

Abstract
Aging is the principal demographic risk factor for Alzheimer disease (AD), the most common neurodegenerative disorder. Klotho is a key modulator of the aging process and, when overexpressed, extends mammalian lifespan, increases synaptic plasticity, and enhances cognition. Whether klotho can counteract deficits related to neurodegenerative diseases, such as AD, is unknown. Here we show that elevating klotho expression decreases premature mortality and network dysfunction in human amyloid precursor protein (hAPP) transgenic mice, which simulate key aspects of AD. Increasing klotho levels prevented depletion of NMDA receptor (NMDAR) subunits in the hippocampus and enhanced spatial learning and memory in hAPP mice. Klotho elevation in hAPP mice increased the abundance of the GluN2B subunit of NMDAR in postsynaptic densities and NMDAR-dependent long-term potentiation, which is critical for learning and memory. Thus, increasing wild-type klotho levels or activities improves synaptic and cognitive functions, and may be of therapeutic benefit in AD and other cognitive disorders.
Keywords: aging, Alzheimer's disease, cognition, klotho, mice, NMDA receptors
Introduction
Alzheimer's disease (AD) is reaching pandemic proportions (Wimo and Prince, 2010; Huang and Mucke, 2012; Matthews et al., 2013). Potential treatments for AD have targeted symptoms and individual pathogenic factors suspected of contributing causally to the disease, but so far no truly effective therapeutics have emerged (Mangialasche et al., 2010; Selkoe, 2012; Giacobini and Gold, 2013). In light of these failures and the urgent need for better treatments, it is important to explore alternate or complementary strategies to counteract the disease. Such approaches may be to delay aging, the primary demographic risk factor for AD, or to enhance the cognitive processes that AD erodes.
Klotho is a pleiotropic protein that delays aging and enhances cognition (Kurosu et al., 2005; Dubal et al., 2014). It circulates throughout the body and brain following cleavage by ADAM 10 and 17 from its transmembrane form (Chen et al., 2007). In brain, klotho is produced primarily by the choroid plexus (German et al., 2012; Kuang et al., 2014) and its brain levels decrease with aging (Duce et al., 2008). Klotho levels in the CSF of humans also decrease with aging and in AD, correlating with Mini-Mental State Exam scores (Semba et al., 2014).
Genetic reduction of klotho in mice reduces lifespan (Kuro-o et al., 1997) and impairs myelination, synaptic integrity, and cognition (Nagai et al., 2003; Shiozaki et al., 2008; Chen et al., 2013), whereas transgenic overexpression extends lifespan (Kurosu et al., 2005) and enhances synaptic plasticity and cognitive functions in mice (Dubal et al., 2014) and protects neuronal cultures against Aβ oligomers and excitotoxicity (Zeldich et al., 2014). In humans, a single allele of a genetic variant of KLOTHO, KL-VS, that increases circulating levels of klotho (Dubal et al., 2014) and modifies its functions in cell culture (Arking et al., 2002; Tucker Zhou et al., 2013), promotes longevity (Arking et al., 2002, 2005; Invidia et al., 2010) and is associated with larger prefrontal cortical regions (Yokoyama et al., 2015) and better cognitive functions (Dubal et al., 2014; Yokoyama et al., 2015) in normal aging populations.
However, it is unknown whether klotho elevation can counteract cognitive disorders, such as AD. To determine whether it can protect against AD-related deficits, we crossed klotho (KL) transgenic mice that overexpress wild-type mouse klotho throughout the body and brain (Kuro-o et al., 1997) with human amyloid precursor protein (hAPP) transgenic mice from line J20, which carry mutations that cause early onset AD in humans (Mucke et al., 2000). Like humans with AD, singly transgenic hAPP-J20 mice have elevated levels of amyloid-β (Aβ) peptides in the brain as well as premature mortality, deficits in cognition, behavioral abnormalities, neural network dysfunction, synaptic impairments, and neuritic amyloid plaques (Palop et al., 2003, 2007; Cheng et al., 2007; Cissé et al., 2011; Roberson et al., 2011; Sanchez et al., 2012; Verret et al., 2012). This model also shares a large number of molecular alterations with the human condition, including decreased expression of calbindin in the dentate gyrus and of reelin in the entorhinal cortex (Palop et al., 2003; Chin et al., 2007), increased expression of collagen VI (Cheng et al., 2009) and metenkephalin (Meilandt et al., 2008) in the hippocampus, and depletion of select voltage-gated sodium channels in the parietal cortex (Verret et al., 2012). Furthermore, treatment with the anti-epileptic drug levetiracetam, which ameliorated network dysfunction in hAPP-J20 mice (Sanchez et al., 2012), also ameliorated network dysfunction in a trial of people with amnestic mild cognitive impairment (n = 34 individuals: 17 Controls, 17 MCI; Bakker et al., 2012).
Here we show that elevating klotho levels in hAPP-J20 mice effectively reduces their premature mortality, cognitive deficits, behavioral abnormalities, synaptic impairments, and network dysfunction, without altering the levels of hAPP, hAPP metabolites, or the microtubule-associated protein tau, on which many Aβ-induced deficits depend (Roberson et al., 2007, 2011; Ittner et al., 2010; Vossel et al., 2010). We also provide evidence that these beneficial effects may be mediated by modulation of NMDA receptor (NMDAR) function.
Materials and Methods
Animals.
Hemizygous hAPP-J20 mice (Mucke et al., 2000), which express an alternatively spliced hAPP minigene encoding hAPP695, hAPP751, and hAPP770 with the Swedish and Indiana familial AD mutations directed by the PDGF promoter (Rockenstein et al., 1995; Mucke et al., 2000), were crossed with hemizygous klotho (KL) transgenic mice (Kuro-o et al., 1997), which express mouse klotho ubiquitously from the EF-1α promoter. Mice were on an incipient congenic C57BL/6 background (N6–N7: B6;C3H). All studies were conducted on age-matched and sex-balanced littermates that included all four genotypes tested in parallel: nontransgenic (NTG), KL, hAPP, and hAPP/KL. Some data obtained from NTG and KL mice in these studies were previously reported (Dubal et al., 2014) as indicated in figure legends. Mice were analyzed in multiple cohorts, as detailed in Table 1. Mice were kept on a 12 h light/dark cycle with ad libitum access to food (Picolab Rodent Diet 20, Labdiet) and water. The studies were approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco and conducted in compliance with NIH guidelines.
Table 1.
Mice analyzed in different experiments
| Figure | Mice per group | Total no. | From cohort(s) | Age at analysis | |
|---|---|---|---|---|---|
| Longevity | |||||
| 2B | NTG, 111 | 366 mice | A–H | 3 weeks to 12.5 months | |
| hAPP, 67 | |||||
| KL, 110 | |||||
| hAPP/KL, 78 | |||||
| EEG | |||||
| 2D | NTG, 3 | 21 mice | B | 2–3.5 months | |
| KL, 4 | |||||
| hAPP, 7 | |||||
| hAPP/KL, 7 | |||||
| Behavior | |||||
| Water maze | 3A–C | NTG, 14 | 63 mice | B, C | 4–7 months |
| KL, 14 | |||||
| hAPP, 18 | |||||
| hAPP/KL, 17 | |||||
| Novel object | 3D | NTG, 10 | 45 mice | C | 5–8 months |
| KL, 11 | |||||
| hAPP, 12 | |||||
| hAPP/KL, 12 | |||||
| Passive avoidance | 3E | NTG, 23 | 72 mice | C | 5–8 months |
| KL, 21 | |||||
| hAPP, 15 | |||||
| hAPP/KL, 13 | |||||
| Open field, elevated plus maze | 3F, G | NTG, 24 | 81 mice | C | 4–7 months |
| KL, 21 | |||||
| hAPP, 20 | |||||
| hAPP/KL, 16 | |||||
| Biochemistry/histology | |||||
| Western | 1 | NTG, 10 | 23 mice | D | 5–7 months |
| hAPP, 13 | |||||
| Western | 2A, 6B–F, 7B, C | NTG, 14–15 | 43–45 mice | E | 3 months |
| KL, 13 | |||||
| hAPP, 7–8 | |||||
| hAPP/KL, 9 | |||||
| Westerns, Aβ ELISA | 4B, E, K, L | hAPP, 7–8 | 15–17 mice | E | 3 months |
| hAPP/KL, 8–9 | |||||
| Aβ oligomer ECL assay | 4F | hAPP, 4 | 8 mice | F | 3–4.5 months |
| hAPP/KL, 4 | |||||
| Western | 4D | hAPP, 4 | 8 mice | F | 3–4.5 months |
| hAPP/KL, 4 | |||||
| Immunohistochemistry | 4H, J | hAPP, 12–13 | 21 mice | C | 10–12.5 months |
| hAPP/KL, 8–9 | |||||
| Immunohistochemistry | 5B, C | NTG, 25 | 75 mice | C | 10–12.5 months |
| KL, 27 | |||||
| hAPP, 14 | |||||
| hAPP/KL, 9 | |||||
| Electrophysiology | |||||
| EPSP (LTP) | 8A, B | NTG 3 slices/3 mice | 23 slices, 16 mice | G | 3.5–4.5 months |
| KL 6 slices/5 mice | |||||
| hAPP 7 slices/5 mice | |||||
| hAPP/KL 7 slices/3 mice | |||||
| fEPSP (I/O curves) | 8C | NTG 9 slices/3 mice | 39 slices, 16 mice | H | 3.5–4.5 months |
| KL 10 slices/5 mice | |||||
| hAPP 9 slices/5 mice | |||||
| hAPP/KL 11 slices/3 mice | |||||
Cohorts are independent groups of mice, some or all of which were analyzed at one or more time points/ages in one or more paradigms.
Behavioral testing.
Arenas, objects, or chambers were cleaned with 70% alcohol between testing sessions in all behavioral testing except water maze.
Morris water maze.
The water maze pool was 122 cm in diameter and contained whitened, opaque water at a temperature of 21°± 1°C. A square, 14 cm2 platform was submerged 2 cm below the water surface. Mice undertook two pretraining trials by swimming through a channel and mounting a hidden platform. During hidden platform training, the platform location remained in the same location between trials while the drop location varied. Mice underwent two hidden platform training sessions, comprising two trials each, every day for 5 d. Each trial allotted a maximum time of 60 s. During the probe trial that followed the hidden platform training, the platform was taken away and mice were permitted to swim for 60 s. After the probe trial, mice underwent further testing to determine their ability to find the platform marked with a clearly visible cue (a 15 cm pole on the platform) in two consecutive training sessions.
Novel object recognition.
Mice were acclimated to the testing room for 1 h before testing, which was performed in a square white chamber (40 × 40 cm) under dim lighting. On the first day, mice were habituated to the arena for 10 min. Twenty-four hours later, mice were presented with two objects placed equidistant from each other and from the surrounding chamber walls. During this 10 min training session, mice showed a similar preference for each of the objects (data not shown). For the test session 24 h later, one of the objects was replaced with an unfamiliar object of a different shape and texture, and mice were allowed to explore for 10 min. Frequency of object interactions and time of object exploration were manually scored from videos and analyzed.
Passive avoidance.
