

Abstract
Poly(ADP-ribosyl)ation (PAR) has been implicated in various aspects of the cellular response to DNA damage and genome stability. Although 17 human poly(ADP-ribose) polymerase (PARP) genes have been identified, a single poly(ADP-ribosyl) glycohydrolase (PARG) mediates PAR degradation. Here we investigated the role of PARG in the replication of human chromosomes. We show that PARG depletion affects cell proliferation and DNA synthesis, leading to replication-coupled H2AX phosphorylation. Furthermore, PARG depletion or inhibition per se slows down individual replication forks similarly to mild chemotherapeutic treatment. Electron microscopic analysis of replication intermediates reveals marked accumulation of reversed forks and single-stranded DNA (ssDNA) gaps in unperturbed PARG-defective cells. Intriguingly, while we found no physical evidence for chromosomal breakage, PARG-defective cells displayed both ataxia-telangiectasia-mutated (ATM) and ataxia-Rad3-related (ATR) activation, as well as chromatin recruitment of standard double-strand-break-repair factors, such as 53BP1 and RAD51. Overall, these data prove PAR degradation to be essential to promote resumption of replication at endogenous and exogenous lesions, preventing idle recruitment of repair factors to remodeled replication forks. Furthermore, they suggest that fork remodeling and restarting are surprisingly frequent in unperturbed cells and provide a molecular rationale to explore PARG inhibition in cancer chemotherapy.
INTRODUCTION
Cellular responses are crucial for the adaptability and survival of a cell exposed to different types of endogenous and exogenous stress. The DNA damage response (DDR) consists of one such defense mechanism in response to different types of insults to the DNA. Poly(ADP)ribosylation of proteins is one of the quickest cellular responses to DNA damage and is brought about by proteins of the poly(ADP) ribose polymerase family (PARP), mostly PARP1 (1). Upon being recruited to sites of the DNA damage, NAD+ is used as a substrate by PARP to synthesize negatively charged poly(ADP-ribosyl)ation (PAR) polymers onto itself and also its target proteins (1). Through this posttranslational modification, PARP targets a variety of nuclear proteins to facilitate the recruitment of DNA repair factors to sites of damage (2, 3). Accordingly, PARP-1 or PARP-2-deficient mice and mouse embryonic fibroblasts show chromosomal aberrations and various DNA repair defects (4–6).
Inhibition of PARP has become a promising therapeutic approach for the treatment of certain types of cancer (7). It was shown that PARP inhibitors could selectively kill homologous recombination (HR)-deficient cancer cells (8, 9). The reason behind the sensitivity of HR-deficient cells to PARP inhibition is thought to be the accumulation of single-stranded DNA (ssDNA) breaks in the absence of PAR synthesis, leading to replication fork collapse and double-stranded breaks (DSBs), which would then require HR factors for repair (8, 10). Recently, PARP activity has also been reported to play a role in the control of replication fork reversal upon topoisomerase 1 poisoning (11). Upon auto-PARylation, PARP1 interacts with the RecQ1 helicase and inhibits its specific fork restart activity, thereby transiently preventing restart of the reversed forks until repair of the damage has occurred (12).
PAR synthesis is a highly dynamic process and is counteracted by fast degradation by poly(ADP-ribosyl) glycohydrolase (PARG), an enzyme with both endo- and exoglycosidase activities (13, 14). PARG has 4 different isoforms in the cells: 99-kDa and 102-kDa isoforms, which localize to the cytoplasm, a 110-kDa isoform, which localizes to the nucleus, and a 60-kDa isoform, which localizes to the mitochondria (15). PARG activity has been previously associated with the control of various cellular processes, including response to oxidative stress and apoptosis (16, 17). Depleting all isoforms of PARG in mice results in embryonic lethality (18). However, a hypomorphic mutant for the nuclear isoform is viable but is highly sensitive to treatments with alkylating agents and ionizing radiation, implicating nuclear PARG in the maintenance of genome stability (19). Accordingly, PARG is recruited to DNA repair sites through PARP- and PCNA-dependent pathways (20, 21) and prevents mitotic catastrophe upon treatments with ionizing irradiation (22). It was also reported recently that BRCA2-deficient cells are exquisitely sensitive to PARG inhibition, suggesting that PARG functionally assists PARPs in preventing deleterious events at the replication fork (23). However, the molecular mechanisms underlying PARG involvement in DNA replication and repair have remained elusive, limiting also the possible clinical applications of PARG inactivation in cancer therapy.
Most recently, cells experiencing a stable downregulation of PARG were reported to display various phenotypes, such as increased lethality and defective HR-dependent recovery of collapsed forks, upon prolonged nucleotide depletion (24). However, the cell biological assays used in that study failed to reveal any specific defect associated with PAR accumulation during unperturbed replication or mild replication stress. In this work, using the same cellular system, we show that PARG depletion significantly interferes with the DNA replication process, even in the absence of exogenous genotoxic stress. PARG-depleted cells display DDR activation in S-phase, which is associated with slow fork progression, accumulation of abnormal DNA replication intermediates, and chromatin recruitment of DSB repair factors in the absence of detectable DSB. All these phenotypes are exacerbated by mild chemotherapeutic treatments. Our results indicate that replication fork reversal and restart—tightly controlled by PAR metabolism—are remarkably frequent events at endogenous lesions and/or difficult-to-replicate sequences. Furthermore, they provide mechanistic insight into the potential use of PARG inactivation in combination with DNA-damaging agents.
MATERIALS AND METHODS
Materials.
The following antibodies were used: ataxia-telangiectasia-mutated (ATM) p1981 (Epitomics, catalog no. 2152-1), ATM (2C1; Gene Tex, no. GTX70103), γH2AX (Millipore, no. 05-636), 53BP1 (Santa Cruz, no. sc-22760), CHK1 pS345 (Cell Signaling, no. 2348), CHK1 (Santa Cruz, no. sc-8408), KAP1-pS824 (Bethyl, no. A300-767A), KAP1 (Bethyl, no. A300-274A), Phospho-RPA32 (S4/S8) (Bethyl Laboratories, Inc., no. A300-245A), RPA32 (Calbiochem, no. NA19L), Rad51 (Santa Cruz, no. sc-8349), TFIIH (Santa Cruz, no. sc-293), and β-tubulin (Santa Cruz, no. sc-5274). Camptothecin (CPT) was purchased from Sigma Chemicals. Gallotannin (GLTN) was purchased from Enzo Life Sciences (no. ALX-270-418-G001) and added at a final concentration of 20 μM for 24 h.
