

Abstract
Perturbations in fast-spiking parvalbumin (PV) interneurons are hypothesized to be a major component of various neuropsychiatric disorders; however, the mechanisms regulating PV interneurons remain mostly unknown. Recently, cyclin-dependent kinase 5 (Cdk5) has been shown to function as a major regulator of synaptic plasticity. Here, we demonstrate that genetic ablation of Cdk5 in PV interneurons in mouse brain leads to an increase in GABAergic neurotransmission and impaired synaptic plasticity. PVCre;fCdk5 mice display a range of behavioral abnormalities, including decreased anxiety and memory impairment. Our results reveal a central role of Cdk5 expressed in PV interneurons in gating inhibitory neurotransmission and underscore the importance of such regulation during behavioral tasks. Our findings suggest that Cdk5 can be considered a promising therapeutic target in a variety of conditions attributed to inhibitory interneuronal dysfunction, such as epilepsy, anxiety disorders, and schizophrenia.
Keywords: anxiety, axon initial segment, cognition, cyclin-dependent kinase 5, fast-spiking (FS) parvalbumin (PV)-interneurons
Introduction
Parvalbumin (PV)-expressing interneurons constitute a major (up to 50%) component of inhibitory GABAergic neurons in the cortex (Kubota et al., 1994; Wonders and Anderson, 2006), as well as in other brain regions. For example, ∼30% of all interneurons in the hippocampus (including CA1 region) express PV (Kosaka et al., 1987; Katsumaru et al., 1988; Bezaire and Soltesz, 2013). PV interneurons have unique fast-spiking action potential properties that allow for very efficient communication within neuronal networks (Bartos et al., 2007). PV-interneuronal activity is both necessary and sufficient for the generation of gamma oscillations in vivo (Cardin et al., 2009; Sohal et al., 2009). Gamma rhythms were shown to be involved in the regulation of perception, attention, and memory (Uhlhaas and Singer, 2010). Dysfunctions in PV interneurons have been implicated in the pathogenesis of epilepsy, anxiety disorders, and schizophrenia (Wonders and Anderson, 2006). Despite strong interest, the mechanisms regulating the PV interneuron-dependent inhibitory neurotransmission and the relevance of these mechanisms to behavior remain unclear.
Cyclin-dependent kinase 5 (Cdk5) is a proline-directed serine/threonine kinase expressed primarily in the CNS in mammals. Postsynaptic Cdk5 plays a critical role in homeostatic synaptic plasticity by controlling spine-associated RapGAP (SPAR) phosphorylation and degradation of SPAR protein (Pak et al., 2001; Seeburg et al., 2008). Presynaptically, Cdk5 also plays multiple roles, including regulation of synaptic vesicle exocytosis by modulating interactions between syntaxin1 and Sept5 (Taniguchi et al., 2007) and by phosphorylation of Munc18 (Shuang et al., 1998) and Pctaire1 (Cheng et al., 2002), as well as endocytosis by phosphorylating multiple dephosphin proteins (Matsubara et al., 1996; Cousin et al., 2001; Lee et al., 2004). Importantly, a recent study (Kim and Ryan, 2010) demonstrated that a balance of the activities of Cdk5 and protein phosphatase calcineurin is a key regulatory mechanism determining the size of recycling vesicle pool and controlling neurotransmission. Altogether, Cdk5 is considered to be a major regulator of signaling that orchestrates plasticity at the synapse (Su and Tsai, 2010; Mitra et al., 2012). The critical presynaptic role of Cdk5 led us to hypothesize that removal of this enzyme from PV interneurons may allow to manipulate their synaptic properties and behavioral outputs. This, in turn, should provide an insight into the regulation of PV interneurons and the roles of these cells in animal behavior.
Here, we demonstrate that selective ablation of Cdk5 from PV interneurons leads to an increase in the number of synaptic vesicles in PV cells and enhanced GABA release and causes alterations in emotional and cognitive behavior. Our results demonstrate that Cdk5 gates GABAergic function of PV interneurons regulating their behavioral outputs and thus can be considered a promising molecular target for impairments attributed to PV-interneuronal dysfunction, such as epilepsy, anxiety disorders, and schizophrenia.
Materials and Methods
All experiments were performed according to the Guide for the Care and Use of Laboratory Animals and were approved by the National Institutes of Health and the Committee on Animal Care at the Massachusetts Institute of Technology.
Subjects.
We generated PVCre;fCdk5 mice by crossing PVCre mice (B6;129P2-Pvalbtm1(cre)Arbr/J; The Jackson Laboratory) with mice carrying floxed alleles of Cdk5 (fCdk5) [B6.129S4(Cg)-Cdk5tm1.1Lht/J; The Jackson Laboratory]. Both strains were maintained on the inbred C56BL/6J background for multiple generations. Three- to 4-month-old congenic PVCre;fCdk5 (homozygous mutant) and littermate fCdk5 (control) mice from the F2 generation were used for experiments. For consistency purposes, only male mice were used in all experiments.
Histology and immunohistochemistry.
Mice were transcardially perfused with PBS, followed by 4% paraformaldehyde (PFA) in 1× PBS. The brains were dissected and postfixed in 4% PFA in PBS at 4°C overnight. Free-floating vibratome coronal sections (40 μm) were incubated in a blocking solution of 10% normal donkey serum, 3% bovine serum albumin, 0.2% Triton X-100, and 0.02% sodium azide in 1× PBS for 1–2 h at room temperature (RT). Sections were then incubated with primary antibodies in the blocking solution overnight at 4°C, followed by the appropriate Cy3-congugated (1:1000; The Jackson Laboratory) and Alexa Fluor 488 (1:1000; Invitrogen) secondary antibodies for 2 h at RT. Twenty minute incubations with Hoechst dye (Invitrogen) at RT were performed to label cell nuclei. The following primary antibodies were used: PV-positive interneurons were identified with anti-PV rabbit polyclonal PV-25 antibody at 1:2000 [catalog #PV 25; Resource Research Identifier (RRID) AB10000344; Swant] and mouse monoclonal PV-235 antibody at 1:1000 (catalog #PV 235; RRID AB10000343; Swant); rabbit polyclonal antibody against Cux I at 1:200 (M 22; catalog #sc-13024; RRID AB2261231; Santa Cruz Biotechnology) was used as a cortical layer marker; and Cdk5 immunoreactivity was identified with C8 rabbit polyclonal antibody against Cdk5 at 1:100 (catalog #sc-173; RRID AB631224, Santa Cruz Biotechnology). The axon initial segment (AIS) was identified with rabbit polyclonal H-212 antibody against ankyrin-G (AnkG) at 1:400 (catalog #sc-9167; RRID AB2140663; Santa Cruz Biotechnology) and mouse monoclonal S8809 antibody against pan-Nav at 1:200 (catalog #S8809; RRID AB477552; Sigma-Aldrich). Images were acquired using high-resolution multichannel scanning confocal microscopy (Zeiss LSM 510). PV cell numbers in the mutant and control mice were assessed by counting PV-positive cells in the hippocampus and cingulate cortex in every fourth section throughout the brain. The cortical layer distribution of PV cells was assessed by counting their relative numbers in layers II–IV and V–VI of every fourth section through the motor cortex. All the cell counts were performed blind to the genotype of the mice. Labeling of the AIS was performed with anti-AnkG and anti-pan-Nav antibodies. The evaluation of AIS length was performed using an antibody against AnkG and PV. Approximately 100 neurons were compared in corresponding sections of the CA1 area of the hippocampus from three pairs of littermate PVCre;fCdk5 and fCdk5 mice. All PV-positive cells were excluded from AIS scoring. Scoring was performed blind to the genotype of the mouse. All the images were acquired using high-resolution multichannel scanning confocal microscopy (Zeiss LSM 510) and quantified using NIH ImageJ software.
Immunogold labeling transmission electron microscopy.
