the purified oligonucleotides were quantified by UV absorbance at 260 nm, and characterized by MALDI-TOF MS.
Acknowledgments
This work was supported by the MRC (Project No. MC_UP_A024_1009) and the EPSRC. We thank Drs. Stefan Freund and Trevor Rutherford for assistance with NMR spectroscopy.
Supporting Information Available
1H and 13C NMR spectra for all new compounds, MADLI-TOF mass spectra, and HPLC traces of synthesized partially 2′ or 3′-O-acetylated RNA oligonucleotides and UV melting curves of duplexes. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Gilbert W. Nature 1986, 319, 618. [Google Scholar]
- Powner M. W.; Gerland B.; Sutherland J. D. Nature 2009, 459, 239. [DOI] [PubMed] [Google Scholar]
- Verlander M. S.; Orgel L. E. J. Mol. Evol. 1974, 3, 115. [DOI] [PubMed] [Google Scholar]
- Tapiero C. M.; Nagyvary J. Nature 1971, 231, 42. [DOI] [PubMed] [Google Scholar]
- Usher D. A.; McHale A. H. Science 1976, 192, 53. [DOI] [PubMed] [Google Scholar]
- Bolli M.; Micura R.; Pitsch S.; Eschenmoser A. Helv. Chim. Acta 1997, 80, 1901. [Google Scholar]
- Bowler F. R.; Chan C. K. W.; Duffy C. D.; Gerland B.; Islam S.; Powner M. W.; Sutherland J. D.; Xu J. Nat. Chem. 2013, 5, 383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki A. M.; Casper M. D.; Freier S. M.; Lesnik E. A.; Zounes M. C.; Cummins L. L.; Gonzalez C.; Cook P. D. J. Med. Chem. 1993, 36, 831. [DOI] [PubMed] [Google Scholar]
- Freier S. M.; Altmann K. H. Nucleic Acids Res. 1997, 25, 4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoharan M. Biochim. Biophys. Acta 1999, 1489, 117. [DOI] [PubMed] [Google Scholar]
- Reese C. B.; Trentham D. R. Tetrahedron Lett. 1965, 6, 2467. [DOI] [PubMed] [Google Scholar]
- Griffin B. E.; Reese C. B. Tetrahedron Lett. 1964, 5, 2925. [DOI] [PubMed] [Google Scholar]
- Rozners E.; Renhofa R.; Petrova M.; Popelis J.; Kumpins V.; Bizdena E. Nucleosides Nucleotides 1992, 11, 1579. [Google Scholar]
- Kempe T.; Chow F.; Sundquist W. I.; Nardi T. J.; Paulson B.; Peterson S. M. Nucleic Acids Res. 1982, 10, 6695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackey J. G.; Sabatino D.; Damha M. J. Org. Lett. 2007, 9, 789. [DOI] [PubMed] [Google Scholar]
- Lackey J. G.; Mitra D.; Somoza M. M.; Cerrina F.; Damha M. J. J. Am. Chem. Soc. 2009, 131, 8496. [DOI] [PubMed] [Google Scholar]
- Usman N.; Ogilvie K. K.; Jiang M. Y.; Cedergren R. J. J. Am. Chem. Soc. 1987, 109, 7845. [Google Scholar]
- Wincott F.; Direnzo A.; Shaffer C.; Grimm S.; Tracz D.; Workman C.; Sweedler D.; Gonzalez C.; Scaringe S.; Usman N. Nucleic Acids Res. 1995, 23, 2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride L. J.; Kierzek R.; Beaucage S. L.; Caruthers M. H. J. Am. Chem. Soc. 1986, 108, 2040. [Google Scholar]
- Zhu Q.; Delaney M. O.; Greenberg M. M. Bioorg. Med. Chem. Lett. 2001, 11, 1105. [DOI] [PubMed] [Google Scholar]
- Schulhof J. C.; Molko D.; Teoule R. Nucleic Acids Res. 1987, 15, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merk C.; Reiner T.; Kvasyuk E.; Pfleiderer W. Helv. Chim. Acta 2000, 83, 3198. [Google Scholar]
- Eritja R.; Robles J.; Aviñó A.; Alberico F.; Pedroso E. Tetrahedron 1992, 48, 4171. [Google Scholar]
- Westman E.; Stromberg R. Nucleic Acids Res. 1994, 22, 2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesler W. T.; Caruthers M. H. J. Org. Chem. 1996, 61, 4272. [DOI] [PubMed] [Google Scholar]
- Schirmeister H.; Himmelsbach F.; Pfleiderer W. Helv. Chim. Acta 1993, 76, 385. [Google Scholar]
- Lang H.; Gottlieb M.; Schwarz M.; Farkas S.; Schulz B. S.; Himmelsbach F.; Charubala R.; Pfleiderer W. Helv. Chim. Acta 1999, 82, 2172. [Google Scholar]