Cell lines and culture conditions.
Short hairpin control (shCtrl) and shPARG SilenciX HeLa cell lines were purchased from Tebu Bio SAS, Le Perray En Yvelines, France (product no. 00301-00085). Cell cultures were maintained at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium containing 10% fetal calf serum and standard antibiotics (150 μg/ml). Hygromycin was added to the medium to provide selection pressure in the SilenciX cell lines. Effective PARG downregulation by small interfering RNA (siRNA) was achieved by double transfection (at 24 h) of U-2 OS (U2OS) cells with 40 nM siGENOME human PARG (8505) siRNA (SMARTpoolAlias: no. M011488020005). All experiments were performed 72 h after the first transfection.
Proliferation curves and clonogenic assays.
Cells (3 × 105) were seeded in 10-cm-diameter dishes at day 0. After 1, 2, 3, and 4 days, the cells were collected by trypsinization and counted using a Neubauer chamber. The proliferation rate was plotted as the fold change in total cell number with respect to the number of cells seeded at day 0 using Graphpad Prism software. For clonogenic assays, 4,000 cells were seeded in 10-cm-diameter dishes. Colonies were fixed for 15 min with 4% formaldehyde 9 days after seeding, stained with crystal violet, and counted.
Flow cytometry.
For flow cytometric analysis of γH2AX, EdU, and DAPI (4′,6-diamidino-2-phenylindole), cells were labeled for 30 min with 10 μM EdU, harvested, and fixed for 10 min with 4% formaldehyde–phosphate-buffered saline (PBS). Cells were washed with 1% bovine serum albumin (BSA)–PBS (pH 7.4), permeabilized with 0.5% saponin–1% BSA–PBS, and stained with anti-γH2AX antibody (Millipore, no. 05-636) for 2 h, followed by incubation with a suitable secondary antibody for 30 min. Incorporated EdU was labeled according to the manufacturer's instructions (Invitrogen, no. C35002). DNA was stained with 1 μg/ml DAPI, and samples were measured on a Cyan ADP flow cytometer (Beckman Coulter) and analyzed with Summit software v4.3.
DNA fiber analysis.
Asynchronous cells were labeled with 30 μM CldU, washed, and exposed to 250 μM IdU (with or without camptothecin [CPT]) before collection and resuspension in PBS. Cells were then lysed and DNA fibers stretched onto glass slides, as described previously (25). The fibers were denatured with 2.5 M HCl for 1 h, washed with PBS, and blocked with 2% BSA–phosphate-buffered saline–Tween 20 for 30 min. The newly replicated CldU and IdU tracks were revealed with anti-BrdU antibodies recognizing CldU and IdU, respectively. The secondary antibodies used were anti-mouse antibody–Alexa Fluor 488 and anti-rat antibody–Cy3. Microscopy was done using a Leica DMRB microscope equipped with a Leica DFC360 FX camera. Images were taken at ×63 magnification, using Leica Application Suite 3.3.0. Statistical analysis was carried out using GraphPad Prism.
Immunofluorescence staining and confocal microscopic analysis.
Cells were pre-extracted for 10 min on ice using 25 mM HEPES (pH 7.4), 50 mM NaCl, 1 mM EDTA, 3 mM MgCl2, 300 mM sucrose, and 0.5% Triton X-100 and then fixed using 4% formaldehyde. The cells were then stained with 53BP1, Rad51, RPA, and γH2AX antibodies, detected by appropriate secondary antibodies, and mounted with Vectashield (Vector Laboratories). Cells were imaged with a Leica TCS Sp5 microscope. Images were taken at ×63 magnification, using Leica Application Suite Advanced Fluorescence software. At least 100 cells were analyzed for the statistical analysis.
Western blot analysis.
Whole-cell extracts were prepared in Laemmli buffer (120 mM Tris-Cl [pH 6.8], 4% SDS, 20% glycerol); proteins were resolved by SDS-PAGE and transferred to nitrocellulose membranes. Immunoblots were carried out using the appropriate antibodies.
DSB detection by PFGE.
DSB detection by pulsed-field gel electrophoresis (PFGE) was performed as reported previously (11). Briefly, cells were embedded in a 0.8% agarose plug (2.5 × 105 cells/plug), digested in lysis buffer (100 mM EDTA, 1% [wt/vol] sodium lauryl sarcosine, 0.2% [wt/vol] sodium deoxycholate, 1 mg/ml proteinase K) at 37°C for 48 h, and washed in 10 mM Tris-HCl (pH 8.0)–100 mM EDTA. Electrophoresis was performed at 14°C in 0.9% (wt/vol) pulse field-certified agarose (Bio-Rad) containing Tris-borate-EDTA buffer in a Bio-Rad Chef DR III apparatus (9 h, 120°, 5.5 V/cm, and 30- to 18-s switch time; 6 h, 117°, 4.5 V/cm, and 18- to 9-s switch time; and 6 h, 112°, 4 V/cm, and 9- to 5-s switch time). The gel was stained with ethidium bromide and imaged on an Alpha Innotech imager.
EM analysis of genomic DNA mammalian cells.
In vivo psoralen cross-linking, isolation of total genomic DNA, and enrichment of the replication intermediates and their electron microscopy (EM) visualization were carried out as described previously (26). Briefly, cells were harvested, and genomic DNA was cross-linked by two rounds of incubation in 10 μM 4,5′,8-trimethylpsoralen and 2 min of irradiation with 366 nm UV light. Cells were lysed; genomic DNA was isolated from the nuclei by proteinase K digestion and phenol-chloroform extraction. Purified DNA was digested with PvuII, and replication intermediates were enriched on a BND cellulose column. EM samples were prepared by spreading the DNA on carbon-coated grids and visualized by platinum rotary shadowing. Images were acquired on a Philips CM 100 microscope and analyzed with ImageJ.
RESULTS
PARG depletion affects cell proliferation and interferes with unperturbed DNA replication.