PVCre;fCdk5 and control mice (three animals per group) were transcardially perfused with PBS, followed by 4% PFA and 0.1% glutaraldehyde in PBS. The brains were dissected and postfixed in 4% PFA in PBS at 4°C overnight. Free-floating coronal sections (100 μm) were cut using a vibratome (Leica) and incubated in permeabilizing/blocking in solution for 1 h at 4°C. Preembedding labeling with anti-PV antibody was conducted overnight at 4°C. Brain sections were washed and incubated with rabbit anti-mouse IgG (1:100; Cappel) for 1 h at RT. The section were then incubated with Protein A–gold 5 nm particles (Utrecht University Medical Center, Utrecht, The Netherlands) for 1 h at RT, washed, and fixed in 1% glutaraldehyde/PBS for 10 min. The sections were washed in 0.1 m cacodylate buffer and postfixed with 0.5% osmium tetroxide for 30 min, washed in water, and dehydrated in graded alcohols. The samples were then embedded in TAAB Epon (Marivac Canada) between two sheets of Aclar film (order #50425; EMSdiasum.com) and polymerized at 60°C for 48 h. Areas containing the CA1 region of the hippocampus were cut out using a stereo microscope and mounted for sectioning. Before trimming the block for thin sectioning (60 nm), a 1-μm-thick section was taken for each sample, stained with 1% toluidine blue, and examined by light microscopy to ensure that comparable areas were selected for each animal. Ultrathin hippocampal sections (60 nm) were cut on a Reichert Ultracut-S microtome (Leica), picked up onto copper grids, stained with uranyl acetate and lead citrate, and examined in a JEOL 1200EX transmission electron microscope. Images were recorded with an AMT 2k CCD camera (Advanced Microscopy Techniques) at 30,000× magnification. Imaging was conducted on three grids per animal in the stratum pyramidale and stratum radiatum areas of the hippocampal CA1 regions. Ultrastructural analysis was performed for ∼100 unequivocally detected PV/Protein A–gold-labeled boutons identified in the CA1 stratum pyramidale and stratum radiatum in the corresponding hippocampal sections from three pairs of control and PVCre;fCdk5 mice. The analyzed PV boutons were not selected for the target specificity. The diameter of the presynaptic area (two-dimensional area; two straight line measurements across the diameter per bouton) was calculated using AMT Image Capture Version 600.214 software (Advanced Microscopy Techniques). Scoring the number of synaptic vesicles was performed in the same PV boutons using NIH ImageJ software.
Mouse behavior.
All the behavior experiments were performed using groups of 8–11 3-to 4-month-old littermate male PVCre;fCdk5 (mutant) and fCdk5 (control) mice. Mice were housed in groups of three to five animals. Mice were left to acclimate in the testing rooms for 45 min before the experiments. If the same groups of mice were used in different behavioral experiments, the tests were separated by 1 week. The most stressful tests, the fear conditioning and Morris water maze paradigms, were performed last. All the experiments were done during the light phase, the second part of the day, with experimenters that were blind to the genotype and treatment of the mice.
Open field.
Activity in a novel environment was measured in the 40 × 40 cm Plexiglas VersaMax chambers (Accuscan Instruments) using sets of 16 photobeam arrays. During each 60 or 10 min session, the number of beam breaks (activity) was measured automatically.
Elevated plus maze.
Anxiety-like behavior was assessed using the elevated plus-maze paradigm. The maze consisted of the central platform (10 × 10 cm) with two opposing open arms (30 × 10 × 0.5 cm) and two arms enclosed by Plexiglas walls (30 × 10 × 30 cm) elevated 50 cm from the floor. The animal's position was automatically determined, and the mouse was tracked for 10 min with VersaMax software (TSE Systems).
Light/dark exploration.
The light/dark apparatus consisted of a transparent Plexiglas open-field box (40 × 40 cm) containing a black Plexiglas box (20 × 20 cm) occupying half of the area. Experiments were conducted in a room with the overhead light off and a bright 120 W lamp directed at the light part of the open-field area. A mouse was placed in the black Plexiglas box, and its behavior was video recorded for 10 min. The frequency of exits to the bright area and the time spent in the bright and the dark areas were scored.
Morris water maze.
Spatial reference memory testing was performed in a circular tank (1.2 m) filled with opaque water, 22°C. The walls surrounding the tank contained bright and contrasting shapes that served as reference cues. The tank contained a fixed platform (10 cm diameter) in a target quadrant. On day 1 of the experiment, mice were placed into the water, guided toward a visible platform, and kept on the platform for 15 s. This procedure was repeated one more time. On the following days, the platform was submerged, and the mice were place into the maze at one of four points randomly facing the wall of the tank. Mice were allowed to search for a platform for 60 s, and, if they did not find a platform, they were gently guided to it and kept on the platform for 15 s. Two trials a day were conducted with 2 min intertrial intervals. Mouse behavior was video recorded and analyzed using Videomot 2 software (TSE Systems). On day 6 of the experiment, the platform was removed, and a probe trial was conducted. The time spent in each of the four quadrants was recorded automatically.
Prepulse inhibition.
Testing was conducted using the Startle Monitor System (SM100; Hamilton Kinder). For the first 2 d, animals were habituated to the startle monitor apparatus. On day 3, six different types of trials were then performed on randomized intervals: no acoustic stimulus trials, trials with acoustic stimulus (120 dB, 40 ms) alone, and trials with four different prepulse stimuli (70, 75, 80, and 85 dB, 20 ms each) 100 ms later, followed by an acoustic startle stimulus (120 dB). Prepulse trials were performed in blocks of six in a pseudorandom order with intertrial intervals of 20–30 s. The prepulse inhibition (PPI) level was calculated as [100 − (response amplitude for prepulse stimulus with the startle stimulus/amplitude for the startle stimulus) × 100].
Slice electrophysiology.
Transverse hippocampal slices were prepared from 2- to 3-month-old male littermates. The brain was rapidly removed and transferred to ice-cold, oxygenated (95% O2 and 5% CO2) low-Ca2+/high-Mg2+ cutting solution containing the following (in mm): 211 sucrose, 3.3 KCl, 1.3 NaH2PO4, 0.5 CaCl2, 10 MgCl2, 26 NaHCO3, and 11 glucose. Hippocampal slices were cut with a Leica VT1000S vibratome (Leica) and transferred for recovery to a holding chamber containing oxygenated artificial CSF (ACSF) consisting of 124 mm NaCl, 3.3 mm KCl, 1.3 mm NaH2PO4, 2.5 mm CaCl2, 1.5 mm MgCl2, 26 mm NaHCO3, and 11 mm glucose at 32–34°C for 30 min and then allowed to return to RT for at least another 60 min before recording. CA1 field EPSP (fEPSP) evoked by Schaffer–collateral (SC) stimulation were measured at 28–30°C as described previously (Oh et al., 2010). Long-term potentiation (LTP) was induced by four episodes of theta burst stimulation (TBS) with 10 s intervals. TBS consisted of 10 brief bursts of stimuli delivered at 5 Hz; each burst contains four pulses at 100 Hz. Whole-cell recordings were performed in CA1 pyramidal neurons as described previously (Morales et al., 2002; Deng et al., 2011), and all data were acquired using a MultiClamp 700B amplifier and a Digidata 1440A analog-to-digital converter (Molecular Devices). For mEPSC or mIPSC measurement, cells were held at −70 mV, and the internal solution contained the following (in mm): 145 CsCl, 5 NaCl, 10 HEPES-CsOH, 10 EGTA, 4 MgATP, and 0.3 Na2GTP. TTX (1 μm) and picrotoxin (100 μm) were added to ACSF for mEPSC measurements, and picrotoxin was replaced with CNQX (10 μm) and d-APV (50 μm) for mIPSC measurements. For recording evoked current, SC fibers were stimulated, and whole-cell recordings were performed in CA1 pyramidal neurons or PV interneurons using pipettes filled with the following (in mm): 125 Cs-methanesulfonate, 20 CsCl, 10 NaCl, 10 HEPES, 0.2 EGTA, 2 MgATP, 0.3 Na2GTP, and 2.5 QX-314 as described previously (Kreple et al., 2014). PV interneurons were confirmed by filling with 0.1 mm Alexa Fluor 488 hydrazide (Life Technologies) combined with anti-PV antibody staining. To determine NMDAR/AMPAR ratio, picrotoxin was added to ACSF, and EPSCs were recorded at a holding potential of −70 mV (for AMPAR-mediated currents) or at a holding potential of +40 mV (for NMDAR-mediated currents). The amplitude for NMDAR-mediated current was measured 50 ms after the stimulus artifact.
Statistics.