The apparatus consisted of a two-chamber light/dark box separated by a guillotine door (Gemini, Avoidance System, San Diego Instruments). For the acquisition trial, each mouse was placed in the lit chamber. After 15 s, the door separating the light and dark chambers opened and the latency to enter the dark chamber was recorded. After the mouse entered the dark chamber, the door was closed and an electric foot shock (0.35 mA, 2 s) was delivered by the floor grids. Ten seconds later, the mouse was removed from the dark chamber and returned to its cage. Five days later, the entrance latency into the dark chamber was measured and recorded up to a maximum of 500 s.
Open field.
The apparatus was a clear plastic chamber (41 × 30 cm) with two rows of photobeams. Total activity in this open field was detected by beam breaks and measured with an automated Flex-Field/Open Field Photobeam Activity System (San Diego Instruments). Before testing, mice were acclimated to the room for 30 min. They were then allowed to explore the open field for 10 min.
Elevated plus maze.
The apparatus (Hamilton-Kinder) consisted of open and closed arms for exploration. After habituation to dim lighting in the room, mice were placed at the junction between open and closed arms of the maze and permitted to explore for 5 min. The percentage time spent in open arms was measured.
Protein extraction.
Total protein lysates were obtained by microdissection of brain regions from mice or humans followed immediately by homogenization in ice-cold lysis buffer [1× PBS, pH 7.4, 1 mm DTT, 0.5 mm EDTA, 0.5% Triton, 0.1 m phenylmethyl sulfonyl fluoride (PMSF), protease inhibitor mixture (Roche), and phosphatase inhibitors 2 and 3 (Sigma-Aldrich)]. Samples were centrifuged at 10,000 rpm for 10 min at 4°C. Supernatant from each sample was then collected for protein concentration measurements by Bradford assay and Western blot analyses.
Aβ ELISAs.
Hippocampus was dissected, homogenized, and sonicated in ice-cold lysis buffer [1× PBS, pH 7.4, 1 mm DTT, 0.5 mm EDTA, 0.5% Triton, 0.1 m PMSF, protease inhibitor mixture (Roche), and phosphatase inhibitors 2 and 3 (Sigma-Aldrich)]. Following the addition of guanidine (final concentration 5 m), samples were rehomogenized and analyzed for levels of human Aβ 1-x and Aβ 1-42 by ELISA as described previously (Johnson-Wood et al., 1997; Mucke et al., 2000).
Aβ oligomer measurements.
Frozen hippocampal homogenates were thawed on ice and analyzed in duplicate. Oligomer levels were quantified with a electrochemiluminescent immunosorbent assay (ECL) on the Meso Scale Discovery platform following the manufacturer's instructions and published protocols (Yang et al., 2013). Oligomers were captured with the 3D6 antibody (Elan) and detected with biotinylated 3D6. Streptavidin conjugated to SULFO-TAG (Meso Scale Discovery) was then added, and light emitted by the SULFO-TAG following activation at the electrode surface was quantified (Sector Imager 2400, Meso Scale Discovery). Oligomeric Aβ 1-42 was generated by incubating monomeric Aβ 1-42 (synthesized in-house) at 100 μm in 50 mm PBS for 4 h at room temperature. The standard curve was then generated by serially diluting the sample in 1% blocking buffer (Meso Scale Discovery). Because this approach to oligomer quantification does not yield an absolute value of oligomers, levels were expressed in relative units. In control experiments, the assay detected ∼50% lower Aβ oligomer levels in hAPP mice from line J9 than in hAPP mice from line J20 (data not shown), which matches the differences in hAPP/Aβ expression between these lines (Mucke et al., 2000).
Western blotting.
For electrophoresis, 15 μg of protein was loaded into each well of a 4–12% gradient SDS-PAGE gel. Gels were transferred to nitrocellulose membranes and immunoblotted with antibodies against hAPP (8E5, 1:1500, Elan Pharmaceuticals), tau (total, 1:5000, from Gerald Hart, Johns Hopkins University or PHF-1, 1:2000, from Peter Davies, Albert Einstein College of Medicine), klotho (KM2076, 1:1000, TransGenic), GluN1 (1:200, Santa Cruz Biotechnology), GluN2A (1:1000, Millipore), GluN2B (1:1000, Millipore), GluA1 (1:1000, Millipore), GluA2 (1:4000, Millipore), GAPDH (1:5000, Abcam), or actin (1:3000, Sigma-Aldrich). For each experiment, loading controls (GAPDH or actin) were quantified independently to ensure that they did not differ among groups (data not shown). Quantifications were performed as described previously (Palop et al., 2003).
Enrichment of synaptic membranes.
Synaptic membrane fractions were separated as described previously (Goebel-Goody et al., 2009; Li et al., 2011; Dubal et al., 2014). The protocol enriches for postsynaptic density (PSD)-associated synaptic membranes and largely excludes intracellular sources of membranes (Goebel-Goody et al., 2009).
Immunohistochemistry.
Immunohistochemistry was performed as described previously (Palop et al., 2003) on floating 30 μm sections obtained with a sliding microtome. Aβ deposits were stained with a biotinylated monoclonal antibody (3D6, 1:500, Elan Pharmaceuticals) using avidin-biotin/peroxidase (Vector Laboratories). Sections were then incubated with an avidin-biotin complex (Vector Laboratories) before development with 3,3′-diaminobenzidine tetrahydrochloride (Sigma-Aldrich). Images were captured with a digital microscope (Axiocam, Carl Zeiss) and percentage area calculations were performed with the Bioquant software package (BIOQUANT Image Analysis) as described previously (Palop et al., 2003). Briefly, the average percentage area of the hippocampus occupied by Aβ-immunoreactive (IR) deposits was determined in three coronal sections 500 μm apart (approximate distances from bregma: −1.0, −1.5, and −2.0 mm) per mouse.
For double-labeling of dystrophic neurites associated with fibrillar amyloid plaques, floating sections were stained with monoclonal anti-hAPP antibody 8E5 (1:2000, Elan Pharmaceuticals), mounted on glass slides, and stained with 0.015% thioflavin-S. Images were collected with a fluorescence microscope (Keyence BZ-9000) and quantified as described previously (Tsai et al., 2004).
Dendritic spines were immunolabeled and quantified as described previously (Masliah et al., 2011; Talantova et al., 2013) using a rabbit polyclonal antibody against spinophilin (1:10,000, Millipore, affinity purified polyclonal) and a mouse monoclonal antibody against the dendritic marker MAP2 (1:200, Millipore) on 30 μm microtome sections. Spinophilin-IR spines were detected with the Tyramide Signal Amplification-Direct (Red) system (NEN Life Sciences) and MAP2 was detected with a horse anti-mouse IgG fluorescein isothiocyanate antibody (Vector Laboratories). Colabeled sections were imaged by laser scanning confocal microscopy and analyzed with Image 1.43 (NIH) as described previously (Masliah et al., 2011; Vázquez-Roque et al., 2014). For each mouse, two sections were analyzed and for each section, four fields in the molecular layer of the dentate gyrus were examined. Sections were imaged with a Zeiss 63× microscope (N.A. 1.4).
EEG recordings.
Mice were implanted for video-electroencephalography (EEG) monitoring following anesthesia with avertin (tribromoethanol, 250 mg/kg, by i.p. injection). Teflon-coated silver wire electrodes (0.125 mm diameter) soldered to a multichannel electrical connector were implanted under the skull, over the left frontal cortex (coordinates relative to bregma were M/L, −1 mm; A/P, +1 mm) and the left and right parietal cortex (M/L, ±2 mm, A/P, −2 mm). The left frontal cortex electrode was used as a reference. All EEG recordings were performed at least 2 weeks after surgery on freely moving mice in a recording chamber. Digital EEG activity with video was recorded with Harmonie software, v5.0b (Stellate Systems). Epileptiform spikes were automatically detected and scored by the Gotman spike and seizure detectors from Harmonie (Gotman and Gloor, 1976). Spikes were defined as a brief (<100 ms) high-voltage deflection on the EEG eightfold greater than the average baseline amplitude measured during the 5 s preceding the deflection, and the number of spikes per hour on subdural EEG recordings was used as the main outcome measure. EEG traces and videos were systematically inspected for detection of false spikes by an investigator blinded to the genotype of the mice. Spike frequency was measured during 24 h EEG recording in each mouse.
Acute brain slices and field recording.
Transverse brain slices of 400 μm from 3- to 4-month-old mice were generated as described previously (Dubal et al., 2014). Briefly, mice were anesthetized by isoflurane inhalation and killed by decapitation. The brains were immediately placed in an ice-cold solution containing the following (in mm): 234 sucrose, 2.5 KCl, 1.25 NaH2PO4, 10 MgSO4, 26 NaCO3, 11 glucose, and 1.3 ascorbic acid and sliced on a Leica VS100 vibroslicer (Leica). Slices were incubated for at least 1 h in oxygenated artificial CSF (aCSF) at room temperature before transfer to a submerged recording chamber. Slices were equilibrated for 10–20 min before recording. Field EPSP (fEPSP) recordings of the medial perforant pathway were performed as described previously (Dubal et al., 2014). Briefly, aCSF-filled glass pipettes were used for both stimulation and recording with the stimulating electrode positioned halfway between the end of the granule cell layer and the vertex of the two blades of the dentate gyrus and recording electrode at the same distance from the granule cell layer, but 300–350 μm closer to CA3. Medial perforant path stimulation was confirmed by paired-pulse depression and a 15 min stable baseline of fEPSP was established before long-term potentiation (LTP) induction. LTP was induced by a modified theta-burst protocol (4 trains of 10 bursts at 5 Hz delivered every 20 s, with each burst consisting of four pulses at 200 Hz), while the stimulus pulse width was increased to 2× of baseline EPSP recording. Bicuculline (2.5 μm, Tocris Bioscience) was included in aCSF to facilitate induction of LTP. Electrophysiological recordings were filtered, digitized, and acquired by WinLTP (University of Bristol). Analysis was performed with WinLTP and Original pro 8.0 (Origin Laboratories).
Statistical Analyses.
Experimenters were blinded to the genotypes of mice. Statistical analyses were performed with GraphPad Prism (v5.0) for t tests and log-rank tests for survival analyses. R (nmle package; R Development Core Team, 2011) was used for ANOVAs, post hoc tests, and proportional hazard testing. Differences between two means were assessed by t tests or Welch's t tests (for paired data with unequal variance). Differences among multiple means for unpaired data were assessed by two-way (genotypes: hAPP and KL), between-subjects ANOVAs. A mixed-model ANOVA was used for analyses of Morris water maze, LTP, and I/O curves, (factors: genotype and day, time, or intensity) and included effects of repeated measures as described previously (Young et al., 2009). Using this model, we accounted for correlated responses from repeated measures from each mouse. For LTP analyses, time was treated as a continuous variable. For I/O analyses, intensity was treated as a continuous variable. Unless indicated otherwise, t tests were one-tailed because we hypothesized that klotho decreased abnormalities in hAPP mice. Only significant p values were stated for two-way ANOVA results. Unless indicated otherwise, multiple comparisons of post hoc t tests were corrected for with the Bonferroni–Holm (stepwise Bonferroni) procedure (Holm, 1979) to control for familywise error rate at a level of α = 0.05. Log-rank tests were used for survival analyses. Then, proportional hazard testing (Grambsch and Therneau, 1994) was performed on the survival curves to determine whether genotype effects on survival were dependent on age. Error bars represent SEM. Null hypotheses were rejected below a p value of 0.05.
