

Abstract
Sustained Akt activation induces cardiac hypertrophy (LVH), which may lead to heart failure. This study tested the hypothesis that Akt activation contributes to mitochondrial dysfunction in pathological LVH. Akt activation induced LVH and progressive repression of mitochondrial fatty acid oxidation (FAO) pathways. Preventing LVH by inhibiting mTOR failed to prevent the decline in mitochondrial function, but glucose utilization was maintained. Akt activation represses expression of mitochondrial regulatory, FAO, and oxidative phosphorylation genes in vivo that correlate with the duration of Akt activation in part by reducing FOXO-mediated transcriptional activation of mitochondrion-targeted nuclear genes in concert with reduced signaling via peroxisome proliferator-activated receptor α (PPARα)/PGC-1α and other transcriptional regulators. In cultured myocytes, Akt activation disrupted mitochondrial bioenergetics, which could be partially reversed by maintaining nuclear FOXO but not by increasing PGC-1α. Thus, although short-term Akt activation may be cardioprotective during ischemia by reducing mitochondrial metabolism and increasing glycolysis, long-term Akt activation in the adult heart contributes to pathological LVH in part by reducing mitochondrial oxidative capacity.
INTRODUCTION
Mitochondrial metabolism of fatty acids (FA) and, to a lesser extent, glucose, lactate, and ketone bodies generates ATP to sustain cardiac contractile function. Myocardial metabolism is a flexible process that adapts to various stimuli, including substrate supply, hormonal and growth factor stimulation, and cardiac hypertrophy (LVH). In physiological hypertrophy (e.g., after exercise), FA and glucose oxidation are both increased in the heart (1). Pathological hypertrophy, as occurs following pressure overload leading to heart failure, is associated with increased glucose utilization but mitochondrial dysfunction (2). Although increased glucose utilization may be an adaptive response, persistent pathological stimulation ultimately limits cardiac metabolic flexibility, which may contribute to heart failure. Acute activation of Akt in the heart in vitro or in vivo increases glucose uptake and protects the heart from ischemia/reperfusion injury (3, 4). In contrast, long-term activation of Akt results in cardiac hypertrophy (LVH) that is associated with a range of functional outcomes from increased contractility to heart failure, due in part to the level of overexpression or subcellular localization of Akt (5, 6). Persistent Akt signaling may be deleterious to the heart due to feedback inhibition of insulin receptor substrate (IRS) and phosphatidylinositol 3-kinase (PI3K) signaling or GLUT4-mediated glucose uptake (7–9). Although short-term activation of Akt may induce LVH with preserved cardiac function, sustained Akt activation precipitates heart failure due in part to a mismatch between cardiac hypertrophy and angiogenesis (7, 10). Cardiac failure is also associated with significant changes in myocardial substrate energy metabolism (1). Thus, the possibility exists that long-term Akt activation in cardiomyocytes contributes to heart failure by impairing myocardial mitochondrial substrate utilization and ATP production. The present study was designed to directly determine if Akt activation in the heart induces mitochondrial dysfunction.
MATERIALS AND METHODS
Animals.
Adult age- and sex-matched mice harboring the following transgenes were studied: constitutively active Akt1 (T308D/S473D) (caAkt), inducible myristoylated Akt1 (IND-Akt and tON-Akt), and wild-type (WT) or single transgenic controls. Mice with cardiomyocyte-restricted expression of a constitutively active Akt1 (caAkt) mutant (T308D/S473D) were generated in the laboratory of Seigo Izumo and have been previously described (6). Cardiomyocyte-restricted tetracycline-inducible myristoylated Akt1 mice (IND-Akt) were generated in the laboratory of Kenneth Walsh and have been previously described (9, 10). These animals contain a myristoylated Akt1 transgene downstream of a tetracycline operon (TetAkt) and a tetracycline-sensitive transcription factor (Tet-Off) transgene expressed in cardiomyocytes by the α-myosin heavy chain (α-MHC) promoter (tTA). Unless specified, control values were the averages of wild-type and single transgenic TetAkt or tTA mice. The mice were fed normal rodent chow diet supplemented with 1 g/kg of body weight doxycycline (DOX) until the time of transgene induction. A second model of cardiomyocyte-restricted tetracycline-inducible myristoylated Akt1 mice (tON-Akt), which phenocopies the IND-Akt mice, was also used. In this model, the second transgene is a codon-optimized reverse tetracycline transactivator (tON), regulated by α-MHC, and mice were fed 1 mg/kg DOX chow at the time of transgene induction. Mice were housed in temperature-controlled facilities with a 12-h light and 12-h dark cycle (lights on at 6:00 A.M.). Experiments were conducted in accordance with guidelines approved by Institutional Animal Care and Use Committees of the University of Utah and the Carver College of Medicine, University of Iowa.
TAC-induced pressure overload hypertrophy.
Aortic banding was performed as described by us previously (2). Mice 6 to 8 weeks of age were anesthetized and placed in the supine position on a heating pad (37°C). Following a horizontal skin incision of ∼1 cm in length made at the level of the suprasternal notch, an ∼3-mm longitudinal cut was made in the proximal portion of the sternum. Transverse aortic constriction (TAC) was implemented by placement of a metal clip calibrated to a 27-gauge (27G-TAC; mild)- or 30-gauge (30G-TAC; severe)-diameter needle between the innominate artery and the left common carotid artery. The sham procedure was identical except that the aortic arch was not constricted. Mice were followed for 4 weeks, and tissue was harvested for hypertrophy and protein measures.
Hemodynamic measures following LV catheterization.
Invasive left ventricular (LV) hemodynamic measurements were performed with a temperature-calibrated 1.0-French (Fr) micromanometer-tipped catheter (Millar Instruments, Houston, TX) inserted through the right carotid artery in anesthetized mice and analyzed as described by us previously (2).
Adenoviral injection of IND-Akt mice.
Adenovirus harboring a constitutively active FOXO1 (Ad-FOXO1 AAA; generously provided by Pere Puigserver, Dana-Farber Cancer Institute) and green fluorescent protein (GFP) was directly injected into cardiac tissue of IND-Akt mice. Animals were withdrawn from DOX on the day of adenoviral injection. After 10 to 14 days of expression, hearts were excised and cardiomyocytes were isolated as previously described (11). Individual cardiomyocytes were manually separated to 10 cells per tube dependent on GFP fluorescence and analyzed using the CellsDirect two-step reverse transcription-quantitative PCR (qRT-PCR) kit with SYBR green (Life Technologies, Carlsbad, CA) according to the manufacturer's instructions and using primers detailed in Table S1 in the supplemental material.
Substrate metabolism in isolated working hearts.
Glycolysis, glucose oxidation, and palmitate oxidation were measured in isolated working hearts as previously described (11). Hearts were perfused with 0.4 mM palmitate and 5 mM glucose without insulin. Data are corrected to dry heart weight determined after perfusion.
Measurement of tissue triglyceride content.
Triglyceride content in cardiac tissue was measured after chloroform-ethanol extraction as described previously (12).
Mitochondrial respiration in permeabilized cardiac fibers.
Mitochondrial oxygen consumption and ATP production were measured in permeabilized cardiac fibers using techniques that have been previously described (13, 14). Briefly, fibers were prepared from left ventricular tissue (2 to 5 mg) and permeabilized with 50 μg/ml saponin. The respiratory rates of saponin-permeabilized fibers were determined using a fiber-optic oxygen sensor (Ocean Optics, Orlando, FL) in 2 ml KCl buffer at 25°C as previously described (13). Studies were performed with three independent substrates: (i) 5 mM glutamate and 2 mM malate, (ii) 10 mM pyruvate and 5 mM malate, or (iii) 0.02 mM palmitoyl-carnitine (PC) (which bypasses carnitine palmitoyltransferase 1 [CPT 1] and enters the mitochondria via CPT 2) and 5 mM malate. Maximally stimulated respiratory rates of permeabilized fibers following the addition of 1 mM ADP are defined as VADP.
ATP production in permeabilized cardiac fibers.
ATP concentration was determined as described previously (13). Briefly, saponin-permeabilized cardiac fibers were allowed to equilibrate for 2 min in 2 ml of buffer B at 25°C. After addition of 1 mM ADP, 10-μl samples of incubation buffer were obtained every 10 s for 1 min and were placed directly onto 190 μl of frozen dimethyl sulfoxide (DMSO). ATP was quantified by a bioluminescent assay based on the luciferin/luciferase reaction using the Enliten ATP assay system (Promega) in a Wallac 1480 Trilux scintillation and luminescence counter. ATP/O was calculated as the ratio of ATP synthesis rate divided by the VADP rate of respiration.
EM.
Samples were prepared and processed at the Core Research Microscopy Facility at the University of Utah. Briefly, small pieces of endocardial and subendocardial tissue from the left ventricle were fixed in 2.5% glutaraldehyde and 1% paraformaldehyde in 0.1 M sodium cacodylate, with 2.4% sucrose and 8 mM CaCl2 (pH 7.4) for at least a day. Samples were postfixed in 2% osmium tetroxide in 0.1 M sodium cacodylate, en bloc stained with aqueous uranyl acetate, and dehydrated through a graded series of ethanol washes (50% up to 100%). Fixed samples were then infiltrated with and embedded in Spurr's plastic and processed for electron microscopy (EM). Mitochondrial morphology was assessed at ×3,500, ×18,000, and ×70,000 magnifications. Mitochondrial volume density was determined as the mitochondrion-containing fraction of a 32-by-32 grid placed on pictures of ×3,500 magnification (n = 3 hearts and 2 pictures per heart). Mitochondrial number was determined in identical-size pictures of ×18,000 magnification (n = 3 hearts and 4 pictures per heart).
Mitochondrial isolation and activity assays.
Mitochondria were isolated from fresh heart tissue by differential centrifugation as previously described (15). Hearts were excised and immediately placed in ice-cold STE1 buffer (250 mM sucrose, 5 mM Tris-HCl, 2 mM EGTA, pH 7.4). Two hearts were pooled, minced, incubated in 2.5 ml STE2 buffer (STE1 containing 0.5% [wt/vol] bovine serum albumin [BSA], 5 mM MgCl2, 1 mM ATP, and 2.5 U/ml protease type VIII from Bacillus licheniformis) for 4 min, diluted with 2.5 ml STE1 buffer, and homogenized using a Teflon pestle in a Potter-Elvehjem glass homogenizer. The homogenate was further diluted with 5 ml STE1 buffer containing 1 tablet Complete Mini protease inhibitor cocktail (Roche, Indianapolis, IN) and centrifuged at 8,000 × g for 10 min. The resulting pellet was resuspended in STE1 buffer and centrifuged at 700 × g for 10 min. The resulting supernatant was centrifuged twice at 8,000 × g for 10 min, the pellet was resuspended in 1 ml buffer B (250 mM sucrose, 1 mM EDTA, 10 mM Tris-HCl, pH 7.4), and protein was quantified using the micro-bicinchoninic acid (micro-BCA) kit (Pierce) with BSA as a standard. All centrifugation steps were carried out at 4°C. CPT activity was determined in isolated mitochondria as previously described (16). Briefly, 25 μg of fresh mitochondrial protein was used to measure total CPT activity at 412 nm in 1 ml of reaction buffer (pH 7.4 at 25°C) containing 20 mM HEPES, 1 mM EGTA, 220 mM sucrose, 40 mM KCl, 0.1 mM 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB), 0.04 mM palmitoyl coenzyme A (palmitoyl-CoA), and 1 mM carnitine (omitted in blank) using an Ultrospec 3000 spectrophotometer. CPT 2 activity was measured as described above with addition of 30 μM malonyl-coenzyme A to the reaction mixture to inhibit CPT 1. CPT 1 activity was calculated as the difference between total CPT and CPT 2 activity. Aconitase activity was measured in isolated mitochondria as previously described (17). Briefly, 10 to 20 μg of frozen mitochondrial protein was used to measure aconitase activity at 240 nm in 1 ml of reaction buffer (pH 7.5 at 25°C) containing 50 mM Tris-HCl and 0.2 mM cis-aconitate (omitted in blank) using an Ultrospec 3000 spectrophotometer. Citrate synthase (CS) and 3′-hydroxyacyl-CoA dehydrogenase (HADH) activity were determined spectrophotometrically using whole-heart homogenates as previously described (13, 18, 19).
Comparative mitochondrial proteomics.
