

Abstract
Synaptic vesicle protein 2A (SV2A) is a ubiquitous component of synaptic vesicles (SVs). It has roles in both SV trafficking and neurotransmitter release. We demonstrate that Casein kinase 1 family members, including isoforms of Tau–tubulin protein kinases (TTBK1 and TTBK2), phosphorylate human SV2A at two constellations of residues, namely Cluster-1 (Ser42, Ser45, and Ser47) and Cluster-2 (Ser80, Ser81, and Thr84). These residues are also phosphorylated in vivo, and the phosphorylation of Thr84 within Cluster-2 is essential for triggering binding to the C2B domain of human synaptotagmin-1. We show by crystallographic and other analyses that the phosphorylated Thr84 residue binds to a pocket formed by three conserved Lys residues (Lys314, Lys326, and Lys328) on the surface of the synaptotagmin-1 C2B domain. Finally, we observed dysfunctional synaptotagmin-1 retrieval during SV endocytosis by ablating its phospho-dependent interaction with SV2A, knockdown of SV2A, or rescue with a phosphorylation-null Thr84 SV2A mutant in primary cultures of mouse neurons. This study reveals fundamental details of how phosphorylation of Thr84 on SV2A controls its interaction with synaptotagmin-1 and implicates SV2A as a phospho-dependent chaperone required for the specific retrieval of synaptotagmin-1 during SV endocytosis.
Keywords: CK1, SV2A, synaptotagmin
Introduction
Synaptic vesicle protein 2 (SV2A, SV2B, and SV2C) isoforms are all integral 12 transmembrane domain synaptic vesicle (SV) proteins. SV2A is the most widely distributed SV2 isoform and is ubiquitously expressed in the CNS, whereas SV2B is brain specific with SV2C the minor isoform (Buckley and Kelly, 1985; Bajjalieh et al., 1994).
SV2A knock-out mice are normal at birth but fail to grow and experience severe seizures before premature death after 3 weeks attributable to multiple neural and endocrine deficits (Crowder et al., 1999), indicating that SV2A plays key roles in postnatal brain function. It has been proposed to function as a positive effector of SV exocytosis, potentially by binding calcium (Mendoza-Torreblanca et al., 2013). All SV2 isoforms possess an N-terminal cytoplasmic domain that interacts with another integral SV protein, synaptotagmin-1 (Syt1; Schivell et al., 1996; Schivell et al., 2005; Lazzell et al., 2004). Synaptotagmin-1 is the key Ca2+ sensor for evoked synchronous neurotransmitter release at the synapse (Brose et al., 1992; Geppert et al., 1994), and thus its efficient incorporation into newly formed SVs during endocytosis is essential to maintain the fidelity of neurotransmission. Disruption of SV2A retrieval during SV endocytosis results in accumulation of synaptotagmin-1 on the plasma membrane of central nerve terminals (Yao et al. 2010), suggesting that SV2A may be required for efficient packaging of this essential calcium-sensing protein into SVs.
Relatively little is understood regarding how SV2 isoforms are regulated. Global phosphorylation site mass spectrometry studies have revealed that SV2A is potentially phosphorylated on at least 10 residues within its N-terminal cytoplasmic region (http://www.phosphosite.org/proteinAction.do?id=8509&showAllSites=true). To our knowledge, only one other investigation has analyzed the regulation of SV2A by phosphorylation (Pyle et al., 2000). This study identified an SV-associated kinase that phosphorylated the N-terminal cytoplasmic domain of SV2A. In gel kinase assays and the use of a Casein kinase 1 (CK1) inhibitor (CK1-7) suggested that this kinase was a member of the CK1 group of eukaryotic protein kinases (Pyle et al., 2000). Intriguingly, in vitro phosphorylation of a recombinant N-terminal fragment of SV2A comprising the cytoplasmic domain by CK1 increased its ability to interact with recombinant synaptotagmin-1 approximately threefold (Pyle et al., 2000).
To date, no studies have determined the site(s) phosphorylated by CK1 isoforms within the SV2A N-terminal cytoplasmic domain and how phosphorylation of these residues stimulates recruitment of synaptotagmin-1 and/or influences SV trafficking. In this study, we demonstrate that CK1 and Tau–tubulin protein kinase (TTBK) isoforms efficiently phosphorylate the N-terminal cytoplasmic domain of SV2A within two constellations, namely Cluster-1 (Ser42, Ser45, and Ser47) and Cluster-2 (Ser80, Ser81, and Thr84). We demonstrate that phosphorylation of Thr84 in Cluster-2 is key in triggering binding to synaptotagmin-1. Crystallographic analysis revealed that the phosphorylated Thr84 residue specifically bound to a pocket formed by three conserved Lys residues (Lys314, Lys326, and Lys328) on the surface of the synaptotagmin-1 C2B domain. Finally, we present evidence that phosphorylation of SV2A at Cluster-2 is essential for the efficient retrieval of synaptotagmin-1 during SV endocytosis.
Materials and Methods
Materials.
Synaptotagmin-1–pHluorin (Syt1–pHluorin) and synaptophysin–pHluorin constructs were provided by Prof. V. Haucke (Leibniz Institute of Molecular Pharmacology, Berlin, Germany) and Prof. L. Lagnado (University of Sussex, Sussex, UK). Neurobasal media, B-27 supplement, penicillin/streptomycin, minimal essential medium (MEM), Lipofectamine 2000, and anti-rabbit Alexa Fluor 568 were obtained from Invitrogen. Recombinant human CK1 family kinases TTBK2[1–316] (DU (Dundee University) number 38313), TTBK1[1–1321, full length] (DU number 34496), Vaccinia-related kinase 1 (VRK1)[1–396, full length] (DU number 34413), CK1α1[1–337, full length] (DU number 329), CK1δ[1–415, full length] (DU number 19064), CK1ε[1–416, full length] (DU number 5127), CK1γ1[1–422, full length] (DU number 31197), SV2A[1–160] (DU number 38732), SV2A[1–160] Cluster-1 mutant (DU number 39656), SV2A[1–160] Cluster-1 mutant (DU number 44015), and SV2A[1–160] Cluster-1 + Cluster2 mutant (DU number 40838) were all expressed in Escherichia coli with the indicated N-terminal glutathione S-transferase (GST) or maltose binding protein (MBP) tag. The SV2A full-length proteins [wild type (WT) or eight Ser/Thr to A mutants] with GST tag were expressed and purified from HEK293 cells transfected with corresponding constructs [WT, DU number 38880; eight S(T)/A, DU number 44610]. The WT and phosphorylation site(s) mutants full-length SV2A proteins were expressed and purified from HEK293 cells for analyzing the phosphorylation on SV2A in vivo by mass spectrometry. All other reagents were obtained from Sigma-Aldrich. All recombinant proteins, plasmids, and antibodies generated for the present study are available on request and are described in additional detail on our reagents website (https://mrcppureagents.dundee.ac.uk/).
General methods.