To elucidate the functional relevance of PARG activity for chromosome replication, we analyzed a previously established cellular system for stable PARG depletion in HeLa cells (22) (see Fig. S1A in the supplemental material). Our single-cell immunostainings revealed PAR accumulation upon PARG depletion, even in the absence of genotoxic treatments. However, PARG-depleted cells showed small punctate PAR foci distinct from the typical pannuclear staining observed upon treatment with the genotoxic agent H2O2 (Fig. 1A), suggesting that PARG depletion leads to PAR accumulation specifically at sites of endogenous DNA damage. Cell proliferation assays revealed that PAR accumulation in shPARG cells is associated with decreased cell proliferation compared to shCtrl cells (Fig. 1B). This is mostly due to a prolonged division time, as clonogenic assays showed markedly reduced colony size and marginal effects on clonogenic potential in PARG-depleted cells (Fig. 1C). We next investigated if reduced proliferation in PARG-deficient cells reflected problems in chromosomal replication by assessing DNA content (DAPI), DNA synthesis (EdU incorporation), and DNA damage accumulation (H2AX phosphorylation) by flow cytometry (fluorescence-activated cell sorter [FACS]) analysis. PARG depletion leads to mild accumulation of S-phase cells (data not shown), accompanied by a small but reproducible reduction in EdU incorporation compared to control cells (Fig. 1D), suggesting that PAR accumulation interferes with the replication process under unperturbed conditions. Accordingly, H2AX phosphorylation—reportedly higher upon PARG inactivation (23)—was specifically increased in cells with intermediate DNA content, i.e., those undergoing chromosomal replication (Fig. 1E; see also Fig. S1B in the supplemental material). Overall, the effects of PARG depletion on EdU incorporation and H2AX phosphorylation were comparable to those induced by mild treatment (25 nM) with the prototypical Top1 poison camptothecin (CPT) (Fig. 1D and E), previously shown to interfere with replication fork progression and to induce fork reversal in the absence of detectable chromosomal breakage (11). Indeed, PAR accumulation—which is cell cycle independent upon H2O2 treatment—occurred specifically in S-phase (EdU-positive [EdU+]) cells upon PARG depletion (Fig. 1F) and was frequently associated with H2AX phosphorylation in the same cell (Fig. 1G). Taken together, these data suggest that PARG depletion affects cell proliferation and induces DNA damage by interference with the replication process.
FIG 1.

PARG depletion results in reduced proliferation and accumulation of DNA damage in replicating human cells. (A) Immunofluorescence analysis of shCtrl and shPARG HeLa cells for formation of poly(ADP) ribosylation (PAR). H2O2 (1 mM) treatment (10 min) was used as a positive control. (B) Proliferation curves of shCtrl and shPARG HeLa cells. The proliferation rate has been plotted as fold change in cell number with respect to the number of cells seeded at day 0. Median values and standard deviations of the results from three independent experiments are indicated. (C) Clonogenic assay of the indicated cell lines, with median values and standard deviations of the results from three independent experiments. Representative images are shown to indicate the marked effect of PARG depletion on colony size. (D) Flow cytometric analysis of DNA synthesis (EdU) in shCtrl and shPARG HeLa cells with mock (NT) or camptothecin (CPT) (25 nM) treatment. EdU-negative cells represent G1 and G2/M cells. Appreciable differences were observed upon PARG depletion and/or CPT treatment in EdU-positive (S-phase) cells. (E) Quantification of γH2AX-positive cells in shCtrl and shPARG cells with mock or CPT treatment. The percentage of γH2AX-positive cells is indicated in parentheses. (F) Quantification of PAR (green)- and/or EdU-positive (red) shCtrl and shPARG HeLa cells, optionally treated with H2O2. (G) Quantification of PAR (green)- and/or γH2AX-positive (red) shCtrl and shPARG HeLa cells, optionally treated with H2O2. In panels F and G gray indicates double-negative cells and yellow indicates double-positive cells.
PARG depletion results in slow replication fork progression even in the absence of genotoxic treatments.
These effects on bulk DNA replication in PARG-depleted cells prompted us to test the effect of PARG depletion on the progression of individual replication forks, using a well-established “DNA fiber spreading” assay (25). Both shCtrl and shPARG cells were pulse labeled with the thymidine analog CldU (detected as red tracks) for 30 min and then for 30 min with a second thymidine analog (IdU, detected as green tracks). Mild CPT treatment was optionally applied or not applied during the second labeling. In agreement with previous results (11), we confirmed that mild CPT treatments are sufficient to significantly slow down replication fork progression in control HeLa cells (Fig. 2). Surprisingly, replication fork progression was slowed down to a similar extent by PARG depletion, even in the absence of exogenous genotoxic treatments. Furthermore, CPT treatment in PARG-depleted cells led to marginal further effects on the progression of individual forks (Fig. 2). Taken together, these data suggest that interfering with PAR catabolism by itself results in marked replication stress and fork slowdown comparable to the effects of mild chemotherapeutic treatments.
FIG 2.

PARG depletion slows down replication fork progression. (A) Schematic experimental conditions for DNA replication track analysis. shCtrl and shPARG cells were labeled with CldU and IdU as indicated. Red and green identify CldU- and IdU-containing tracks, respectively. CPT (25 nM) was optionally added concomitantly with the second label. Representative DNA fiber tracks from shCtrl and shPARG cells with or without CPT treatment are shown below the schematic. Scale bar, 5 μm. (B) Statistical analysis of IdU tract length measurements from shCtrl or shPARG cells. Data represent relative lengths of IdU tracts (green) synthesized after mock (NT) or CPT (25 nM) treatment. At least 125 tracks were scored for each data set. Whiskers indicate the 10 and 90 percentiles. Statistical tests were performed using Mann-Whitney analysis: *, P = 0.0206; ***, P < 0.0001. Very similar results were obtained in two independent experiments.
PARG prevents the accumulation of reversed replication forks and postreplicative ssDNA gaps.
We next investigated whether slow replication fork progression upon PARG depletion was accompanied by altered architecture of replication intermediates, using a combination of in vivo psoralen cross-linking and transmission electron microscopy (EM) (26). Interestingly, our EM analysis revealed that depletion of PARG in unperturbed cells resulted in a substantial accumulation of reversed replication forks (25% versus 6% in control cells) (Fig. 3A and B; see also Fig. S2A in the supplemental material). The frequency of reversed forks in untreated PARG-depleted cells is close to that observed with mild CPT treatments in control HeLa cells (36%) (Fig. 3B) and that reported upon Top1 poisoning in yeast, Xenopus egg extracts, U2OS cells, and mouse embryonic fibroblasts (15 to 40%) (11). Furthermore, PARG depletion increased the frequency of reversed forks upon CPT treatments only marginally (from 36% to 42%) (Fig. 3B), mirroring our observations on fork progression examined by DNA fiber analysis (Fig. 2B). In addition to fork reversal, approximately 50% of the replication forks in PARG-depleted cells exhibited ssDNA gaps on replicated duplexes, compared to about 20% of the forks in control cells (Fig. 3C and D; see also Fig. S2B in the supplemental material). A significant fraction of replication intermediates from PARG-depleted cells displayed 2 or more ssDNA gaps, detectable on both replicated duplexes (Fig. 3D; see also Fig. S2B). As shown for fork slowing and fork reversal, CPT treatment did not markedly increase ssDNA gap accumulation in PARG-depleted cells (Fig. 3D). Overall, these EM data show that PARG depletion in unperturbed cells results in alterations of replication fork structure similar to those seen with mild chemotherapeutic treatments.