As described previously (20), mitochondrial isolates were loaded on a Percoll gradient (2.2 ml 2.5 M sucrose, 6.55 ml Percoll, 12.25 ml TE [10 mM Tris-HCl, 1 mM EDTA, pH 7.4]) and centrifuged at 60,000 × g for 45 min at 4°C. The lower layer was resuspended in 5 ml of buffer B and centrifuged three times at 10,000 × g for 10 min at 4°C. The pellet was resuspended in 100 μl 10 mM Tris-HCl, pH 8.5, and freeze-thawed three times (5 min of liquid nitrogen/5-min 37°C water bath). Fractionation was achieved by centrifuging the isolate at 40,000 × g for 20 min at 4°C. Centrifugation was repeated for the respective supernatant (matrix) and pellet (membrane) fractions to reduce membrane or matrix protein cross-contamination. Protein concentrations were determined using the micro-BCA protein assay kit (Pierce, Rockford, IL). Following isolation, in-solution tryptic digestion was performed; 5 μl of 0.2% RapidGest (Waters, Manchester, United Kingdom) was added to 20 μg of membrane protein sample in 15 μl H2O. The mixed solution was heated at 80°C for 20 min, and the protein mixtures were tryptically digested as described by the Waters Protein Expression System manual (66). After addition of NH4HCO3 and treatment with dithiothreitol and iodoacetamide, 4 μl of 0.11-μg/μl trypsin in 25 mM NH4HCO3 was added to the protein sample. Samples were incubated at 37°C overnight and then incubated with 1% formic acid for 30 min at 37°C and centrifuged at 10,000 × g for 10 min. The supernatant was used to determine the proteome. Digested protein samples (3 μl each) were introduced into a Symmetry C18 trapping column (180 μm by 20 mm) with the NanoACQUITY sample manager (Waters) and washed with H2O for 2 min at 10 ml/min. Using solvent A (99.9% H2O and 0.1% formic acid) and solvent B (99.9% acetonitrile and 0.1% formic acid), the peptides were eluted from the trapping column over a 100-μm by 100-mm BEH 130 C18 column with a 140-min gradient (1 to 4% B for 0.1 min, 4 to 25% B for 89.9 min, 25 to 35% B for 5 min, 35 to 85% B for 2 min, 85% B for 13 min, 85 to 95% B for 8 min, 95 to 1% B for 2 min, and 1% B for 20 min) at an 0.8-μl/min flow rate using a NanoACQUITY ultraperformance liquid chromatograph (UPLC) (Waters). The Premier (Waters) quadrupole time of flight (Q-TOF) mass spectrometer (MS) was set to a parallel fragmentation mode with scan times of 1.0 s. The low-fragmentation energy was 5 V, and the high-fragmentation energy was 17 to 45 V. Fibrinopeptide B (GLU1) was used as the external calibration standard with LockSpray. Enolase was used as the internal control. MS spectra were analyzed by Waters ProteinLynx Global Server (PLGS) 2.3. The following default settings were used for protein identification: minimum peptide matches per protein, 1; minimum fragment ion matches per peptide, 3; minimum fragment ion matches per protein, 7; and protein false-positive discovery rate, 4. Statistical analysis of proteomic data was performed using the Waters ProteinLynx Global Server version 2.3 software using a clustering algorithm, which chemically clusters peptide components by mass and retention time for all injected samples and performs binary comparisons for each experimental condition to generate an average normalized intensity ratio for all matched AMRT (accurate mass, retention time) components.
Microarray analysis.
Total myocardial RNA was labeled using the Affymetrix GeneChip One-Cycle target labeling and control kit as described in the Affymetrix eukaryotic RNA one-cycle cDNA synthesis and labeling manual using an MJ Research DNA Engine Tetrad 2 thermocycler (Bio-Rad Laboratories, Hercules, CA). Hybridization was performed using a GeneChip Hybridization Oven 640 (Affymetrix, Santa Clara, CA). Washing and staining were performed using an Affymetrix GeneChip Fluidics Station 650, Affymetrix murine expression U430 2.0 chips were scanned using an Affymetrix GeneChip Scanner 3000, and images were interpreted and monitored using GCOS version 1.2. Data were analyzed by standard methods using R (21) and Bioconductor (22) (open-source program; http://www.bioconductor.org) for all statistical analyses. Canonical pathway analysis was performed using Ingenuity Pathways Analysis (IPA) (Ingenuity Systems, Redwood City, CA), further examined by gene ontology terms (GO [23]), and visualized using Matrix2png (24).
Gene expression.
mRNA was quantified by real-time PCR (RT-PCR) as previously described (25). Total RNA was extracted from heart tissue with TRIzol reagent (Life Technologies) and purified with the RNeasy kit (Qiagen Inc., Valencia, CA). Equal amounts of RNA were used to synthesize cDNA with Superscript III (Life Technologies). RT-PCR was performed using an ABI Prism 7900HT instrument (Applied Biosystems, Foster City, CA) in a 384-well plate format with SYBR green I chemistry and 6-carboxyl-X-rhodamine (ROX) internal reference (Life Technologies). Actb was used as a template normalizer in Tet-Off Akt studies. Because Actb levels were significantly increased in caAkt samples, vascular endothelial growth factor A (Vegfa), which was unchanged between groups, was used as a template normalizer for caAkt studies. As both Actb and Vegfa were regulated in the in vitro studies, H3 histone, family 3A (H3f3a), was used as a template normalizer for cell culture studies. Primer sequences are listed in Table S1 in the supplemental material.
Western blot analysis.
Total proteins were extracted from frozen hearts as previously described (13). Proteins were resolved by SDS-PAGE and electrotransferred onto a polyvinylidene difluoride (PVDF) membrane (Millipore Corp., Bedford, MA). The following antibodies were used: phospho-Akt (Ser473), phospho-Akt (Thr308), Akt, phospho-p70 S6-kinase (Thr389), p70 S6-kinase, phospho-FOXO1 (Thr24), FOXO1, phospho-FOXO3 (Ser318/321), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and phospho-glycogen synthase kinase 3β (Ser9) (Cell Signaling Technology, Danvers, MA) and glycogen synthase kinase 3β and FOXO3 (Santa Cruz Biotechnology, Santa Cruz, CA). Protein detection was carried out with the appropriate IRDye 800CW–anti-mouse (Li-Cor, Lincoln, NE) or Alexa Fluor–anti-rabbit 680 (Life Technologies) secondary antibodies, and fluorescence was quantified using the Li-Cor Odyssey imager.
ChIP for transcription factor binding.
FOXO1 and FOXO3 promoter occupancy was determined by chromatin immunoprecipitation (ChIP) of nuclear extracts from hearts of caAkt and wild-type (WT) control mice as previously described (26). Freshly harvested ventricular tissue was pooled (100 to 150 mg) and homogenized, after which the nuclear pellet was subjected to 10 min of 1% formaldehyde cross-linking. The reaction was stopped by addition of 125 μM glycine. Following nuclear lysis, DNA was sheared by sonication; enrichment for 500-bp fragments was determined by agarose gel electrophoresis. Sheared chromatin complexes were precleared with preimmune rabbit serum, followed by dilution of an aliquot in immunoprecipitation dilution buffer (20 mM Tris-HCl, pH 8.0, 2 mM EDTA, 150 mM NaCl, 10% Triton X-100 [TX-100], 0.1% SDS, 10 mM n-butyric acid, 300 μM phenylmethylsulfonyl fluoride [PMSF], 1× Halt protease/phosphatase inhibitor) and incubated overnight at 4°C with immunoglobulin G (IgG) negative control, anti-FOXO1 (generously provided by Anne Brunet, Stanford University), anti-FOXO3 (Santa Cruz; sc-11381 X), or anti-RNA polymerase II (RPB1; Covance; MMS-126R) positive control. Antibody-chromatin complexes were bound with Dynabeads immobilized protein A/G (Pierce), followed by isolation and purification. Primer sequences for qPCR quantification of genomic regions are listed in Table S2 in the supplemental material, and corresponding candidate response elements are listed in Table S5 in the supplemental material. Quantitative PCR values of the immunoprecipitation are presented relative to a 2% input control and normalized to WT mouse heart values (=100%).
Primary NRVCMs and siRNA Foxo knockdown.
Primary neonatal rat ventricular cardiomyocytes (NRVCMs) were prepared as previously described (27). The biventricular portion of hearts from 1- to 3-day-old rat pups was mechanically and enzymatically (collagenase/pancreatin) separated, and cardiomyocytes were Percoll gradient purified and plated on gelatin-coated 6-well plates at a density of 106 cells in 2 ml per well. Following a 24-h recovery, medium was refreshed and cells were transfected with small interfering RNA (siRNA) for Foxo1 (RSS331474), Foxo3 (RSS334413), or control (12935-400) using Lipofectamine 2000 (Life Technologies) according to the manufacturer's instructions. Medium was refreshed daily for 5 days, at which time cells were harvested and RNA purified. qPCR was performed as described above, and primer sequences are listed in Table S1 in the supplemental material.
C2C12 myotubes and adenoviral FOXO1 expression.
C2C12 myoblasts were maintained at 37°C under 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) containing 4.5 g/liter glucose supplemented with 10% fetal bovine serum. When cells were confluent, cell medium was changed to DMEM supplemented with 2% horse serum for myotube differentiation. At the time of medium change, adenovirus for GFP or FOXO1-AAA (described above) was added at a multiplicity of infection sufficient to infect >95% of the cells based on the GFP fluorescence with minimal cell death. Cells were maintained for an additional 5 days prior to harvest and RNA purification. qPCR was performed as described above, and primer sequences are listed in Table S1 in the supplemental material.
caAkt retroviral transformation of C2C12 myotubes.
Plasmid for cDNA of a hemagglutinin (HA)-tagged constitutively active AKT1 (HA-myrAKT1; generously provided by Kenneth Walsh, Boston University School of Medicine) was digested with EcoRI and subcloned into pBABE-puro (Addgene, Cambridge, MA). Retrovirus packaging was performed by cotransfection of pCMV-gag-pol, pCMV-VSV-env, and pBABE-HA-myrAKT1 vectors into 293T cells. The resulting virus was used to infect C2C12 myoblasts, and positive cells were selected by 3-μg/ml puromycin treatment for 3 days. Positive cell lines were then maintained in 2 μg/ml puromycin. Cells were differentiated as described above and analyzed as follows. qPCR was performed as described above, and primer sequences are listed in Table S1 in the supplemental material. Additional sets of cells were used for cellular respiration with the XF24 extracellular flux (XF) bioanalyzer (Seahorse Bioscience, North Billerica, MA). Cells were plated at a density of 20 × 103 per well and differentiated as described above. At the time of medium change, adenovirus for GFP, FOXO1-AAA (described above), or PGC-1α (generously provided by Daniel P. Kelly, Sanford-Burnham Medical Research Institute [28]) was added at a multiplicity of infection sufficient to infect >95% of the cells based on the GFP fluorescence with minimal cell death. Prior to analysis, medium was changed to XF-DMEM and cells were kept in a non-CO2 incubator for 30 min. Basal oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured in XF-DMEM followed by these additional conditions: oligomycin (1 μg/ml), carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) (1 μM), rotenone (1 μM), and antimycin A (10 μM). Data are normalized to protein and represent the mean ± standard error of the mean (SEM), with n of >5 biological replicates per group. Detailed calculations are described elsewhere (29, 30). In parallel, cells were plated at 160 × 103 per well in 6-well plates and treated identically as described above. Following treatment, cells were harvested, and high-performance liquid chromatography (HPLC) separation and measurement of adenine nucleotides were performed as described previously (31).
Statistical analysis.
Data are expressed as mean ± SEM. Unpaired Student's t test was used to analyze data sets between two groups unless otherwise stated. Data sets with more than two groups were analyzed by analysis of variance (ANOVA), and significance was assessed by Fisher's protected least significant difference test. For all analyses, a P value of <0.05 was accepted as indicating a significant difference. Statistical calculations were performed using the StatView 5.0.1 software package or the JMP PRO 9.0 software package (SAS Institute, Cary, NC).
Microarray data accession number.
Microarray data may be found on the Gene Expression Omnibus (GEO) website under accession number GSE63848 (http://www.ncbi.nlm.nih.gov/geo).
RESULTS
Transition from compensated cardiac hypertrophy to heart failure is associated with Akt activation and FOXO inhibition.
We examined phosphorylation of Akt and its targets the transcription factors FOXO1 and FOXO3 in murine models of LVH following TAC with and without compensation (Fig. 1A). Mice with compensated LVH developed a 28.8% increase in the ratio of heart weight to body weight (HW/BW) in the sham group, 4.65 ± 0.10 mg/g, versus the 27G-TAC group, 5.99 ± 0.27 mg/g (n = 10; P < 0.01), and cardiac function was maintained, as evidenced by preserved fractional shortening (FS; 26.3% ± 1.0% for sham versus 24.1% ± 1.1% for 27G-TAC). Protein levels of Akt, FOXO1, and FOXO3 and phosphorylation status revealed no significant changes (Fig. 1). Mice with decompensated LVH (30G-TAC) had a 54.2% increase in HW/BW in the sham group, 4.51 ± 0.15 mg/g, versus the 30G-TAC group, 6.95 ± 0.47 mg/g (n = 8; P < 0.01), with a 19% reduction in FS (28.4% ± 1.8% for sham versus 22.9% ± 0.5% for 30G-TAC [n = 5; P < 0.05]). Akt phosphorylation increased (Fig. 1A to C). Although total FOXO1 declined, it was not significantly phosphorylated (Fig. 1D and F). FOXO3 phosphorylation was modestly but significantly increased (Fig. 1E).
FIG 1.
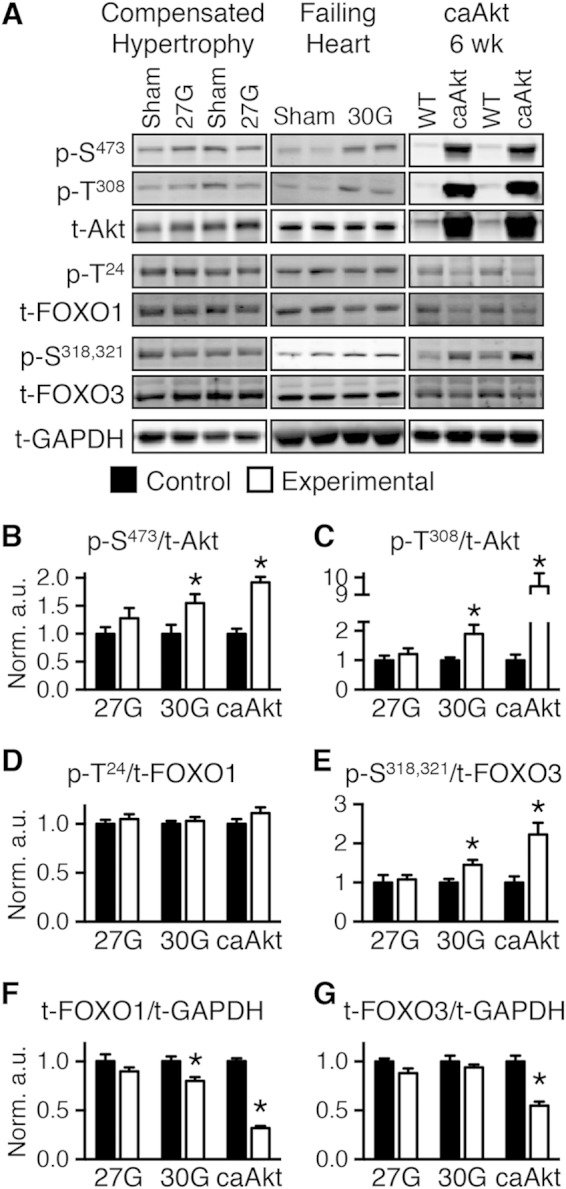
Transition from compensated cardiac hypertrophy to heart failure is associated with Akt activation and FOXO inhibition. (A) Representative Western blots of whole-heart extract from mice banded for 4 weeks at 27G-TAC (left) and 30G-TAC (middle) and 6- to 7-week-old caAkt mice (right). (B to E) Quantification of Western blotting results for phosphorylated to total protein: Akt at Ser473 (p-S473) (B) and Thr308 (p-T308) (C), FOXO1 at Thr24 (p-T24) (D), and FOXO3 at Ser318 and Ser321 (p-S318,321) (E) (n = 6 to 8). (F and G) Quantification of Western blotting results for total FOXO1 (F) and FOXO3 (G) normalized to total GAPDH (n = 6 to 8). Data shown as mean ± SEM. *, P < 0.05 versus group control. a.u., arbitrary units.