Student's two-tailed t tests were used for analysis of two experimental groups. One-way ANOVA with Tukey's post hoc test was used when more than two groups were compared. The graphs represent mean values, with error bars indicating SEM. All the statistical analysis was performed using GraphPad Prism (GraphPad Software).
Results
Selective ablation of Cdk5 from PV interneurons
To examine the role of Cdk5 in PV interneurons, we ablated Cdk5 specifically in PV cells by generating PVCre;fCdk5 mice obtained by crossing PVCre+/− animals (Hippenmeyer et al., 2005) with mice carrying loxP alleles of Cdk5 (Samuels et al., 2007; Guan et al., 2011). PVCre;fCdk5 mice are viable, fertile, bred according to Mendelian ratios, do not exhibit developmental disadvantages or anatomical abnormalities, and have normal general health (data not shown). Immunohistochemical examination demonstrated the complete loss of Cdk5 from all observed PV interneurons in various cortical and hippocampal areas in young adult mice (Fig. 1A,B). Characterization of the general brain architecture of the mutant animals did not reveal any obvious abnormalities. Survival, migration, and differentiation of PV interneurons were unaffected by the loss of Cdk5 (Fig. 1C–E).
Figure 1.
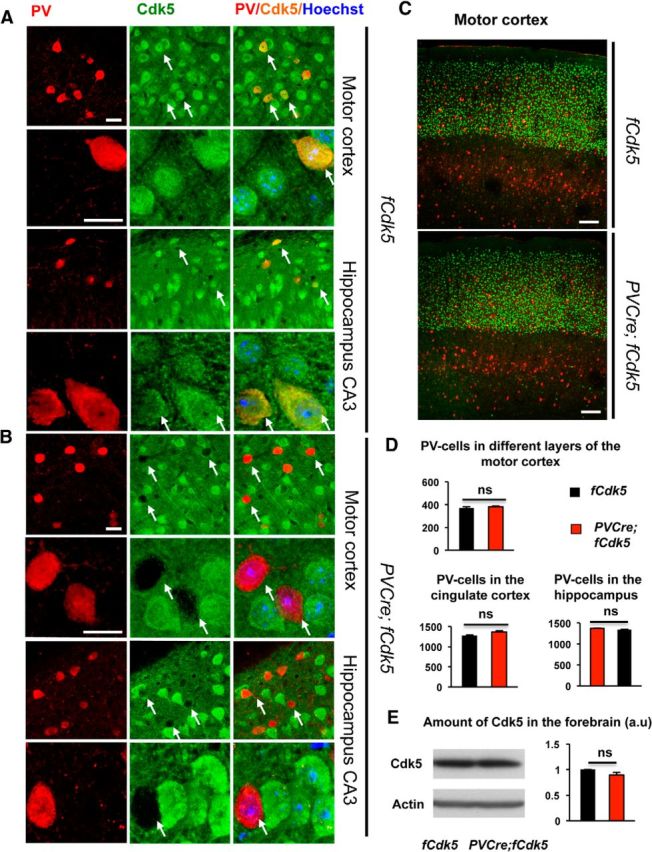
Genetic ablation of Cdk5 specifically in PV interneurons. A, B, Immunoreactivity for PV and Cdk5 in the motor cortex in 3.5-month-old fCdk5 (control) and PVCre;fCdk5 (mutant) littermate mice. The expression of Cdk5 (green) is present in all PV-positive (red) cells (arrows) in control mice (A). The complete ablation of Cdk5 is observed in PV interneurons of the PVCre;fCdk5 mice (B). Scale bars, 15 μm. C, D, Immunoreactivity for PV interneurons in different brain areas and layers in the cortex in control and PVCre;fCdk5 mice. C, PV interneurons (red) in different layers of the motor cortex in control and PVCre;fCdk5 mice, with Cux I (green) immunoreactivity labeling cortical layers II–IV. Scale bars, 100 μm. D, Bar graphs represent the relative numbers of PV interneurons in the cingulate cortex, hippocampus, and different layers of the motor cortex in 3.5-month-old control and PVCre;fCdk5 littermate mice. E, Representative Western blot and related quantification of Cdk5 protein in control and PVCre;fCdk5 (red) brains. Black, Control mice; red, PVCre;fCdk5 mice; ns, not significant. p > 0.05, error bars indicate ±SEM.
Decreased anxiety-related behavior in PVCre;fCdk5 mice
We performed a general battery of tests to analyze behavior of the PVCre;fCdk5 mice. All the tests were conducted using groups of 3- to 4-month-old PVCre;fCdk5 and control fCdk5 male littermates, 8–11 animals per group. Examination of the novelty-induced locomotion and in the open-field arena revealed an increase in horizontal activity (p < 0.05; Fig. 2A) and total distance traveled by PVCre;fCdk5 mice (p < 0.05; Fig. 2A) compared with control animals.
Figure 2.
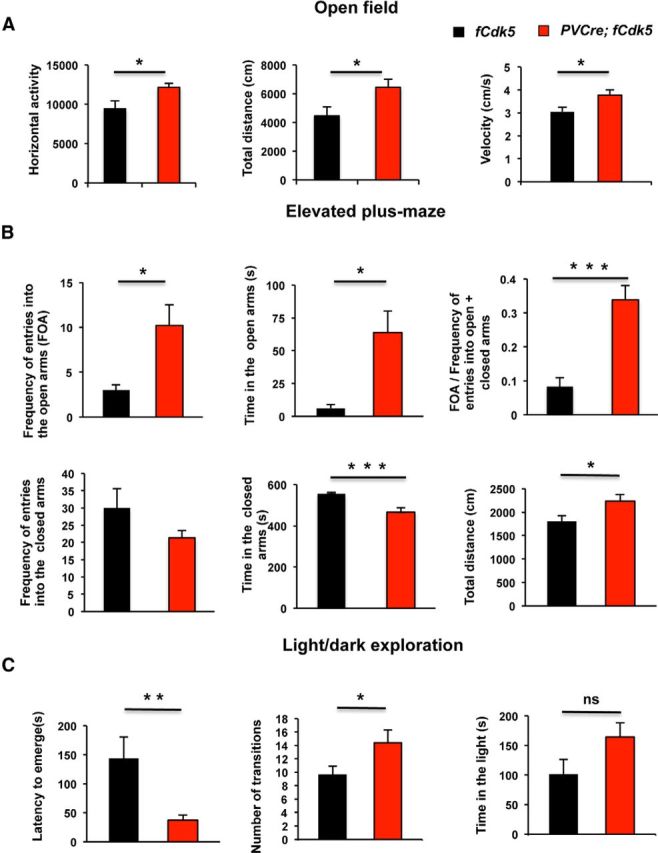
PVCre;fCdk5 mice exhibit decreased anxiety and hyperactivity. A, Horizontal activity and total distance traveled in the open field; movement velocity in the elevated plus maze. B, Frequency of entries into open arms (FOA), time spent in the open arms, frequency of entries to the open arms (FOA)/(frequency of entries to the open + closed arms), frequency of entries to the closed arms, time spent in the closed arms, and distance traveled during 10 min exploration in the elevated plus maze. C, Latency to emerge, the number of dark/light transitions, and the time spent in the light area in the light/dark exploration test. Black, Control mice; red, PVCre;fCdk5 mice. *p < 0.05, **p < 0.01, ***p < 0.001; error bars indicate ±SEM.
To examine the influence of Cdk5 ablation on anxiety-related behavior, we conducted an elevated plus-maze test (Dawson and Tricklebank, 1995). Remarkably, PVCre;fCdk5 mice showed a significantly increased frequency of entries into the open arms (10.2 ± 2.4) and spent significantly more time in the open arms (63.9 ± 16.4 s) and less time in the closed arms (466. 4 ± 21.9 s) than the control mice (3.0 ± 0.6, 6.2 ± 2.7, and 555.8 ± 23.7 s, respectively; p < 0.01, p < 0.05, and p < 0.001 respectively; Fig. 2B). These behaviors indicate decreased anxiety in the PVCre;fCdk5 mice.
To extend the observation of anxiety phenotype in the PVCre;fCdk5 mice, the light/dark exploration test was performed (Crawley and Goodwin, 1980; Crawley, 2007). Although the total time that PVCre;fCdk5 mice spent in the light area was not significantly different from the controls (164.2 ± 24.5 vs 100.6 ± 25.3 s; p = 0.09), PVCre;fCdk5 animals exhibited a significantly shortened latency to enter the brightly lit area (36.8 ± 8.9 vs 143.6 ± 37.4 s; p < 0.001) and an increased number of transition to the light (14.4 ± 1.3 vs 9.6 ± 1.9; p < 0.05; Fig. 2C).