Results
Hippocampal klotho levels are decreased in hAPP mice and increased in KL mice
We determined klotho protein levels in the hippocampus of hAPP-J20 transgenic and NTG mice by Western blot analysis. Klotho levels in the dentate gyrus were on average 62% lower in hAPP mice than in NTG controls (Fig. 1).
Figure 1.
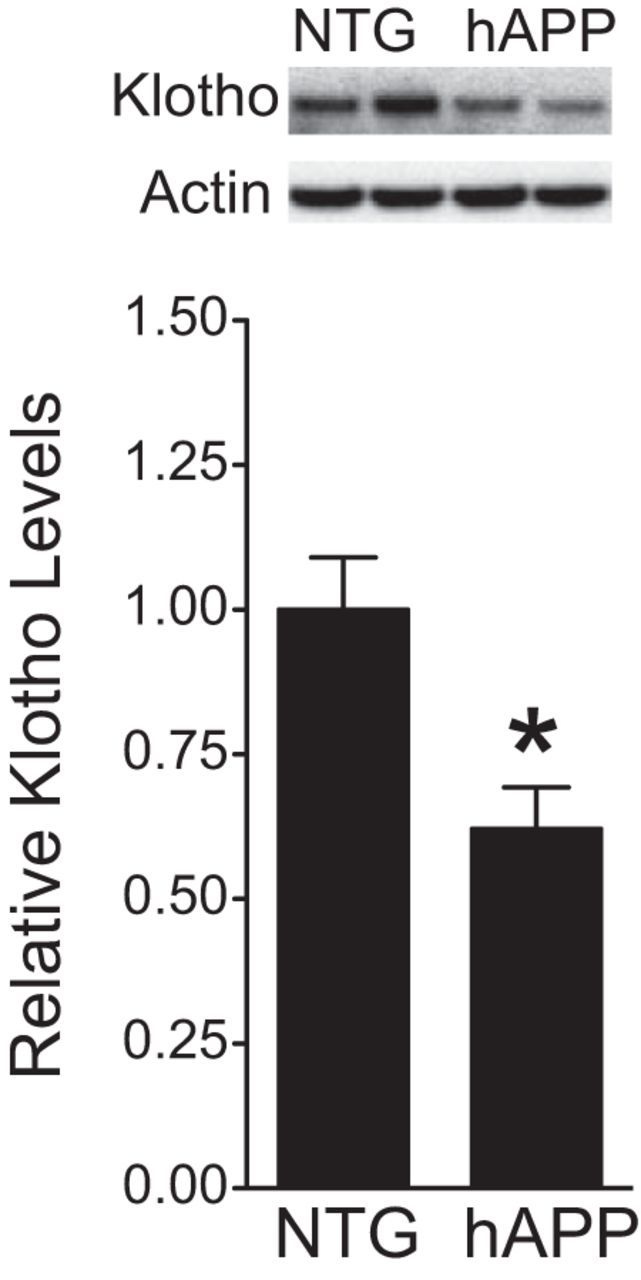
Klotho is decreased in the DG of hAPP mice. Klotho protein levels in dentate gyrus homogenates from NTG and hAPP mice (n = 10–13 mice per genotype, age 5–7 months) were determined by Western blot analysis. Actin was used as a loading control. Representative Western blots (top) and relative levels of mouse klotho determined by densitometric quantitations of Western blot signals (bottom). Mean levels in controls were arbitrarily defined as 1.0; *p < 0.05 (t test). Bar graphs represent mean ± SEM.
To test whether elevating klotho can counteract impairments in hAPP mice, we crossed hemizygous hAPP-J20 mice with hemizygous KL mice. The resulting offspring included the following genotypes: NTG, singly transgenic for hAPP or KL, and hAPP/KL doubly transgenic. Compared with NTG controls, KL and hAPP/KL mice had comparable 3.0- to 3.5-fold elevations in hippocampal klotho levels (Fig. 2A).
Figure 2.
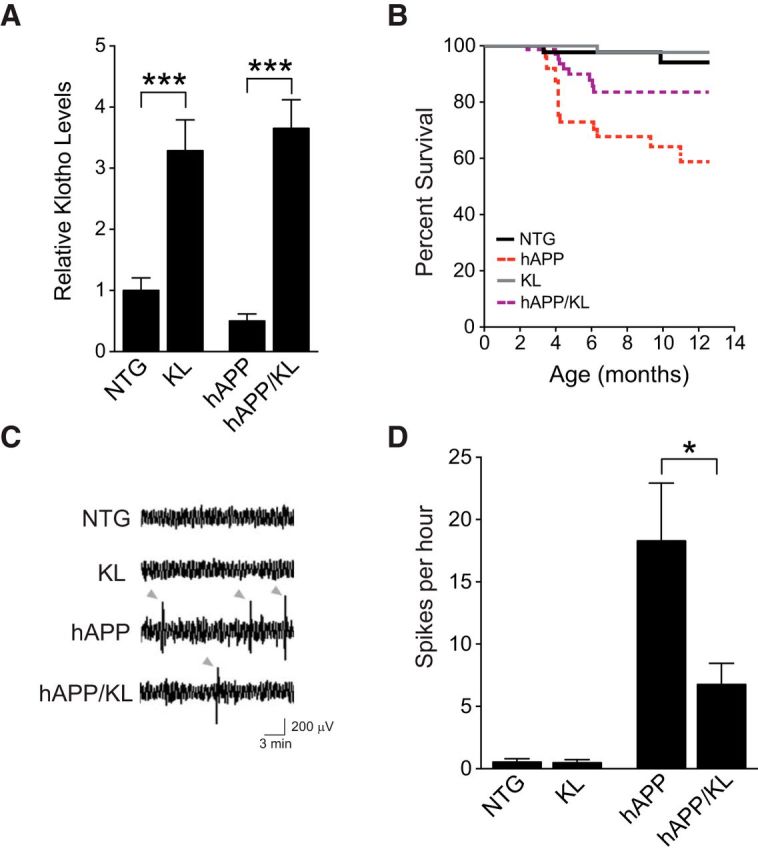
Elevating klotho reduces mortality and improves network dysfunction in hAPP mice. A, Hippocampal klotho levels in KL and hAPP/KL mice determined by Western blot analysis and expressed relative to mean levels in NTG controls (n = 7–14 mice per genotype, age 3 months). KL effect p < 0.0001 by two-way ANOVA. B, Kaplan–Meier curves showing differences in survival between weaning and 12.5 months of age among the four genotypes indicated (n = 366 mice: 111 NTG, 67 hAPP, 110 KL, and 78 hAPP/KL; p < 0.0001 by log rank test; hAPP vs all other groups p < 0.05). Proportional hazard testing revealed the KL effect in hAPP mice to be independent of age (p = 0.5). C, Representative EEG traces recorded over the parietal cortex depicted with a compressed time scale. Gray arrowheads indicate abnormal spikes. D, Frequency of spikes recorded during 24 h measured from uncompressed, raw EEG traces (n = 3–7 mice per genotype, age 2–3.5 months). Two-way ANOVA: hAPP effect p < 0.01; *p < 0.05, ***p < 0.001 by Bonferroni-Holm test (A) or Welch's t test (D). Bar graphs represent mean ± SEM.
Elevating klotho reduces premature mortality and improves network dysfunction in hAPP mice
Many lines of APP transgenic mice, including hAPP-J20 mice (Roberson et al., 2007, 2011; Sanchez-Mejia et al., 2008), show premature mortality. Klotho overexpression improved survival in hAPP mice, and this effect was independent of age or time by proportional hazard analysis (Fig. 2B). Premature mortality in hAPP mice may be caused by epileptic activity (Palop and Mucke, 2010), which in turn may relate to the increased incidence of seizures in patients with AD (Hauser et al., 1986; Amatniek et al., 2006; Palop and Mucke, 2009; Vossel et al., 2013). Abnormal epileptiform spike discharges or seizures have been observed in multiple lines of hAPP mice (Palop et al., 2007; Minkeviciene et al., 2009; Harris et al., 2010b; Palop and Mucke, 2010; Roberson et al., 2011; Vogt et al., 2011; Sanchez et al., 2012; Verret et al., 2012). We measured the frequency of epileptiform spikes over both parietal cortices by intracranial EEG. As expected, hAPP-J20 mice showed an increased spike frequency (Fig. 2C,D). Klotho elevation in hAPP mice decreased the occurrence of these abnormal events by ∼60% (Fig. 2D). Mice without hAPP did not show abnormal EEG activities (Fig. 2C,D). Thus, klotho elevation reduces premature mortality and network dysfunction in hAPP mice.
Klotho elevation ameliorates cognitive deficits and behavioral abnormalities in hAPP mice
To determine whether klotho elevation in hAPP mice can decrease deficits in learning and memory, we tested mice in the Morris water maze. As expected based on previous findings (Palop et al., 2003; Roberson et al., 2007; Cissé et al., 2011; Dubal et al., 2014), hAPP mice were impaired and KL mice showed better learning and memory relative to NTG controls (Fig. 3A,B). hAPP/KL mice learned better than hAPP mice (Fig. 3A). In a probe trial following the hidden-platform training task, hAPP/KL mice found the target faster than hAPP mice, and in contrast to hAPP mice, spent significantly more time in the target quadrant than the other quadrants (Fig. 3B,C).
Figure 3.

Klotho elevation improves cognitive and behavioral abnormalities in hAPP mice. A–C, NTG, KL, hAPP, and hAPP/KL mice (n = 14–18 per genotype) were tested in the Morris water maze at 4–7 months. A, Spatial learning curves (platform hidden). Data points represent the daily average of total distance traveled to reach the platform in four trials. Mixed-model ANOVA: hAPP effect p < 0.001, KL effect p < 0.0001, hAPP by KL interaction p < 0.05; hAPP versus all other groups p < 0.001, NTG versus KL p = 0.06 (Bonferroni-Holm test). B, C, Probe trial (platform removed) results obtained 24 h after completion of hidden-platform training. B, Latency to reach the original platform location. Two-way ANOVA: hAPP effect p < 0.05, KL effect p < 0.001, hAPP by KL interaction p = 0.05; *p < 0.05 versus NTG, **p < 0.01 versus hAPP (Bonferroni-Holm test). C, Percentage of time mice spent in the target quadrant (black) versus the average percentage of time they spent in the other three quadrants (white); **p < 0.01, *p < 0.05 versus 25% (two-tailed, one-sample t tests). Dashed gray line indicates performance expected based on chance. Water maze data from some of the NTG and KL mice were reported previously (Dubal et al., 2014). D, Object recognition memory as reflected by the percentage of time mice spent exploring a novel object (n = 11–12 mice per genotype, age 5–8 months); *p < 0.05, **p < 0.01 versus 50% (two-tailed, one-sample t tests). Dashed gray line is average time spent with object during training, which was equivalent in all groups (data not shown). E, Passive avoidance memory, as reflected by latency to enter the dark chamber during the test session (n = 13–23 mice per genotype, age 5–8 months). Two-way ANOVA: KL effect p < 0.05, hAPP by KL interaction p < 0.05; *p < 0.05 versus all other groups (Bonferroni-Holm test). Dashed gray line is average latency to enter chamber during training, which was similar in all groups (data not shown). F, Total number of movements during exploration of an open field (n = 16–24 mice per genotype, age 4–7 months). Two-way ANOVA: hAPP effect p < 0.01; KL effect p < 0.05; *p < 0.05 versus NTG (Bonferroni-Holm test), op < 0.05 (Newman–Keuls test). G, Percentage of time mice spent exploring the open arms of an elevated plus maze (n = 16–24 mice per genotype, age 4–7 months). Two-way ANOVA: hAPP effect p < 0.001, hAPP by KL interaction p = 0.06; ***p < 0.001 versus NTG (Bonferroni-Holm test). Data represent mean ± SEM.