FIG 3.

PARG-depleted cells accumulate reversed forks and ssDNA gaps on replicated duplexes. (A) Representative electron micrograph of a reversed replication fork observed on genomic DNA from non-shPARG-treated cells. The arrow points to the four-way junction at the replication fork, indicative of fork reversal. (B) Frequency of fork reversal in shCtrl and shPARG HeLa cells treated with or without CPT (25 nM). The percentage values are indicated on top of the bars. The numbers of analyzed molecules are indicated in parentheses. (C) Representative electron micrograph of a replication fork observed on genomic DNA from non-shPARG-treated cells. The arrows point to ssDNA gaps along the replicated duplexes, detectable by locally reduced thickness of the DNA filament (26). (D) Statistical distribution of the number of ssDNA gaps observed in the populations of molecules analyzed in panel B. Values very similar to those shown in panels B and D have been obtained in another independent EM experiment.
PARG prevents recruitment of DSB repair factors to replicating chromatin.
Since PARG depletion results in profound structural alterations of replication intermediates and H2AX phosphorylation, we next tested whether this was accompanied by detectable chromatin recruitment of other DDR factors, previously reported to respond to DSB. Our immunofluorescence (IF)-based confocal imaging experiments confirmed our results (Fig. 1E) and the previously reported accumulation of γH2AX foci in unperturbed PARG-depleted cells (23) (Fig. 4A). Surprisingly, we noticed that, upon PARG depletion, a large fraction of γH2AX-positive cells were also positive for the DSB repair factor 53BP1 in both the absence and the presence of exogenous genotoxic stress (CPT; Fig. 4A to C). Besides increasing the fraction of 53BP1-positive cells (cells with more than five foci), PARG depletion clearly increased the number of γH2AX and 53BP1 foci detected in the positive-testing cells, leading to a dense, punctuated immunostaining pattern that resembles control cells treated with mild CPT doses (Fig. 4A and B). Importantly, these small 53BP1 foci detected by confocal microscopy are clearly distinguishable from the intense larger 53BP1 foci observed in infrared (IR)-treated cells (see Fig. S3A and B in the supplemental material) and may escape detection by epifluorescence microscopy (11). One possible interpretation of these data is that PARG depletion, similarly to low CPT doses, leads to mild and transient chromosomal breakage, marked by local 53BP1 recruitment. In line with this hypothesis, one additional marker typically recruited to DSB upon end resection—i.e., RAD51—displayed similar trends of accumulation upon PARG depletion (Fig. 4D; see also Fig. S3C). As for 53BP1 recruitment, PARG depletion per se was sufficient to induce RAD51 recruitment at levels similar to those seen with mild CPT treatments, albeit only in a subpopulation of the γH2AX-positive cells (Fig. 4D; see also Fig. S3C). Taken together, these data indicate that DNA replication interference by PARG depletion is associated with recruitment of DDR and DNA repair factors to DSB or other replication-associated DNA structures.
FIG 4.

PARG depletion leads to chromatin accumulation of typical DSB markers. (A and B) Representative images from confocal immunofluorescence analysis of shCtrl and shPARG cells treated with or without 25 nM CPT and stained for γH2AX, 53BP1, and RAD51. In panel B, to better reveal the increased numbers of 53BP1 and RAD51 foci in positive-testing shPARG cells versus shCtrl cells, fewer untreated cells are shown at higher magnification. (C and D) Quantification plots show the percentages of γH2AX-positive cells also positive for 53BP1 and RAD51, respectively.
PARG downregulation and inhibition lead to similar phenotypic consequences.
In order to test whether the described phenotypes are specifically and reproducibly induced by PARG inactivation, we reverted to the Rb/p53-proficient osteosarcoma cell line U-2 OS (U2OS) and induced either PARG downregulation by siRNA transfection or PARG inhibition by gallotannin (GLTN) (27). Importantly, despite different basal levels of endogenous stress in U2OS and HeLa cells, the two independent strategies for PARG inactivation in U2OS cells led to similar phenotypic consequences (Fig. 5), which were largely comparable to those already described in stably downregulated HeLa cells (Fig. 2 to 4). Specifically, PARG downregulation or inhibition led to significantly slower replication fork progression (Fig. 5A), to 3-to-5-fold accumulation of reversed replication forks (Fig. 5B), and to marked accumulation of γH2AX foci, frequently associated with chromatin recruitment of the DNA repair factors 53BP1 and RAD51 (Fig. 5C and D). Thus, the peculiar phenotypic consequences of PARG downregulation during unperturbed replication can be reproduced in a different human cell line and can be recapitulated by treatment with the PARG inhibitor gallotannin.
FIG 5.