PVCre;fCdk5 mice exhibit impaired learning and memory
To evaluate cognitive function in the PVCre;fCdk5 mice, we performed pavlovian cued and contextual fear conditioning (Phillips and LeDoux, 1992). Both hippocampus- and amygdala-dependent contextual fear memory (62 ± 8 vs 41 ± 6.5% freezing events; p < 0.05) and amygdala-dependent cued fear memory (58 ± 7 vs 37 ± 4% freezing events; p < 0.01) were significantly decreased in PVCre;fCdk5 mice compared with control animals (Fig. 3A).
Figure 3.
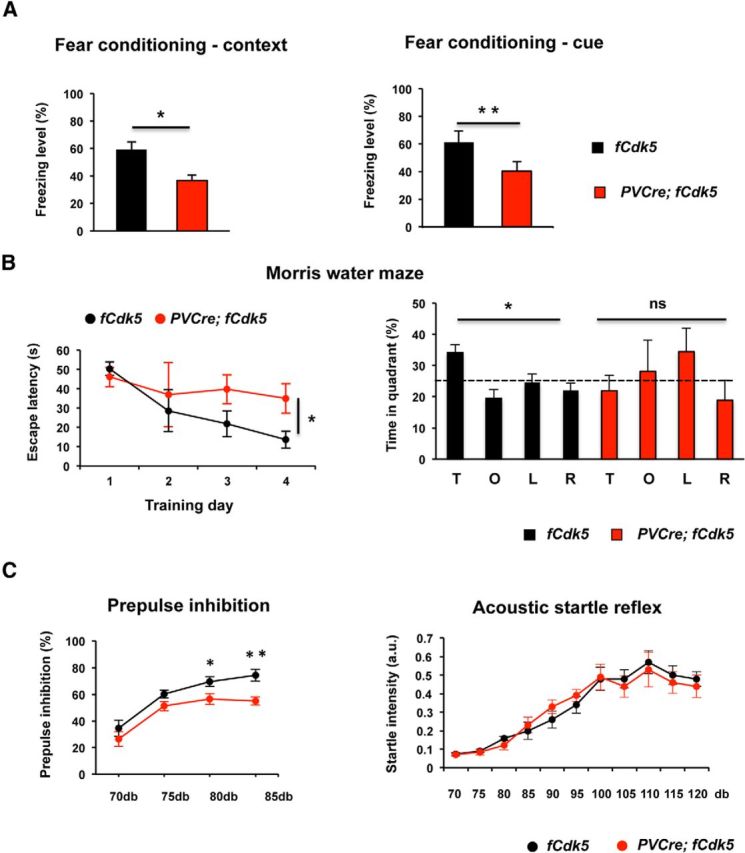
PVCre;fCdk5 mice exhibit cognitive impairment and deficient sensorimotor gating. A, Freezing levels in contextual and cued pavlovian fear conditioning paradigms. B, The Morris water-maze test. The escape latency and time spent in the target (T), opposite (O), right (R), and left (L) quadrants during probe trials. C, The percentage of PPI of acoustic startle reflex at four different prepulse intensities (70, 75, 80, and 85dB). Acoustic startle response across 70–120 dB amplitude range. Black, Control mice; red, PVCre;fCdk5 mice; ns, not significant. *p < 0.05, **p < 0.01; error bars indicate ±SEM.
To extend our observations of memory impairment in PVCre;fCdk5 mice, we used the Morris water-maze paradigm to evaluate hippocampus-dependent spatial reference memory (Vorhees and Williams, 2006). During 5 d of training, control mice demonstrated a reduction in their escape latency from an average 50.33 ± 4.9 to 13.6 ± 8.7 s (Fig. 3B). In contrast, the PVCre;fCdk5 mice showed poor learning, with less reduction in their escape latency (46.06 ± 9.2 to 35 ± 17.6 s), which was significantly worse than in control mice (p = 0.023). A probe trial conducted 24 h after the last training session also revealed significant differences in target versus opposite quadrant preference between the mutant and control groups of animals (p < 0.05 for fCdk5 mice and p > 0.05 for PVCre;fCdk5 mice; Fig. 3B). Control experiments indicated that there was no significant difference in swim speed between fCdk5 and PVCre;fCdk5 mice (p > 0.05; data not shown).
Deficient PPI of acoustic startle reflex in PVCre;fCdk5 mice
PPI of the startle reflex is a physiological measure of sensorimotor gating, i.e., the ability to process and filter sensory information. PPI was shown to significantly correlate with an operational measure of thought disorder in psychiatric patients and proposed to reflect cognitive fragmentation. An examination of PPI revealed a significant decrease of the inhibition at different prepulse intensities in the PVCre;fCdk5 mice compared with controls (p < 0.05; Fig. 3C). We found no differences in the amplitude of the acoustic startle reflex across the whole range of acoustic startle stimulus intensities (70–120 dB) between PVCre;fCdk5 and control mice, which indicates that the PVCre;fCdk5 mice have normal hearing and motor reflexes (Fig. 3C).
Impaired LTP and decreased basal synaptic transmission in PVCre;fCdk5 mice
To investigate the potential neurophysiological basis of cognitive impairment in PVCre;fCdk5 animals, we tested LTP in the hippocampus of PVCre;fCdk5 and control mice. CA1 fEPSPs were evoked by SC stimulation, and LTP was induced by four episodes of TBS with 10 s intervals. This stimulus induced robust LTP in control mice (148.4 ± 11.5%); however, LTP induction was significantly impaired in the PVCre;fCdk5 animals (108.1 ± 10.6%; p < 0.05; Fig. 4A).
Figure 4.
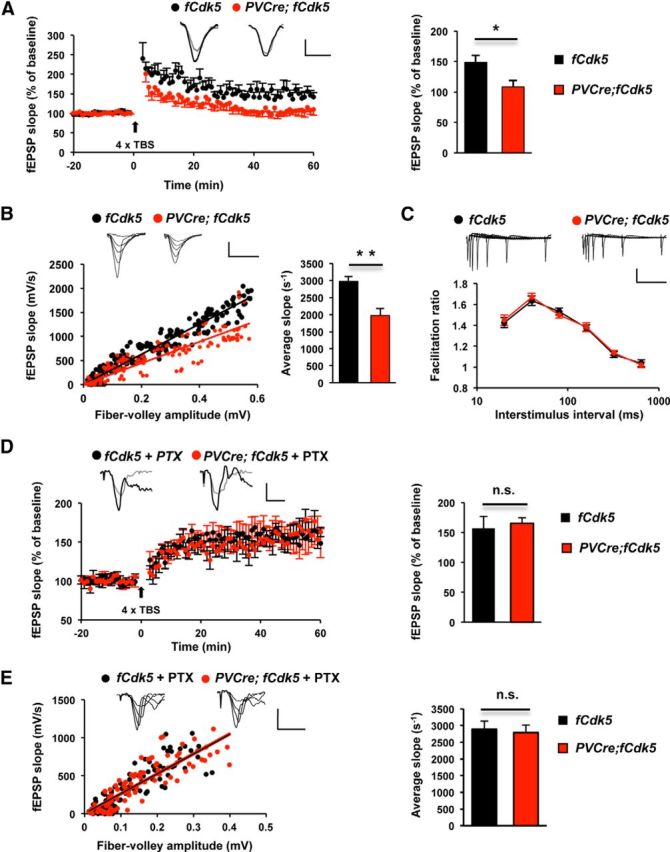
Impaired LTP and decreased basal synaptic transmission in PVCre;fCdk5 mice. A, LTP in CA1 in acute hippocampal slices from 3-month-old control (fCdk5; 5 animals, 8 slices) and littermate PVCre;fCdk5 (5 animals, 10 slices) mice. fEPSP slope is shown as percentage of baseline. The traces were taken 1 min before TBS (gray) and at the end of the recording (black). Calibration: 1 mV, 10 ms. A bar graph shows the average of fEPSP as a percentage of baseline during the last 10 min of recording. B, C, CA1 fEPSPs evoked by SC pathway stimulation in acute hippocampal slices. B, The input–output curve was obtained by plotting the slopes of fEPSP against fiber valley amplitudes. Calibration: 1 mV, 10 ms. C, PPF ratio was plotted against interstimulus interval (control, 3 animals, 11 slices; PVCre;fCdk5, 3 animals, 9 slices). Calibration: 1 mV, 250 ms. D, LTP in CA1 in acute hippocampal slices from 3-month-old control (4 animals, 8 slices) and littermate PVCre;fCdk5 (4 animals, 9 slices) mice in the presence of picrotoxin (PTX; 100 μm). The traces were taken 1 min before TBS (gray) and at the end of the recording (black). Calibration: 1 mV, 10 ms. A bar graph shows the average of fEPSP as a percentage of baseline during the last 10 min of recording. E, CA1 fEPSPs evoked by SC pathway stimulation in the presence of picrotoxin. The input–output curve was obtained by plotting the slopes of fEPSP against fiber valley amplitudes. Calibration: 1 mV, 10 ms. Black, Control mice; red, PVCre;fCdk5 mice. *p < 0.05, **p < 0.01; error bars indicate ±SEM.