Recombinant DNA procedures were performed using standard protocols. Mutagenesis was performed using the QuikChange site-directed mutagenesis kit (Stratagene) with KOD polymerase (Novagen). DNA constructs were purified from E. coli DH5α using a maxi prep kit (Qiagen) according to the instructions of the manufacturer. Verification of the constructs was performed by the Sequencing Service [Medical Research Council Protein Phosphorylation Unit (MRC–PPU), College of Life Sciences, University of Dundee, Dundee, Scotland, UK; ]. DNA for bacterial protein expression was transformed into E. coli BL21-CodonPlus (DE3)-RIL cells (Stratagene).
Plasmid generation.
Mouse SV2A was amplified from IMAGE EST6493509 using KOD Hot Start DNA Polymerase (Merck Millipore), cloned into pSC-b (Agilent), and sequenced to completion. This was then ligated into the BglII NotI sites in pCMV mCerulean (mCer) N1 (Anggono et al., 2006). Mutations and shRNA-resistant versions were created using the Quick Change method (Agilent) but using KOD DNA Hot Start polymerase. SV2A–pHluorin was created in a Clontech EGFP–C1 backbone by replacing EGFP with human SV2A using XhoI and AgeI restriction enzymes. The fluorescent pHluorin protein was then inserted between amino acids 197 and 198 at a PclI restriction enzyme site (DU number 42587).
SV2A knockdown was achieved using the published oligonucleotide sequence (GAATTGGCTCAGCAGTATGttcaagagaCATACTGCTGAGCCAATTC) against the rat sequence of SV2A that is identical to the mouse sequence (shRNA1; Dong et al. 2006). Oligonucleotides were ligated into the BglII HindIII sites of pSUPER mCer (Clayton et al., 2010). The SYN1 promoter-driven pHluorin–rSYT1–BGH pA cassette was amplified by adding flanking BglII and SmaI restriction sites and ligated into either vectors pSuper.Neo mCer or pSuper.Neo mCer mSV2A shRNA1 after digestion.
Antibodies.
The following antibodies were raised in sheep by the Division of Signal Transduction Therapy at the University of Dundee and affinity purified against the indicated antigens: anti-SV2A (S290D, first bleed; raised against residues 1–160 of human SV2A), anti-phospho-SV2A T84 (S679D, third bleed; raised against residues 77–91 of human SV2A: RRGGASSDApTEGHDEDDRR), and anti-phospho-SV2A S42, 45, and 47 (S686D, third bleed; raised against residues 36–54 of human SV2A: CKKRVQDEYpSRRpSYpSRFEEEDDKK). Anti-SV2A (ab32942) and anti-GST [horseradish peroxidase (HRP); ab58626] antibodies were purchased from Abcam, and anti-synaptotagmin-1 (catalog #3347) was from Cell Signaling Technology. Secondary antibodies coupled to HRP were obtained from Thermo Fisher Scientific.
Identification of phosphorylation sites.
HEK293 cells were transfected with a construct encoding WT and the indicated mutants of full-length Flag–SV2A. Thirty-six hours after transfection, cells were lysed, and Flag–SV2A was purified by immunoprecipitation and incubated with recombinant WT TTBK2[1–316] or kinase inactive (KD) TTBK2[D163A, 1–316] and Mg2–[-32P]ATP for 60 min. Samples were subjected to electrophoresis on a polyacrylamide gel that was stained with colloidal Coomassie blue and autoradiographed. Phosphorylated SV2A was digested with trypsin, and the peptides were separated on a reverse-phase HPLC Vydac C18 column (catalog #218TP5215; Separations Group) equilibrated in 0.1% (v/v) trifluoroacetic acid, and the column developed with a linear acetonitrile gradient at a flow rate of 0.2 ml/min. Fractions (0.1 ml each) were collected and analyzed for 32P radioactivity by Cerenkov counting. Phosphopeptides were analyzed by liquid chromatography (LC)-MS/MS using a Thermo U3000 RSLC nano liquid chromatography system (Thermo Fisher Scientific) coupled to a Thermo LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher Scientific). Data files were searched using Mascot (www.matrixscience.com) run on an in-house system against a database containing the appropriate SV2A sequences, with a 10 ppm mass accuracy for precursor ions, a 0.6 Da tolerance for fragment ions, and allowing for Phospho (ST), Phospho (Y), Oxidation (M), and Dioxidation (M) as variable modifications. Individual MS/MS spectra were inspected using Xcalibur 2.2 (Thermo Fisher Scientific), and Proteome Discoverer with phosphoRS 3.1 (Thermo Fisher Scientific) was used to assist with phosphosite assignment. The site of phosphorylation of 32P-labeled peptides was determined by solid-phase Edman degradation on a Shimadzu PPSQ33A Sequencer of the peptide coupled to Sequelon-AA membrane (Applied Biosystems) as described previously (Campbell and Morrice, 2002). The above LC-MS/MS approach was also used for the analysis of phosphosites on mouse brain SV2A (Table 1) and on WT and mutant SV2A overexpressed in HEK293 cells (Table 2).
Table 1.
Identification of phosphorylation sites on mouse brain SV2A
| Peptides identified by mass spectrometry | Mass | Mascot score | Possible P sites | % Probability (PD1.4 pRS3.1) |
|---|---|---|---|---|
| >150 kDa | ||||
| R.GGLSDGEGPPGGR.G + P (ST) | 1234.4310 | 64 | S127 | 100 |
| R.GGLSDGEGPPGGRGEAQR.R + P (ST) | 1775.6966 | 35 | S127 | 100 |
| R.GEGAQDEEEGGASSDATEGHDEDDEIYEGEYQGIPR.A + 2 P (ST) | 4000.3955 | 66 | S80, 81 | 98, 98 |
| R.GEGAQDEEEGGASSDATEGHDEDDEIYEGEYQGIPR.A + 3 P (ST) | 4080.3606 | 32 | S80, S81, T84 | 100, 100, 100 |
| 75–150 kDa | ||||
| R.VFSVTHIK.T + P (ST) | 1009.4332 | 44 | S393 | 99 |
| R.GGLSDGEGPPGGR.G + P (ST) | 1234.4322 | 72 | S127 | 100 |
| R.GGLSDGEGPPGGRGEAQR.R + P (ST) | 1775.6939 | 50 | S127 | 100 |
| R.GEGAQDEEEGGASSDATEGHDEDDEIYEGEYQGIPR.A + P (ST) | 3920.4297 | 90 | S81 | 88 |
| R.GEGAQDEEEGGASSDATEGHDEDDEIYEGEYQGIPR.A + 2 P (ST) | 4000.3966 | 60 | S81 and either of S80 or T84 | 53, 92, 53 |
| R.GEGAQDEEEGGASSDATEGHDEDDEIYEGEYQGIPR.A + 3 P (ST) | 4080.3698 | 66 | S80, S81, T84 | 100, 100, 100 |
Endogenous SV2A was immunoprecipitated from mouse brain extract (8 mg). The immunoprecipitates were electrophoresed on a polyacrylamide gel, and after Colloidal blue staining, bands corresponding to SV2A were excised and digested with trypsin. Phosphorylated peptides were identified by LC-MS/MS analysis. The data analysis was as in Materials and Methods. The sequence of each peptide is shown with possible phosphorylation sites underlined. Also shown are the mass of the peptide, the Mascot score [of the peptide identification, in which individual ion scores >21 indicate identity or extensive homology (p < 0.05)], and the percentage probability of the phosphosite assignment, as determined by the phosphoRS 3.1 component of the Proteome Discoverer 1.4 software.