Reduced function and substrate metabolism in hearts with constitutive activation of Akt.
To determine if persistent Akt activation in the heart could precipitate pathological LVH and mitochondrial dysfunction, we investigated mice with cardiomyocyte-restricted constitutively activated Akt1 (caAkt). Hearts from caAkt mice were previously reported to exhibit decreased FS by 14 weeks of age (6). We therefore analyzed an earlier time point in 6-week-old mice. By noninvasive echocardiography, trends toward dysfunction were present even at this younger age (Table 1). Using more sensitive measures by LV catheterization and organ weights, we found that significant LVH, contractile dysfunction, and pulmonary edema were present as early as 6 weeks of age (Table 2). Total and phosphorylated Akt were increased (Fig. 1A to C), with no change in phosphorylation of FOXO1 (Fig. 1D) and a significant increase in phosphorylation of FOXO3 (Fig. 1E). Total FOXO1 and FOXO3 proteins were decreased (Fig. 1F and G).
TABLE 1.
Echocardiography for in vivo cardiac function in WT and caAkt mice (6 weeks)
| Parametera | Valueb |
|
|---|---|---|
| WT | caAkt | |
| LVDd (mm) | 3.38 ± 0.21 | 3.68 ± 0.27 |
| LVDs (mm) | 2.10 ± 0.19 | 2.47 ± 0.25 |
| IVSd (mm) | 0.74 ± 0.07 | 0.86 ± 0.04 |
| IVSs (mm) | 1.01 ± 0.09 | 1.06 ± 0.08 |
| LVPWd (mm) | 1.03 ± 0.10 | 1.04 ± 0.07 |
| LVPWs (mm) | 1.42 ± 0.09 | 1.34 ± 0.12 |
| FS (%) | 38.8 ± 2.3 | 31.4 ± 3.6 |
| EF (%) | 56.1 ± 2.1 | 47.8 ± 5.9 |
| HR (bpm) | 356 ± 12 | 302 ± 27 |
LVDd and LVDs, left ventricular cavity diameter at diastole and systole, respectively; IVSd and IVSs, interventricular septum diameter at diastole and systole, respectively; LVPWd and LVPWs, LV posterior wall thickness at diastole and systole, respectively; FS, fractional shortening; EF, ejection fraction; HR, heart rate; bpm, beats per minute.
n = 5 to 8.
TABLE 2.
LV catheterization for hemodynamic parameters and organ weights in WT and caAkt mice (6 weeks)
| Parametera | Value |
|
|---|---|---|
| WT | caAktb | |
| Arterial SP (mm Hg) | 103.8 ± 2.4 | 65.9 ± 4.9* |
| Arterial DP (mm Hg) | 76.3 ± 2.0 | 48.1 ± 6.0* |
| LV SP (mm Hg) | 98.2 ± 3.0 | 62.1 ± 4.4* |
| LV EDP (mm Hg) | 11.6 ± 1.8 | 23.6 ± 2.0* |
| LV DevP (mm Hg) | 98.5 ± 2.6 | 53.0 ± 3.4* |
| +dP/dt (mm Hg/s) | 9,381 ± 921 | 2,670 ± 216* |
| −dP/dt (mm Hg/s) | −8,227 ± 562 | −3,002 ± 207* |
| HR (beats/min) | 528 ± 6 | 482 ± 17* |
| Body wt (g) | 21.6 ± 0.6 | 20.8 ± 0.7 |
| Heart wt (mg) | 96.2 ± 2.6 | 182.4 ± 1.5* |
| HW/BW | 4.45 ± 0.07 | 8.81 ± 0.15* |
| LW/BW | 7.31 ± 0.27 | 9.00 ± 0.39* |
SP, systolic pressure; DP, diastolic pressure; LV, left ventricular; EDP, end-DP; DevP, developed pressure; +dP/dt, peak rate of LV pressure rise; −dP/dt, peak rate of LV pressure decline; HR, heart rate; HW, heart weight; BW, body weight; LW, lung weight.
*, P < 0.05 versus wild type (WT); n = 7.
We also determined metabolism and cardiac function in isolated perfused working hearts from 18-week-old mice. Dry heart weight-to-body weight ratio (DHW/BW) was increased by 2.1-fold relative to controls (Fig. 2A), and function was decreased (Table 3 and Fig. 2B), as were rates of glycolysis, glucose oxidation, palmitate oxidation, and myocardial oxygen consumption (MVO2) (Fig. 2C to F). Despite the strong trend toward reduced fatty acid oxidation (FAO), triglyceride content decreased from 17.0 ± 2.3 μmol/g in WT control hearts to 10.7 ± 0.4 μmol/g in caAkt hearts (n = 7 to 8 per group, P < 0.05). The decline in mitochondrial oxidative capacity was independent of changes in mitochondrial volume density or number at either 6 weeks or 18 weeks of age (Fig. 2G to I).
FIG 2.

Impaired function and substrate metabolism in hearts from mice with constitutive activation of Akt in cardiomyocytes (caAkt). (A) Dry heart weight-to-body weight ratio (DHW/BW) in caAkt and WT mice (n = 4). (B to F) Cardiac power (B), glycolysis (C), glucose oxidation (D), palmitate oxidation (P = 0.08) (E), and MVO2 (F) in isolated working hearts from 18-week-old WT and caAkt mice (n = 4). (G) Representative electron micrographs of cardiac tissue from 18-week-old WT and caAkt mice. (H and I) Mitochondrial volume density (H) and mitochondrial number (I) per high-power field (hpf) in electron micrographs of 6- and 18-week-old WT and caAkt hearts (n = 3 or 4). Data are shown as mean ± SEM. *, P < 0.05 versus WT of same age.
TABLE 3.
Cardiac function in isolated working hearts from WT and caAkt mice (18 weeks)
| Parametera | Value |
|
|---|---|---|
| WT | caAktb | |
| HR (beats/min) | 305 ± 11 | 276 ± 24* |
| Aortic SP (mm Hg) | 61.1 ± 1.3 | 54.2 ± 0.8* |
| Aortic DP (mm Hg) | 31.7 ± 0.6 | 35.0 ± 0.8* |
| DevP (mm Hg) | 29.5 ± 1.6 | 19.2 ± 0.7* |
| Coronary flow (ml/min) | 4.11 ± 0.09 | 4.52 ± 0.45 |
| Aortic flow (ml/min) | 10.33 ± 0.56 | 2.91 ± 0.20* |
| CO (ml/min) | 14.43 ± 0.59 | 7.43 ± 0.52* |
HR, heart rate; aortic SP, aortic systolic pressure; aortic DP, aortic diastolic pressure; DevP, developed pressure; CO, cardiac output.
*, P < 0.05 versus wild type (WT); n = 4.
Constitutive activation of Akt leads to progressive mitochondrial dysfunction.
Akt activation was associated with a decline in mitochondrial oxygen consumption with palmitoyl-carnitine (PC) as a substrate as early as 6 weeks of age (Fig. 3A). In contrast, mitochondrial respiration with pyruvate-malate (PM) was relatively preserved (Fig. 3B), with glutamate-malate (GM) declining gradually only at 18 weeks of age (Fig. 3C). Defective mitochondrial ATP synthesis was present with all substrates as early as 6 weeks of age and declined progressively as duration of transgene activation increased (Fig. 3D to F). The ratio of ATP synthesis rates to ADP-stimulated (VADP) maximal respirations (ATP/O) provides an index of mitochondrial coupling. ATP/O ratios were significantly reduced at 6 weeks and 18 weeks of age regardless of substrate (Fig. 3G to I).
FIG 3.
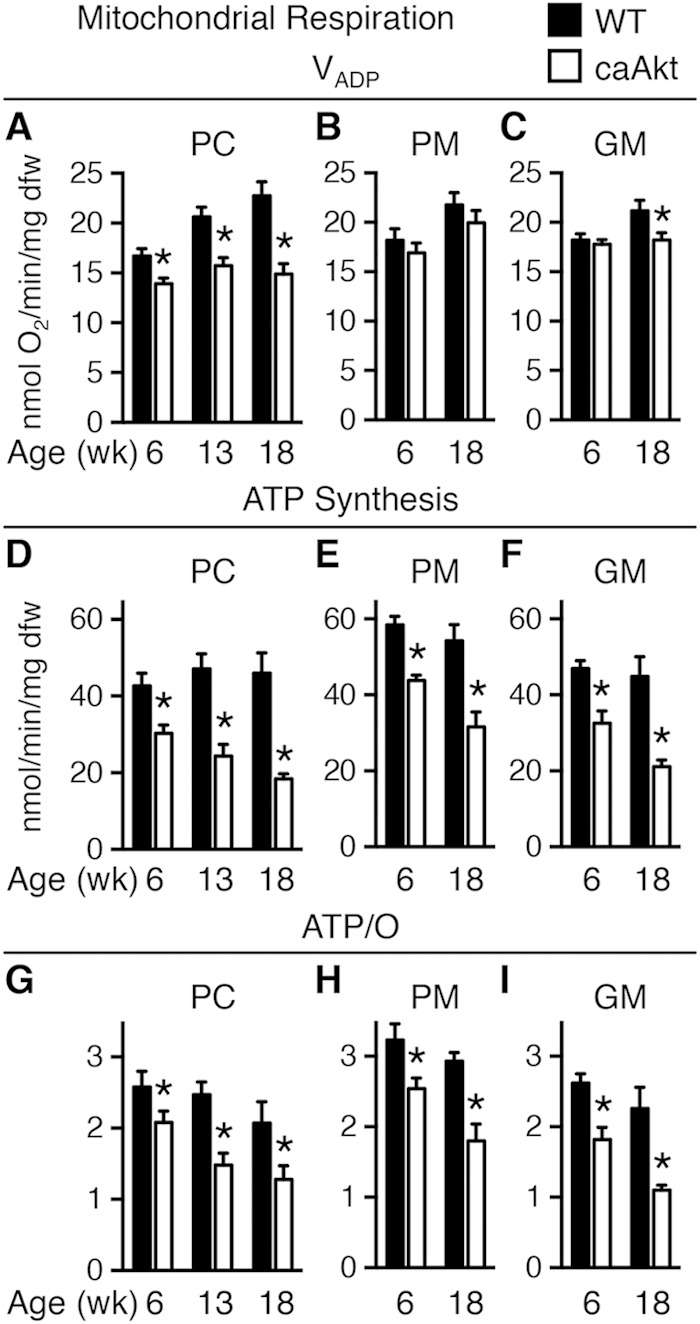
Age-related impairment of mitochondrial function in saponin-permeabilized cardiac fibers from mice with constitutive activation of cardiac Akt (caAkt) at 6, 13, or 18 weeks of age. (A to C) Changes in maximal ADP-stimulated mitochondrial respiration (VADP) with the following substrates: palmitoyl-carnitine (PC) (A), pyruvate-malate (PM) (B), or glutamate-malate (GM) (C). (D to F) ATP synthesis rates in similarly treated saponin-permeabilized cardiac fibers with PC (D), PM (E), or GM (F) as the substrate. (G to I) ATP/O ratios with PC (G), PM (H), or GM (I) as the substrate. dfw, dry fiber weight. n = 4 to 6 per group. Data are shown as mean ± SEM. *, P < 0.05 versus WT of the same age and substrate.
Despite decreased ATP synthesis, AMP-activated protein kinase (AMPK) phosphorylation was lower in caAkt hearts at 6 weeks of age (Fig. 4A). Oxidative stress was suggested by reduced levels of mitochondrial aconitase (Fig. 4B). Additional indices of mitochondrial dysfunction were evidenced by reduced activity levels of two important regulators of mitochondrial β-oxidation. Total carnitine palmitoyltransferase (CPT) activity (Fig. 4C) and hydroxyacyl-CoA dehydrogenase (HADH) activity were reduced in hearts from 6-week-old caAkt mice (Fig. 4D). HADH declined further by 18 weeks of age (Fig. 4D). Citrate synthase (CS) activity, a rate-limiting step in the citric acid cycle, was reduced in both 6- and 18-week-old caAkt hearts (Fig. 4E).
FIG 4.