Next, we evaluated basal synaptic transmission using hippocampal slices. The input–output curve was obtained by plotting the slopes of fEPSP against fiber volley amplitudes to calculate basal synaptic transmission. Short-term presynaptic release probability was assessed using the paired-pulse facilitation (PPF) ratio, which was plotted against various interstimulus intervals. We found a significant decrease in the average slope of fEPSP (p < 0.01; Fig. 4B), which suggests a downregulation of synaptic transmission. However, there was no difference in PPF between control and PVCre;fCdk5 mice (p > 0.05; Fig. 4C), which suggests normal presynaptic glutamate release probability.
To evaluate the potential contribution of inhibitory neurotransmission to the LTP impairment observed in PVCre;fCdk5 mice, we tested LTP induction in the hippocampus of PVCre;fCdk5 and control animals in the presence of picrotoxin, a noncompetitive antagonist of GABAA receptors. LTP was induced in hippocampal slices using the protocol described previously. Remarkably, application of picrotoxin (100 μm) resulted in complete rescue of the LTP (165.4 ± 9.3 vs 157.1 ± 19.4% in controls; p > 0.05; Fig. 4D) and basal synaptic transmission (Fig. 4E) deficit in PVCre;fCdk5 mice, suggesting that these impairments in PVCre;fCdk5 animals were attributable to an abnormal increase in GABAergic function.
Increase in GABAergic neurotransmission in PVCre;fCdk5 mice
GABAergic PV interneurons provide a main source of inhibitory control in the mammalian brain by exerting a hyperpolarizing influence over pyramidal neurons (Glickfeld et al., 2009). To directly examine inhibitory neurotransmission in PVCre;fCdk5 and control animals, we conducted whole-cell recordings in CA1 pyramidal neurons. Whereas there was no significant difference in mEPSC between PVCre;fCdk5 and control mice (Fig. 5A,B), we observed that mIPSC frequency was approximately twofold greater in PVCre;fCdk5 (1.8 ± 0.3 Hz) than in control littermates (0.94 ± 0.2 Hz; p < 0.05; Fig. 5C,D), which suggests an increased GABA release.
Figure 5.
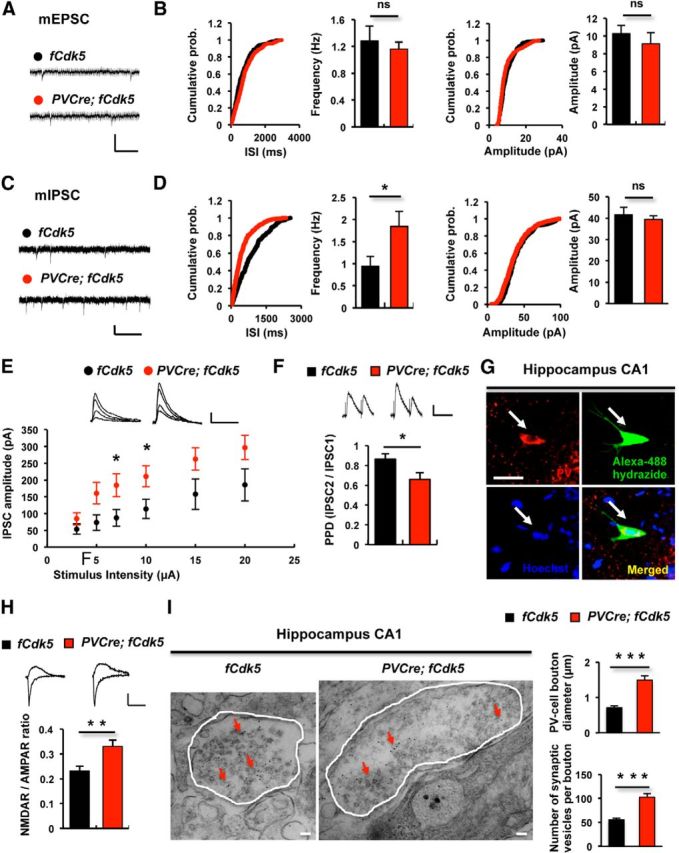
PVCre;fCdk5 mice display increased inhibitory neurotransmission. A–F, Whole-cell recordings from pyramidal cells in the CA1 area of acute hippocampal slices from control (fCdk5) and PVCre;fCdk5 mice. A, Representative traces of mEPSCs from control and PVCre;fCdk5 mice. Calibration: 20 pA, 200 ms. B, Cumulative probability distribution histograms and bar graphs showing average values of mEPSC frequency or amplitude from control (3 animals, 9 cells) and PVCre;fCdk5 mice (3 animals, 9 cells) C, Representative traces of mIPSCs from control and PVCre;fCdk5 mice. Calibration: 25 pA, 500 ms. D, Cumulative probability distribution histograms and bar graphs showing average values of mIPSC frequency and amplitude in control (4 animals, 7 cells) and PVCre;fCdk5 (5 animals, 9 cells) mice. E, IPSCs recording from control (3 animals, 9 cells) and PVCre;fCdk5 (3 animals, 11 cells) mice. IPSCs amplitude plotted against stimulus intensity is shown. Calibration: 50 pA, 50 ms. F, Paired-pulse depression (PPD = IPSC2/IPSC1; interstimulus interval, 100 ms) of IPSCs was measured in control (3 animals, 5 slices) and PVCre;fCdk5 (3 animals, 7 slices) mice. Calibration: 20 pA, 100 ms. G, PV interneurons were identified based on their morphological features and later confirmed by filling with Alexa Fluor 488 hydrazide combined with anti-PV antibody labeling. Scale bar, 15 μm. H, NMDAR- or AMPAR-mediated current was measured at PV interneurons in hippocampal CA1 region in control (3 animals, 8 cells) and PVCre;fCdk5 (3 animals, 9 cells) mice. Calibration: 50 pA, 50 ms. I, Synaptic boutons of PV interneurons were identified in control and PVCre;fCdk5 mice by preembedding immunogold labeling transmission electron microscopy in ultrathin hippocampal sections. Diameter of all unequivocally identified PV boutons and number of synaptic vesicles per bouton were measured in corresponding hippocampal sections in CA1 stratum pyramidale and stratum radiatum from control and PVCre;fCdk5 mice (3 animals and ∼100 boutons per group). Scale bars, 100 nm. Red arrows, PV/Protein A gold particles; white contours, PV bouton outlines; black, control mice; red, PVCre;fCdk5 mice; ns, not significant. *p < 0.05, **p < 0.01, ***p < 0.001; error bars indicate SEM.
To further strengthen our conclusion of an increased inhibition in PVCre;fCdk5 mice, we measured IPSCs in CA1 pyramidal neurons during SC stimulation. To isolate IPSCs, the responses of cells were recorded at 0 mV in the presence of CNQX and APV. We observed a significant increase in IPSCs in PVCre;fCdk5 mice (Fig. 5E) compared with control animals, indicating that activity-dependent inhibitory transmission was also increased in these mice compared with controls. Furthermore, increased paired-pulse depression of IPSCs in PVCre;fCdk5 mice compared with controls (0.66 ± 0.07 vs 0.86 ± 0.05; p < 0.05; Fig. 5F) suggests enhanced presynaptic GABA release in these mice.