The four genotypes showed no significant differences in gross motor function or swim speeds (data not shown). Furthermore, after hidden-platform training and probe testing, all groups of mice were able to locate the target platform when it was identified by a visible cue (data not shown).
We observed less robust differences in task acquisition and probe trial performance between hAPP and hAPP/KL mice when the cued-platform training preceded the hidden-platform training (data not shown), possibly because this protocol helps hAPP mice overcome procedural learning deficits before the spatial training.
In a novel object recognition test, which measures perirhinal cortex-dependent nonspatial learning and memory, only hAPP mice failed to spend more time exploring the novel object, indicating poor memory for the familiar object, whereas KL and hAPP/KL mice performed at NTG control levels (Fig. 3D). In passive avoidance testing, which measures hippocampus- and amygdala-dependent fear memory, hAPP mice reentered the dark chamber, where they had received a shock during training, more quickly than all other groups, whereas KL and hAPP/KL mice performed at NTG control levels (Fig. 3E).
We then tested whether elevation of klotho also improves other behavioral abnormalities in hAPP mice. In the open field test, which measures exploration and locomotor activity, only hAPP mice were hyperactive, whereas KL and hAPP/KL mice performed at NTG control levels (Fig. 3F). In the elevated plus maze, which measures exploration and anxiety-related behavior, hAPP mice, but not hAPP/KL mice, spent significantly more time in the open arms than NTG and KL mice (Fig. 3G). Thus, elevating klotho in hAPP mice prevented or reduced abnormalities in multiple cognitive and behavioral domains.
Elevation of klotho in hAPP mice does not alter hAPP, β-CTFs, Aβ, tau, or phospho-tau levels or plaque-related neuritic dystrophy
Because multiple functional deficits in hAPP mice depend on levels of hAPP/Aβ and endogenous murine tau (Chin et al., 2005; Roberson et al., 2007, 2011; Ittner et al., 2010), we investigated whether overexpression of klotho altered the levels of these proteins or of β-C-terminal fragments (β-CTFs) that result from hAPP processing by β-secretase. Klotho elevation in hAPP mice did not significantly change hippocampal levels of hAPP, β-CTFs, soluble Aβ 1-x and Aβ 1-42 (Fig. 4A–E), Aβ oligomers (Fig. 4F), or the Aβ 1-42/Aβ 1-x ratio (data not shown) at 3–5 months of age. Further, it did not change the extent of Aβ deposition or the percentage of hippocampal plaques with dystrophic neurites at 10–12.5 months of age (Fig. 4G–J). hAPP and hAPP/KL mice also had comparable hippocampal levels of total and phosphorylated tau at 3 months of age (Fig. 4A,K,L). Thus, it is unlikely that klotho elevation protects hAPP mice against functional deficits by altering the levels of hAPP, β-CTFs, Aβ, tau, or neuritic amyloid plaques.
Figure 4.
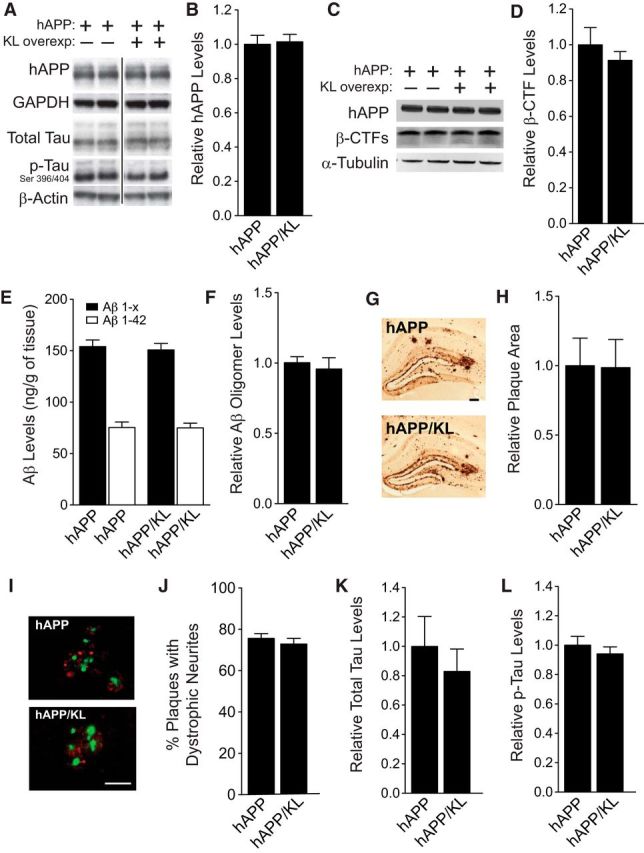
Elevation of klotho in hAPP mice does not alter levels of hAPP, Aβ, β-CTFs, Aβ oligomers, total tau, or phospho-tau and does not prevent neuritic dystrophy. A, Representative Western blots showing hippocampal levels of hAPP, total mouse tau, phosphorylated (p) mouse tau (PHF-1 antibody, Ser 396/404), and β-actin. Images were captured from the same gel. B, Relative hAPP levels determined by quantitation of Western blot signals (n = 8–9 mice per genotype, age 3 months). GAPDH was used as a loading control. C, Representative Western blots showing hippocampal levels of hAPP, β-CTFs and α-tubulin. D, Relative β-CTF levels determined by quantitation of Western blot signals (n = 4 mice per genotype, age 3–4.5 months). E, Hippocampal Aβ1-x and Aβ1-42 levels determined by ELISA (n = 7–9 mice per genotype, age 3 months). F, Hippocampal Aβ oligomer levels determined by ECL assay (n = 4 mice per genotype, age 3–4.5 months). G, Immunostaining of hippocampal Aβ deposits in coronal brain sections from an hAPP (top) and an hAPP/KL mouse (bottom). Scale bar, 200 μm. H, Relative levels of hippocampal Aβ deposition (n = 12–14 mice per genotype, age 10–12.5 months). The percentage area covered by Aβ deposits in hAPP mice was arbitrarily defined as 1.0. I, Representative double-labeling of hippocampus for dystrophic neurites (antibody 8E5, red) and amyloid plaques (thioflavin-S, green) in hAPP and hAPP/KL mice (age 10–12.5 months; n = 10–12 per genotype). Scale bar, 50 μm. J, Quantification of dystrophic neurites expressed as a percentage of thioflavin-S-labeled plaques with surrounding neuritic dystrophy. K, L, Quantitation of total (K) and phosphorylated (L) mouse tau levels relative to levels found in hAPP mice (n = 8–9 mice per genotype, age 3 months). Actin was used as a loading control. Bar graphs represent mean ± SEM.
Elevation of klotho in hAPP mice prevents spine loss
Dendritic spines provide the postsynaptic elements of many synapses, fulfill important functions in synaptic plasticity (Segal, 2002), and are major targets of degeneration in AD (Spires and Hyman, 2004; Akram et al., 2008; Wu et al., 2012) and hAPP mice (Lanz et al., 2003; Spires et al., 2005). To determine whether klotho elevation prevents dendritic spine loss in hAPP mice, we measured spinophilin, a marker of spines (Feng et al., 2000), by immunohistochemistry. Compared with NTG controls, hAPP mice had reduced levels of spinophilin-immunoreactive structures in the DG (Fig. 5A–C) and CA1 region (data not shown) of the hippocampus. Klotho elevation prevented this abnormality in hAPP/KL mice (Fig. 5A–C). However, it did not increase spinophilin levels in KL and hAPP/KL mice above those found in NTG controls (Fig. 5A–C).
Figure 5.
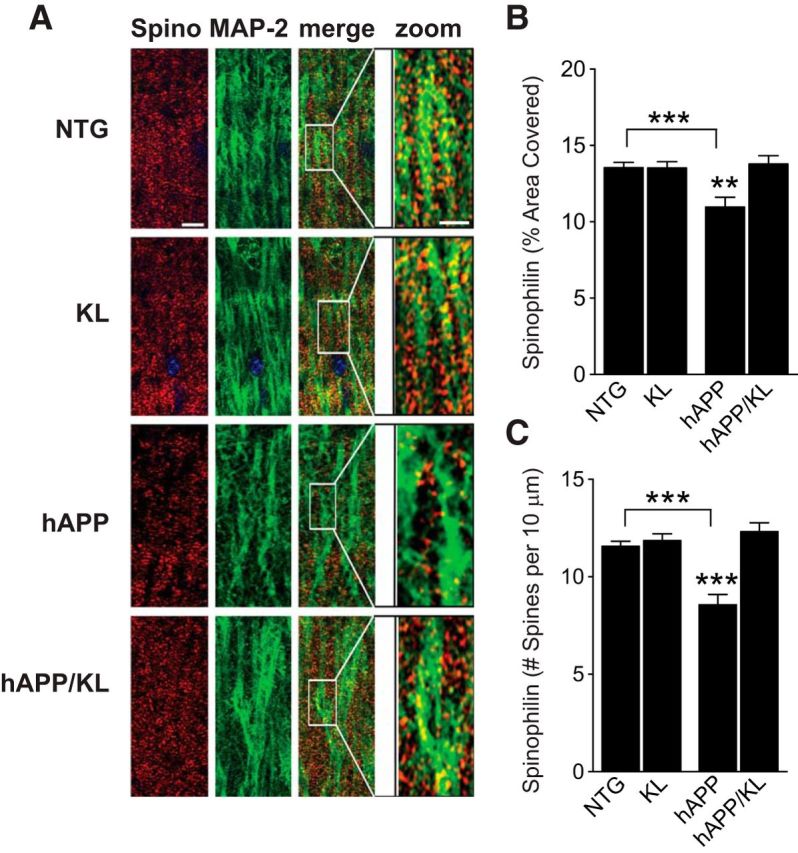
Elevation of klotho prevents loss of spinophilin in the hippocampus of hAPP mice. A, Representative immunostaining of spinophillin (spino, red) and MAP-2 (green) in the hippocampus of NTG, KL, hAPP, and hAPP/KL mice. Scale bars: Spino, MAP-2, and merge, 5 μm; zoom, 10 μm. B, C, Quantification of spinophilin-immunoreactive spines in the DG (n = 10–27 mice per group, age 10–12.5 months) expressed as percentage area of MAP-2-positive structures covered (B) and number of punctae per 10 μm of MAP-2-positive dendrite (C). Two-way ANOVA: hAPP effect p < 0.05 (B) and p < 0.01 (C), KL effect p < 0.05 (B), and p < 0.0001 (C), hAPP by KL interaction p < 0.01 (B) and p < 0.0001 (C); **p < 0.01, ***p < 0.001 versus hAPP/KL or as indicated by brackets (Bonferroni-Holm test). Bar graphs represent mean ± SEM.