- Matsuda A.; Shinozaki M.; Suzuki M.; Watanabe K.; Miyasaka T. Synthesis 1986, 385. [Google Scholar]
- Fromageot H. P.; Griffin B. E.; Reese C. B.; Sulston J. E. Tetrahedron 1967, 23, 2315. [DOI] [PubMed] [Google Scholar]
- Murata M.; Bhuta P.; Owens J.; Zemlicka J. J. Med. Chem. 1980, 23, 781. [DOI] [PubMed] [Google Scholar]
- Robins M. J.; Mengel R.; Jones R. A.; Fouron Y. J. Am. Chem. Soc. 1976, 98, 8204. [DOI] [PubMed] [Google Scholar]
- Dahl B. H.; Nielsen J.; Dahl O. Nucleic Acids Res. 1987, 15, 1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanghvi Y. S.; Guo Z. Q.; Pfundheller H. M.; Converso A. Org. Process Res. Dev. 2000, 4, 175. [Google Scholar]
- Vargeese C.; Carter J.; Yegge J.; Krivjansky S.; Settle A.; Kropp E.; Peterson K.; Pieken W. Nucleic Acids Res. 1998, 26, 1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welz R.; Muller S. Tetrahedron Lett. 2002, 43, 795. [Google Scholar]
- Berner S.; Mühlegger K.; Seliger H. Nucleic Acids Res. 1989, 17, 853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakimelahi G. H.; Proba Z. A.; Ogilvie K. K. Can. J. Chem. 1982, 60, 1106. [Google Scholar]
- Damha M. J.; Giannaris P. A.; Zabarylo S. V. Nucleic Acids Res. 1990, 18, 3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pon R. T.; Yu S. Y. Nucleic Acids Res. 1997, 25, 3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan H.; Greenberg M. M. J. Org. Chem. 1996, 61, 525. [DOI] [PubMed] [Google Scholar]
- McMinn D. L.; Hirsch R.; Greenberg M. M. Tetrahedron Lett. 1998, 39, 4155. [Google Scholar]
- McMinn D. L.; Greenberg M. M. Tetrahedron 1996, 52, 3827. [Google Scholar]
- Greenberg M. M.; Gilmore J. L. J. Org. Chem. 1994, 59, 746. [Google Scholar]
- Johnsson R.; Lackey J. G.; Bogojeski J. J.; Damha M. J. Bioorg. Med. Chem. Lett. 2011, 21, 3721. [DOI] [PubMed] [Google Scholar]
- Avino A.; Garcia R. G.; Diaz A.; Albericio F.; Eritja R. Nucleosides Nucleotides 1996, 15, 1871. [Google Scholar]
- Eritja R.; Robles J.; Fernandezforner D.; Albericio F.; Giralt E.; Pedroso E. Tetrahedron Lett. 1991, 32, 1511. [Google Scholar]
- Ajayaghosh A.; Pillai V. N. R. Tetrahedron 1988, 44, 6661. [Google Scholar]
- Ford L.; Atefi F.; Singer R. D.; Scammells P. J. Eur. J. Org. Chem. 2011, 942. [Google Scholar]
- Chen T. Q.; Fu J. S.; Greenberg M. M. Org. Lett. 2000, 2, 3691. [DOI] [PubMed] [Google Scholar]
- Zhou C.; Pathmasiri W.; Honcharenko D.; Chatterjee S.; Barman J.; Chattopadhyaya J. Can. J. Chem. 2007, 85, 293. [Google Scholar]
- Schroeder S. J.; Turner D. H. Methods Enzymol. 2009, 468, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mergny J. L.; Lacroix L. Oligonucleotides 2003, 13, 515. [DOI] [PubMed] [Google Scholar]
- Marky L. A.; Breslauer K. J. Biopolymers 1987, 26, 1601. [DOI] [PubMed] [Google Scholar]
- Mathis G.; Bourg S.; Aci-Seche S.; Truffert J.-C.; Asseline U. Org. Biomol. Chem. 2013, 11, 1345. [DOI] [PubMed] [Google Scholar]
- Egli M.; Portmann S.; Usman N. Biochemistry 1996, 35, 8489. [DOI] [PubMed] [Google Scholar]
- Adamiak D. A.; Milecki J.; Adamiak R. W.; Rypniewski W. New J. Chem. 2010, 34, 903. [Google Scholar]
- Lesnik E. A.; Guinosso C. J.; Kawasaki A. M.; Sasmor H.; Zounes M.; Cummins L. L.; Ecker D. J.; Cook P. D.; Freier S. M. Biochemistry 1993, 32, 7832. [DOI] [PubMed] [Google Scholar]
- Giannaris P. A.; Damha M. J. Nucleic Acids Res. 1993, 21, 4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng J.; Li L.; Engelhart A. E.; Gan J.; Wang J.; Szostak J. W. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szostak J. W. J. Syst. Chem. 2012, 3, 2. [Google Scholar]
- Usher D. A.; McHale A. H. Proc. Natl. Acad. Sci. U.S.A. 1976, 73, 1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.