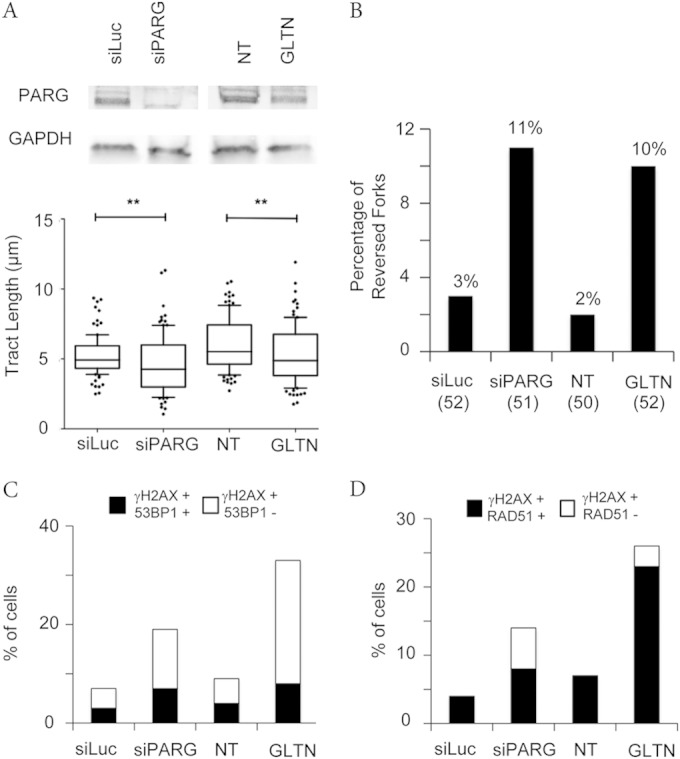
PARG inactivation by different methods leads to replication interference and DDR marks. (A) PARG levels after siRNA-mediated depletion were detected by immunoblotting. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used as the loading control. DNA fiber experiments were performed as in Fig. 2A (no CPT treatment), but with a 20-min labeling time. Data represent the results of statistical analysis of IdU tract (green in Fig. 2A) length measurements in U2OS cells 72 h after siLuc or siPARG transfection or upon mock treatment (NT) or 20 μM gallotannin (GLTN) treatment for 24 h. At least 100 tracks were scored for each data set. Whiskers indicate the 10 and 90 percentiles. Statistical tests were performed using Mann-Whitney analysis; **, P < 0.001. (B) Frequency of fork reversal in U2OS cells 72 h after siLuc or siPARG transfection or upon mock treatment (NT) or 20 μM gallotannin (GLTN) treatment for 24 h. The numbers of analyzed molecules are indicated in parentheses. (C and D) Quantification plots to show the percentage of γH2AX-positive cells also positive for 53BP1 and RAD51, respectively, in U2OS cells 72 h after siLuc or siPARG transfection or upon mock treatment (NT) or 20 μM gallotannin (GLTN) treatment for 24 h.
Depletion of PARG results in checkpoint activation uncoupled from detectable DSB formation.
In line with the hypothesis that unusual DNA structures resembling DSB may accumulate upon PARG depletion, promoting the recruitment of repair factors, we observed that PARG-depleted cells display basal activation of ataxia-Rad3-related (ATR)/Chk1 and ATM/Kap1 pathways in the absence of exogenous damage. Upon mild CPT treatment, ATM and ATR activation in PARG-depleted cells becomes comparable to that seen in IR-treated cells (Fig. 6A). Using an optimized pulsed-field gel-electrophoresis (PFGE) protocol, which can detect <100 DSB per cell (11, 12), we verified that ATM and ATR activation upon IR treatment is associated with marked accumulation of DSB (Fig. 6B) and with the expected phosphorylation of RPA32 on S4/S8 (Fig. 6A), a typical DSB marker (28). However, PARG depletion, even in combination with mild CPT treatments, was associated neither with detectable chromosomal breakage over background levels nor with any detectable RPA32-S4/S8 phosphorylation (Fig. 6). These surprising results uncouple DSB formation from ATM/ATR signaling and 53BP1/RAD51 recruitment and strongly suggest that checkpoint activation and recruitment of repair factors upon PARG inactivation conditions reflect accumulation of unusual DNA structures different from DSB.
FIG 6.
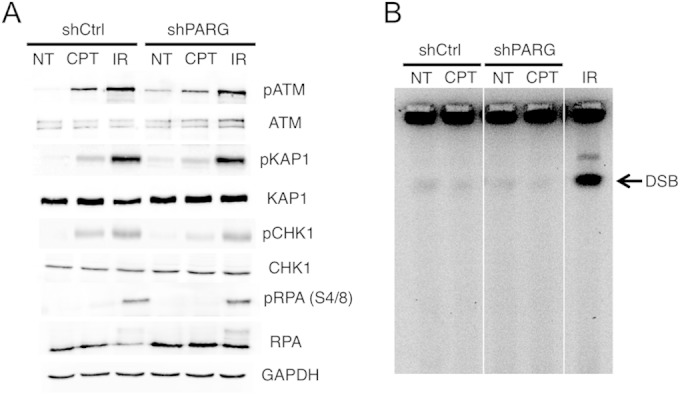
Checkpoint activation upon PARG depletion can be uncoupled from DSB formation. (A) Western blot analysis of ATR checkpoint activation (CHK1 phosphorylation) and ATM checkpoint activation (ATM and KAP1 phosphorylation, respectively) in shCtrl and shPARG HeLa cells optionally treated with CPT (25 nM) or 20 Gy IR. IR was used as a positive control for ATM/ATR checkpoint activation. RPA32 phosphorylation in S4/S8 is used as a marker for DSB formation. Total ATM, KAP1, CHK1, and RPA32 levels are displayed, and GAPDH was used as loading control. (B) PFGE analysis of DSB formation in shCtrl and shPARG HeLa cells optionally treated with CPT (25 nM). IR (20 Gy) treatment in shCtrl cells was used as a positive control for DSB formation.
DISCUSSION
In this report, we show that depletion or inhibition of PARG in human cells results in reduced cell proliferation associated with impairment of the DNA replication process. This is accompanied by detection of several recognized markers of replication stress, such as H2AX hyperphosphorylation in S phase, impaired progression of individual replication forks, and checkpoint activation. Notably, impaired replication fork progression is accompanied by a widespread alteration of the fork structure, such as accumulation of postreplicative ssDNA gaps and reversed replication forks. Accumulation of these unusual structures during replication is accompanied by chromatin recruitment of DDR and DSB repair factors, in the absence of detectable chromosomal breakage. Importantly, albeit exacerbated by genotoxic treatments, all these phenotypes are already clearly detectable in unperturbed PARG-depleted cells, clearly showing that PAR catabolism is of crucial importance to assist complete and effective replication during unperturbed S phase.