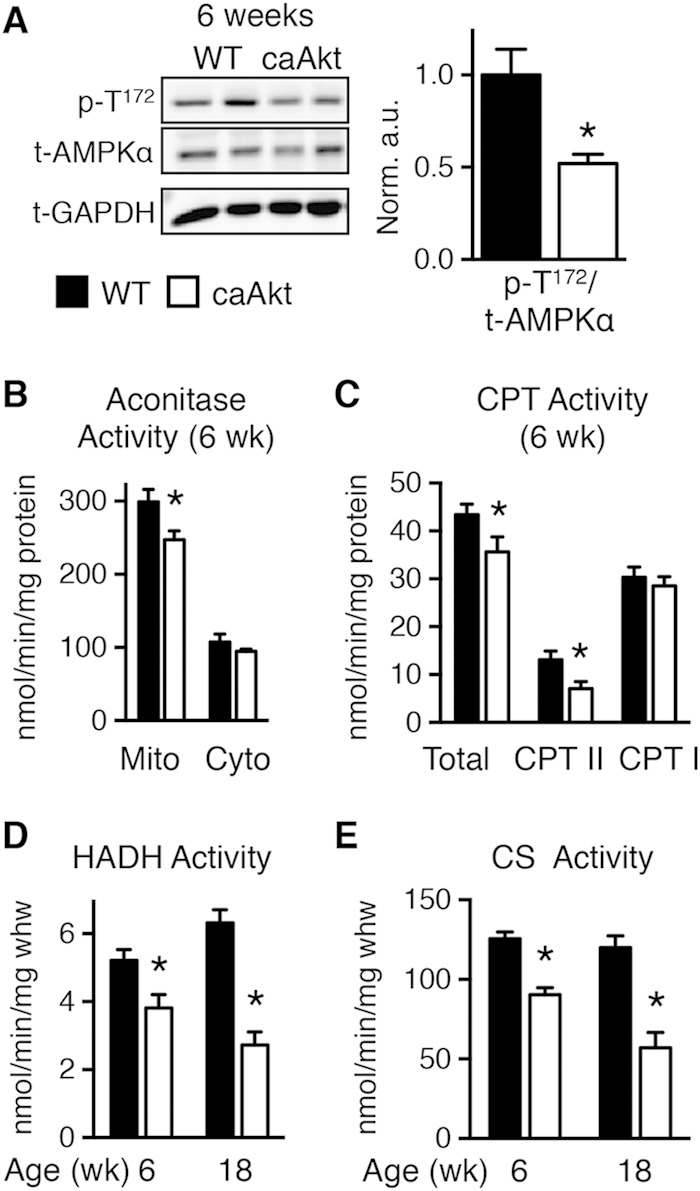
Impaired energy signaling and mitochondrial enzymatic activities in hearts of 6- and 18-week-old caAkt mice. (A) Western blot analysis of whole-heart protein extract from wild-type (WT) and caAkt mice at 6 weeks of age (left). Quantification of Western blot analysis for phosphorylated AMPKα at Thr172 (p-T172) to total AMPKα (n = 6). (B) Aconitase enzymatic activity in mitochondrial and cytosolic fractions of cardiac tissue from 6-week-old WT and caAkt mice. (C) Carnitine palmitoyltransferase (CPT) enzymatic activities in isolated mitochondria from hearts of 6-week-old WT and caAkt mice (n = 4 to 6). (D) Hydroxyacyl-CoA dehydrogenase (HADH) in hearts from 6- and 18-week-old WT and caAkt mice (n = 4 to 6). (E) Citrate synthase (CS) enzymatic activities in WT and caAkt hearts (n = 4 to 6). Data shown as mean ± SEM. *, P < 0.05 versus age-matched WT.
Short-term activation of an inducible Akt1 transgene in the heart increases glycolysis and impairs mitochondrial FAO.
To determine if mitochondrial dysfunction is a direct effect of Akt activation or is secondary to cardiac hypertrophy or failure, we studied mice with inducible activation of a myristoylated Akt (IND-Akt). These hearts exhibit a time-dependent activation of Akt and LVH (Fig. 5A and B); as early as 7 days, there is a significant increase, and by 10 days, the heart weight has increased by 61.3% ± 9.9% (Fig. 5B). Prior reports suggested that cardiac function was not decreased following 4 weeks of transgene induction (10). However, we were unable to maintain cardiac function in isolated perfused hearts after 21 days of Akt activation. Thus, we determined myocardial substrate metabolism in working hearts after 14 days of doxycycline withdrawal. There was a reduction in cardiac output and cardiac power (Table 4) that accompanied cardiac hypertrophy (Fig. 5C). Rates of glycolysis were increased 2.3-fold at 14 days of induction in IND-Akt hearts (Fig. 5D), but glucose oxidation rates were unchanged (Fig. 5E). Palmitate oxidation rates were reduced after 14 days in IND-Akt hearts (Fig. 5F).
FIG 5.
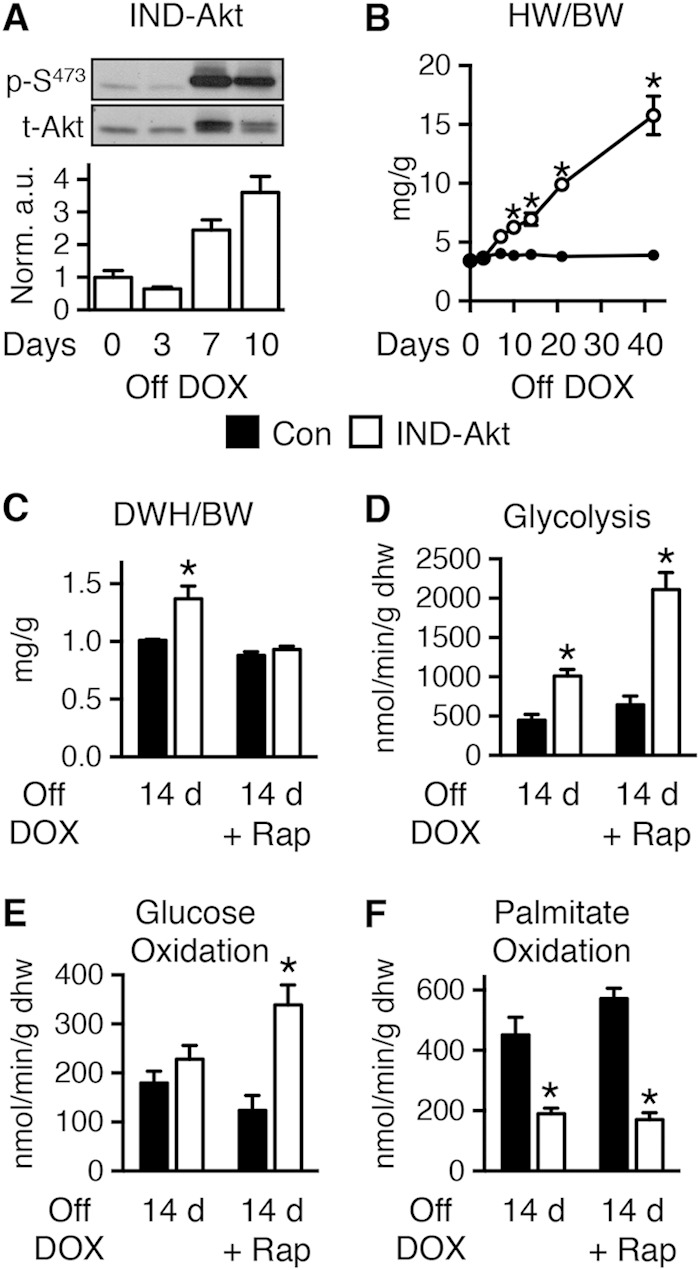
Short-term activation of Akt increased glycolysis and reduced FAO in the heart, independently of hypertrophy. (A) Representative immunoblots and ratios of phosphorylated Akt at Ser473 (p-S473) to total Akt in hearts from IND-Akt mice withdrawn from doxycycline (DOX) (n = 3). (B) Heart weight-to-body weight ratio (HW/BW) changes with time zero (n = 3 or 4) and 3 (n = 3), 7 (n = 3 or 4), 10 (n = 9), 14 (n = 3), 21 (n = 6), and 42 (n = 4 or 8) days in control mice (Con) and IND-Akt mice, respectively. (C) Dry heart weight (DHW)-to-body weight ratios (DHW/BW) in control and IND-Akt mice withdrawn from DOX for 14 days and treated daily with rapamycin (Rap) were obtained after isolated working heart perfusions (n = 9). (D to F) Glycolysis (n = 4) (D), glucose oxidation (n = 4) (E), and palmitate oxidation (n = 5) (F) in isolated working hearts from control and IND-Akt mice withdrawn from DOX for 14 days and treated daily with rapamycin (Rap). Data shown as mean ± SEM. *, P < 0.05 versus induction and treatment-matched control.
TABLE 4.
Cardiac function in isolated working hearts from control and IND-Akt mice (14 days)
| Parametera | Value for the group by treatmentb |
|||
|---|---|---|---|---|
| Vehicle |
Rapamycin |
|||
| Control | IND-Akt | Control | IND-Akt | |
| HR (beats/min) | 259 ± 11 | 226 ± 14 | 233 ± 9 | 224 ± 16 |
| Aortic SP (mm Hg) | 75.6 ± 1.5 | 79.3 ± 1.6 | 74.2 ± 1.0 | 74.9 ± 1.0 |
| Aortic DP (mm Hg) | 27.5 ± 1.3 | 25.4 ± 1.4 | 24.7 ± 1.8 | 29.1 ± 1.5 |
| DevP (mm Hg) | 48.1 ± 1.8 | 53.9 ± 2.0 | 49.5 ± 2.1 | 45.9 ± 1.8 |
| Coronary flow (ml/min) | 4.0 ± 0.1 | 4.6 ± 0.2* | 3.4 ± 0.1 | 3.4 ± 0.2 |
| Aortic flow (ml/min) | 8.0 ± 0.5 | 5.8 ± 0.3* | 6.3 ± 0.4 | 6.7 ± 0.3 |
| CP (mW/g) | 31.9 ± 1.7 | 23.5 ± 1.2* | 34.2 ± 1.8 | 29.2 ± 1.9* |
| CO (ml/min) | 12.0 ± 0.6 | 10.3 ± 0.3* | 9.8 ± 0.4 | 10.1 ± 0.4 |
HR, heart rate; aortic SP, aortic systolic pressure; aortic DP, aortic diastolic pressure; CP, cardiac power; CO, cardiac output; DevP, developed pressure.
*, P < 0.05 versus control for same treatment; n = 3 to 5.
Blocking cardiac hypertrophy does not prevent Akt-induced changes in mitochondrial function.
To distinguish between direct effects of Akt activation and potential independent contributions of LVH and LV dysfunction to mitochondrial dysfunction, we treated IND-Akt mice following transgene induction with rapamycin. Rapamycin treatment prevented hypertrophy in IND-Akt mice (Fig. 5C) and mostly preserved cardiac function (Table 4). After 14 days of rapamycin treatment and DOX withdrawal, rates of glycolysis and glucose oxidation were increased and rates of palmitate oxidation remained decreased in IND-Akt hearts (Fig. 5D to F).
To further distinguish between Akt-induced changes in LVH and mitochondrial function, we examined a cohort of animals that were induced for 10, 21, or 42 days of DOX withdrawal. Mitochondrial respiration and ATP production rate were preserved at 10 days of induction (Fig. 6A and B). However, by 21 days there was a significant decline in mitochondrial oxygen consumption and a proportionate reduction in ATP synthesis, so that ATP/O ratios were unchanged (Fig. 6A to C). The decline continued out to 42 days, and reduced ATP/O ratios suggested that mitochondria had become uncoupled with this longer induction (Fig. 6A to C). Activity levels of HADH tended to decline, and CS was already significantly reduced in IND-Akt mouse hearts after 10 days of DOX withdrawal, with no further decline at day 21 (Fig. 6D and E).
FIG 6.

Mitochondrial function is impaired following 10, 21, or 42 days of Akt induction. (A to C) Changes in maximal ADP-stimulated mitochondrial respiration (VADP) (A), ATP synthesis rates (B), and ATP/O ratios (C) in saponin-permeabilized cardiac fibers treated with palmitoyl-carnitine (PC) as the substrate from control (Con) or IND-Akt mice withdrawn from DOX for 10 days (n = 8 or 7), 21 days (n = 6 or 8), 21 days with rapamycin (Rap) (n = 8 or 9), or 42 days (n = 8 or 9), respectively. (D and E) Hydroxyacyl-CoA dehydrogenase (HADH) enzymatic activity (D) and citrate synthase (CS) enzymatic activity (E) from hearts of Con or IND-Akt mice withdrawn from DOX for 10 days (n = 6), 21 days (n = 4), or 21 days plus Rap (n = 3), respectively. (F) Heart weight-to-body weight ratio (HW/BW) of Con or IND-Akt mice withdrawn from DOX for 10 days (n = 7 or 10), 21 days (n = 6 or 9), or 21 days with Rap (n = 8 or 9), respectively. (G to I) Rapamycin alters signaling in IND-Akt hearts withdrawn from DOX. Ratio of phosphorylated p70 S6 kinase at Thr389 (p-T389) to total S6K (H) and ratio of phosphorylated glycogen synthase kinase 3β at Ser9 (p-S9) to total GSK-3β (I) in hearts from control and IND-Akt mice treated daily with rapamycin or vehicle. dfw, dry fiber weight; whw, wet heart weight; n.s., nonspecific band loading control. Data shown as mean ± SEM. *, P < 0.05 versus induction and treatment-matched control; #, P < 0.05 versus vehicle-treated animals of the same genotype.