Klotho elevation alters NMDAR, but not AMPAR, subunit levels in hAPP mice
NMDAR and AMPA receptor (AMPAR) functions are essential to learning and memory (Nakazawa et al., 2004; Lee and Silva, 2009) and disrupted by Aβ (Hsieh et al., 2006; Palop and Mucke, 2010; D'Amelio et al., 2011; Li et al., 2011; Chang et al., 2012; Huang and Mucke, 2012). We therefore examined whether klotho elevation modulates these receptors. We first measured the levels of receptor subunits in hippocampal homogenates. Compared with NTG controls, hAPP mice had decreased hippocampal levels of NMDAR subunits GluN1, GluN2A, and GluN2B (Fig. 6A–D), consistent with decreased NMDAR levels found in other hAPP mice (Zhang et al., 2010), primary neurons treated with Aβ (Snyder et al., 2005; Cissé et al., 2011; Li et al., 2011; Roberson et al., 2011; Zhang et al., 2011), and postmortem brain tissues from humans with AD (Sze et al., 2001; Mishizen-Eberz et al., 2004; Amada et al., 2005). Klotho elevation prevented GluN1 and GluN2A depletions in hAPP/KL mice (Fig. 6A–C). Remarkably, GluN2B levels in hAPP/KL mice were even higher than those in NTG controls (Fig. 6A,D). In contrast to these NMDAR alterations, hAPP mice had no changes in GluA1 and GluA2 levels, and klotho elevation did not change the levels of these AMPAR subunits in NTG or hAPP mice (Fig. 6A,E,F).
Figure 6.
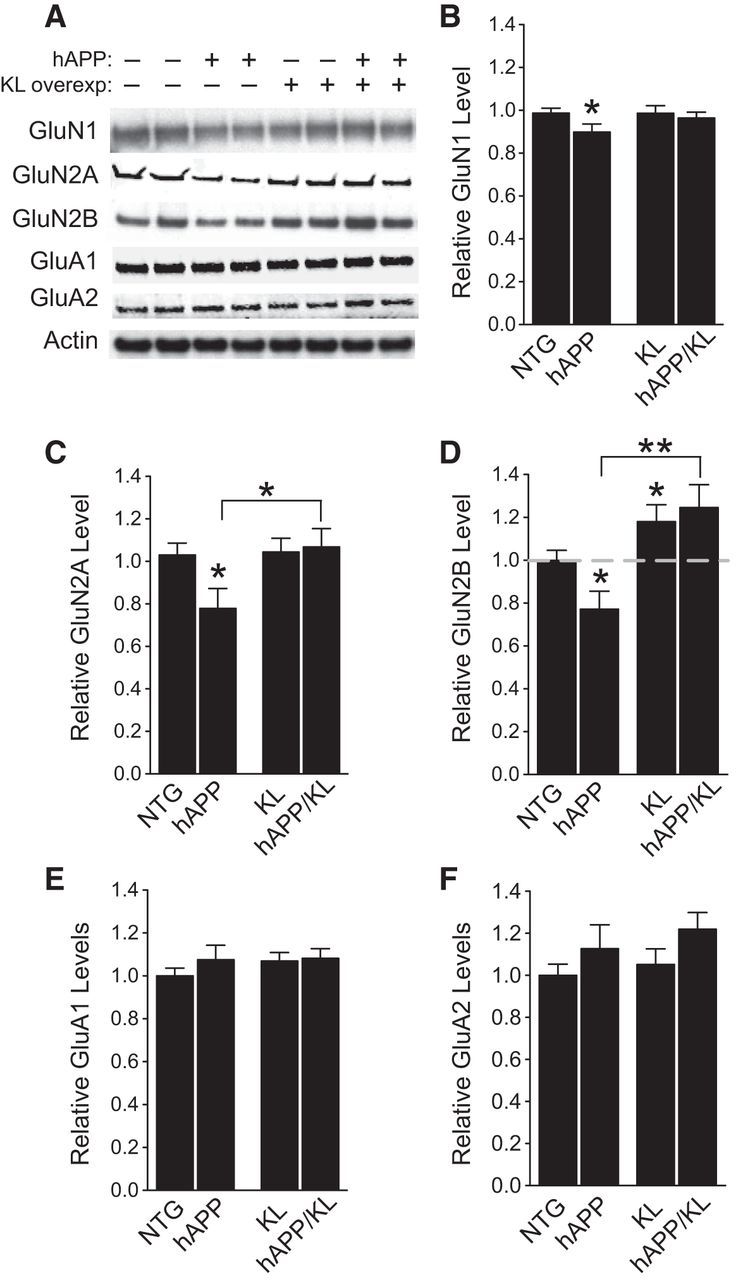
Klotho elevation prevents depletion of NMDAR subunits GluN1 and GluN2A, and increases hippocampal levels of GluN2B in hAPP mice. A, Representative Western blots showing hippocampal levels of NMDAR subunits GluN1, GluN2A, and GluN2B, and AMPAR subunits GluA1 and GluA2. Actin served as a loading control. B–F, Quantitation of Western blot signals relative to mean levels found in NTG mice (n = 7–15 mice per genotype, age 3 months). B, GluN1 levels. Two-way ANOVA: hAPP effect p < 0.05. C, GluN2A levels. Two-way ANOVA: KL effect p < 0.05; hAPP by KL interaction p < 0.05. D, GluN2B levels. Two-way ANOVA: KL effect p < 0.001. Dashed gray line is level of GluN2B in NTG mice. E, GluA1 levels. F, GluA2 levels; *p < 0.05, **p < 0.01 versus NTG or as indicated by brackets (Bonferroni-Holm test). Levels of NMDAR and AMPAR subunits in the NTG and KL mice were reported previously (Dubal et al., 2014). Bar graphs represent mean ± SEM.
Klotho elevation enriches GluN2B in postsynaptic densities of hAPP mice
The biological consequences of NMDAR stimulation are critically dependent on whether the receptors are located in synaptic or extrasynaptic sites (Hardingham and Bading, 2010). We therefore examined the levels of GluN2B in synaptosome fractions enriched for PSD or for non-PSD components such as extrasynaptic dendritic membranes and presynaptic specializations. Compared with NTG controls, hAPP mice had increased levels of GluN2B in non-PSD fractions, and klotho elevation did not prevent this abnormality (Fig. 7A,B). However klotho elevation increased GluN2B levels in PSD fractions by ∼2-fold in mice with or without hAPP expression (Fig. 7A,C), more than doubling the ratio of PSD to non-PSD levels of GluN2B in hAPP mice (Fig. 7D). These effects could counteract Aβ-induced synaptic dysfunction, which involves overactivation of GluN2B-containing NMDARs in extrasynaptic sites (Li et al., 2011; Talantova et al., 2013; Gomes et al., 2014; Molokanova et al., 2014).
Figure 7.

Klotho elevation increases GluN2B levels in hippocampal PSD fractions of hAPP mice. Non-PSD (1) and PSD (2) synaptic membrane fractions were isolated from hippocampal homogenates from mice of the indicated genotypes (n = 6–14 per genotype, age 3 months). A, Representative Western blots. B, Quantitation of GluN2B Western blot signals in the non-PSD fraction relative to NTG levels. Two-way ANOVA: hAPP effect p < 0.01; *p < 0.05 versus NTG, #p = 0.06 versus KL (Bonferroni-Holm test). C, Quantitation of GluN2B signals in PSD fraction relative to NTG levels. Two-way ANOVA: KL effect p < 0.05; *p < 0.05 versus NTG or as indicated by bracket (Bonferroni-Holm test). D, Ratio of PSD/non-PSD GluN2B levels in hAPP and hAPP/KL mice relative to ratio in hAPP mice; *p < 0.05 (t test). Bar graphs represent mean ± SEM.
Klotho elevation enhances NMDAR-dependent synaptic plasticity in the hippocampus of hAPP mice
To determine whether the klotho-dependent enrichment of GluN2B in the PSD is associated with an improvement in synaptic functions in hAPP mice, we assessed LTP in acute hippocampal slices obtained from the four groups of mice at 3.5–4.5 months of age. LTP provides a measure of synaptic plasticity and is a cellular substrate of learning and memory (Morris et al., 1986; Nakazawa et al., 2004; Nabavi et al., 2014). LTP elicited at the medial perforant path to granule cell synapse in the DG was severely impaired in hAPP mice, consistent with previous findings (Palop et al., 2007; Sun et al., 2008; Harris et al., 2010a; Cissé et al., 2011; Roberson et al., 2011), and this deficit was ameliorated by klotho elevation (Fig. 8A,B). Klotho elevation also prevented hAPP/Aβ-dependent deficits in basal synaptic strength measured at the Schaffer collateral to pyramidal cell synapse in CA1 (Fig. 8C). Thus, klotho elevation decreases synaptic dysfunction in hAPP mice.
Figure 8.

Klotho elevation ameliorates synaptic dysfunction in the hippocampus of hAPP mice. Field EPSPs were recorded from acute hippocampal slices obtained from 3.5- to 4.5-month-old mice. A, LTP induction and decay in the DG were monitored for 45–50 min following theta burst stimulation of the medial perforant pathway. Mixed-model ANOVA: hAPP effect, KL effect, and hAPP by KL interaction p < 0.001; NTG versus KL p < 0.05; hAPP versus all other groups p < 0.001 (Bonferroni-Holm test). B, Mean of the last 10 min of LTP recordings. Mixed-model ANOVA: hAPP effect p < 0.001, KL effect p < 0.001; **p < 0.01 versus all other groups or as indicated by brackets (Bonferroni-Holm test). The fEPSP results in NTG and KL slices were reported previously (Dubal et al., 2014). C, Input/output curves reflecting AMPAR-mediated synaptic transmission at Schaffer collateral to CA1 pyramidal cell synapses. Mixed-model ANOVA: hAPP effect p < 0.001, KL effect <0.05, hAPP by KL interaction p < 0.001; hAPP versus all other groups p < 0.001 (Bonferroni-Holm test). Number of slices/number of mice: NTG 3/3, KL 6/5, hAPP 7/5, hAPP/KL 7/3 (A, B), and NTG 9/3, KL 10/5, hAPP 9/5, hAPP/KL 11/3 (C). Data represent mean ± SEM.
Discussion
This study demonstrates that klotho effectively protects hAPP mice against a range of potentially AD-related deficits. Elevating klotho levels, which are reduced in the hippocampus of hAPP mice, decreased premature mortality, network and synaptic dysfunction, as well as cognitive and behavioral deficits in hAPP mice, without altering levels of soluble Aβ, Aβ deposition, plaque-associated neuritic dystrophy, β-CTFs, or tau. Based on the biochemical and electrophysiological data we obtained, we offer synaptic enrichment of GluN2B-containing NMDARs as a potential mechanism by which klotho may enhance synaptic and cognitive functions and counteract adverse effects of pathologically elevated levels of Aβ. Our study was not designed to exclude contributions of additional mechanisms, which deserve to be explored in future studies. Together, our findings suggest that enhancing the expression or activities of klotho could be of therapeutic benefit in AD.