It has been shown that Cdk5 negatively regulates surface levels of NMDAR subunit NR2B (Zhang et al., 2008) and that Cdk5 knock-out in the brain leads to an increase in surface NR2B accumulation (Hawasli et al., 2007). We asked whether ablation of Cdk5 results in functional changes of NMDARs in PV interneurons. To address the question, we performed whole-cell recording at PV interneurons in hippocampal CA1 regions by stimulating SC fibers and measuring the ratio of NMDAR/AMPAR-mediated currents. We observed a significantly increased NMDAR/AMPAR ratio in PVCre;fCdk5 mice compared with controls (0.33 ± 0.03 vs 0.23 ± 0.02; p < 0.01; Fig. 5G,H).
To further evaluate potential ultrastructural alterations that may lead to an increased GABA release in PV+Cdk5− interneurons, we performed electron microscopy analysis using transverse hippocampal floating brain sections. Remarkably, the measurements revealed an approximately twofold increase in average PV+ bouton diameter in PVCre;fCdk5 brains compared with controls (1.49 ± 0.12 vs 0.72 ± 0.04 μm; p < 0.0001; Fig. 5H). Estimation of the total number of synaptic vesicles in all PV/Protein A–gold-labeled boutons detected unequivocally in the CA1 area of the hippocampus showed significantly increased number of vesicles in PVCre;fCdk5 mice compared with controls (102.74 ± 7.38 vs 55.67 ± 3.08 vesicles; p < 0.0001; Fig. 5I).
Increased length of AIS in excitatory neurons in PVCre;fCdk5 mice
The synapses formed by PV interneurons onto excitatory neurons are mostly located perisomatically or at the AIS. Recent findings demonstrate that the size of the AIS can be regulated by presynaptic activity and that this variability serves as a component of synaptic plasticity that fine-tunes neuronal excitability (Grubb and Burrone, 2010; Kuba et al., 2010). To determine the potential effect of an increased inhibitory synaptic activity on AIS parameters, we calculated the average length of the AIS in excitatory neurons in the CA1 area of the hippocampus of PVCre;fCdk5 and control mice. Because AnkG is a main protein component located at the AIS and is required for its maintenance (Hedstrom et al., 2008), we used AnkG immunoreactivity to identify the AIS in neuronal populations. The scoring revealed a highly significant increase in the size of the AIS in PVCre;fCdk5 animals compared with control littermates (p < 0.0001; Fig. 6). Calculating the length of AnkG-delineated AIS in the cingulate cortex produced the results very similar to our hippocampal findings: a significant increase in the length of AIS in pyramidal neurons in PVCre;fCdk5 animals compared with controls (data not shown).
Figure 6.

An increase in the length of the AIS in pyramidal neurons in the hippocampus of PVCre;fCdk5 mice. A, AnkG (green) colocalizes with pan-Nav (red) immunoreactivity at the AIS. B, AnkG immunoreactivity (green) as a marker of AIS of the pyramidal neurons in the corresponding hippocampal sections from control (fCdk5) and PVCre;fCdk5 animals (blue, Hoechst). Scale bars: A, 10 μm; B, 50 μm. C, Quantification of the length (micrometers) of the AIS in the neurons in the CA1 area of the hippocampus revealed by AnkG immunoreactivity. Magnification: main images, 20×; insets, 100×. Approximately 100 neurons were scored in the corresponding hippocampal sections from three pairs of control and PVCre;fCdk5 3.5-month-old littermate mice. Note a significant increase in the average length of the AIS in PVCre;fCdk5 mice compared with controls. Black, Control mice; red, PVCre;fCdk5 mice. ***p < 0.001; error bars indicate ±SEM.
Discussion
Here we show that reduction of Cdk5, an important component of synaptic function, selectively in PV interneurons affects neurophysiological processes at cellular, network, and behavioral levels. PVCre;fCdk5 mice exhibit increased GABAergic neurotransmission, decreased anxiety, and cognitive impairment.
Evaluation of the potential mechanism of an enhanced GABAergic function in PVCre;fCdk5 animals indicated that Cdk5 ablation leads to an increased GABA release. Our results are in good agreement with recent findings that Cdk5 acts presynaptically as a major negative constraint on neurotransmission via regulating synaptic vesicle pools and release probability in excitatory neurons (Kim and Ryan, 2010, 2013), but our study is the first one to demonstrate such a role for Cdk5 specifically in PV interneurons. Furthermore, examination of the ultrastructural basis for an enhanced GABAergic function in PVCre;fCdk5 mice revealed a significant increase in the diameter of synaptic boutons and, importantly, the number of synaptic vesicles in PV+Cdk5− cells compared with controls. Such an increase in the number of vesicles may explain an observed increase in GABAergic neurotransmission and demonstrate a structural mechanism underlying the function of Cdk5 in neurotransmitter release in PV interneurons (Pierce and Lewin, 1994; Nüsser et al., 1998; Mackenzie et al., 1999; Kubota and Kawaguchi, 2000). We should note that neurotransmitter release could also be facilitated by presynaptic NMDARs (Berretta and Jones, 1996; Corlew et al., 2007). Therefore, our demonstration of an increased PV-interneuronal NMDAR/AMPAR ratio in PVCre;fCdk5 mice suggests that Cdk5 could control GABA release at least in part via regulating surface NMDARs in PV interneurons.
We showed that stimulation-induced inhibitory synaptic transmission is increased in PVCre;fCdk5 mice compared with controls. A significant decrease in the average slope of CA1 fEPSP, which is rescued by picrotoxin, and normal PPF and mEPSC in PVCre;fCdk5 mice suggest that the decrease in basal synaptic transmission is not attributable to an alteration in excitatory neurotransmission but is a result of an increased inhibition in these animals. The observed decrease in hippocampal LTP and rescue of this deficit by picrotoxin suggests that the main reason for an impaired LTP in PVCre;fCdk5 mice is an increased inhibition.
We demonstrated that PVCre;fCdk5 mice display specific emotional and cognitive deregulation. The most pronounced phenotype was a decrease in anxiety-related behaviors, which phenocopies the effect of the GABAA agonist diazepam (Fraser et al., 2010) and can be explained by increased GABAergic neurotransmission in these animals. The observed PPI deficit in PVCre;fCdk5 mice is also likely a result of a hyperinhibition. The cognitive impairments observed in PVCre;fCdk5 mice appear to reflect a decreased hippocampus-dependent LTP.
It may be interesting to compare our current behavioral findings with our previous report (Su et al., 2013), describing ablation of Cdk5 in the forebrain excitatory neurons using αCaMKIICre (Cdk5f/f/CW2 mice). The most pronounced behavioral abnormality exhibited by Cdk5f/f/CW2 mice was a strong hyperactivity evident throughout multiple paradigms. Additionally, Cdk5f/f/CW2 animals displayed an impaired performance in fear conditioning and Morris water-maze tasks, light/dark exploration, forced swim, and acoustic startle tests. Although PVCre;fCdk5 animals also display a decrease in anxiety and cognitive impairment, there are some obvious behavioral dissimilarities between two transgenic strains. They include deficient PPI and normal startle reflex in PVCre;fCdk5 animals but normal PPI and startle hyper-reactivity and decreased behavioral despair in Cdk5f/f/CW2 animals. Although it is not easy to directly compare the mutant strains analyzed because of partially non-overlapping brain areas affected by the Cre lines used, we speculate that some behavioral domains, such as learning and memory, may require normal Cdk5 expression and function in multiple neuronal types. Therefore, a loss of Cdk5 from either excitatory neurons or PV interneurons may lead to a cognitive abnormality. The differences in PPI between two strains could be attributable to a differential contribution of excitatory forebrain neurons and inhibitory PV interneurons to a sensorimotor gating, which may be of particular relevance for disorders such as schizophrenia. Deregulation of pathways associated with mania could be responsible for mania-like features, such as hyper-reactivity and decreased behavioral despair in Cdk5/f/fCW2 mice (Prickaerts et al., 2006; Su et al., 2013).