Rapamycin treatment during 21-day transgene induction reduced cardiac hypertrophy (Fig. 6F) and prevented activation of mechanistic target of rapamycin (mTOR) and phosphorylation of S6 kinase (S6K) (Fig. 6G and H), without altering phosphorylation of glycogen synthase kinase-3β (GSK 3β) (Fig. 6G and I). The reversal of cardiac hypertrophy did not prevent defects in mitochondrial respiration and ATP synthesis rates with PC as the substrate in cardiac fibers from rapamycin-treated IND-Akt mice (Fig. 6A to C). Unlike the whole-heart metabolism and mitochondrial respiration results, HADH and CS enzymatic activities were normalized in rapamycin-treated IND-Akt mice (Fig. 6D and E).
Constitutive activation of Akt selectively represses protein levels of TCA cycle, OXPHOS, and FAO pathways.
To define the molecular mechanisms mediating the Akt-induced changes, we examined levels of mitochondrial proteins to determine if there was a common pathway being regulated. We performed proteomics analysis on isolated and fractionated mitochondria from 8-week-old caAkt and WT mouse hearts from 6 mice pooled in pairs for an n value of 3 per group. These analyses identified 174 proteins with 59 of significant or unique expression (see Table S3a in the supplemental material). The regulated proteins were enriched in tricarboxylic acid (TCA) cycle, oxidative phosphorylation (OXPHOS), and FAO proteins as identified by ingenuity pathway analysis (IPA) (see Table S3b in the supplemental material). The significantly regulated proteins from these pathways are shown by a heat map (Fig. 7A).
FIG 7.

Activation of Akt in the heart alters protein levels and gene expression of mitochondrion-related targets. (A) Heat maps of top canonical pathways of changes in mitochondrial proteins by ingenuity pathway analysis (IPA) (n = 3). (B) Microarray results of mRNA levels of the same three statistically changed proteomics canonical pathways in hearts of WT and caAkt mice (n = 3). (C) qPCR quantification of mRNA levels of OXPHOS and FAO genes in hearts from 6- and 15-week-old caAkt mice (n = 6). (D) qPCR quantification of mRNAs for OXPHOS and FAO genes in hearts from tON-Akt mice following 10 days of transgene induction (n = 6). (E and F) qPCR quantification of mRNAs for transcriptional regulators in hearts from caAkt (E) and tON-Akt (F). (G) qPCR quantification of mRNA levels measured in control and IND-Akt hearts induced for 21 days and treated with/without rapamycin (Rap). Data shown as mean ± SEM. Gene names are described in Table S1 in the supplemental material. *, P < 0.05 versus control. Veh, vehicle.
Gene expression profiling in caAkt hearts revealed global repression of genes for metabolic pathways.
We next used microarray analysis to determine if transcriptional mechanisms contributed to the Akt-induced mitochondrial dysfunction and proteome remodeling. The number of transcripts significantly regulated by constitutive activation of Akt was 2,385 (see Table S4a in the supplemental material). IPA revealed that a number of metabolic and signaling pathways were significantly regulated (see Table S4b). Genes for the same pathways identified by proteomics are shown by a heat map (Fig. 7B).
A subset of genes identified by microarray or with known involvement in OXPHOS, FAO, and transcriptional regulation was measured by qPCR in hearts of 6- and 15-week-old WT and caAkt mice. Expression of genes involved in OXPHOS was reduced in caAkt hearts relative to WT as early as 6 weeks of age and continued to decline at 15 weeks of age (Fig. 7C). Similarly, expression of a number of FAO genes was reduced in caAkt hearts at either age examined (Fig. 7C). To address potential differences between altered signaling from birth and induced Akt activation in the adult heart, we examined an independent model of inducible Akt activation. These DOX-inducible, tON-Akt mice were examined following 10 days of induction, a time point that phenocopies the 21-day IND-Akt model (Table 5). In contrast to caAkt hearts, a broad downregulation of OXPHOS and FAO gene expression levels was not observed at 10 days in tON-Akt hearts, but some changes were evident at this time point, such as those in Cyct, Suclg2, and Hadhb (Fig. 7D).
TABLE 5.
LV catheterization for hemodynamic parameters and organ weights in control and tON-Akt mice (7 to 10 days)
| Parametera | Valueb |
|
|---|---|---|
| Control | tON-Akt | |
| Arterial SP (mm Hg) | 98.0 ± 5.2 | 71.5 ± 5.5* |
| Arterial DP (mm Hg) | 72.4 ± 4.0 | 52.0 ± 4.2* |
| LV SP (mm Hg) | 93.8 ± 3.6 | 76.5 ± 6.0* |
| LV EDP (mm Hg) | 13.9 ± 2.4 | 20.2 ± 10.0* |
| LV DevP (mm Hg) | 92.8 ± 3.5 | 69.1 ± 9.4* |
| +dP/dt (mm Hg/s) | 9,148 ± 867 | 5,257 ± 849* |
| −dP/dt (mm Hg/s) | −7,905 ± 458 | −4,048 ± 427* |
| HR (beats/min) | 497 ± 28 | 440 ± 14 |
| Body wt (g) | 21.6 ± 0.6 | 21.7 ± 0.6 |
| Heart wt (mg) | 101.1 ± 2.6 | 308.5 ± 12.5* |
| HW/BW | 4.70 ± 0.15 | 14.32 ± 0.75* |
| LW/BW | 7.47 ± 0.22 | 9.22 ± 0.36* |
SP, systolic pressure; DP, diastolic pressure; LV, left ventricular; EDP, end-DP; DevP, developed pressure; +dP/dt, peak rate of LV pressure rise; −dP/dt, peak rate of LV pressure decline; HR, heart rate; HW, heart weight; BW, body weight; LW, lung weight.
*, P < 0.05 versus control; n = 6 to 9.
To define potential pathways regulating these changes in gene expression, we examined the expression of transcriptional regulators identified on the array or those with known contributions to metabolic regulation. The most repressed transcriptional gene on the array is Ppargc1a, along with a number of its DNA-binding partners, including genes for peroxisome proliferator-activated receptor α (PPARα) and FOXO1, which were decreased (see Table S4a in the supplemental material). This reduction of mRNA for the transcriptional regulator Ppargc1a was found as early as 6 weeks in caAkt mice (Fig. 7E). A number of known PGC-1α transcription factor binding partners were also regulated at this time point, including Foxo1, Nef2l2 (also known as Nrf2), and Ppara, but not their related family members Foxo3, Nrf2, and Ppargc1b. There was a similar pattern of expression at 15 weeks in caAkt mice (data not shown). Interestingly, Gata4 and Hif1a expression was induced at 6 weeks in caAkt hearts (Fig. 7E). There was an overlapping pattern of changes in gene expression found in day 10 tON-Akt adult mouse hearts, including those in Foxo1, Hif1a, Nef2l2, Ppara, and Ppargc1a (Fig. 7F). Additionally, Esrrg and Foxo3 were repressed following 10 days of Akt activation (Fig. 7F). To test the hypothesis that Akt activation could repress mitochondrial target genes independently of hypertrophy, we examined the expression of the two most consistent changes across all models, those in Ppara and Ppargc1a. The mRNA levels of these two remained repressed following rapamycin treatment in IND-Akt (21-day) hearts compared to controls (Fig. 7G). This was at a time when mitochondrial dysfunction was evident (Fig. 6).
Thus, Ppara and Ppargc1a are similarly repressed in three independent models of Akt induction. Similarly to the other models of Akt induction (Fig. 1 and 4), inducing Akt for 10 days in the adult heart in the tON-Akt mice did not change FOXO1 phosphorylation, increased FOXO3 phosphorylation, and impaired AMPK phosphorylation (Fig. 8A to D). This was in the presence of decreased total FOXO1 and FOXO3 with no change in total AMPK (Fig. 8E to G).
FIG 8.

Short-term transgenic activation of Akt is associated with inhibition of FOXO and AMPK. (A) Representative Western blot analysis of whole-heart extract from Con or tON-Akt mice following 10 days of DOX treatment. (B to G) Quantification of Western blot analysis for ratios of phosphorylated to total protein or total protein to loading control (GAPDH): FOXO1 at Thr24 (p-T24), FOXO3 at Ser318,321 (p-S318,321), AMPKα at Thr172 (p-T172) to total protein and total FOXO1, FOXO3, and AMPKα to GAPDH (n = 6). Data shown as mean ± SEM normalized to control (=1.0). *, P < 0.05 versus control.
Role of FOXO transcription factors in Akt-induced changes in mitochondrial function.
To further define a mechanism for altered OXPHOS gene expression in caAkt hearts, we examined the promoter regions for FOXO regulatory elements (FOXO-REs) (TTGTTTAC [32]), as FOXO is a known target of Akt-mediated regulation (33). We used the Transcriptional Regulatory Element Database to identify candidate FOXO-REs (34). All 44 downregulated OXPHOS gene promoters contained at least one candidate FOXO-RE (see Table S5 in the supplemental material). To determine FOXO promoter occupancy, we performed chromatin immunoprecipitation (ChIP) experiments using antibodies to either FOXO1 or FOXO3. As Ppargc1a and Foxo1 were previously identified as FOXO transcriptional targets (35, 36), we also examined their promoter regions. None of the candidate promoters exhibited statistically different occupancy of FOXO3 in caAkt compared to WT hearts (data not shown). However, Cyct, Ppargc1a, and Suclg2 all revealed significantly less FOXO1 occupancy in caAkt hearts than in WT hearts (Fig. 9A).
FIG 9.
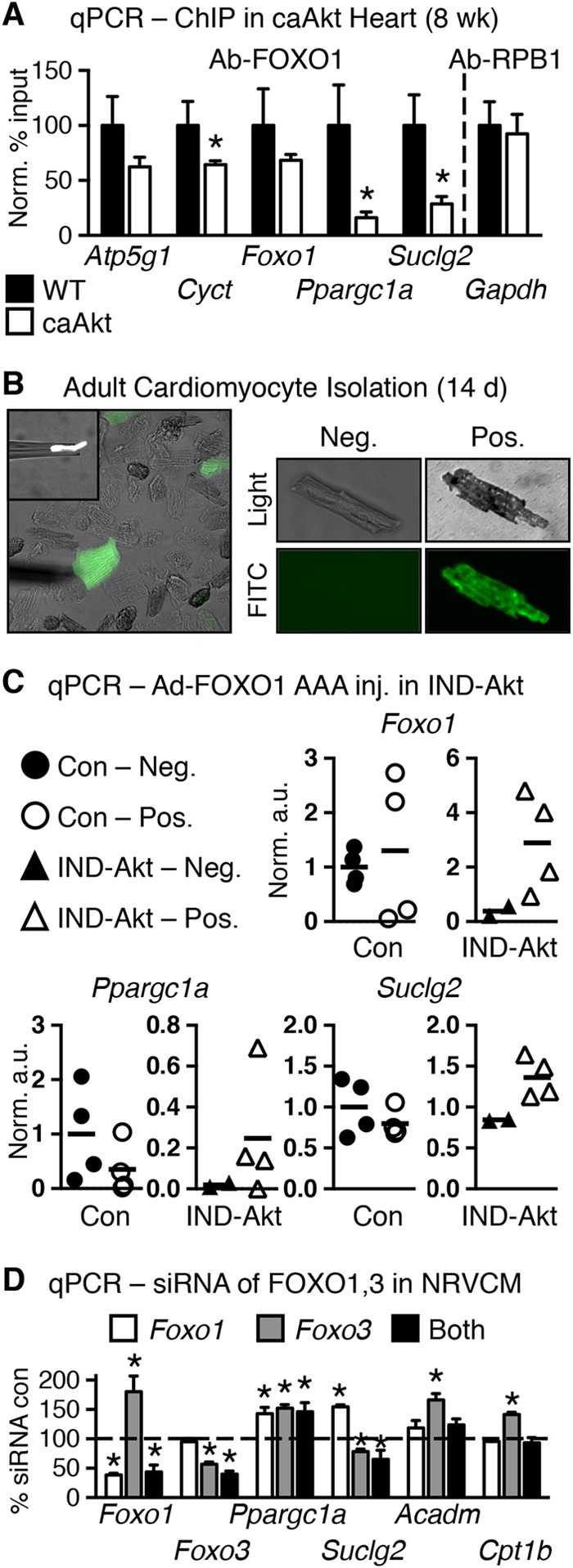
FOXO transcription factors may regulate Akt-mediated modulation of mitochondrial gene expression. (A) qPCR quantification of DNA promoter occupancy following chromatin immunoprecipitation (ChIP) in control and 8-week-old caAkt mouse hearts for antibody to FOXO1 (Ab-FOXO1) or positive-control RNA polymerase II (Ab-RPB1) (n = 3). Data shown as mean ± SEM. *, P < 0.05 versus WT. Candidate response elements are defined in Table S5 in the supplemental material. (B) Representative pictures of cardiomyocytes isolated from IND-Akt (14-day) hearts after in vivo adenoviral injection of GFP and constitutively active FOXO1 (Ad-FOXO1 AAA). FITC, fluorescein isothiocyanate. (C) qPCR from GFP-negative and GFP-positive cells; arbitrary units normalized to GFP-negative Con cells (=1.0) (n = 2 to 4 independent harvests of 10 cells per sample). (D) qPCR from primary neonatal ventricular cardiomyocytes (NRVCMs) following Foxo1 and/or Foxo3 siRNA knockdown relative to scrambled siRNA control (=100%) and normalized to GAPDH (n = 6 to 9). Data shown as mean ± SEM. *, P < 0.05 versus Con.