Table 2.
Identification of phosphorylation sites on wild-type and mutant SV2A overexpressed in HEK293 cells
| Peptides identified by mass spectrometry | Mass | Mascot score | Possible P sites | % Probability (PD1.4 pRS3.1) |
|---|---|---|---|---|
| SV2A WT | ||||
| 150–250 kDa | ||||
| R.GGLSDGEGPPGGR.G + P (ST) | 1234.4970 | 79 | S127 | 100 |
| K.TIHQEDELIEIQSDTGTWYQR.W + P (ST) | 2641.1762 | 21 | S411 | 99 |
| R.SYSRFEEEDDDDDFPAPSDGYYR.G + P (ST) | 2854.0594 | 42 | Either S45 or Y46 or S47 | 33, 33, 33 |
| R.GEGTQDEEEGGASSDATEGHDEDDEIYEGEYQGIPR.A + P (ST) | 3950.5028 | 72 | Either S80 or S81 | 49, 49 |
| R.GEGTQDEEEGGASSDATEGHDEDDEIYEGEYQGIPR.A + 2 P (ST) | 4030.4774 | 54 | S80, T84, (S81) | 86, 97, (16) |
| R.GEGTQDEEEGGASSDATEGHDEDDEIYEGEYQGIPR.A + 3 P (ST) | 4110.4389 | 47 | S80, S81, T84 | 100, 100, 100 |
| 75–150 kDa | ||||
| R.VFSVTHIK.T + P (ST) | 1009.4900 | 30 | Either S393 or T395 | 50, 50 |
| R.GGLSDGEGPPGGR.G + P (ST) | 1234.4972 | 83 | S127 | 100 |
| K.TIHQEDELIEIQSDTGTWYQR.W + P (ST) | 2641.1706 | 36 | S411 | 99 |
| R.SYSRFEEEDDDDDFPAPSDGYYR.G + P (ST) | 2854.0568 | 47 | Either S45 or Y46 or S47 | 33, 33, 33 |
| R.RSYSRFEEEDDDDDFPAPSDGYYR.G + P (ST) | 3010.1636 | 76 | S45 | 99 |
| R.GEGTQDEEEGGASSDATEGHDEDDEIYEGEYQGIPR.A + P (ST) | 3950.5028 | 101 | S81, (S80) | 84, (14) |
| R.GEGTQDEEEGGASSDATEGHDEDDEIYEGEYQGIPR.A + 2 P (ST) | 4030.4763 | 58 | Either of S80, S81 and T84 | 53, 53, 92 |
| R.GEGTQDEEEGGASSDATEGHDEDDEIYEGEYQGIPR.A + 3 P (STY) | 4110.4447 | 60 | S80, S81, T84 | 100, 100, 100 |
| R.GEGTQDEEEGGASSDATEGHDEDDEIYEGEYQGIPR.A + 4 P (STY) | 4190.4062 | 26 | T71, S80, S81 | 100, 100, 100, 100 |
| SV2A T84A | ||||
| 150–250 kDa | ||||
| R.GGLSDGEGPPGGR.G + P (ST) | 1234.4978 | 63 | S127 | 100 |
| R.SYSRFEEEDDDDDFPAPSDGYYR.G + P (ST) | 2854.0609 | 24 | Either S45 or Y46 or S47 | 33, 33, 33 |
| R.GEGTQDEEEGGASSDAAEGHDEDDEIYEGEYQGIPR.A + P (STY) | 3920.4970 | 64 | Either S80 or S81 | 50, 50 |
| R.GEGTQDEEEGGASSDAAEGHDEDDEIYEGEYQGIPR.A + 2 P (STY) | 4000.4665 | 56 | S80, S81 | 100, 100 |
| R.GEGTQDEEEGGASSDAAEGHDEDDEIYEGEYQGIPR.A + 3 P (STY) | 4080.4425 | 45 | T71, S80, S81, T84 | 100, 100, 100 |
| 75–150 kDa | ||||
| R.VFSVTHIK.T + P (ST) | 1009.4989 | 22 | Either S393 or T395 | 50, 50 |
| R.GGLSDGEGPPGGR.G + P (ST) | 1234.4968 | 56 | S127 | 100 |
| R.SYSRFEEEDDDDDFPAPSDGYYR.G + P (ST) | 2854.0609 | 45 | Either S45 or Y46 or S47 | 33, 33, 33 |
| R.RSYSRFEEEDDDDDFPAPSDGYYR.G + P (ST) | 3010.1631 | 51 | S45 | 99 |
| R.GEGTQDEEEGGASSDAAEGHDEDDEIYEGEYQGIPR.A + P (ST) | 3920.5014 | 90 | S81, (S80) | 87, (13) |
| R.GEGTQDEEEGGASSDAAEGHDEDDEIYEGEYQGIPR.A + 2 P (STY) | 4000.4705 | 58 | S80, S81 | 100, 100 |
| R.GEGTQDEEEGGASSDAAEGHDEDDEIYEGEYQGIPR.A + 3 P (STY) | 4080.4261 | 57 | T71, S80, S81 | 100, 100, 100 |
| SV2A S80, S81A | ||||
| 150–250 kDa | ||||
| R.GGLSDGEGPPGGR.G + P (ST) | 1234.4972 | 57 | S127 | 100 |
| K.TIHQEDELIEIQSDTGTWYQR.W + P (ST) | 2641.1698 | 37 | S411 | 100 |
| R.SYSRFEEEDDDDDFPAPSDGYYR.G + P (ST) | 2854.0585 | 46 | Either S45 or Y46 or S47 | 33, 33, 33 |
| 75–150 kDa | ||||
| R.GGLSDGEGPPGGR.G + P (ST) | 1234.4972 | 60 | S127 | 100 |
| K.TIHQEDELIEIQSDTGTWYQR.W + P (ST) | 2641.1697 | 57 | S411 | 100 |
| R.SYSRFEEEDDDDDFPAPSDGYYR.G + P (ST) | 2854.0555 | 69 | Either S45 or Y46 or S47 | 33, 33, 33 |
HEK293 cells were transfected with a construct encoding full-length WT or indicated mutant of Flag–SV2A. Thirty-six hours after transfection, cells were lysed and Flag–SV2A immunoprecipitated from 4 mg of cell extract, and the immunoprecipitates were electrophoresed on a polyacrylamide gel. After colloidal Coomassie blue staining, the bands corresponding to SV2A were excised and digested with trypsin. Phosphorylated peptides were identified by LC-MS/MS analysis. The data analysis was as in Materials and Methods. The sequence of each peptide is shown with possible phosphorylation sites underlined. Also shown are the mass of the peptide, the Mascot score [of the peptide identification, in which individual ion scores >17 indicate identity or extensive homology (p < 0.05)], and the percentage probability of the phosphosite assignment, as determined by the phosphoRS 3.1 component of the Proteome Discoverer 1.4 software.