PARP activity was recently reported to mediate accumulation of reversed replication forks upon Top1 inhibition (11), by transiently inhibiting the fork restart activity of the RecQ1 helicase (12), in order to coordinate fork restart with repair of the damaged template. While PARG inactivation was thus expected to induce reversed fork accumulation upon CPT treatment, the drastic accumulation of reversed forks and the global reduction of fork speed in untreated PARG-deficient cells are important unexpected observations. These data strongly suggest that remarkably frequent endogenous lesions and/or alternative DNA structures induce transient reversal of replication forks in unperturbed S phase, rendering active PAR degradation essential to ensure continued replication fork progression once DNA synthesis can resume (Fig. 7). This is in agreement with the recent finding that repetitive DNA sequences with a propensity to form non-B DNA structures induce reversal of traversing replication forks at remarkably high frequencies (29). Considering the high number of endogenous DNA lesions (30) and the abundance of non-B-forming structures in the human genome (31), it is conceivable that fine-tuning of PAR synthesis and degradation at a number of chromosomal locations plays a pivotal role in assisting complete and faithful replication of the human genome. These observations may contribute to explain the mitotic defects previously associated with the essential role of PARG in development, as deletion of all isoforms of PARG leads to embryonic lethality in mice (29).
FIG 7.
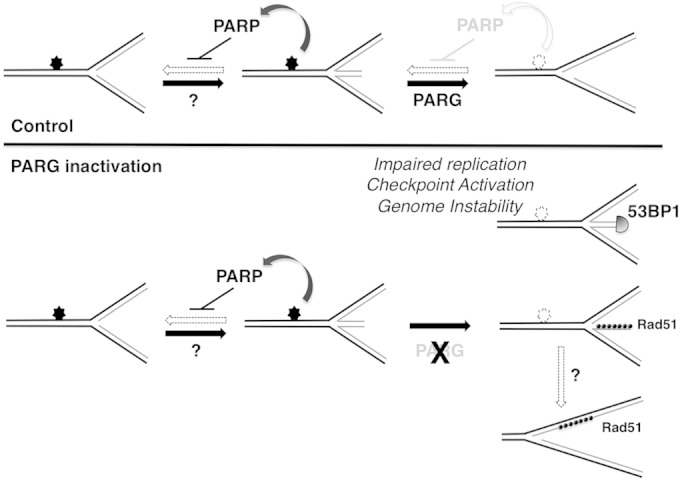
Model suggesting the role of PARG in continued DNA replication under conditions of endogenous and exogenous genotoxic stress. In control cells, replication forks encountering endogenous lesions or non-B DNA structures (filled stars) undergo dynamic fork reversal, promoted by as-yet-unknown factors and stabilized by PARP-mediated transient inhibition of fork restart. Once the lesion/structure is repaired/resolved (empty stars), PARG locally overcomes PARP inhibitory activity and mediates fork restart, ensuring replication completion and genome integrity. In the absence of PARG, forks reversed at endogenous lesions/non-B DNA structures fail to restart due to a lack of PAR degradation. Persistence and/or processing of reversed forks leads to unscheduled recruitment of DNA repair factors resulting in pathological DNA structures, impaired replication, checkpoint activation, and genome instability. Accumulation of postreplicative ssDNA gaps may arise from alternative pathways of fork restart or by independent molecular mechanisms. The molecular defects associated with PARG inactivation are exacerbated by exogenous sources of genotoxic stress, increasing the number of potential fork-blocking lesions on chromosomal DNA.
PARG inhibition was shown to kill homologous recombination (HR)-defective cells, via a replication-dependent mechanism (23). Although the effect of PARG inhibition on the replication process was not directly investigated, the authors postulated that replication fork collapse by PARG inhibition may lead to DSB formation and thus the HR requirement to restart collapsed forks. Analogously, PAR accumulation was recently reported to affect recovery of replication forks collapsed by prolonged nucleotide depletion, possibly by interfering with RPA loading on the resulting ssDNA (24). These studies seemed to suggest that PAR catabolism assists the replication process only under the conditions of extensive genotoxic stress associated with fork breakage. However, taking advantage of single-molecule analysis of the replication process, we now show that PARG inactivation affects the progression of all replication forks and alters the molecular architecture of a significant fraction of replication intermediates, in the absence of detectable chromosomal breakage. We also show that accumulation of these unusual replication intermediates is accompanied by DDR activation and chromatin recruitment of DSB repair factors, suggesting the conceptually attractive hypothesis that the formation of DNA ends (regressed arms) by fork reversal could contribute to checkpoint signaling. It should be noted, however, that other experimental conditions recently reported to induce similar levels of reversed replication forks have not been associated with DDR activation (11, 32–34), excluding the possibility that fork reversal per se is sufficient to induce checkpoint activation and chromatin recruitment of DSB repair factors. Checkpoint activation upon PAR accumulation may directly reflect the recently reported physical association of PAR with checkpoint factors, such as CHK1 (35). Alternatively, the persistence of reversed forks upon impairment of PAR degradation may lead to recruitment of cellular factors usually recruited at DSB (53BP1 and RAD51), either before or after nucleolytic processing of the stalled reversed forks (Fig. 7). Furthermore, recruitment of these factors and ATR/ATM checkpoint activation may be linked to minor changes in the molecular architecture of reversed forks associated with their persistence, which may escape systematic EM detection. In this scenario, upon PARG depletion and reversed fork accumulation, HR and other DSB repair factors may become essential to drive alternative, RecQ1-independent pathways for the restart of reversed replication forks. This may provide an alternative explanation for the reported requirement of HR for cell survival upon PARG inhibition (23). Detection of ssDNA gaps has been linked in model systems to repriming events across DNA lesions (36, 37). Although we cannot directly link the observed accumulation of postreplicative ssDNA gaps to the persistence of reversed forks, a tantalizing alternative hypothesis is that ssDNA gaps may accumulate on replicated duplexes as a consequence of RecQ1-independent replication fork restart (Fig. 7). These restart events may entail nucleolytic degradation rather than branch migration of the reversed forks (12), in agreement with the nucleolytic processing of reversed forks previously reported in yeast (38, 39). Regardless of their source, postreplicative ssDNA gaps can certainly contribute to explain the observed accumulation of RAD51 in PARG-depleted cells. Intriguingly, RAD51 was previously reported to limit ssDNA accumulation at yeast and Xenopus replication forks, especially in response to genotoxic stress (40). Furthermore, RAD51 itself and several HR and Fanconi anemia factors were shown to prevent excessive degradation of newly synthesized DNA in response to replication stress (41, 42). Whether the role of HR factors in the face of endogenous or exogenous replication stress is related to replication fork remodelling will be the subject of intense studies in the near future.