Akt regulates FOXO1 transcriptional activity by an inhibitory phosphorylation that leads to nuclear exclusion. To directly determine if FOXO1 regulates gene expression in the face of constitutive Akt activation, we performed adenoviral gene therapy by injecting an adenovirus expressing green fluorescent protein (GFP) and a constitutively active (nonphosphorylatable) FOXO1 (Ad-FOXO1 AAA) directly into the hearts of Akt transgenic mice. We first attempted caAkt animals; however, they did not survive surgery. So, we returned to the IND-Akt mice and injected them at the time of DOX withdrawal. At 14 days postinjection, hearts were excised and individual cardiomyocytes were manually separated into GFP-positive and -negative cells of control and IND-Akt mouse hearts (Fig. 9B). Groups of 10 cells were pooled, and gene expression was measured by qPCR (Fig. 9C). Ad-FOXO1 AAA had relatively no effect on gene expression in cells from control hearts (Fig. 9C). Maintaining nuclear FOXO1 signaling by expressing Ad-FOXO1 AAA in cardiomyocytes of IND-Akt transgenic mice reversed the repression of a small subset of candidate genes, by increasing Ppargc1a by 6-fold and Suclg2 expression by 50% relative to GFP-negative Akt transgenic myocytes (Fig. 9C).
Although these findings support a role for FOXO1 in the regulation of Ppargc1a, we wanted to determine if FOXO signaling was sufficient to regulate Ppargc1a and expression of its target genes. Using primary neonatal rat ventricular cardiomyocytes (NRVCMs), we performed siRNA knockdown of Foxo1 and Foxo3 individually and together (Fig. 9D). In this cell culture model, Ppargc1a was induced when Foxo1 and Foxo3 expression levels were individually or concurrently reduced by >50% (Fig. 9D). Despite this, knockdown of Foxo3 repressed Suclg2 expression (Fig. 9D). Other targets identified from the in vivo models, such as Acadm and Cpt1b, were not repressed in cell culture by loss of FOXO (Fig. 9D).
Akt activation in cultured cells differentially regulates metabolism-related gene expression.
To determine if Akt activation could impair mitochondrial function in a cell-autonomous fashion in vitro, we generated a stably transformed line of C2C12 myoblasts that overexpressed a retroviral caAkt (Fig. 10A) and differentiated into myotubes (Fig. 10B). We observed equivalent repression of Ppara and Ppargc1a and activation of Hif1a genes in caAkt-expressing cells (Fig. 10C), as was seen in the caAkt overexpression mouse hearts (Fig. 7). In contrast to in vivo observations, Akt activation modestly but significantly increased expression of Esrra, Esrrg, and Mef2a (Fig. 10C), and most OXPHOS/FAO genes were induced in vitro, with the exception of Cpt1b, which was repressed by caAkt overexpression (Fig. 10D). Expression of the constitutively active FOXO1-AAA mutant in control (GFP-infected cells) had negligible effects on OXPHOS or FAO genes (Fig. 10D), consistent with the in vivo study (Fig. 9C). Only Ppargc1b expression reached a significant change (Fig. 10C). In contrast, FOXO1-AAA expression in caAkt-transduced cells reversed the Akt-mediated induction of most of the OXPHOS and FAO genes (Fig. 10D). The caAkt-mediated repression of Ppara and Ppargc1a was not reversed by FOXO1-AAA (Fig. 10C). Using human FOXO1-specific primers, we confirmed expression, and this transcript was found only in Ad-FOXO1 AAA-infected cells (Fig. 10E). Thus, in cultured myocytes the repression of Ppara and Ppargc1a is FOXO independent, whereas the regulation of OXPHOS and FAO genes is FOXO dependent.
FIG 10.

In vitro expression of caAkt alters gene expression in myotubes. (A) Western blot analysis of whole-cell lysate following selective passaging to confirm overexpression of caAkt in retrovirus-transformed C2C12 myotubes. (B) Representative images of myotube formation in control and caAkt-transformed C2C12 cells following 5 days of differentiation. (C and D) qPCR analysis of mRNA from C2C12 myotubes 5 days following Ad-FOXO1 AAA infection relative to Ad-GFP control (=100%) and normalized to control H3f3a for genes of transcriptional regulators (C) or oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO) (D). (E) qPCR for human FOXO1 cDNA as encoded by the Ad-FOXO1 AAA adenovirus. n = 5 to 6. n.d., not detected. Data shown as mean ± SEM. *, P < 0.05 versus Ad-GFP-infected control cells.
Akt activation in cultured cells induces defects in mitochondrial bioenergetics.
Mitochondrial bioenergetics and metabolic flux were determined using the Seahorse XF analyzer. Basal, ATP-linked, proton leak, maximal, reserve capacity, and nonmitochondrial (other) oxygen consumption rates (OCRs) were calculated. In general, OCR was increased in caAkt cells (Fig. 11A). Extracellular acidification rate (ECAR), a measure of glycolytic flux, was also increased with caAkt (Fig. 11B). Despite these changes, total cellular ATP levels were decreased and AMP and ADP levels were increased, resulting in a significant reduction in the ATP/ADP ratio (Fig. 11C). Thus, persistent Akt activation in culture impairs mitochondrial bioenergetics as evidenced by uncoupled respiration and a switch toward glycolytic metabolism.
FIG 11.
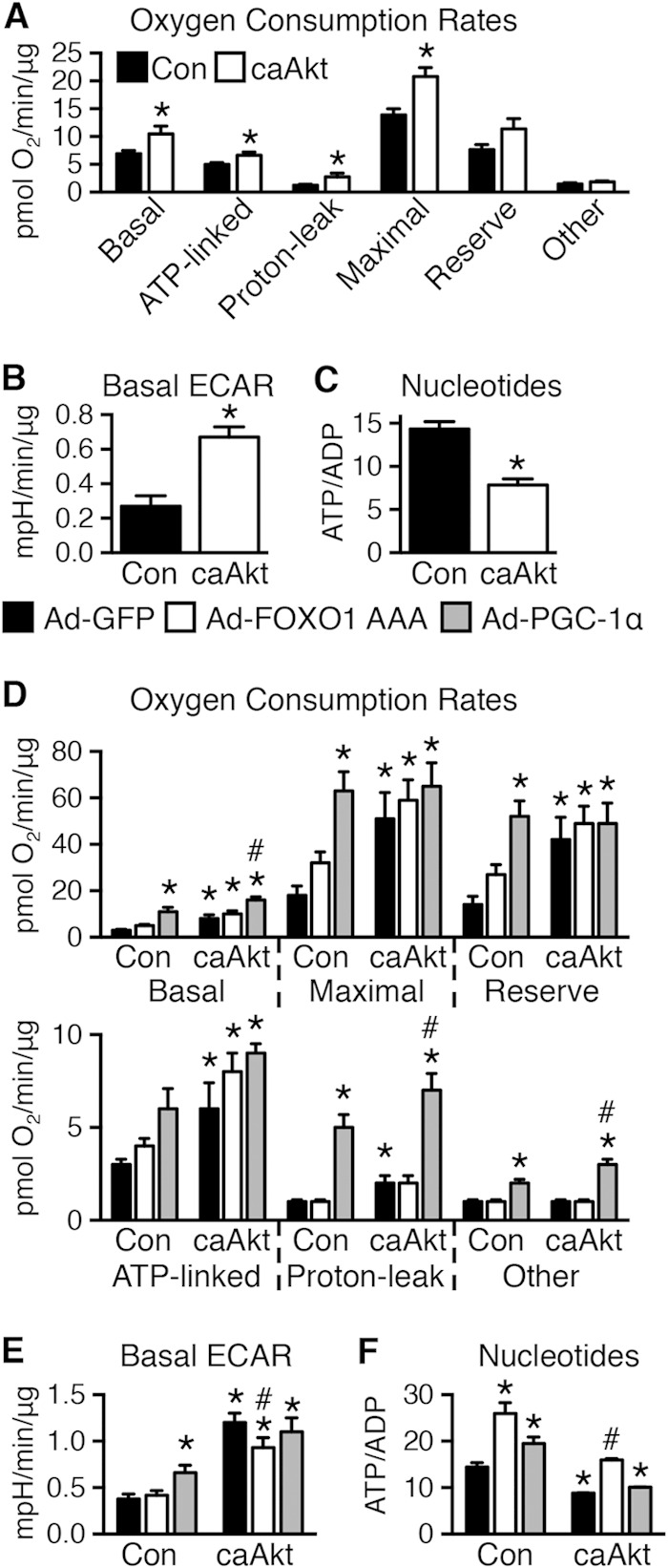
In vitro expression of caAkt alters cellular bioenergetics in myotubes. (A and B) Seahorse cellular bioenergetics analysis of oxygen consumption rates (OCR) (A) (further details in Materials and Methods) and extracellular acidification rates (ECAR) (B) in control or caAkt-transduced C2C12 myotubes as in Fig. 10 (n = 10 to 11). (C) HPLC quantification of the nucleotides ATP and ADP in control or caAkt-transduced C2C12 myotubes (n = 6). Data shown as mean ± SEM. *, P < 0.05 versus Con. (D and E) Seahorse cellular bioenergetics analysis of OCR (D) and ECAR (E) in C2C12 myotubes as described above following Ad-PGC-1α or Ad-FOXO1 AAA infection compared to Ad-GFP (n ≥ 4). (F) HPLC quantification of the nucleotides ATP and ADP in C2C12 myotubes as described for panels D and E (n = 3). Data shown as mean ± SEM. *, P < 0.05 versus Ad-GFP Con. #, P < 0.05 versus Ad-GFP caAkt.
As Ppargc1a expression levels were decreased in these cells, we sought to determine if mitochondrial function could be restored by increasing PGC-1α levels using an adenoviral PGC-1α (Ad-PGC-1α). In control cells, PGC-1α increased basal and uncoupled OCR and increased ECAR and ATP/ADP ratios. In contrast, Ad-FOXO1 AAA had little impact on OCR or ECAR, but it increased ATP/ADP ratios, suggesting increased mitochondrial coupling (Fig. 11D to F). In caAkt cells, PGC-1α further increased basal and uncoupled respirations without changing ATP-linked respirations or ECAR. Moreover, ATP/ADP ratios were not normalized. In contrast, although FOXO1 transfection did not alter OCR, the elevated ECAR was reduced and ATP/ADP ratios were restored to levels of control cells (Fig. 11D to F).
DISCUSSION
In the present study, we show that the transition from compensated LVH to heart failure is associated with increased Akt signaling and reduced levels of FOXO1. Similar changes were observed in multiple in vivo models and time points of transgenic Akt activation with some overlap with in vitro studies as summarized in Table 6. These changes are associated with mitochondrial dysfunction and repression of Ppara and Ppargc1a, which were also replicated in cultured myocytes in vitro (Fig. 7 and 10). We previously reported that in pressure overload-induced heart failure, mitochondrial dysfunction develops and expression of mitochondrial regulatory genes and nucleus-encoded FAO and OXPHOS genes was repressed (2, 37, 38). Although the mechanism for repression of mitochondrial function in pressure overload-induced heart failure is likely multifactorial, we sought to explore the potential contribution of long-term Akt activation. We now show that sustained Akt activation alters cardiac substrate metabolism and precipitates progressive mitochondrial dysfunction as a function of transgene duration that is independent of the developmental stage at which Akt is activated. Mitochondrial dysfunction occurs without changes in mitochondrial architecture or morphology, but mRNA levels of genes critical to FAO, OXPHOS, and their transcriptional regulators, Foxo1, Ppara, and Ppargc1a, are markedly reduced. ATP/O ratios are reduced after 6 weeks of Akt activation, indicating that mitochondrial uncoupling occurs in concert with the transition to heart failure in this model.
TABLE 6.
Summary of models, time points, and findings of caAkt-mediated regulation
| Parameter | Direction of change for:a |
|||||
|---|---|---|---|---|---|---|
|
In vivo models |
In vitro model, myotubes, 5 days | |||||
| caAkt developmental |
IND-Akt/tON-Akt adult |
|||||
| 6–8 wk | 3–4 mo | 10 days | 2–3 wk | 6 wk | ||
| PPARα/PGC-1α expression and signaling | ↓/↓ | ↓/↓ | ↓/↓ | ↓/↓ | ND | ↓/↓ |
| FOXO1/FOXO3 expression and signaling | =/↓ | ↓/= | =/↓ | ND | ND | =/= |
| OXPHOS/FAO gene expression | ↓/↓ | ↓/↓ | =/↓ | ND | ND | =/↑ |
| Mitochondrial function | ↓ | ↓ | = | ↓ | ↓ | ↓ |
| Nucleotide levels or synthesis | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ |
| Contractile function | = | ↓ | ↓ | ↓ | ↓ | NA |
ND, not determined; NA, not applicable; =, unchanged.
We observed that promoters of many OXPHOS and FAO genes were enriched for FOXO binding sites and confirmed reduced promoter occupancy by FOXO1 on Ppargc1a and representative OXPHOS genes. Introducing a nucleus-localized FOXO1 to Akt transgenic hearts in vivo restored Ppargc1a and Suclg2 expression. These observations suggest that reduced FOXO1 transcriptional activity could, by repressing PGC-1α, contribute to mitochondrial dysfunction in decompensated cardiac hypertrophy in vivo. In vitro analyses in Akt-overexpressing cells suggest that FOXO1 might also modulate mitochondrial function via mechanisms that are independent of PGC-1α. Although we did not observe increased FOXO1 phosphorylation, we believe that this could be due to a technical limitation to detect FOXO1 phosphorylation in cardiac tissue. Other posttranslational mechanisms are also known to regulate FOXO activity, as has been recently reviewed (39). These include acetylation (40) and GlcNAcylation (41), which should be explored in future studies. We also cannot rule out a FOXO3 mechanism, given that FOXO3 was also changed in some of our endpoints.