Isothermal titration calorimetry.
Binding of SV2A peptides to synaptotagmin-1 was assayed by isothermal titration calorimetry (ITC) using a VP-ITC microcalorimeter (MicroCal). Unphosphorylated or phosphorylated peptides (0.8 mm) of SV2A (residues 76–90) were titrated into synaptotagmin-1 C2B domain (residues 270–422, 0.04 mm), and enthalpic changes for each injection (5 μl) were recorded while stirring at 300 rpm at a constant temperature of 25°C for 180 s. Injections were performed until a molar peptide/protein ratio of at least 2.5:1 was reached. The integrated binding isotherms for each injection were obtained from the enthalpic changes versus the ratios between peptide concentration and total protein concentration using data analysis software Origin 7.0 (including MicroCal add-on; OriginLab). Single or multiple independent site models were then fitted to the data by nonlinear regression in Origin 7.0 with the MicroCal ITC patch installed. The model with the lowest χ2 fit by least-squares analysis was used to obtain the Kd apparent. The first injection (2 μl) was excluded from the analysis. In all cases, an independent single sites model provided the best fit with an overall stoichiometry of 1 ± 0.3.
Protein purification and crystallization of synaptotagmin-1 C2B domain.
Recombinant human synaptotagmin-1 C2B domain (residues 270–422) was overexpressed in E. coli BL21 (DE3; Studier and Moffatt, 1986) with an N-terminal GST tag and TEV-protease cleavage site from construct DU number 44151 (MRC–PPU). Bacterial cells were grown in 50 ml of Luria–Bertani (LB) broth supplemented with 100 mg/ml ampicillin (Amp) at 37°C for 16 h and subsequently used to inoculate 950 ml of LB broth (with Amp) in baffled 2 L flasks. Cultures were grown at 37°C for ∼5 h with vigorous shaking at 180 rpm until an optical density of ∼0.6 at 600 nm was reached, and overexpression was subsequently induced with 0.25 mm isopropyl β-d-1-thiogalactopyranoside at 19°C for 16 h. Bacterial cells were harvested by centrifugation at 5000 × g and 4°C for 15 min, and the cell pellet was resuspended in 50 mm HEPES, pH7.5, and 250 mm NaCl, supplemented with protease inhibitor cocktail (Calbiochem). Cells were lysed by freeze–thawing at −80°C, followed by sonication on ice, and the cell extract was cleared from intact cells and debris by centrifugation at 30,000 × g and 4°C for 20 min.
GST-tagged protein was purified by affinity chromatography in a 16/20 XK column (GE Healthcare) packed with 10 ml of glutathione Sepharose 4B (GE Healthcare) in 50 mm HEPES, pH 7.5, and 250 mm NaCl, and bound protein was eluted with 40 mm reduced glutathione, 50 mm HEPES, pH 7.5, and 250 mm NaCl. Purified protein was cleaved with TEV protease (DU6811; MRC–PPU) in 50 mm HEPES, pH 7.5, 250 mm NaCl, and 40 mm reduced glutathione on ice overnight and subsequently desalted and eluted into 50 mm HEPES, pH 7.5, using a HiPrep 26/10 desalting column (GE Healthcare). Cleaved protein was further purified by ion exchange chromatography using a 16/20 XK column packed with 10 ml of Source 15S matrix (GE Healthcare), which was equilibrated in 50 mm HEPES, pH 7.5, and bound protein was eluted in a linear concentration gradient from 0 to 50% in five column volumes of 50 mm HEPES, pH 7.5, and 1 m NaCl. Fractions containing cleaved synaptotagmin-1 C2B domain fragment were finally purified by size exclusion chromatography in a HiLoad 16/600 Superdex 200 PG column (GE Healthcare) in 50 mm HEPES, pH 7.5, and 50 mm NaCl. Protein purity and quality was monitored by SDS-PAGE, followed by staining with Coomassie Brilliant Blue, and protein concentration was estimated with a NanoDrop photospectrometer (Thermo Fisher Scientific) using the theoretical extinction coefficient and molecular mass of the protein sequence using the PROTPARAM web server (Gasteiger et al., 2005).
Purified human synaptotagmin-1 C2B domain (residues 270–422) was crystallized with the synthetic peptide of SV2A (residues 76–90), which was phosphorylated at Thr84. Commercially available crystallization screens were used to find preliminary conditions for protein crystallization, and a Mosquito Crystal liquid handling robot (TTP Labtech) was used to spot 0.2 μl of protein/peptide solutions (molar ratio of 1:3) and 0.2 μl of reservoir solution into 96-well MRC plates (Hampton Research). Initial protein crystals were obtained by sitting drop vapor diffusion at 291 K in Index HT, Crystal Screen HT, and PEG/Ion HT crystallization screens (Hampton Research) and were subsequently manually optimized. Final crystallization using hanging-drop vapor diffusion at 291 K was set up by co-crystallizing 0.5 mm synaptotagmin-1 C2B with 1.4 mm SV2A peptide [EGGASSDA(pT)EGHDED], which was combined with reservoir solution consisting of 0.1 m HEPES, pH 7.5, 18% polyethylene glycol (PEG) 3350, and 50 mm CaCl2 in a 1:1 ratio.
x-Ray data collection and structure determination and refinement crystals were soaked in 25% glycerol, 20% PEG 3350, 100 mm, and 50 mm CaCl2 for 5 min and flash frozen in liquid nitrogen. Diffraction data was obtained at 100 K using a Rigaku MicroMax-007 rotating anode x-ray generator (1.54 A(Angstrom)) coupled to a Saturn 944 CCD detector and processed using the XDS program package (Evans, 2006). Diffraction images of two reflection sets from one crystal were scaled and merged using Aimless version 0.2.17, and Laue groups were determined with Pointless version 1.8.17 (Evans and Murshudov, 2013) within the CCP4 program suite (Kabsch, 2010). The crystals were packed in the monoclinic space group P1211 and crystallized to 1.87 Å resolution, which was cut to 1.95 Å to obtain >90% completeness for the outer shell.
The synaptotagmin-1 C2B domain structure was solved using PHENIX Phaser version 2.5.5 (McCoy et al., 2007) by molecular replacement with Protein Data Bank (PDB) entry 1TJX (Cheng et al., 2004) as input template. The model was refined by iterative operations using PHENIX Refine version 1.8.3 (Adams et al., 2010) in combination with manual model building in COOT (Emsley and Cowtan, 2004). Residues 81–90 of the SV2A peptide and solvent molecules were added manually to the model according to electron- and difference-density maps. A difference map of the Apo protein of Syt1 was created in Phenix Refine after initial refinements without the SV2A peptide chain included in the model. Illustrations were prepared with PyMol (http://www.pymol.org/), and electrostatics representations were calculated using the APBS plugin (Baker et al., 2001).