Klotho extends lifespan and protects against network dysfunction in hAPP mice
Multiple lines of hAPP mice, including the hAPP-J20 line (Roberson et al., 2007, 2011; Sanchez-Mejia et al., 2008; Verret et al., 2012), show premature mortality. Although the underlying causes remain to be fully elucidated, epileptic activity could be a key factor (Palop et al., 2007; Minkeviciene et al., 2009; Palop and Mucke, 2010; Roberson et al., 2011; Verret et al., 2012). This network dysfunction may relate closely to the increased incidence of epileptic seizures in humans with AD, which is most evident in aggressive forms of AD with early onset (Snider et al., 2005; Cabrejo et al., 2006; Palop and Mucke, 2009; Jayadev et al., 2010; Vossel et al., 2013). Klotho elevation markedly decreased epileptiform activity in young hAPP mice, suggesting that it may decrease mortality in this model by preventing network hyperexcitability rather than through its known aging-related mechanisms (Kurosu et al., 2005; Liu et al., 2007; Kurosu and Kuro, 2009). How klotho decreases aberrant network activity, and how this might relate to its effects on cognition, remain important questions. Answering these questions will likely require a better understanding of how pathologically elevated levels of Aβ cause network dysfunction in the first place; diverse mechanisms appear to be involved (Roberson et al., 2007, 2011; Sanchez-Mejia et al., 2008; Ittner et al., 2010; Palop and Mucke, 2010; Huang and Mucke, 2012; Verret et al., 2012). Notably, suppression of aberrant network activity with the anti-epileptic drug levetiracetam was associated with improvements in cognitive functions in both hAPP mice (Sanchez et al., 2012) and humans with amnestic mild cognitive impairment (Bakker et al., 2012), which often leads to AD (Petersen et al., 1999; Albert et al., 2011). These findings highlight the pathogenic importance of aberrant network activity in the pathogenesis of AD and identify its suppression as a potential therapeutic strategy, in which klotho could have a role.
Klotho improves cognitive functions independently of Aβ accumulation
Klotho elevation in hAPP mice effectively prevented their impairments in spatial and nonspatial learning and memory, as determined by the water maze, novel object recognition, and passive avoidance tests. As discussed above, suppression of aberrant network activity may have contributed to this beneficial effect. However, klotho elevation also improved spatial learning and memory in young mice that did not overexpress hAPP/Aβ and had normal neural network activity (Dubal et al., 2014; and this study). These results broaden the potential applications of klotho-related therapeutic strategies and suggest a novel mechanism that is independent of the aging process per se and does not directly target Aβ or one of its essential copathogens. Consistent with this interpretation, klotho elevation did not affect hippocampal levels of hAPP, β-CTFs, Aβ, Aβ deposition, total tau, or phospho-tau. However, because klotho elevation counteracted neuronal dysfunction in hAPP mice (this study) and cell death in Aβ-treated primary neuronal cultures (Zeldich et al., 2014), it is likely that the downstream cascades these pathogenic proteins trigger converge upon processes klotho is able to modulate.
Klotho modulates the distribution and function of NMDARs
Our study revealed that the function of NMDARs, which is critical for normal cognition and impaired by Aβ and tau (Nakazawa et al., 2004; Lee and Silva, 2009; Hoover et al., 2010; Ittner et al., 2010; Palop and Mucke, 2010; Cissé et al., 2011; Li et al., 2011), represents a potential convergence point at which klotho may counteract the adverse effects of these pathogens. Klotho elevation prevented the depletion of NMDAR subunits GluN1, GluN2A, and GluN2B and ameliorated impairments of NMDAR-dependent synaptic plasticity in the hippocampus of hAPP mice. GluN2B stood out among these subunits in that its PSD levels in hAPP/KL mice were increased above NTG control levels.
Several lines of evidence suggest that the ratio of synaptic to extrasynaptic NMDAR activation has a critical impact on neuronal activity and survival (Hardingham et al., 2002; Hardingham and Bading, 2010) and that accumulation of Aβ oligomers around the synaptic cleft may impair synaptic functions by diminishing this ratio (Li et al., 2009, 2011). It is interesting in this regard, that klotho elevation markedly increased GluN2B levels in hippocampal PSD fractions, but not in non-PSD fractions, resulting in a twofold increase in the PSD (“synaptic”) to non-PSD (“extrasynaptic”) ratio of GluN2B in hAPP mice. Klotho elevation also increased GluN2B levels in PSD, but not non-PSD, fractions from mice without hAPP/Aβ expression (Dubal et al., 2014; and this study). These data suggest that klotho enriches GluN2B-containing NMDARs in the PSD and may enhance their synaptic functions in the normal and diseased brain.
Interestingly, transgenic overexpression of GluN2B (Tang et al., 1999; Cao et al., 2007; Wang et al., 2009) or increasing the surface expression of GluN2B by disrupting its phosphorylation by cyclin-dependent kinase 5 (Hawasli et al., 2007; Plattner et al., 2014) in mice or rats enhanced both synaptic and cognitive functions, possibly by delaying deactivation of NMDARs (Wang et al., 2008) and increasing the summation of synaptic current and calcium influx (Yashiro and Philpot, 2008; Wang et al., 2009; Foster et al., 2010). These studies suggest that the GluN2B subunit gates and optimizes coincidence detection by NMDARs, a process that leads to enhanced synaptic potentiation (Tsien, 2000). Therefore, klotho-mediated synaptic enrichment of GluN2B may explain the enhanced LTP and improved learning and memory we observed in hAPP/KL mice.
Our study does not exclude additional mechanisms, such as effects of klotho on insulin signaling (Kurosu et al., 2005), carbohydrate metabolism (Chang et al., 2005; Cha et al., 2008), growth factor signaling (Urakawa et al., 2006), regulation of members of the redox system (Zeldich et al., 2014), and trafficking of other ion channels (Chang et al., 2005; Imura et al., 2007). Additional studies are needed to explore these possibilities, to determine the mechanisms by which klotho enriches GluN2B in the PSD, and to further evaluate its therapeutic potential in relation to age-related cognitive decline, AD and other neurological disorders. Increasing the level or activities of klotho (Abraham et al., 2012; King et al., 2012; Dubal et al., 2014) might be an effective strategy for enhancing cognition in health and disease.
Footnotes
This work was supported by NIH Grants NS041787, NS065780, and NS088532 (L.M.), AG034531 (D.B.D.), AG18440 and AG010435 (E.M.), AG000001 (C.R.A.), AG019712 (M.K.), a MetLife/AFAR Award (L.M.), gifts from the S. D. Bechtel Jr Foundation (L.M.), the Weeks-Coulter Foundation (D.B.D.), the Bakar Family Foundation (D.B.D.), and NIH National Center for Research Resources Grant RR18938-01 to the Gladstone Institutes. We thank Drs Gerald Hart and Peter Davies for tau antibodies; Elan Pharmaceuticals for Aβ antibodies; X. Wang, W. Guo, C. Wang, and S. Cheung for technical assistance; J. Palop, M. Morris, J. Harris, M. Cisse, and B. Djukic for discussions; K. Eilertson for assistance with statistics; and M. Dela Cruz for administrative support.
References
- Abraham CR, Chen C, Cuny GD, Glicksman MA, Zeldich E. Small-molecule Klotho enhancers as novel treatment of neurodegeneration. Future Med Chem. 2012;4:1671–1679. doi: 10.4155/fmc.12.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akram A, Christoffel D, Rocher AB, Bouras C, Kövari E, Perl DP, Morrison JH, Herrmann FR, Haroutunian V, Giannakopoulos P, Hof PR. Stereologic estimates of total spinophilin-immunoreactive spine number in area 9 and the CA1 field: relationship with the progression of Alzheimer's disease. Neurobiol Aging. 2008;29:1296–1307. doi: 10.1016/j.neurobiolaging.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, Snyder PJ, Carrillo MC, Thies B, Phelps CH. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the national institute on aging-Alzheimer's association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:270–279. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amada N, Aihara K, Ravid R, Horie M. Reduction of NR1 and phosphorylated Ca2+/calmodulin-dependent protein kinase II levels in Alzheimer's disease. Neuroreport. 2005;16:1809–1813. doi: 10.1097/01.wnr.0000185015.44563.5d. [DOI] [PubMed] [Google Scholar]
- Amatniek JC, Hauser WA, DelCastillo-Castaneda C, Jacobs DM, Marder K, Bell K, Albert M, Brandt J, Stern Y. Incidence and predictors of seizures in patients with Alzheimer's disease. Epilepsia. 2006;47:867–872. doi: 10.1111/j.1528-1167.2006.00554.x. [DOI] [PubMed] [Google Scholar]
- Arking DE, Krebsova A, Macek M, Sr, Macek M, Jr, Arking A, Mian IS, Fried L, Hamosh A, Dey S, McIntosh I, Dietz HC. Association of human aging with a functional variant of klotho. Proc Natl Acad Sci U S A. 2002;99:856–861. doi: 10.1073/pnas.022484299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arking DE, Atzmon G, Arking A, Barzilai N, Dietz HC. Association between a functional variant of the KLOTHO gene and high-density lipoprotein cholesterol, blood pressure, stroke, and longevity. Circ Res. 2005;96:412–418. doi: 10.1161/01.RES.0000157171.04054.30. [DOI] [PubMed] [Google Scholar]
- Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, Stark CE, Yassa MA, Bassett SS, Shelton AL, Gallagher M. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron. 2012;74:467–474. doi: 10.1016/j.neuron.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrejo L, Guyant-Maréchal L, Laquerrière A, Vercelletto M, De la Fournière F, Thomas-Antérion C, Verny C, Letournel F, Pasquier F, Vital A, Checler F, Frebourg T, Campion D, Hannequin D. Phenotype associated with APP duplication in five families. Brain. 2006;129:2966–2976. doi: 10.1093/brain/awl237. [DOI] [PubMed] [Google Scholar]
- Cao X, Cui Z, Feng R, Tang YP, Qin Z, Mei B, Tsien JZ. Maintenance of superior learning and memory function in NR2B transgenic mice during ageing. Eur J Neurosci. 2007;25:1815–1822. doi: 10.1111/j.1460-9568.2007.05431.x. [DOI] [PubMed] [Google Scholar]
- Cha SK, Ortega B, Kurosu H, Rosenblatt KP, Kuro-O M, Huang CL. Removal of sialic acid involving Klotho causes cell-surface retention of TRPV5 channel via binding to galectin-1. Proc Natl Acad Sci U S A. 2008;105:9805–9810. doi: 10.1073/pnas.0803223105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang PK, Verbich D, McKinney RA. AMPA receptors as drug targets in neurological disease: advantages, caveats, and future outlook. Eur J Neurosci. 2012;35:1908–1916. doi: 10.1111/j.1460-9568.2012.08165.x. [DOI] [PubMed] [Google Scholar]
- Chang Q, Hoefs S, van der Kemp AW, Topala CN, Bindels RJ, Hoenderop JG. The beta-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science. 2005;310:490–493. doi: 10.1126/science.1114245. [DOI] [PubMed] [Google Scholar]