Comparison of Akt overexpression and FOXO1 rescue in vivo with those in vitro revealed some similarities but also important differences. In both models, Ppara and Ppargc1a expression levels were repressed. However, expression of a constitutively active FOXO1 restored Ppargc1a in vivo but did not influence Ppara and Ppargc1a expression in vitro. Also, whereas Akt overexpression repressed OXPHOS and FAO genes in the adult heart, a phenomenon that was also observed in skeletal muscle in vivo (42), Akt overexpression in cultured cells induced OXPHOS and FAO genes. Some of these differences between adult tissues and cultured cells in response to Akt activation could reflect fundamental differences in baseline substrate utilization. Specifically, the adult heart generates nearly 70% of its energy needs from fatty acids, whereas cultured cells rely predominantly on glycolytic metabolism. Regardless, the reversal of Akt-induced induction of OXPHOS and FAO genes in cultured cells following expression of FOXO1-AAA supports the hypothesis that regulation of mitochondrial gene expression by Akt is in part FOXO dependent. The differences in the direction of the changes in FOXO-regulated gene expression in cultured cells versus cardiomyocytes in vivo are incompletely understood but underscore the complexity of FOXO signaling that may involve differential interactions with corepressors or coactivators in a cell-type- or differentiation-dependent context.
Various in vivo models could potentially be used to decipher the mechanisms downstream of Akt that mediate mitochondrial dysfunction in pathological LVH. We previously reported that haploinsufficiency of Akt1 could prevent pressure overload-induced heart failure, strongly supporting the concept that Akt hyperactivation is deleterious in pathological LVH (43). Transgenic modulation of FOXO or PGC-1α signaling could potentially be used to test their relative contributions to Akt-mediated mitochondrial dysfunction. However, inducible overexpression of PGC-1α leads to uncontrolled mitochondrial proliferation and heart failure (44). We also reported that maintaining physiological levels of PGC-1α did not preserve cardiac or mitochondrial function in mice with TAC-induced LVH and heart failure (37). Thus, normalizing PGC-1α expression in models of pathological LVH might not counteract other molecular mechanisms that may impair mitochondrial function in the failing heart. Mice with inducible constitutive activation of nuclear FOXO3 in cardiomyocytes develop dramatic loss of mitochondria and heart failure, rendering this model unsuitable as a tool to rescue Akt transgenic mice (45). Mice with cardiomyocyte deficiency of FOXO might phenocopy Akt-mediated mitochondrial dysfunction. However, nonstressed FOXO1/FOXO3-deficient hearts do not develop LVH or LV dysfunction, underscoring additional FOXO-independent roles for Akt signaling in the pathophysiology of heart failure (46). Importantly, following permanent coronary artery ligation, these animals developed exaggerated LVH and accelerated heart failure, indicating an important role for nuclear FOXO signaling in the cardiac adaptations to hemodynamic stress. Mitochondrial function remains to be determined in these mice, representing an important question to be addressed in future studies.
Reduced expression of Ppargc1a is consistent with reports that nuclear exclusion of FOXO1 by insulin signaling to Akt may repress Ppargc1a expression in liver and skeletal muscle (35). However, our studies in cultured myocytes suggest that regulation of PGC-1α by FOXO1 might be cell type or differentiation dependent. Decreased PGC-1α levels could also contribute to Akt-induced cardiac dysfunction in the presence of ongoing cardiac hypertrophy, consistent with increased susceptibility to heart failure in PGC-1α-deficient hearts following TAC (47). Contractile dysfunction and reduced myocardial ATP content are also observed in PGC-1α null mice prior to any changes in mitochondrial morphology or number (48). Thus, activation of Akt may limit cardiac metabolic flexibility in part by repressing Ppara and Ppargc1a expression.
Our studies also identified a number of other transcriptional regulators with differential expression following induction of Akt. These included Esrra, Esrrg, Hif1a, Gata4, and Mef2a. Hif1a was induced under all conditions examined and warrants future investigation as it has previously been shown to play a role in cardiac hypertrophy (49), while Gata4 and Mef2a were not consistently regulated in all the models examined. The regulation of the genes Esrra and Esrrg, encoding ERRα and ERRγ, respectively, were also not consistently regulated across the different models. Further investigation into these additional targets may highlight some of the more subtle attributes of Akt-mediated mitochondrial regulation, as each has established roles in regulation of genes involved in mitochondrial function, cardiac hypertrophy, and heart failure (50–52). Both ERRα and ERRγ transcript levels are repressed in the human failing heart and partially restored by mechanical unloading (53). However, we found Esrrg to be significantly repressed only in tON-Akt hearts. Additionally, ERRα and ERRγ have been shown to bind to a number of OXPHOS and FAO gene promoters (54). Their binding specificities are overlapping (e.g., Acadm, Cpt1, and Atp5g1), leaving open the possible contribution of ERR-dependent mechanisms, although our data would suggest the existence of ERR-independent pathways.
Whether Akt-induced cardiac hypertrophy is physiological or pathological is complex (7). Activation of PI3K/Akt1 signaling is required for exercise-induced LVH (55, 56). However, long-term overexpression of activated Akt induces heart failure, and Akt activation is also observed in the failing myocardium (8, 43, 57). Shiojima et al. reported that short-term Akt activation induced compensated LVH (10). However, we show that short-term Akt activation (14 days) impaired cardiac function, diminished palmitate oxidation, and increased glycolysis in isolated working hearts, a metabolic pattern that mimics those associated with pathological hypertrophy. Moreover, short-term Akt activation reduced mRNA levels of Ppara and Ppargc1a, whose expression is also reduced in response to pressure overload (2, 37). Thus, these changes are in contrast with the metabolic phenotype of exercise-induced LVH (58). Furthermore, Akt activation in the adult heart (IND-Akt ± rapamycin) or in vitro (caAkt C2C12 myotubes) increases glycolytic flux without increasing mitochondrial oxidative capacity. Thus, even when cardiac function is relatively preserved in the context of Akt-induced cardiac growth, a metabolic signature characteristic of pressure overload LVH is already present.
Changes in myocardial metabolism are intrinsic to Akt activation and not secondary to LVH, as prevention of LVH with rapamycin did not restore Ppara and Ppargc1a or mitochondrial oxidative capacity or reverse increased glucose utilization. Although increased glucose utilization in the face of mitochondrial dysfunction could be due to activation of AMPK (59), we did not observe AMPK activation. This supports prior studies demonstrating that Akt activation directly inhibits the activation of AMPK (60, 61). Thus, increased glucose utilization likely occurs by an Akt-mediated increase in GLUT4 translocation, glucose transport, and glycogen accumulation (3, 4). Reduced AMPK activation could also inhibit FAO by decreasing the inhibitory phosphorylation of acetyl-CoA carboxylase, leading to increased malonyl-CoA (62). Reduced levels of tissue triglycerides despite decreased FAO also raise the possibility of decreased FA uptake or esterification. Thus, even in the absence of Akt-induced cardiac growth, repression of PPARα/PGC-1α and inhibition of AMPK impose metabolic limitations on the heart. Other pathways not explored here could also be involved. Recently, it was shown that mTORC1 and Akt increase mitochondrial volume and activity through translational regulation (63, 64). Given that Akt activation consistently decreased mitochondrial function and FAO, we focused on transcriptional mechanisms in the current study. We previously reported that exercise-induced LVH requires Akt activation via class 1A PI3K, but the mitochondrial and metabolic adaptations of PI3K activation are independent of Akt (27, 65). Thus, in the case of long-term Akt activation there is no additional or equivalent activation of PI3K-dependent signaling pathways, leading to an imbalance in cellular signaling that favors growth pathways at the expense of pathways that will enhance mitochondrial energetics, which may also contribute to cardiac dysfunction.
In conclusion, Akt activation promotes a metabolic switch toward glucose utilization but impairs mitochondrial oxidative capacity independently of cardiac hypertrophy. Activation of Akt-mediated survival pathways might be particularly suited to circumstances such as ischemia when oxygen or metabolic substrates might be limiting. Under these conditions, increased glycolysis and reduced mitochondrial FAO might maintain cardiac function and protect mitochondrial integrity. However, Akt signaling becomes maladaptive if it persists, because these metabolic changes cannot maintain cardiac energy requirements in the face of continuing Akt-mediated LVH. Thus, prosurvival Akt signaling should be rapidly deactivated once the underlying stress is removed or ameliorated because persistent Akt activation promotes mitochondrial and metabolic adaptations characteristic of pathological LVH.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants R01HL070070, R01DK092065, and U01HL70525 from the National Institutes of Health (NIH) to E.D.A., an Established Investigator of the American Heart Association (AHA). B.T.O. was supported by a physician scientist-training award from the American Diabetes Association; A.R.W. was supported by a postdoctoral fellowship from the JDRF (10-2009-672) and NIH K99R00 HL111322. H.B. and C.R. were supported by postdoctoral fellowships from the German Research Foundation (DFG), P.C. was supported by a predoctoral fellowship from the Spanish Ministry of Education and Science, and R.O.P. was supported by a postdoctoral fellowship from the AHA, Western Affiliates, and T32 HL007576 from the NIH.
David K. Crossman (bioinformatics director at the University of Alabama at Birmingham Heflin Center for Genomics), Ellis B. Jensen, Valentina Parra, Christopher K. Rodesch (director of the University of Utah Microscopy Core), and Heather A. Theobald provided technical support and data collection.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01109-14.
REFERENCES
- 1.Abel ED, Doenst T. 2011. Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc Res 90:234–242. doi: 10.1093/cvr/cvr015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Riehle C, Wende AR, Zaha VG, Pires KM, Wayment B, Olsen C, Bugger H, Buchanan J, Wang X, Moreira AB, Doenst T, Medina-Gomez G, Litwin SE, Lelliott CJ, Vidal-Puig A, Abel ED. 2011. PGC-1β deficiency accelerates the transition to heart failure in pressure overload hypertrophy. Circ Res 109:783–793. doi: 10.1161/CIRCRESAHA.111.243964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matsui T, Nagoshi T, Hong EG, Luptak I, Hartil K, Li L, Gorovits N, Charron MJ, Kim JK, Tian R, Rosenzweig A. 2006. Effects of chronic Akt activation on glucose uptake in the heart. Am J Physiol Endocrinol Metab 290:E789–E797. doi: 10.1152/ajpendo.00564.2004. [DOI] [PubMed] [Google Scholar]
- 4.Matsui T, Tao J, del Monte F, Lee KH, Li L, Picard M, Force TL, Franke TF, Hajjar RJ, Rosenzweig A. 2001. Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation 104:330–335. doi: 10.1161/01.CIR.104.3.330. [DOI] [PubMed] [Google Scholar]
- 5.Matsui T, Li L, Wu JC, Cook SA, Nagoshi T, Picard MH, Liao R, Rosenzweig A. 2002. Phenotypic spectrum caused by transgenic overexpression of activated Akt in the heart. J Biol Chem 277:22896–22901. doi: 10.1074/jbc.M200347200. [DOI] [PubMed] [Google Scholar]
- 6.Shioi T, McMullen JR, Kang PM, Douglas PS, Obata T, Franke TF, Cantley LC, Izumo S. 2002. Akt/protein kinase B promotes organ growth in transgenic mice. Mol Cell Biol 22:2799–2809. doi: 10.1128/MCB.22.8.2799-2809.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O'Neill BT, Abel ED. 2005. Akt1 in the cardiovascular system: friend or foe? J Clin Invest 115:2059–2064. doi: 10.1172/JCI25900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagoshi T, Matsui T, Aoyama T, Leri A, Anversa P, Li L, Ogawa W, del Monte F, Gwathmey JK, Grazette L, Hemmings BA, Kass DA, Champion HC, Rosenzweig A. 2005. PI3K rescues the detrimental effects of chronic Akt activation in the heart during ischemia/reperfusion injury. J Clin Invest 115:2128–2138. doi: 10.1172/JCI23073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu Y, Pereira RO, O'Neill BT, Riehle C, Ilkun O, Wende AR, Rawlings TA, Zhang YC, Zhang Q, Klip A, Shiojima I, Walsh K, Abel ED. 2013. Cardiac PI3K-Akt impairs insulin-stimulated glucose uptake independent of mTORC1 and GLUT4 translocation. Mol Endocrinol 27:172–184. doi: 10.1210/me.2012-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. 2005. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest 115:2108–2118. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mazumder PK, O'Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, Boudina S, Abel ED. 2004. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes 53:2366–2374. doi: 10.2337/diabetes.53.9.2366. [DOI] [PubMed] [Google Scholar]
- 12.Sloan C, Tuinei J, Nemetz K, Frandsen J, Soto J, Wride N, Sempokuya T, Alegria L, Bugger H, Abel ED. 2011. Central leptin signaling is required to normalize myocardial fatty acid oxidation rates in caloric-restricted ob/ob mice. Diabetes 60:1424–1434. doi: 10.2337/db10-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boudina S, Sena S, O'Neill BT, Tathireddy P, Young ME, Abel ED. 2005. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation 112:2686–2695. doi: 10.1161/CIRCULATIONAHA.105.554360. [DOI] [PubMed] [Google Scholar]