Altogether, our data illustrate that PAR degradation is required for remodeling of replication forks in unperturbed S phase and thus provide mechanistic insight into the essential role of PARG in cell growth and development. At the same time, we provide a molecular basis for the anticipated use of PARG inhibitors to potentiate cancer chemotherapy, by showing that the molecular defects associated with PARG inactivation are exacerbated by mild chemotherapeutic treatments (43). This attractive therapeutic perspective has been hampered thus far by the limited specificity of the currently available PARG inhibitors (14), which will be likely improved based on the recent resolution of the PARG crystal structure (44). At least one additional protein—i.e., the ADP-ribose protein glycohydrolase TARG1—has been recently shown to assist PARG in full removal of PAR chains from target proteins and has been implicated in human disease (45); it will thus be important to test the possible involvement of this and possibly other PAR-degrading enzymes in the maintenance of genome stability during replication.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Center for Microscopy and Image Analysis of the University of Zurich for technical assistance with electron microscopy. We are grateful to all current and past members of the Lopes group for useful discussions.
This work was supported by the Swiss National Science Foundation (grants PP00P3_135292 and 31003A_146924 to M.L. A.R.C. was supported by the Zürcher Universitätsverein (ZUNIV), Fonds zur Förderung des akademischen Nachwuchses (FAN).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01077-14.
REFERENCES
- 1.Gibson BA, Kraus WL. 2012. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol 13:411–424. doi: 10.1038/nrm3376. [DOI] [PubMed] [Google Scholar]
- 2.Bouchard VJ, Rouleau M, Poirier GG. 2003. PARP-1, a determinant of cell survival in response to DNA damage. Exp Hematol 31:446–454. doi: 10.1016/S0301-472X(03)00083-3. [DOI] [PubMed] [Google Scholar]
- 3.Woodhouse BC, Dianov GL. 2008. Poly ADP-ribose polymerase-1: an international molecule of mystery. DNA Repair (Amst) 7:1077–1086. doi: 10.1016/j.dnarep.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 4.de Murcia JM, Niedergang C, Trucco C, Ricoul M, Dutrillaux B, Mark M, Oliver FJ, Masson M, Dierich A, LeMeur M, Walztinger C, Chambon P, de Murcia G. 1997. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci U S A 94:7303–7307. doi: 10.1073/pnas.94.14.7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ménissier de Murcia J, Ricoul M, Tartier L, Niedergang C, Huber A, Dantzer F, Schreiber V, Ame JC, Dierich A, LeMeur M, Sabatier L, Chambon P, de Murcia G. 2003. Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J 22:2255–2263. doi: 10.1093/emboj/cdg206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang ZQ, Stingl L, Morrison C, Jantsch M, Los M, Schulze-Osthoff K, Wagner EF. 1997. PARP is important for genomic stability but dispensable in apoptosis. Genes Dev 11:2347–2358. doi: 10.1101/gad.11.18.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anders CK, Winer EP, Ford JM, Dent R, Silver DP, Sledge GW, Carey LA. 2010. Poly(ADP-ribose) polymerase inhibition: “targeted” therapy for triple-negative breast cancer. Clin Cancer Res 16:4702–4710. doi: 10.1158/1078-0432.CCR-10-0939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. 2005. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 9.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. 2005. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 10.Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. 2010. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell 37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ray Chaudhuri A, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, Cocito A, Costanzo V, Lopes M. 2012. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat Struct Mol Biol 19:417–423. doi: 10.1038/nsmb.2258. [DOI] [PubMed] [Google Scholar]
- 12.Berti M, Ray Chaudhuri A, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, Odreman F, Glatter T, Graziano S, Mendoza-Maldonado R, Marino F, Lucic B, Biasin V, Gstaiger M, Aebersold R, Sidorova JM, Monnat RJ Jr, Lopes M, Vindigni A. 2013. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat Struct Mol Biol 20:347–354. doi: 10.1038/nsmb.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gagné JP, Hendzel MJ, Droit A, Poirier GG. 2006. The expanding role of poly(ADP-ribose) metabolism: current challenges and new perspectives. Curr Opin Cell Biol 18:145–151. doi: 10.1016/j.ceb.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 14.Min W, Wang ZQ. 2009. Poly (ADP-ribose) glycohydrolase (PARG) and its therapeutic potential. Front Biosci 14:1619–1626. doi: 10.2741/3329. [DOI] [PubMed] [Google Scholar]
- 15.Meyer-Ficca ML, Meyer RG, Coyle DL, Jacobson EL, Jacobson MK. 2004. Human poly(ADP-ribose) glycohydrolase is expressed in alternative splice variants yielding isoforms that localize to different cell compartments. Exp Cell Res 297:521–532. doi: 10.1016/j.yexcr.2004.03.050. [DOI] [PubMed] [Google Scholar]
- 16.Erdelyi K, Bai P, Kovacs I, Szabo E, Mocsar G, Kakuk A, Szabo C, Gergely P, Virag L. 2009. Dual role of poly(ADP-ribose) glycohydrolase in the regulation of cell death in oxidatively stressed A549 cells. FASEB J 23:3553–3563. doi: 10.1096/fj.09-133264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng X, Zhou Y, Proctor AM, Hopkins MM, Liu M, Koh DW. 2012. Silencing of apoptosis-inducing factor and poly(ADP-ribose) glycohydrolase reveals novel roles in breast cancer cell death after chemotherapy. Mol Cancer 11:48. doi: 10.1186/1476-4598-11-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koh DW, Lawler AM, Poitras MF, Sasaki M, Wattler S, Nehls MC, Stoger T, Poirier GG, Dawson VL, Dawson TM. 2004. Failure to degrade poly(ADP-ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality. Proc Natl Acad Sci U S A 101:17699–17704. doi: 10.1073/pnas.0406182101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cortes U, Tong WM, Coyle DL, Meyer-Ficca ML, Meyer RG, Petrilli V, Herceg Z, Jacobson EL, Jacobson MK, Wang ZQ. 2004. Depletion of the 110-kilodalton isoform of poly(ADP-ribose) glycohydrolase increases sensitivity to genotoxic and endotoxic stress in mice. Mol Cell Biol 24:7163–7178. doi: 10.1128/MCB.24.16.7163-7178.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fisher AE, Hochegger H, Takeda S, Caldecott KW. 2007. Poly(ADP-ribose) polymerase 1 accelerates single-strand break repair in concert with poly(ADP-ribose) glycohydrolase. Mol Cell Biol 27:5597–5605. doi: 10.1128/MCB.02248-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mortusewicz O, Fouquerel E, Ame JC, Leonhardt H, Schreiber V. 2011. PARG is recruited to DNA damage sites through poly(ADP-ribose)- and PCNA-dependent mechanisms. Nucleic Acids Res 39:5045–5056. doi: 10.1093/nar/gkr099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amé JC, Fouquerel E, Gauthier LR, Biard D, Boussin FD, Dantzer F, de Murcia G, Schreiber V. 2009. Radiation-induced mitotic catastrophe in PARG-deficient cells. J Cell Sci 122:1990–2002. doi: 10.1242/jcs.039115. [DOI] [PubMed] [Google Scholar]