- Chen CD, Podvin S, Gillespie E, Leeman SE, Abraham CR. Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc Natl Acad Sci U S A. 2007;104:19796–19801. doi: 10.1073/pnas.0709805104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CD, Sloane JA, Li H, Aytan N, Giannaris EL, Zeldich E, Hinman JD, Dedeoglu A, Rosene DL, Bansal R, Luebke JI, Kuro-o M, Abraham CR. The antiaging protein Klotho enhances oligodendrocyte maturation and myelination of the CNS. J Neurosci. 2013;33:1927–1939. doi: 10.1523/JNEUROSCI.2080-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng IH, Scearce-Levie K, Legleiter J, Palop JJ, Gerstein H, Bien-Ly N, Puoliväli J, Lesné S, Ashe KH, Muchowski PJ, Mucke L. Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem. 2007;282:23818–23828. doi: 10.1074/jbc.M701078200. [DOI] [PubMed] [Google Scholar]
- Cheng JS, Dubal DB, Kim DH, Legleiter J, Cheng IH, Yu GQ, Tesseur I, Wyss-Coray T, Bonaldo P, Mucke L. Collagen VI protects neurons against Aβ toxicity. Nat Neurosci. 2009;12:119–121. doi: 10.1038/nn.2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J, Palop JJ, Puoliväli J, Massaro C, Bien-Ly N, Gerstein H, Scearce-Levie K, Masliah E, Mucke L. Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2005;25:9694–9703. doi: 10.1523/JNEUROSCI.2980-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J, Massaro CM, Palop JJ, Thwin MT, Yu GQ, Bien-Ly N, Bender A, Mucke L. Reelin depletion in the entorhinal cortex of human amyloid precursor protein transgenic mice and humans with Alzheimer's disease. J Neurosci. 2007;27:2727–2733. doi: 10.1523/JNEUROSCI.3758-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cissé M, Halabisky B, Harris J, Devidze N, Dubal DB, Sun B, Orr A, Lotz G, Kim DH, Hamto T, Ho K, Yu GQ, Mucke L. Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature. 2011;469:47–52. doi: 10.1038/nature09635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amelio M, Cavallucci V, Middei S, Marchetti C, Pacioni S, Ferri A, Diamantini A, De Zio D, Carrara P, Battistini L, Moreno S, Bacci A, Ammassari-Teule M, Marie H, Cecconi F. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer's disease. Nat Neurosci. 2011;14:69–76. doi: 10.1038/nn.2709. [DOI] [PubMed] [Google Scholar]
- Dubal DB, Yokoyama JS, Zhu L, Broestl L, Worden K, Wang D, Sturm VE, Kim D, Klein E, Yu GQ, Ho K, Eilertson KE, Yu L, Kuro-o M, De Jager PL, Coppola G, Small GW, Bennett DA, Kramer JH, Abraham CR, et al. Life extension factor klotho enhances cognition. Cell Rep. 2014;7:1065–1076. doi: 10.1016/j.celrep.2014.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duce JA, Podvin S, Hollander W, Kipling D, Rosene DL, Abraham CR. Gene profile analysis implicates Klotho as an important contributor to aging changes in brain white matter of the rhesus monkey. Glia. 2008;56:106–117. doi: 10.1002/glia.20593. [DOI] [PubMed] [Google Scholar]
- Feng J, Yan Z, Ferreira A, Tomizawa K, Liauw JA, Zhuo M, Allen PB, Ouimet CC, Greengard P. Spinophilin regulates the formation and function of dendritic spines. Proc Natl Acad Sci U S A. 2000;97:9287–9292. doi: 10.1073/pnas.97.16.9287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster KA, McLaughlin N, Edbauer D, Phillips M, Bolton A, Constantine-Paton M, Sheng M. Distinct roles of NR2A and NR2B cytoplasmic tails in long-term potentiation. J Neurosci. 2010;30:2676–2685. doi: 10.1523/JNEUROSCI.4022-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German DC, Khobahy I, Pastor J, Kuro-O M, Liu X. Nuclear localization of Klotho in brain: an anti-aging protein. Neurobiol Aging. 2012;33:1483.e25–e30. doi: 10.1016/j.neurobiolaging.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacobini E, Gold G. Alzheimer disease therapy: moving from amyloid-beta to tau. Nat Rev Neurol. 2013;9:677–686. doi: 10.1038/nrneurol.2013.223. [DOI] [PubMed] [Google Scholar]
- Goebel-Goody SM, Davies KD, Alvestad Linger RM, Freund RK, Browning MD. Phospho-regulation of synaptic and extrasynaptic N-methyl-d-aspartate receptors in adult hippocampal slices. Neuroscience. 2009;158:1446–1459. doi: 10.1016/j.neuroscience.2008.11.006. [DOI] [PubMed] [Google Scholar]
- Gomes GM, Dalmolin GD, Bär J, Karpova A, Mello CF, Kreutz MR, Rubin MA. Inhibition of the polyamine system counteracts beta-amyloid peptide-induced memory impairment in mice: involvement of extrasynaptic NMDA receptors. PLoS One. 2014;9:e99184. doi: 10.1371/journal.pone.0099184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotman J, Gloor P. Automatic recognition and quantification of interictal epileptic activity in the human scalp EEG. Electroencephalogr Clin Neurophysiol. 1976;41:513–529. doi: 10.1016/0013-4694(76)90063-8. [DOI] [PubMed] [Google Scholar]
- Grambsch PM, Therneau TM. Proportional hazards tests and diagnostics based on weighted residuals. Biometrika. 1994;81:515–526. doi: 10.1093/biomet/81.3.515. [DOI] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Harris JA, Devidze N, Halabisky B, Lo I, Thwin MT, Yu GQ, Bredesen DE, Masliah E, Mucke L. Many neuronal and behavioral impairments in transgenic mouse models of Alzheimer's disease are independent of caspase cleavage of the amyloid precursor protein. J Neurosci. 2010a;30:372–381. doi: 10.1523/JNEUROSCI.5341-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JA, Devidze N, Verret L, Ho K, Halabisky B, Thwin MT, Kim D, Hamto P, Lo I, Yu GQ, Palop JJ, Masliah E, Mucke L. Transsynaptic progression of amyloid-β-induced neuronal dysfunction within the entorhinal-hippocampal network. Neuron. 2010b;68:428–441. doi: 10.1016/j.neuron.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser WA, Morris ML, Heston LL, Anderson VE. Seizures and myoclonus in patients with Alzheimer's disease. Neurology. 1986;36:1226–1230. doi: 10.1212/WNL.36.9.1226. [DOI] [PubMed] [Google Scholar]
- Hawasli AH, Benavides DR, Nguyen C, Kansy JW, Hayashi K, Chambon P, Greengard P, Powell CM, Cooper DC, Bibb JA. Cyclin-dependent kinase 5 governs learning and synaptic plasticity via control of NMDAR degradation. Nat Neurosci. 2007;10:880–886. doi: 10.1038/nn1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm S. A simple sequentially rejective multiple test procedure. Scand J Statist. 1979;6:65–70. [Google Scholar]
- Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, Ashe KH, Liao D. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imura A, Tsuji Y, Murata M, Maeda R, Kubota K, Iwano A, Obuse C, Togashi K, Tominaga M, Kita N, Tomiyama K, Iijima J, Nabeshima Y, Fujioka M, Asato R, Tanaka S, Kojima K, Ito J, Nozaki K, Hashimoto N, et al. α-Klotho as a regulator of calcium homeostasis. Science. 2007;316:1615–1618. doi: 10.1126/science.1135901. [DOI] [PubMed] [Google Scholar]
- Invidia L, Salvioli S, Altilia S, Pierini M, Panourgia MP, Monti D, De Rango F, Passarino G, Franceschi C. The frequency of Klotho KL-VS polymorphism in a large Italian population, from young subjects to centenarians, suggests the presence of specific time windows for its effect. Biogerontology. 2010;11:67–73. doi: 10.1007/s10522-009-9229-z. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Jayadev S, Leverenz JB, Steinbart E, Stahl J, Klunk W, Yu CE, Bird TD. Alzheimer's disease phenotypes and genotypes associated with mutations in presenilin 2. Brain. 2010;133:1143–1154. doi: 10.1093/brain/awq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L. Amyloid precursor protein processing and Aβ42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci U S A. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King GD, Chen C, Huang MM, Zeldich E, Brazee PL, Schuman ER, Robin M, Cuny GD, Glicksman MA, Abraham CR. Identification of novel small molecules that elevate Klotho expression. Biochem J. 2012;441:453–461. doi: 10.1042/BJ20101909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang X, Chen YS, Wang LF, Li YJ, Liu K, Zhang MX, Li LJ, Chen C, He Q, Wang Y, Du JR. Klotho upregulation contributes to the neuroprotection of ligustilide in an Alzheimer's disease mouse model. Neurobiol Aging. 2014;35:169–178. doi: 10.1016/j.neurobiolaging.2013.07.019. [DOI] [PubMed] [Google Scholar]
- Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- Kurosu H, Kuro-O M. The Klotho gene family as a regulator of endocrine fibroblast growth factors. Mol Cell Endocrinol. 2009;299:72–78. doi: 10.1016/j.mce.2008.10.052. [DOI] [PubMed] [Google Scholar]
- Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, McGuinness OP, Chikuda H, Yamaguchi M, Kawaguchi H, Shimomura I, Takayama Y, Herz J, Kahn CR, Rosenblatt KP, Kuro-o M. Suppression of aging in mice by the hormone Klotho. Science. 2005;309:1829–1833. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanz TA, Carter DB, Merchant KM. Dendritic spine loss in the hippocampus of young PDAPP and Tg2576 mice and its prevention by the ApoE2 genotype. Neurobiol Dis. 2003;13:246–253. doi: 10.1016/S0969-9961(03)00079-2. [DOI] [PubMed] [Google Scholar]
- Lee YS, Silva AJ. The molecular and cellular biology of enhanced cognition. Nat Rev Neurosci. 2009;10:126–140. doi: 10.1038/nrn2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid β-protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci. 2011;31:6627–6638. doi: 10.1523/JNEUROSCI.0203-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Fergusson MM, Castilho RM, Liu J, Cao L, Chen J, Malide D, Rovira II, Schimel D, Kuo CJ, Gutkind JS, Hwang PM, Finkel T. Augmented Wnt signaling in a mammalian model of accelerated aging. Science. 2007;317:803–806. doi: 10.1126/science.1143578. [DOI] [PubMed] [Google Scholar]
- Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M. Alzheimer's disease: clinical trials and drug development. Lancet Neurol. 2010;9:702–716. doi: 10.1016/S1474-4422(10)70119-8. [DOI] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Mante M, Crews L, Spencer B, Adame A, Patrick C, Trejo M, Ubhi K, Rohn TT, Mueller-Steiner S, Seubert P, Barbour R, McConlogue L, Buttini M, Games D, Schenk D. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS One. 2011;6:e19338. doi: 10.1371/journal.pone.0019338. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Matthews FE, Arthur A, Barnes LE, Bond J, Jagger C, Robinson L, Brayne C. A two-decade comparison of prevalence of dementia in individuals aged 65 years and older from three geographical areas of England: results of the cognitive function and ageing study I and II. Lancet. 2013;382:1405–1412. doi: 10.1016/S0140-6736(13)61570-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meilandt WJ, Yu GQ, Chin J, Roberson ED, Palop JJ, Wu T, Scearce-Levie K, Mucke L. Enkephalin elevations contribute to neuronal and behavioral impairments in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2008;28:5007–5017. doi: 10.1523/JNEUROSCI.0590-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fülöp L, Penke B, Zilberter Y, Harkany T, Pitkanen A, Tanila H. Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. 2009;29:3453–3462. doi: 10.1523/JNEUROSCI.5215-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishizen-Eberz AJ, Rissman RA, Carter TL, Ikonomovic MD, Wolfe BB, Armstrong DM. Biochemical and molecular studies of NMDA receptor subunits NR1/2A/2B in hippocampal subregions throughout progression of Alzheimer's disease pathology. Neurobiol Dis. 2004;15:80–92. doi: 10.1016/j.nbd.2003.09.016. [DOI] [PubMed] [Google Scholar]