- 14.Veksler VI, Kuznetsov AV, Sharov VG, Kapelko VI, Saks VA. 1987. Mitochondrial respiratory parameters in cardiac tissue: a novel method of assessment by using saponin-skinned fibers. Biochim Biophys Acta 892:191–196. doi: 10.1016/0005-2728(87)90174-5. [DOI] [PubMed] [Google Scholar]
- 15.Brand MD, Pakay JL, Ocloo A, Kokoszka J, Wallace DC, Brookes PS, Cornwall EJ. 2005. The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochem J 392:353–362. doi: 10.1042/BJ20050890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodnick KJ, Sidell BD. 1994. Cold acclimation increases carnitine palmitoyltransferase I activity in oxidative muscle of striped bass. Am J Physiol 266:R405–R412. [DOI] [PubMed] [Google Scholar]
- 17.Yan LJ, Levine RL, Sohal RS. 1997. Oxidative damage during aging targets mitochondrial aconitase. Proc Natl Acad Sci U S A 94:11168–11172. doi: 10.1073/pnas.94.21.11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clark RJ, Rodnick KJ. 1998. Morphometric and biochemical characteristics of ventricular hypertrophy in male rainbow trout (Oncorhynchus mykiss). J Exp Biol 201:1541–1552. [DOI] [PubMed] [Google Scholar]
- 19.Rodnick KJ, Sidell BD. 1997. Structural and biochemical analyses of cardiac ventricular enlargement in cold-acclimated striped bass. Am J Physiol 273:R252–R258. [DOI] [PubMed] [Google Scholar]
- 20.Bugger H, Chen D, Riehle C, Soto J, Theobald HA, Hu XX, Ganesan B, Weimer BC, Abel ED. 2009. Tissue-specific remodeling of the mitochondrial proteome in type 1 diabetic Akita mice. Diabetes 58:1986–1997. doi: 10.2337/db09-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.R Core Team. 2012. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 22.Gentleman R, Carey V, Bates D, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini A, Sawitzki G, Smith C, Smyth G, Tierney L, Yang J, Zhang J. 2004. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. 2000. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pavlidis P, Noble WS. 2003. Matrix2png: a utility for visualizing matrix data. Bioinformatics 19:295–296. doi: 10.1093/bioinformatics/19.2.295. [DOI] [PubMed] [Google Scholar]
- 25.Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, Cooksey RC, Litwin SE, Abel ED. 2005. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 146:5341–5349. doi: 10.1210/en.2005-0938. [DOI] [PubMed] [Google Scholar]
- 26.Wende AR, Huss JM, Schaeffer PJ, Giguere V, Kelly DP. 2005. PGC-1α coactivates PDK4 gene expression via the orphan nuclear receptor ERRα: a mechanism for transcriptional control of muscle glucose metabolism. Mol Cell Biol 25:10684–10694. doi: 10.1128/MCB.25.24.10684-10694.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O'Neill BT, Kim J, Wende AR, Theobald HA, Tuinei J, Buchanan J, Guo A, Zaha VG, Davis DK, Schell JC, Boudina S, Wayment B, Litwin SE, Shioi T, Izumo S, Birnbaum MJ, Abel ED. 2007. A conserved role for phosphatidylinositol 3-kinase but not Akt signaling in mitochondrial adaptations that accompany physiological cardiac hypertrophy. Cell Metab 6:294–306. doi: 10.1016/j.cmet.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. 2000. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest 106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dranka BP, Benavides GA, Diers AR, Giordano S, Zelickson BR, Reily C, Zou L, Chatham JC, Hill BG, Zhang J, Landar A, Darley-Usmar VM. 2011. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic Biol Med 51:1621–1635. doi: 10.1016/j.freeradbiomed.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hill BG, Benavides GA, Lancaster JR Jr, Ballinger S, Dell'Italia L, Jianhua Z, Darley-Usmar VM. 2012. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol Chem 393:1485–1512. doi: 10.1515/hsz-2012-0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perez J, Hill BG, Benavides GA, Dranka BP, Darley-Usmar VM. 2010. Role of cellular bioenergetics in smooth muscle cell proliferation induced by platelet-derived growth factor. Biochem J 428:255–267. doi: 10.1042/BJ20100090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Calnan DR, Brunet A. 2008. The FoxO code. Oncogene 27:2276–2288. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- 33.Eijkelenboom A, Burgering BM. 2013. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol 14:83–97. doi: 10.1038/nrm3507. [DOI] [PubMed] [Google Scholar]
- 34.Zhao F, Xuan Z, Liu L, Zhang MQ. 2005. TRED: a transcriptional regulatory element database and a platform for in silico gene regulation studies. Nucleic Acids Res 33:D103–D107. doi: 10.1093/nar/gki004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Daitoku H, Yamagata K, Matsuzaki H, Hatta M, Fukamizu A. 2003. Regulation of PGC-1 promoter activity by protein kinase B and the forkhead transcription factor FKHR. Diabetes 52:642–649. doi: 10.2337/diabetes.52.3.642. [DOI] [PubMed] [Google Scholar]
- 36.Essaghir A, Dif N, Marbehant CY, Coffer PJ, Demoulin JB. 2009. The transcription of FOXO genes is stimulated by FOXO3 and repressed by growth factors. J Biol Chem 284:10334–10342. doi: 10.1074/jbc.M808848200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pereira RO, Wende AR, Crum A, Hunter D, Olsen CD, Rawlings T, Riehle C, Ward WF, Abel ED. 2014. Maintaining PGC-1α expression following pressure overload-induced cardiac hypertrophy preserves angiogenesis but not contractile or mitochondrial function. FASEB J 28:3691–3702. doi: 10.1096/fj.14-253823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pereira RO, Wende AR, Olsen C, Soto J, Rawlings T, Zhu Y, Anderson SM, Abel ED. 2013. Inducible overexpression of GLUT1 prevents mitochondrial dysfunction and attenuates structural remodeling in pressure overload but does not prevent left ventricular dysfunction. J Am Heart Assoc 2:e000301. doi: 10.1161/JAHA.113.000301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boccitto M, Kalb RG. 2011. Regulation of Foxo-dependent transcription by post-translational modifications. Curr Drug Targets 12:1303–1310. doi: 10.2174/138945011796150316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Masui K, Tanaka K, Akhavan D, Babic I, Gini B, Matsutani T, Iwanami A, Liu F, Villa GR, Gu Y, Campos C, Zhu S, Yang H, Yong WH, Cloughesy TF, Mellinghoff IK, Cavenee WK, Shaw RJ, Mischel PS. 2013. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab 18:726–739. doi: 10.1016/j.cmet.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Housley MP, Rodgers JT, Udeshi ND, Kelly TJ, Shabanowitz J, Hunt DF, Puigserver P, Hart GW. 2008. O-GlcNAc regulates FoxO activation in response to glucose. J Biol Chem 283:16283–16292. doi: 10.1074/jbc.M802240200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Izumiya Y, Hopkins T, Morris C, Sato K, Zeng L, Viereck J, Hamilton JA, Ouchi N, LeBrasseur NK, Walsh K. 2008. Fast/glycolytic muscle fiber growth reduces fat mass and improves metabolic parameters in obese mice. Cell Metab 7:159–172. doi: 10.1016/j.cmet.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shimizu I, Minamino T, Toko H, Okada S, Ikeda H, Yasuda N, Tateno K, Moriya J, Yokoyama M, Nojima A, Koh GY, Akazawa H, Shiojima I, Kahn CR, Abel ED, Komuro I. 2010. Excessive cardiac insulin signaling exacerbates systolic dysfunction induced by pressure overload in rodents. J Clin Invest 120:1506–1514. doi: 10.1172/JCI40096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Russell LK, Mansfield CM, Lehman JJ, Kovacs A, Courtois M, Saffitz JE, Medeiros DM, Valencik ML, McDonald JA, Kelly DP. 2004. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor γ coactivator-1α promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ Res 94:525–533. doi: 10.1161/01.RES.0000117088.36577.EB. [DOI] [PubMed] [Google Scholar]
- 45.Cao DJ, Jiang N, Blagg A, Johnstone JL, Gondalia R, Oh M, Luo X, Yang KC, Shelton JM, Rothermel BA, Gillette TG, Dorn GW, Hill JA. 2013. Mechanical unloading activates FoxO3 to trigger Bnip3-dependent cardiomyocyte atrophy. J Am Heart Assoc 2:e000016. doi: 10.1161/JAHA.113.000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sengupta A, Molkentin JD, Paik JH, DePinho RA, Yutzey KE. 2011. FoxO transcription factors promote cardiomyocyte survival upon induction of oxidative stress. J Biol Chem 286:7468–7478. doi: 10.1074/jbc.M110.179242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. 2006. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPARγ coactivator 1α. Proc Natl Acad Sci U S A 103:10086–10091. doi: 10.1073/pnas.0603615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, Ahmad F, Matsui T, Chin S, Wu PH, Rybkin II, Shelton JM, Manieri M, Cinti S, Schoen FJ, Bassel-Duby R, Rosenzweig A, Ingwall JS, Spiegelman BM. 2005. Transcriptional coactivator PGC-1α controls the energy state and contractile function of cardiac muscle. Cell Metab 1:259–271. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 49.Krishnan J, Suter M, Windak R, Krebs T, Felley A, Montessuit C, Tokarska-Schlattner M, Aasum E, Bogdanova A, Perriard E, Perriard JC, Larsen T, Pedrazzini T, Krek W. 2009. Activation of a HIF1alpha-PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab 9:512–524. doi: 10.1016/j.cmet.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 50.Li Y, He L, Zeng N, Sahu D, Cadenas E, Shearn C, Li W, Stiles BL. 2013. Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) signaling regulates mitochondrial biogenesis and respiration via estrogen-related receptor alpha (ERRalpha). J Biol Chem 288:25007–25024. doi: 10.1074/jbc.M113.450353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huss JM, Imahashi K, Dufour CR, Weinheimer CJ, Courtois M, Kovacs A, Giguere V, Murphy E, Kelly DP. 2007. The nuclear receptor ERRalpha is required for the bioenergetic and functional adaptation to cardiac pressure overload. Cell Metab 6:25–37. doi: 10.1016/j.cmet.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 52.Alaynick WA, Kondo RP, Xie W, He W, Dufour CR, Downes M, Jonker Johan W, Giles W, Naviaux RK, Giguère V, Evans RM. 2007. ERRγ directs and maintains the transition to oxidative metabolism in the postnatal heart. Cell Metab 6:13–24. doi: 10.1016/j.cmet.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 53.Gupte AA, Hamilton DJ, Cordero-Reyes AM, Youker KA, Yin Z, Estep JD, Stevens RD, Wenner B, Ilkayeva O, Loebe M, Peterson LE, Lyon CJ, Wong STC, Newgard CB, Torre-Amione G, Taegtmeyer H, Hsueh WA. 2014. Mechanical unloading promotes myocardial energy recovery in human heart failure. Circ Cardiovasc Genet 7:266–276. doi: 10.1161/CIRCGENETICS.113.000404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dufour CR, Wilson BJ, Huss JM, Kelly DP, Alaynick WA, Downes M, Evans RM, Blanchette M, Giguère V. 2007. Genome-wide orchestration of cardiac functions by the orphan nuclear receptors ERRα and γ. Cell Metab 5:345–356. doi: 10.1016/j.cmet.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 55.McMullen JR, Shioi T, Zhang L, Tarnavski O, Sherwood MC, Kang PM, Izumo S. 2003. Phosphoinositide 3-kinase(p110α) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc Natl Acad Sci U S A 100:12355–12360. doi: 10.1073/pnas.1934654100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.DeBosch B, Treskov I, Lupu TS, Weinheimer C, Kovacs A, Courtois M, Muslin AJ. 2006. Akt1 is required for physiological cardiac growth. Circulation 113:2097–2104. doi: 10.1161/CIRCULATIONAHA.105.595231. [DOI] [PubMed] [Google Scholar]
- 57.Haq S, Choukroun G, Lim H, Tymitz KM, del Monte F, Gwathmey J, Grazette L, Michael A, Hajjar R, Force T, Molkentin JD. 2001. Differential activation of signal transduction pathways in human hearts with hypertrophy versus advanced heart failure. Circulation 103:670–677. doi: 10.1161/01.CIR.103.5.670. [DOI] [PubMed] [Google Scholar]
- 58.Burelle Y, Wambolt RB, Grist M, Parsons HL, Chow JC, Antler C, Bonen A, Keller A, Dunaway GA, Popov KM, Hochachka PW, Allard MF. 2004. Regular exercise is associated with a protective metabolic phenotype in the rat heart. Am J Physiol 287:H1055–H1063. doi: 10.1152/ajpheart.00925.2003. [DOI] [PubMed] [Google Scholar]
- 59.Xu Q, Si LY. 2010. Protective effects of AMP-activated protein kinase in the cardiovascular system. J Cell Mol Med 14:2604–2613. doi: 10.1111/j.1582-4934.2010.01179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kovacic S, Soltys CL, Barr AJ, Shiojima I, Walsh K, Dyck JR. 2003. Akt activity negatively regulates phosphorylation of AMP-activated protein kinase in the heart. J Biol Chem 278:39422–39427. doi: 10.1074/jbc.M305371200. [DOI] [PubMed] [Google Scholar]
- 61.Soltys CL, Kovacic S, Dyck JR. 2006. Activation of cardiac AMP-activated protein kinase by LKB1 expression or chemical hypoxia is blunted by increased Akt activity. Am J Physiol Heart Circ Physiol 290:H2472–H2479. doi: 10.1152/ajpheart.01206.2005. [DOI] [PubMed] [Google Scholar]
- 62.Zaha VG, Young LH. 2012. AMP-activated protein kinase regulation and biological actions in the heart. Circ Res 111:800–814. doi: 10.1161/CIRCRESAHA.111.255505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morita M, Gravel SP, Chenard V, Sikstrom K, Zheng L, Alain T, Gandin V, Avizonis D, Arguello M, Zakaria C, McLaughlan S, Nouet Y, Pause A, Pollak M, Gottlieb E, Larsson O, St.-Pierre J, Topisirovic I, Sonenberg N. 2013. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab 18:698–711. doi: 10.1016/j.cmet.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 64.Goo CK, Lim HY, Ho QS, Too H-P, Clement M-V, Wong KP. 2012. PTEN/Akt signaling controls mitochondrial respiratory capacity through 4E-BP1. PLoS One 7:e45806. doi: 10.1371/journal.pone.0045806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim J, Wende AR, Sena S, Theobald HA, Soto J, Sloan C, Wayment BE, Litwin SE, Holzenberger M, LeRoith D, Abel ED. 2008. Insulin-like growth factor I receptor signaling is required for exercise-induced cardiac hypertrophy. Mol Endocrinol 22:2531–2543. doi: 10.1210/me.2008-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Waters. 2006. Protein Expression System manual. Waters, Manchester, United Kingdom. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.