Fluorescence polarization.
Binding of SV2A peptide phosphorylated at residue Thr84 to synaptotagin-1 C2B domain mutants with lysine-to-alanine substitutions K314A, K326A, K328A, or K326A/K328A was tested by fluorescence polarization measurements. SV2A peptides corresponding to residues 76–90 were designed with a N-terminal tetra-cysteine motif and either synthesized without phosphorylation (CCPGCCGGGEGGASSDATEGHDED) or phosphorylated at Thr84 [CCPGCCGGGEGGASSDA(pT)EGHDED] (Pepceuticals). Peptides were labeled with FlAsH–EDT2 [4′,5′-bis(1,3,2-dithioarsolan-2-yl)fluorescein-(1,2-ethanedithiol); Griffin et al., 1998] at the tetra-cysteine motif by incubating 0.02 mm peptide with 0.04 mm FlAsH–EDT2 in a final volume of 0.5 ml of 50 mm HEPES–NaOH, pH 7.5, and 50 mm NaCl at room temperature for 2 h. Labeled peptides were dialyzed against 50 mm HEPES–NaOH, pH 7.5, and 50 mm NaCl at 4°C for 24 h and protected from light.
For fluorescence polarization assays, 0–20 μm protein of synaptotagmin-1 C2B domain (residues 270–422) was titrated into 0.1 μm FlAsH-labeled peptide in 30 μl of 50 mm HEPES–NaOH, pH 7.5, and 50 mm NaCl in black 384-well assay plates (Corning). Fluorescence polarization was monitored at 485 nm excitation and 520 nm emission using a Pherastar FS plate reader (BMG Labtech) and plotted against synaptotagmin-1 C2B concentration. A nonlinear one-site binding model with Hill slope was fitted to the data points using analysis software Prism 6 (GraphPad Software).
Hippocampal neuronal cultures.
Dissociated primary hippocampal enriched neuronal cultures were prepared from both male and female E17.5 C56BL/6J mouse embryos by trituration of isolated hippocampi to obtain a single-cell suspension, which was plated at a density of 5 × 105 cells per coverslip on poly-d-lysine and laminin-coated 25 mm coverslips. Cultures were maintained in Neurobasal media supplemented with B-27, 0.5 mm l-glutamine, and 1% v/v penicillin/streptomycin. After 72 h, cultures were further supplemented with 1 μm cytosine β-d-arabinofuranoside to inhibit glial proliferation. Cells were transfected after 7–8 d in culture with Lipofectamine 2000 as described previously (Gordon et al., 2011). In all experiments, two constructs were coexpressed; either Syt1–pHluorin or synaptophysin–pHluorin was cotransfected with either mCer empty vector or mCer–pSUPER shRNA vectors. In addition, mCer–SV2A was coexpressed with a pSUPER shRNA vector, which expressed Syt1–pHluorin. Neurons were imaged after 13–16 d in culture.
Immunolabeling.
Immunolabeling of transfected cultured hippocampal neurons was performed as described previously (Gordon et al., 2011). Syt1–pHluorin expression was visualized at 480 nm, whereas SV2A expression was visualized at 550 nm. Identically sized regions of interest were placed over transfected Syt1–pHluorin puncta and nontransfected puncta in the same field of view, along with background regions. The level of SV2A expression was calculated by subtracting background autofluorescence before rationing transfected/nontransfected SV2A expression levels.
Fluorescent imaging of pHluorin trafficking.
Hippocampal cultures were mounted in a Warner imaging chamber with embedded parallel platinum wires (RC-21BRFS) and placed on the stage of Zeiss Axio Observer D1 epifluorescence microscope. Neurons transfected with mCer vectors were visualized with a Zeiss Plan Apochromat 40× oil-immersion objective (numerical aperture 1.3) at 430 nm excitation, whereas pHluorin reporters were visualized at 500 nm (both using a dichroic filter >525 nm and long-pass emission filter >535 nm). Cultures were stimulated with a train of 300 action potentials delivered at 10 Hz (100 mA, 1 ms pulse width). Cultures were subjected to continuous perfusion at room temperature (23°C) with imaging buffer (136 mm NaCl, 2.5 mm KCl, 2 mm CaCl2, 1.3 mm MgCl2, 10 mm glucose, and 10 mm HEPES, pH 7.4, supplemented with 10 μm 6-cyano-7-nitroquinoxaline-2,3-dione and 50 μm dl-2-amino-5-phosphonopentanoic acid). After stimulation, cultures were then challenged with alkaline imaging buffer (50 mm NH4Cl substituted for 50 mm NaCl) to reveal total pHluorin fluorescence. Fluorescent images were captured at 4 s intervals using a Hamamatsu Orca-ER digital camera and processed offline using NIH Image J 1.43 software. Regions of interest of identical size were placed over nerve terminals, and the total fluorescence intensity was monitored over time. Only regions that responded to action potential stimulation were selected for analysis. All statistical analyses were performed using Microsoft Excel and Prism software (GraphPad Software). The pHluorin fluorescence change was calculated as ΔF/F0.
Surface fraction of Syt1–pHluorin.
Estimation of surface Syt1–pHluorin was performed by perfusing acidic imaging buffer (substituting 20 mm MES for HEPES, pH 5.5) over cultures for 30 s (to quench surface Syt1–pHluorin), followed by a 1 min perfusion with imaging buffer, pH 7.4. Cultures were then subjected to alkaline imaging buffer for 30 s to reveal total Syt1–pHluorin. The surface fraction of Syt1–pHluorin as a percentage of total was estimated using the following equation: (neutral fluorescence − acidic fluorescence/alkali fluorescence − acidic fluorescence) × 100.
Statistical analysis.
In all cases, n refers to the number of independent experiments performed. All statistical analyses were performed using Prism software (GraphPad Software), and all data were tested by Student's t test with the exception of pHluorin time course experiments (two-way ANOVA) and Syt1–pHluorin surface expression during SV2A knockdown (one-way ANOVA).