- Molokanova E, Akhtar MW, Sanz-Blasco S, Tu S, Piña-Crespo JC, McKercher SR, Lipton SA. Differential effects of synaptic and extrasynaptic NMDA receptors on Aβ-induced nitric oxide production in cerebrocortical neurons. J Neurosci. 2014;34:5023–5028. doi: 10.1523/JNEUROSCI.2907-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Anderson E, Lynch GS, Baudry M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-d-aspartate receptor antagonist, AP5. Nature. 1986;319:774–776. doi: 10.1038/319774a0. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein E, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabavi S, Fox R, Proulx CD, Lin JY, Tsien RY, Malinow R. Engineering a memory with LTD and LTP. Nature. 2014;511:348–352. doi: 10.1038/nature13294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai T, Yamada K, Kim HC, Kim YS, Noda Y, Imura A, Nabeshima Y, Nabeshima T. Cognition impairment in the genetic model of aging klotho gene mutant mice: a role of oxidative stress. FASEB J. 2003;17:50–52. doi: 10.1096/fj.02-0448fje. [DOI] [PubMed] [Google Scholar]
- Nakazawa K, McHugh TJ, Wilson MA, Tonegawa S. NMDA receptors, place cells and hippocampal spatial memory. Nat Rev Neurosci. 2004;5:361–372. doi: 10.1038/nrn1385. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Epilepsy and cognitive impairments in Alzheimer disease. Arch Neurol. 2009;66:435–440. doi: 10.1001/archneurol.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci. 2010;13:812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Jones B, Kekonius L, Chin J, Yu GQ, Raber J, Masliah E, Mucke L. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer's disease-related cognitive deficits. Proc Natl Acad Sci U S A. 2003;100:9572–9577. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol. 1999;56:303–308. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- Plattner F, Hernández A, Kistler TM, Pozo K, Zhong P, Yuen EY, Tan C, Hawasli AH, Cooke SF, Nishi A, Guo A, Wiederhold T, Yan Z, Bibb JA. Memory enhancement by targeting Cdk5 regulation of NR2B. Neuron. 2014;81:1070–1083. doi: 10.1016/j.neuron.2014.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2011. [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid ®-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu GQ, Palop JJ, Noebels JL, Mucke L. Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's disease. J Neurosci. 2011;31:700–711. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein EM, McConlogue L, Tan H, Power M, Masliah E, Mucke L. Levels and alternative splicing of amyloid beta protein precursor (APP) transcripts in brains of APP transgenic mice and humans with Alzheimer's disease. J Biol Chem. 1995;270:28257–28267. doi: 10.1074/jbc.270.47.28257. [DOI] [PubMed] [Google Scholar]
- Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, Cirrito JR, Devidze N, Ho K, Yu GQ, Palop JJ, Mucke L. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer's disease model. Proc Natl Acad Sci U S A. 2012;109:E2895–E2903. doi: 10.1073/pnas.1121081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Mejia RO, Newman JW, Toh S, Yu GQ, Zhou Y, Halabisky B, Cissé M, Scearce-Levie K, Cheng IH, Gan L, Palop JJ, Bonventre JV, Mucke L. Phospholipase A2 reduction ameliorates cognitive deficits in mouse model of Alzheimer's disease. Nat Neurosci. 2008;11:1311–1318. doi: 10.1038/nn.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal M. Dendritic spines: elementary structural units of neuronal plasticity. Prog Brain Res. 2002;138:53–59. doi: 10.1016/S0079-6123(02)38070-1. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Preventing Alzheimer's disease. Science. 2012;337:1488–1492. doi: 10.1126/science.1228541. [DOI] [PubMed] [Google Scholar]
- Semba RD, Moghekar AR, Hu J, Sun K, Turner R, Ferrucci L, O'Brien R. Klotho in the cerebrospinal fluid of adults with and without Alzheimer's disease. Neurosci Lett. 2014;558:37–40. doi: 10.1016/j.neulet.2013.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiozaki M, Yoshimura K, Shibata M, Koike M, Matsuura N, Uchiyama Y, Gotow T. Morphological and biochemical signs of age-related neurodegenerative changes in klotho mutant mice. Neuroscience. 2008;152:924–941. doi: 10.1016/j.neuroscience.2008.01.032. [DOI] [PubMed] [Google Scholar]
- Snider BJ, Norton J, Coats MA, Chakraverty S, Hou CE, Jervis R, Lendon CL, Goate AM, McKeel DW, Jr, Morris JC. Novel presenilin 1 mutation (S170F) causing Alzheimer disease with Lewy bodies in the third decade of life. Arch Neurol. 2005;62:1821–1830. doi: 10.1001/archneur.62.12.1821. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-β. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Spires TL, Hyman BT. Neuronal structure is altered by amyloid plaques. Rev Neurosci. 2004;15:267–278. doi: 10.1515/REVNEURO.2004.15.4.267. [DOI] [PubMed] [Google Scholar]
- Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, Bacskai BJ, Hyman BT. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25:7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Zhou Y, Halabisky B, Lo I, Cho SH, Mueller-Steiner S, Devidze N, Wang X, Grubb A, Gan L. Cystatin C-cathepsin B axis regulates amyloid beta levels and associated neuronal deficits in an animal model of Alzheimer's disease. Neuron. 2008;60:247–257. doi: 10.1016/j.neuron.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze C, Bi H, Kleinschmidt-DeMasters BK, Filley CM, Martin LJ. N-methyl-d-aspartate receptor subunit proteins and their phosphorylation status are altered selectively in Alzheimer's disease. J Neurol Sci. 2001;182:151–159. doi: 10.1016/S0022-510X(00)00467-6. [DOI] [PubMed] [Google Scholar]
- Talantova M, Sanz-Blasco S, Zhang X, Xia P, Akhtar MW, Okamoto S, Dziewczapolski G, Nakamura T, Cao G, Pratt AE, Kang YJ, Tu S, Molokanova E, McKercher SR, Hires SA, Sason H, Stouffer DG, Buczynski MW, Solomon JP, Michael S, et al. Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci U S A. 2013;110:E2518–E2527. doi: 10.1073/pnas.1306832110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- Tsai J, Grutzendler J, Duff K, Gan WB. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat Neurosci. 2004;7:1181–1183. doi: 10.1038/nn1335. [DOI] [PubMed] [Google Scholar]
- Tsien JZ. Linking Hebb's coincidence-detection to memory formation. Curr Opin Neurobiol. 2000;10:266–273. doi: 10.1016/S0959-4388(00)00070-2. [DOI] [PubMed] [Google Scholar]
- Tucker Zhou TB, King GD, Chen C, Abraham CR. Biochemical and functional characterization of the Klotho-VS polymorphism implicated in aging and disease risk. J Biol Chem. 2013;288:36302–36311. doi: 10.1074/jbc.M113.490052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–774. doi: 10.1038/nature05315. [DOI] [PubMed] [Google Scholar]
- Vázquez-Roque RA, Ubhi K, Masliah E, Flores G. Chronic cerebrolysin administration attenuates neuronal abnormalities in the basolateral amygdala induced by neonatal ventral hippocampus lesion in the rat. Synapse. 2014;68:31–38. doi: 10.1002/syn.21718. [DOI] [PubMed] [Google Scholar]
- Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, Mucke L, Palop JJ. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell. 2012;149:708–721. doi: 10.1016/j.cell.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt DL, Thomas D, Galvan V, Bredesen DE, Lamb BT, Pimplikar SW. Abnormal neuronal networks and seizure susceptibility in mice overexpressing the APP intracellular domain. Neurobiol Aging. 2011;32:1725–1729. doi: 10.1016/j.neurobiolaging.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, Mucke L. Tau reduction prevents Aβ-induced impairments in axonal transport. Science. 2010;330:198. doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Beagle AJ, Rabinovici GD, Shu H, Lee SE, Naasan G, Hegde M, Cornes SB, Henry ML, Nelson AB, Seeley WW, Geschwind MD, Gorno-Tempini ML, Shih T, Kirsch HE, Garcia PA, Miller BL, Mucke L. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 2013;70:1158–1166. doi: 10.1001/jamaneurol.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Cui Z, Zeng Q, Kuang H, Wang LP, Tsien JZ, Cao X. Genetic enhancement of memory and long-term potentiation but not CA1 long-term depression in NR2B transgenic rats. PLoS One. 2009;4:e7486. doi: 10.1371/journal.pone.0007486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Stradtman GG, 3rd, Wang XJ, Gao WJ. A specialized NMDA receptor function in layer 5 recurrent microcircuitry of the adult rat prefrontal cortex. Proc Natl Acad Sci U S A. 2008;105:16791–16796. doi: 10.1073/pnas.0804318105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimo A, Prince M. World Alzheimer report 2010: the global economic impact of dementia. London: Alzheimer's Disease International; 2010. [Google Scholar]
- Wu HY, Hudry E, Hashimoto T, Uemura K, Fan ZY, Berezovska O, Grosskreutz CL, Bacskai BJ, Hyman BT. Distinct dendritic spine and nuclear phases of calcineurin activation after exposure to amyloid-beta revealed by a novel fluorescence resonance energy transfer assay. J Neurosci. 2012;32:5298–5309. doi: 10.1523/JNEUROSCI.0227-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Hong S, O'Malley T, Sperling RA, Walsh DM, Selkoe DJ. New ELISAs with high specificity for soluble oligomers of amyloid beta-protein detect natural Aβ oligomers in human brain but not CSF. Alzheimers Dement. 2013;9:99–112. doi: 10.1016/j.jalz.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiro K, Philpot BD. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology. 2008;55:1081–1094. doi: 10.1016/j.neuropharm.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama JS, Sturm VE, Bonham LW, Klein E, Arfanakis K, Yu L, Coppola G, Kramer JH, Bennett DA, Miller BL, Dubal DB. Variation in longevity gene KLOTHO is associated with greater cortical volumes. Ann Clin Transl Neurol. 2015;2:215–230. doi: 10.1002/acn3.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young ME, Clark MH, Goffus A, Hoane MR. Mixed effects modeling Morris water maze data: advantages and cautionary notes. Learn Motiv. 2009;40:160–177. doi: 10.1016/j.lmot.2008.10.004. [DOI] [Google Scholar]
- Zeldich E, Chen CD, Colvin TA, Bove-Fenderson EA, Liang J, Tucker Zhou TB, Harris DA, Abraham CR. The neuroprotective effect of Klotho is mediated via regulation of members of the redox system. J Biol Chem. 2014;289:24700–24715. doi: 10.1074/jbc.M114.567321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Kurup P, Xu J, Carty N, Fernandez SM, Nygaard HB, Pittenger C, Greengard P, Strittmatter SM, Nairn AC, Lombroso PJ. Genetic reduction of striatal-enriched tyrosine phosphatase (STEP) reverses cognitive and cellular deficits in an Alzheimer's disease mouse model. Proc Natl Acad Sci U S A. 2010;107:19014–19019. doi: 10.1073/pnas.1013543107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Kurup P, Xu J, Anderson GM, Greengard P, Nairn AC, Lombroso PJ. Reduced levels of the tyrosine phosphatase STEP block beta amyloid-mediated GluA1/GluA2 receptor internalization. J Neurochem. 2011;119:664–672. doi: 10.1111/j.1471-4159.2011.07450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]