- 23.Fathers C, Drayton RM, Solovieva S, Bryant HE. 2012. Inhibition of poly(ADP-ribose) glycohydrolase (PARG) specifically kills BRCA2-deficient tumor cells. Cell Cycle 11:990–997. doi: 10.4161/cc.11.5.19482. [DOI] [PubMed] [Google Scholar]
- 24.Illuzzi G, Fouquerel E, Ame JC, Noll A, Rehmet K, Nasheuer HP, Dantzer F, Schreiber V. 2014. PARG is dispensable for recovery from transient replicative stress but required to prevent detrimental accumulation of poly(ADP-ribose) upon prolonged replicative stress. Nucleic Acids Res 42:7776–7792. doi: 10.1093/nar/gku505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jackson DA, Pombo A. 1998. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol 140:1285–1295. doi: 10.1083/jcb.140.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neelsen KJ, Chaudhuri AR, Follonier C, Herrador R, Lopes M. 2014. Visualization and interpretation of eukaryotic DNA replication intermediates in vivo by electron microscopy. Methods Mol Biol 1094:177–208. doi: 10.1007/978-1-62703-706-8_15. [DOI] [PubMed] [Google Scholar]
- 27.Formentini L, Arapistas P, Pittelli M, Jacomelli M, Pitozzi V, Menichetti S, Romani A, Giovannelli L, Moroni F, Chiarugi A. 2008. Mono-galloyl glucose derivatives are potent poly(ADP-ribose) glycohydrolase (PARG) inhibitors and partially reduce PARP-1-dependent cell death. Br J Pharmacol 155:1235–1249. doi: 10.1038/bjp.2008.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oakley GG, Patrick SM. 2010. Replication protein A: directing traffic at the intersection of replication and repair. Front Biosci (Landmark ed) 15:883–900. doi: 10.2741/3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Follonier C, Oehler J, Herrador R, Lopes M. 3 March 2013, posting date Friedreich's ataxia-associated GAA repeats induce replication-fork reversal and unusual molecular junctions. Nat Struct Mol Biol doi: 10.1038/nsmb.2520. [DOI] [PubMed] [Google Scholar]
- 30.Akbari M, Krokan HE. 2008. Cytotoxicity and mutagenicity of endogenous DNA base lesions as potential cause of human aging. Mech Ageing Dev 129:353–365. doi: 10.1016/j.mad.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 31.Mirkin SM. 2007. Expandable DNA repeats and human disease. Nature 447:932–940. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- 32.Neelsen KJ, Lopes M. Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol, in press. [DOI] [PubMed] [Google Scholar]
- 33.Neelsen KJ, Zanini IM, Herrador R, Lopes M. 2013. Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J Cell Biol 200:699–708. doi: 10.1083/jcb.201212058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid J, Herrador R, Vindigni A, Lopes M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Min W, Bruhn C, Grigaravicius P, Zhou ZW, Li F, Kruger A, Siddeek B, Greulich KO, Popp O, Meisezahl C, Calkhoven CF, Burkle A, Xu X, Wang ZQ. 2013. Poly(ADP-ribose) binding to Chk1 at stalled replication forks is required for S-phase checkpoint activation. Nat Commun 4:2993. doi: 10.1038/ncomms3993. [DOI] [PubMed] [Google Scholar]
- 36.Heller RC, Marians KJ. 2006. Replication fork reactivation downstream of a blocked nascent leading strand. Nature 439:557–562. doi: 10.1038/nature04329. [DOI] [PubMed] [Google Scholar]
- 37.Lopes M, Foiani M, Sogo JM. 2006. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell 21:15–27. doi: 10.1016/j.molcel.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 38.Cotta-Ramusino C, Fachinetti D, Lucca C, Doksani Y, Lopes M, Sogo J, Foiani M. 2005. Exo1 processes stalled replication forks and counteracts fork reversal in checkpoint-defective cells. Mol Cell 17:153–159. doi: 10.1016/j.molcel.2004.11.032. [DOI] [PubMed] [Google Scholar]
- 39.Hu J, Sun L, Shen F, Chen Y, Hua Y, Liu Y, Zhang M, Hu Y, Wang Q, Xu W, Sun F, Ji J, Murray JM, Carr AM, Kong D. 2012. The intra-S phase checkpoint targets Dna2 to prevent stalled replication forks from reversing. Cell 149:1221–1232. doi: 10.1016/j.cell.2012.04.030. [DOI] [PubMed] [Google Scholar]
- 40.Hashimoto Y, Ray Chaudhuri A, Lopes M, Costanzo V. 2010. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17:1305–1311. doi: 10.1038/nsmb.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. 2011. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145:529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schlacher K, Wu H, Jasin M. 2012. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22:106–116. doi: 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tentori L, Leonetti C, Scarsella M, Muzi A, Vergati M, Forini O, Lacal PM, Ruffini F, Gold B, Li W, Zhang J, Graziani G. 2005. Poly(ADP-ribose) glycohydrolase inhibitor as chemosensitiser of malignant melanoma for temozolomide. Eur J Cancer 41:2948–2957. doi: 10.1016/j.ejca.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 44.Slade D, Dunstan MS, Barkauskaite E, Weston R, Lafite P, Dixon N, Ahel M, Leys D, Ahel I. 2011. The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase. Nature 477:616–620. doi: 10.1038/nature10404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sharifi R, Morra R, Appel CD, Tallis M, Chioza B, Jankevicius G, Simpson MA, Matic I, Ozkan E, Golia B, Schellenberg MJ, Weston R, Williams JG, Rossi MN, Galehdari H, Krahn J, Wan A, Trembath RC, Crosby AH, Ahel D, Hay R, Ladurner AG, Timinszky G, Williams RS, Ahel I. 2013. Deficiency of terminal ADP-ribose protein glycohydrolase TARG1/C6orf130 in neurodegenerative disease. EMBO J 32:1225–1237. doi: 10.1038/emboj.2013.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.