Our observation of increased AnkG-delineated AIS length in pyramidal neurons of PVCre;fCdk5 mice demonstrates, for the first time, an alteration in AIS size as a result of a genetic manipulation in vivo. Such an increase suggests the implementation of a compensatory mechanism by excitatory neurons in reaction to enhanced presynaptic inhibition and also identifies a convenient structural readout of neuronal network adaptation to changes in presynaptic inhibitory neurotransmission. Future studies on the mechanism of AIS regulation should be very informative to our understanding of fine-tuning neuronal excitability.
Growing evidence suggests that Cdk5 regulates neurotransmission via multiple mechanisms (Morabito et al., 2004; Hawasli et al., 2007; Seeburg et al., 2008; Zhang et al., 2008; Kim and Ryan, 2010; 2013; Su et al., 2012). Cdk5 has been shown to regulate synaptic vesicle pathways by phosphorylating the dephosphin proteins, such as dynamin I and synaptojanin, that, in turn, are dephosphorylated by the protein phosphatase calcineurin (Matsubara et al., 1996; Cousin et al., 2001; Lee et al., 2004). Interestingly, Kim and Ryan (2010) have shown that a balance of the activities of Cdk5 and calcineurin serves as a key regulatory mechanism determining the size of recycling vesicle pool and controlling neurotransmission. Based on these findings, we would suggest that gating inhibitory neurotransmission by Cdk5 in PV interneurons may occur via phosphorylation of some of the presynaptic dephosphins. It is also feasible that Cdk5 could determine release probability at least in part through regulating voltage-gated calcium channels (Kim and Ryan, 2013). Additionally, our results suggest that the Cdk5 could negatively regulate GABA release from PV interneurons via downregulating surface NMDARs. Additional studies should help to decipher the network of molecular mechanisms underlying the critical role of Cdk5 in gating GABAergic function of PV cells.
The hypofunction of PV interneurons is among the most consistent findings in patients with schizophrenia, a devastating major psychiatric disorder that is characterized by psychotic symptoms and cognitive impairments (Behrens and Sejnowski, 2009; Lewis et al., 2011). Moreover, decreased inhibition has been implicated in the pathobiology of a number of other neurologic and neuropsychiatric illnesses (Wonders and Anderson, 2006). Our finding that Cdk5 expressed in PV interneurons acts a critical negative regulator of inhibitory neurotransmission may suggest potential Cdk5-based treatment strategies for interneuronopathy-related disorders.
In summary, our results indicate that Cdk5 gates GABAergic function of PV interneurons regulating their behavioral outputs and thus can be considered a promising molecular target for therapeutic interventions in a variety of conditions attributed to PV-interneuronal dysfunction, such as epilepsy, anxiety, and schizophrenia.
Footnotes
A.R. and M.C. were supported by National Alliance for Research on Schizophrenia and Depression Young Investigator Awards. This work was supported by National Institutes of Health Grant RO1-NS051874-16 and partially supported a Simons Foundation Autism Research Initiative grant (L.-H.T.). We thank Tracy Petryshen, Mike Lewis, Al Schroeder, and Erin Berry for the valuable advice on behavioral assays, Alison Mungenast and Alexi Nott for helpful comments on this manuscript, and all members of the Tsai laboratory for advice and discussion.
The authors declare no competing financial interests.
References
- Bartos M, Vida I, Jonas P. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat Rev Neurosci. 2007;8:45–56. doi: 10.1038/nrn2044. [DOI] [PubMed] [Google Scholar]
- Behrens MM, Sejnowski TJ. Does schizophrenia arise from oxidative dysregulation of parvalbumin interneurons in the developing cortex? Neuropharmacology. 2009;57:193–200. doi: 10.1016/j.neuropharm.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berretta N, Jones RS. Tonic facilitation of glutamate release by presynaptic N-methyl-d-aspartate autoreceptors in the entorhinal cortex. Neuroscience. 1996;75:339–344. doi: 10.1016/0306-4522(96)00301-6. [DOI] [PubMed] [Google Scholar]
- Bezaire MJ, Soltesz I. Quantitative assessment of CA1 local circuits: knowledge base for interneuron-pyramidal cell connectivity. Hippocampus. 2013;23:751–785. doi: 10.1002/hipo.22141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardin JA, Carlén M, Meletis K, Knoblich U, Zhang F, Deisseroth K, Tsai LH, Moore CI. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 2009;459:663–667. doi: 10.1038/nature08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng K, Li Z, Fu WY, Wang JH, Fu AK, Ip NY. Pctaire1 interacts with p35 and is a novel substrate for Cdk5/p35. J Biol Chem. 2002;277:31988–31993. doi: 10.1074/jbc.M201161200. [DOI] [PubMed] [Google Scholar]
- Corlew R, Wang Y, Ghermazien H, Erisir A, Philpot BD. Developmental switch in the contribution of presynaptic and postsynaptic NMDA receptors to long-term depression. J Neurosci. 2007;27:9835–9845. doi: 10.1523/JNEUROSCI.5494-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousin MA, Tan TC, Robinson PJ. Protein phosphorylation is required for endocytosis in nerve terminals: potential role for the dephosphins dynamin I and synaptojanin, but not AP180 or amphiphysin. J Neurochem. 2001;76:105–116. doi: 10.1046/j.1471-4159.2001.00049.x. [DOI] [PubMed] [Google Scholar]
- Crawley JN. Behavioral phenotyping of transgenic and knockout mice. Hoboken, NJ: Wiley; 2007. What's wrong with my mouse? [Google Scholar]
- Crawley J, Goodwin FK. Preliminary report of a simple animal behavior model for the anxiolytic effects of benzodiazepines. Pharmacol Biochem Behav. 1980;13:167–170. doi: 10.1016/0091-3057(80)90067-2. [DOI] [PubMed] [Google Scholar]
- Dawson GR, Tricklebank MD. Use of the elevated plus-maze in the search for novel anxiolytic agents. Trends Pharmacol Sci. 1995;16:33–36. doi: 10.1016/S0165-6147(00)88973-7. [DOI] [PubMed] [Google Scholar]
- Deng L, Kaeser PS, Xu W, Südhof TC. RIM proteins activate vesicle priming by reversing autoinhibitory homodimerization of Munc13. Neuron. 2011;69:317–331. doi: 10.1016/j.neuron.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser LM, Brown RE, Hussin A, Fontana M, Whittaker A, O'Leary TP, Lederle L, Holmes A, Ramos A. Measuring anxiety- and locomotion-related behaviors in mice: a new way of using old tests. Psychopharmacology (Berl) 2010;211:99–112. doi: 10.1007/s00213-010-1873-0. [DOI] [PubMed] [Google Scholar]
- Glickfeld LL, Roberts JD, Somogyi P, Scanziani M. Interneurons hyperpolarize pyramidal cells along their entire somatodendritic axis. Nat Neurosci. 2009;12:21–23. doi: 10.1038/nn.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb MS, Burrone J. Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature. 2010;465:1070–1074. doi: 10.1038/nature09160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan JS, Su SC, Gao J, Joseph N, Xie Z, Zhou Y, Durak O, Zhang L, Zhu JJ, Clauser KR, Carr SA, Tsai LH. Cdk5 is required for memory function and hippocampal plasticity via the cAMP signaling pathway. PLoS One. 2011;6:e25735. doi: 10.1371/journal.pone.0025735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawasli AH, Benavides DR, Nguyen C, Kansy JW, Hayashi K, Chambon P, Greengard P, Powell CM, Cooper DC, Bibb JA. Cyclin-dependent kinase 5 governs learning and synaptic plasticity via control of NMDAR degradation. Nat Neurosci. 2007;10:880–886. doi: 10.1038/nn1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom KL, Ogawa Y, Rasband MN. AnkyrinG is required for maintenance of the axon initial segment and neuronal polarity. J Cell Biol. 2008;183:635–640. doi: 10.1083/jcb.200806112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, Arber S. Developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol. 2005;3:e159. doi: 10.1371/journal.pbio.0030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsumaru H, Kosaka T, Heizmann CW, Hama K. Immunocytochemical study of GABAergic neurons containing the calcium-binding protein parvalbumin in the rat hippocampus. Exp Brain Res. 1988;72:347–362. doi: 10.1007/BF00250256. [DOI] [PubMed] [Google Scholar]