Results
CK1 family kinases phosphorylate SV2A at two clusters of sites
The CK1 group of eukaryotic protein kinases are composed of seven CK1 isoforms (CK1α, CK1α2, CK1δ, CK1ε, CK1γ1, CK1γ2, and CK1γ3), two Tau–tubulin kinase isoforms (TTBK1 and TTBK2), and three VRK isoforms (VRK1, VRK2, and VRK3; Manning et al., 2002). We compared the ability of CK1 isoforms (CK1α1, CK1δ, CK1ε, and CK1γ1), TTBK isoforms (TTBK1 and TTBK2), and VRK1 to phosphorylate a fragment of SV2A[1–160] that encompasses the cytoplasmic domain that previous work has suggested represents the region that CK1 phosphorylates (Pyle et al., 2000). Moreover, as mentioned in the Introduction, global phosphorylation site analysis has indicated that SV2A is phosphorylated at numerous sites within its N-terminal 160 residue domain (http://www.phosphosite.org/proteinAction.do?id=8509&showAllSites=true). We observed that, under conditions used, all CK1 and TTBK2 isoforms tested phosphorylated SV2A[1–160] to different extents (Fig. 1). A recombinant fragment of TTBK2 encompassing its catalytic domain (residues 1–316) phosphorylated SV2A[1–160] to the highest stoichiometry, followed by CK1ε, TTBK1, and CK1δ. CK1α1 and CK1γ1 phosphorylated SV2A[1–160] >10-fold less efficiently than TTBK2[1–316] (Fig. 1). Recombinant VRK1 failed to phosphorylate SV2A[1–160] in parallel experiments, although it was judged active because it was capable of autophosphorylation (Fig. 1).
Figure 1.

CK1 family members phosphorylate the N-terminal cytoplasmic domain of SV2A. The indicated CK1 family kinases (TTBK2[1–316], TTBK1[1–1321, full length], VRK1[1–396, full length], CK1α1[1–337, full length], CK1δ[1–415, full length], CK1ε[1–416, full length], and CK1γ1[1–422, full length]) were all expressed in E. coli with the indicated N-terminal GST or MBP tag. The kinases were incubated at a final concentration of 0.5 μm with 2 mm Mg2+, 0.1 mm γ-32P[ATP], and 5 μm WT SV2A[1–160] or indicated SV2A mutants in which either Cluster-1 (Ser42, Ser45, and Ser47; termed C1 Mutant) and/or Cluster-2 (Ser80, Ser81, and Thr84; termed C2 Mutant) phosphorylation sites were mutated to Ala to prevent phosphorylation. Reactions were left to proceed for 30 min at 30°C, subjected to electrophoresis on a polyacrylamide gel, stained with colloidal Coomassie blue, and autoradiographed. The Coomassie blue-stained SV2A[1–160] band was excised, and 32P incorporation was assessed by Cerenkov counting. Relative phosphorylation of WT SV2A[1–160] by CK1 family kinases is indicated in the table. Reactions were also subjected to immunoblot analysis with the indicated phospho-specific antibodies that recognize phosphorylated Cluster-1 or Cluster-2 sites. All data are representative of at least two independent experiments.
To map the phosphorylation sites on SV2A, we used full-length SV2A isolated from a HEK293 cell overexpression system, using TTBK2[1–316] that our in vitro studies suggested was the most efficient CK1 family kinase at phosphorylating SV2A in vitro. Consistent with data shown in Figure 1, we found that WT but not a kinase-inactive fragment of TTBK2 phosphorylated full-length SV2A (Fig. 2A). TTBK2[1–316] 32P-phosphorylated full-length SV2A was digested with trypsin, and the resultant tryptic peptides were analyzed by chromatography on a C18 column (Fig. 2B). The major 32P-labeled phosphopeptides were subjected to mass spectrometry and solid-phase Edman sequencing to identify the phosphorylation sites. This analysis resulted in the identification of 8 phosphorylation sites (Ser42, Ser45, Ser47, Ser62, Ser80, Ser81, Thr84 and Ser127; Fig. 2C). These phosphorylation sites are all located within the N-terminal 160 residue cytoplasmic domain of SV2A (Fig. 2D). Sequence alignment indicates that the evolutionary conserved CK1 phosphorylation sites on SV2A lie within two constellations that we have termed here Cluster-1 (Ser42, Ser45, and Ser47) and Cluster-2 (Ser80, Ser81, and Thr84). The nonconserved Ser62 and Ser127 residues lie outside these clusters (Fig. 2E). Mutation of all eight phosphorylation sites on the full-length SV2A abolished phosphorylation by TTBK2[1–316] (Fig. 2A). We were also able to monitor phosphorylation of SV2A by TTBK2[1–316] using phosphospecific antibodies that we generated that recognize SV2A after phosphorylation of either Cluster-1 or Cluster-2 residues (Fig. 2A). This revealed that full-length SV2A isolated from HEK293 cells was detectably phosphorylated at Cluster-2 but not Cluster-1. After incubation with WT but not kinase-inactive TTBK2[1–316], phosphorylation of Cluster-1 sites became detectable and phosphorylation of Cluster-2 sites markedly increased.
Figure 2.
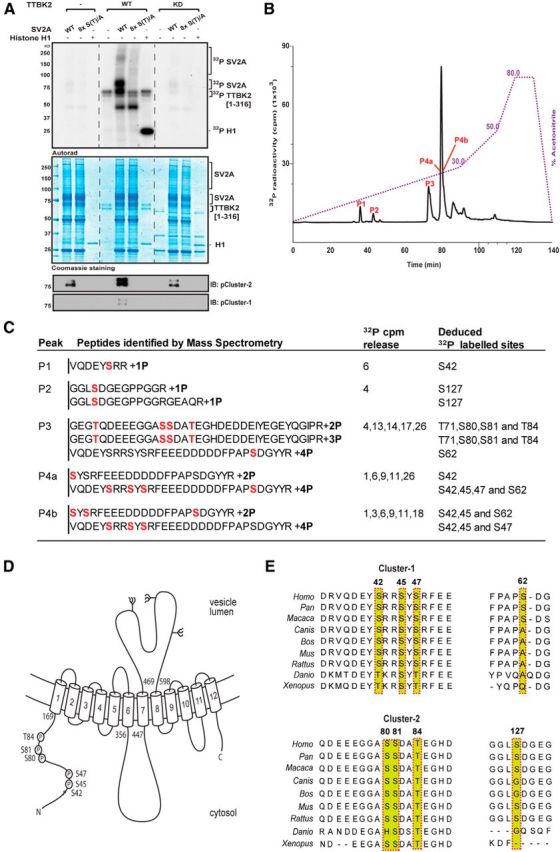
Identification of TTBK2 phosphorylation sites on SV2A. A, HEK293 cells were transfected with a construct encoding WT Flag–SV2A or a mutant Flag–SV2A in which all eight phosphorylation sites identified in C, namely Ser42, Ser45, Ser47, Ser62, Ser80, Ser81, Thr84, and Ser127, are mutated to Ala [termed 8× S(T)/A]. Thirty-six hours after transfection, cells were lysed, and WT and mutant Flag–SV2A was immunoprecipitated and incubated with recombinant WT TTBK2[1–316] or kinase inactive (KD) TTBK2[D163A, 1–316] and Mg2–[-32P]ATP for 60 min. Samples were subjected to electrophoresis on a polyacrylamide gel that was stained with colloidal Coomassie blue and autoradiographed. Reactions were also subjected to immunoblot analysis with the indicated phospho-specific antibodies that recognize phosphorylated Cluster-1 or Cluster-2 sites. It should be noted that a low level of unidentified kinase(s) copurifies with Flag–SV2A from HEK293 cells, which phosphorylates it to a low level in the absence of TTBK2. B, Full-length Flag–SV2A was incubated with WT TTBK2[1–316] in the presence of Mg2+–[γ-32P]ATP for 60 min as in A. Phosphorylated SV2A was digested with trypsin, and peptides were separated by reverse-phase HPLC on a C18 column. The peaks containing the 32P-labeled phosphopeptides are marked as in the subdivisions of C. C, Summary of the mass spectrometry and solid-phase Edman sequencing data obtained after analysis of the peak fractions. The deduced amino acid sequence of each peptide is shown, and the deduced 32P-labeled site is indicated in red. Note that, as SV2A was isolated from HEK293 cells, certain peptides are pre-phosphorylated by endogenous kinase(s). D, Domain structure of SV2A with the position of eight residues phosphorylated by TTBK2 indicated. All numbering refers to the human sequence. E, Sequence alignment of the residues of SV2A that encompass the TTBK2 phosphorylation sites from the indicated species. Residues highlighted indicated in red comprise the phosphorylation sites.