- Kim SH, Ryan TA. CDK5 serves as a major control point in neurotransmitter release. Neuron. 2010;67:797–809. doi: 10.1016/j.neuron.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Ryan TA. Balance of calcineurin Aα and CDK5 activities sets release probability at nerve terminals. J Neurosci. 2013;33:8937–8950. doi: 10.1523/JNEUROSCI.4288-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka T, Katsumaru H, Hama K, Wu JY, Heizmann CW. GABAergic neurons containing the Ca2+-binding protein parvalbumin in the rat hippocampus and dentate gyrus. Brain Res. 1987;419:119–130. doi: 10.1016/0006-8993(87)90575-0. [DOI] [PubMed] [Google Scholar]
- Kreple CJ, Lu Y, Taugher RJ, Schwager-Gutman AL, Du J, Stump M, Wang Y, Ghobbeh A, Fan R, Cosme CV, Sowers LP, Welsh MJ, Radley JJ, LaLumiere RT, Wemmie JA. Acid-sensing ion channels contribute to synaptic transmission and inhibit cocaine-evoked plasticity. Nat Neurosci. 2014;17:1083–1091. doi: 10.1038/nn.3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuba H, Oichi Y, Ohmori H. Presynaptic activity regulates Na(+) channel distribution at the axon initial segment. Nature. 2010;465:1075–1078. doi: 10.1038/nature09087. [DOI] [PubMed] [Google Scholar]
- Kubota Y, Kawaguchi Y. Dependence of GABAergic synaptic areas on the interneuron type and target size. J Neurosci. 2000;20:375–386. doi: 10.1523/JNEUROSCI.20-01-00375.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota Y, Hattori R, Yui Y. Three distinct subpopulations of GABAergic neurons in rat frontal agranular cortex. Brain Res. 1994;649:159–173. doi: 10.1016/0006-8993(94)91060-X. [DOI] [PubMed] [Google Scholar]
- Lee SY, Wenk MR, Kim Y, Nairn AC, De Camilli P. Regulation of synaptojanin 1 by cyclin-dependent kinase 5 at synapses. Proc Natl Acad Sci U S A. 2004;101:546–551. doi: 10.1073/pnas.0307813100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Fish KN, Arion D, Gonzalez-Burgos G. Perisomatic inhibition and cortical circuit dysfunction in schizophrenia. Curr Opin Neurobiol. 2011;21:866–872. doi: 10.1016/j.conb.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie PJ, Kenner GS, Prange O, Shayan H, Umemiya M, Murphy TH. Ultrastructural correlates of quantal synaptic function at single CNS synapses. J Neurosci. 1999;19:RC13. doi: 10.1523/JNEUROSCI.19-12-j0003.1999. (1–7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara M, Kusubata M, Ishiguro K, Uchida T, Titani K, Taniguchi H. Site-specific phosphorylation of synapsin I by mitogen-activated protein kinase and Cdk5 and its effects on physiological functions. J Biol Chem. 1996;271:21108–21113. doi: 10.1074/jbc.271.35.21108. [DOI] [PubMed] [Google Scholar]
- Mitra A, Mitra SS, Tsien RW. Heterogenous reallocation of presynaptic efficacy in recurrent excitatory circuits adapting to inactivity. Nat Neurosci. 2012;15:250–257. doi: 10.1038/nn.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morabito MA, Sheng M, Tsai LH. Cyclin-dependent kinase 5 phosphorylates the terminal domain of the postsynaptic density protein PSD-95 in neurons. J Neurosci. 2004;24:865–876. doi: 10.1523/JNEUROSCI.4582-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales B, Choi SY, Kirkwood A. Dark rearing alters the development of GABAergic transmission in visual cortex. J Neurosci. 2002;22:8084–8090. doi: 10.1523/JNEUROSCI.22-18-08084.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nüsser Z, Hájos N, Somogyi P, Mody I. Increased number of synaptic GABA(A) receptors underlies potentiation at hippocampal inhibitory synapses. Nature. 1998;395:172–177. doi: 10.1038/25999. [DOI] [PubMed] [Google Scholar]
- Oh D, Han S, Seo J, Lee JR, Choi J, Groffen J, Kim K, Cho YS, Choi HS, Shin H, Woo J, Won H, Park SK, Kim SY, Jo J, Whitcomb DJ, Cho K, Kim H, Bae YC, Heisterkamp N, Choi SY, Kim E. Regulation of synaptic Rac1 activity, long-term potentiation maintenance, and learning and memory by BCR and ABR Rac GTPase-activating proteins. J Neurosci. 2010;30:14134–14144. doi: 10.1523/JNEUROSCI.1711-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak DT, Yang S, Rudolph-Correia S, Kim E, Sheng M. Regulation of dendritic spine morphology by SPAR, a PSD-95-associated RapGAP. Neuron. 2001;31:289–303. doi: 10.1016/S0896-6273(01)00355-5. [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037/0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- Pierce JP, Lewin GR. An ultrastructural size principle. Neuroscience. 1994;58:441–446. doi: 10.1016/0306-4522(94)90071-X. [DOI] [PubMed] [Google Scholar]
- Prickaerts J, Moechars D, Cryns K, Lenaerts I, van Craenendonck H, Goris I, Daneels G, Bouwknecht JA, Steckler T. Transgenic mice overexpressing glycogen synthase kinase 3beta: a putative model of hyperactivity and mania. J Neurosci. 2006;26:9022–9029. doi: 10.1523/JNEUROSCI.5216-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels BA, Hsueh YP, Shu T, Liang H, Tseng HC, Hong CJ, Su SC, Volker J, Neve RL, Yue DT, Tsai LH. Cdk5 promotes synaptogenesis by regulating the subcellular distribution of the MAGUK family member CASK. Neuron. 2007;56:823–837. doi: 10.1016/j.neuron.2007.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeburg DP, Feliu-Mojer M, Gaiottino J, Pak DT, Sheng M. Critical role of CDK5 and Polo-like kinase 2 in homeostatic synaptic plasticity during elevated activity. Neuron. 2008;58:571–583. doi: 10.1016/j.neuron.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuang R, Zhang L, Fletcher A, Groblewski GE, Pevsner J, Stuenkel EL. Regulation of Munc-18/syntaxin 1A interaction by cyclin-dependent kinase 5 in nerve endings. J Biol Chem. 1998;273:4957–4966. doi: 10.1074/jbc.273.9.4957. [DOI] [PubMed] [Google Scholar]
- Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702. doi: 10.1038/nature07991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su SC, Tsai LH. Cyclin-dependent kinases in brain development and disease. Annu Rev Cell Dev Biol. 2011;27:465–491. doi: 10.1146/annurev-cellbio-092910-154023. [DOI] [PubMed] [Google Scholar]
- Su SC, Seo J, Pan JQ, Samuels BA, Rudenko A, Ericsson M, Neve RL, Yue DT, Tsai LH. Regulation of N-type voltage-gated calcium channels and presynaptic function by cyclin-dependent kinase 5. Neuron. 2012;75:675–687. doi: 10.1016/j.neuron.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su SC, Rudenko A, Cho S, Tsai LH. Forebrain-specific deletion of Cdk5 in pyramidal neurons results in mania-like behavior and cognitive impairment. Neurobiol Learn Mem. 2013;105:54–62. doi: 10.1016/j.nlm.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi M, Taoka M, Itakura M, Asada A, Saito T, Kinoshita M, Takahashi M, Isobe T, Hisanaga S. Phosphorylation of adult type Sept5 (CDCrel-1) by cyclin-dependent kinase 5 inhibits interaction with syntaxin-1. J Biol Chem. 2007;282:7869–7876. doi: 10.1074/jbc.M609457200. [DOI] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W. Abnormal neural oscillations and synchrony in schizophrenia. Nat Rev Neurosci. 2010;11:100–113. doi: 10.1038/nrn2774. [DOI] [PubMed] [Google Scholar]
- Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1:848–858. doi: 10.1038/nprot.2006.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wonders CP, Anderson SA. The origin and specification of cortical interneurons. Nat Rev Neurosci. 2006;7:687–696. doi: 10.1038/nrn1954. [DOI] [PubMed] [Google Scholar]
- Zhang S, Edelmann L, Liu J, Crandall JE, Morabito MA. Cdk5 regulates the phosphorylation of tyrosine 1472 NR2B and the surface expression of NMDA receptors. J Neurosci. 2008;28:415–424. doi: 10.1523/JNEUROSCI.1900-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]