We also compared the ability of TTBK and CK1 isoforms to phosphorylate mutants of SV2A[1–160] in which Cluster-1 (Ser42, Ser45, and Ser47) and/or Cluster-2 (Ser80, Ser81, and Thr84) residues are mutated to Ala to ablate phosphorylation. These studies revealed that all CK1 family kinases tested phosphorylated SV2A[1–160] at both Cluster-1 and Cluster-2 residues to differing extents, because phosphorylation of SV2A[1–160] was significantly suppressed after mutation of either Cluster-1 or Cluster-2 (Fig. 1). Moreover, mutation of both Cluster-1 and Cluster-2 residues virtually abolished phosphorylation of SV2A by this group of kinases, indicating that these sites do indeed comprise the major phosphorylation sites by the CK1 family. We were also able to monitor phosphorylation of SV2A by CK1 and TTBK isoforms that phosphorylated SV2A to the highest stoichiometry by using phosphospecific antibodies that we generated that recognize SV2A after phosphorylation of either Cluster-1 or Cluster-2 residues (Fig. 1).
Phosphorylation of SV2A at Ser80/Ser81 and Thr84 in vivo
We next analyzed the phosphorylation of SV2A in vivo. Endogenous SV2A immunoprecipitated from mouse brain was resolved in SDS-PAGE (data not shown) from which two major bands corresponding to SV2A were excised and subjected to mass spectrometry analysis (one band migrated beyond 150 kDa maker, whereas the other was between the 75 and 100 kDa markers). Peptides matching mouse SV2A sequence were identified from both excised bands, indicating these two distinguishable bands from SDS-PAGE correspond to SV2A. The significant mass difference between the product ions from modified and unmodified peptides (data not shown) allowed us to identify the phosphorylated peptides from SV2A (Table 1). Other than Ser393, all of the identified phosphorylation sites (Table 1) accumulated in the N-terminal cytoplasmic region (residue 1–169) of SV2A, predominantly in the Cluster-2 phosphorylation sites (Fig. 1).
CK1 isoforms are known to preferentially phosphorylate a Ser/Thr residue in a protein that is “primed” by previous phosphorylation at the −3 position (corresponding to Ser81 relative to Thr84 in SV2A; Flotow et al., 1990). In addition, a −4 position acidic or phosphorylated residue (corresponding to Ser80 relative to Thr84 in SV2A) has also been reported to facilitate phosphorylation (Meggio et al., 1991). Therefore, it is possible that phosphorylation of Ser80/Ser81 might prime phosphorylation of Thr84. To test this possibility in vivo, we overexpressed WT or Ser/Thr to Ala mutant SV2A in HEK293 cells and assessed their phosphorylation state. As shown in Table 2, similar to what was observed in mouse brain extract, WT SV2A was phosphorylated at the Cluster-2 sites (Ser81, Ser82, and Thr84; Table 2), validating the utility of this overexpression system for studying SV2A phosphorylation in vivo. More importantly, no phosphorylated peptide covering Thr84 was identified in the absence of phosphorylation on either Ser80 or Ser81, whereas absence of phosphorylation of Thr84 had no significant effect on the phosphorylation of Ser80 and Ser81. Mutation of Ser80 and Ser81 to Ala also prevented phosphorylation of Thr84.
Cluster-2 phosphorylation promotes the ability of SV2A to bind synaptotagmin-1
To explore whether CK1-mediated phosphorylation of SV2A directly controlled its association with other protein(s), we generated biotinylated peptides that encompassed either the dephosphorylated or triply phosphorylated Cluster-1 (Ser42, Ser45, and Ser47) or Cluster-2 (Ser80, Ser81, and Thr84) residues of SV2A. These were conjugated to streptavidin–Sepharose and used in affinity purification studies to identify protein(s) that associated with the phosphorylated but not dephosphorylated peptides in mouse brain extract. Mass spectrometry analysis revealed that the major hit that associated with the phosphorylated peptide to a much greater extent than the dephosphorylated Cluster-2 peptide are the highly related synaptotagmin-1 and synaptotagmin-2 proteins. As mentioned in the Introduction, synaptotagmin-1 interacts with SV2A through its N-terminal domain (Schivell et al., 1996, 2005; Lazzell et al., 2004; Fig. 3A). Immunoblot analysis confirmed that synaptotagmin-1 interacted with the Cluster-2 phosphorylated peptide but not with the dephosphorylated peptide (Fig. 3B). No clear-cut specific interactors to the Cluster-1 peptide were identified in parallel experiments (data not shown).
Figure 3.

Phosphorylation of SV2A at Cluster-2 promotes the interaction with synaptotagmin-1. Biotinylated dephosphorylated or triply phosphorylated (Ser80, Ser81, and Thr84) peptides that encompassed the Cluster-2 region of SV2A were conjugated to streptavidin–Sepharose and incubated in mouse brain extract. Peptide–streptavidin–Sepharose conjugates were washed, electrophoresed, and analyzed by mass spectrometry (A) or immunoblot analysis with the indicated antibody (B). In A, proteins that were not present in the control biotin sample are listed. We also excluded three proteins (40S protein S19, 40S ribosomal protein S7, and elongation factor 1-α 1) that are known frequent contaminants of mass spectrometry experiments (Mellacheruvu et al., 2013). MW, Molecular weight; SN, supernatant; PD, pull-down. C, Endogenous SV2A was immunoprecipitated from mouse brain extract, and the immunoprecipitates were treated in the presence (+) or absence (−) of lambda phosphatase and/or EDTA that inhibits the activity of lambda phosphatase. The immunoprecipitates were washed and subjected to immunoblot analysis with the indicated antibodies. IP, Immunoprecipitated; IB, immunoblot. D, Schematic representation of the domain architecture of human synaptotagmin-1. To establish which region of synaptotagmin-1 interacts with the phosphorylated Cluster-2 peptide, we expressed in E. coli the indicated fragments of synaptotagmin-1. These proteins were incubated with biotinylated dephosphorylated or triply phosphorylated peptides used in A conjugated to streptavidin–Sepharose. The peptide–streptavidin–Sepharose conjugates were washed, electrophoresed, and subjected to immunoblot analysis with an anti-GST antibody. Similar results were obtained in at least two separate experiments for all data shown in this figure.
To obtain additional evidence that the interaction between SV2A and synaptotagmin-1 was promoted by phosphorylation, we immunoprecipitated SV2A from mouse brain and treated the immunoprecipitates in the presence or absence of lambda phosphatase. This revealed that treatment with lambda phosphatase lowered the level of synaptotagmin-1 associated with immunoprecipitated SV2A, and this effect was blocked by the addition of EDTA, which inhibits lambda phosphatase catalytic activity (Fig. 3C).
Phosphorylated Cluster-2 SV2A peptide interacts with synaptotagmin-1 C2B domain
Human synaptotagmin-1 is a 422 residue protein possessing a transmembrane membrane region at the N terminus (residues 58–80) that anchors it to SV membranes, which is followed by two Ca2+-binding C2 domains termed C2A (residues 157–245) and C2B (residues 287–382). Synaptotagmin-1 operates as the key Ca2+ sensor in SV trafficking and neurotransmitter release at the synapse (Brose et al., 1992; Geppert et al., 1994). To establish which region of synaptotagmin-1 interacts with the phosphorylated Cluster-2 peptide, we expressed fragments of synaptotagmin-1 and tested their ability to interact with biotinylated phosphorylated Cluster-2 peptide. This revealed that a fragment encompassing isolated C2B domain interacted robustly with the phosphorylated Cluster-2 peptide under conditions in which fragments lacking the C2B domain failed to bind (Fig. 3D).
We next performed isothermal calorimetric-binding analysis to assess the interaction of the isolated synaptotagmin-1 C2B domain (residues 270–422) with different combinations of Cluster-2 phosphorylated residues (Ser80, Ser81, and Thr84; Fig. 4). This revealed that the key phosphorylation site mediating binding between the SV2A C2B domain and synaptotagmin-1 was Thr84. The Cluster-2 peptide phosphorylated only at Thr84 interacted with synaptotagmin-1 C2B domain with high affinity (Kd of ∼300 nm), whereas all peptides phosphorylated at Ser80 and/or Ser81, but not Thr84, failed to bind to the synaptotagmin-1 C2B domain. SV2A peptides phosphorylated at Thr84 as well as Ser80 and/or Ser81 also interacted with the C2B domain of synaptotagmin-1, but the binding affinities were lower than with the peptide phosphorylated at Thr84 alone (Fig. 4). In addition, phosphorylation at Ser81 together with phosphorylated Thr84 seems to decrease the enthalpic nature of the interaction, and this may indicate a destabilizing effect on binding of the phosphorylation at Ser81. This destabilizing effect was also reflected in an increased binding entropy of peptides phosphorylated at pSer81 + pThr84 and pSer80 + pSer81 + pThr84, which was estimated at ∼20 cal · mol−1 · K−1. We also calculated the dissociation constant of the interactions, and the peptide phosphorylated at pSer81 + pThr84 had a higher Kd of ∼900 nm than the peptide phosphorylated at pSer80 + pThr84. However, it should be noted that the increased dependency on entropy in the interaction between synaptotagmin-1 and peptides containing phosphorylated Ser81 increased the signal-to-noise ratio and hence error in the calculated Kd. Thus, phosphorylation of SV2A on Thr84 is both necessary and sufficient for its interaction with the C2B domain of synaptotagmin.
Figure 4.

Phosphorylation of Thr84 of SV2A is critical in promoting the interaction with synaptotagmin-1. Quantitative ITC-binding assay to assess the interaction of the isolated synaptotagmin-1 C2B domain (residues 270–422) with a peptide encompassing the Cluster-2 region of SV2A (residues 76–90) in which combinations of Ser80, Ser81, and Thr84 were phosphorylated as indicated. SV2A peptides (0.8 mm), with phosphorylated residues as indicated on top of each panel, were titrated into purified synaptotagmin-1 C2B domain (0.04 mm) at 25°C using a VP-ITC microcaloriter (MicroCal). The titration thermogram representing enthalpic heat release in microcalories per second is shown at the top, and the integrated binding isotherms are shown at the bottom of each panel. When applicable, the best-fit curve, reaction stoichiometry (N), and dissociation constant (Kd) were calculated. Similar results were obtained in at least two separate experiments for all data shown in this figure.
Structure of Cluster-2 phosphorylated peptide bound to synaptotagmin-1 C2B domain
To probe the mechanism by which phosphorylated SV2A binds to synaptotagmin-1, we crystallized the Thr84 phosphorylated Cluster-2 peptide (residues 76–90) with the C2B domain (residues 270–422) of synaptotagmin-1 (PDB entry 4v11; for crystallographic statistics, see Table 3). This revealed that the C2B domain adopted the same overall topology as the previously described nuclear magnetic resonance (NMR; Fernandez et al., 2001) and x-ray crystallography (Cheng et al., 2004; Honigmann et al., 2013) structures, composed of two layers of antiparallel β sheets, along with two short α helices (Fig. 5A). Electron density of the Thr84 phosphorylated Cluster-2 peptide from residues 81–90 was clearly resolved, interacting with a positively charged surface region of synaptotagmin-1 that has been referred to as the “polybasic region” (Fernandez et al., 2001; Fig. 5B). The phosphorylated Thr84 residue formed hydrogen bonds with three highly conserved surface-exposed Lys residues on the synaptotagmin-1 C2B domain (Lys314, Lys326, and Lys328; Fig. 5C). These Lys residues are distributed on two anti-parallel β sheets and form a specific binding pocket that precisely accommodates the phosphorylated Thr84 residue (Fig. 5C). The importance of these residues is emphasized by the finding that individual mutations of Lys314, Lys326, or Lys328 inhibited binding of the Cluster-2 Thr84 phosphorylated peptide to the synaptotagmin-1 C2B domain at least sixfold, with mutation of Lys328 having the largest effect (Fig. 5D). Mutation of Lys326 and Lys328 together reduced binding of the SV2A peptide with phosphorylated Thr84 to levels similar to the nonphosphorylated peptide binding to WT synaptotagmin-1 (Fig. 5D).
Table 3.
Crystallographic statistics for synaptotagmin-1 with bound SV2A peptide phosphorylated at Thr84
| Structure/PDB code | SYT1–SV2A peptide/4v11 |
|---|---|
| Data collection | |
| Space group | P 1 21 1 |
| Wavelength (Å) | 1.54178 |
| Unit-cell parameters | |
| a, b, c (Å) | 46.05, 41.43, 48.01 |
| α, β, γ (°) | 90.00, 96.59, 90.00 |
| Resolution range (Å)a | 31.28–1.95 (2.02–1.95) |
| n reflections | 25874 (2156) |
| Unique reflections | 13172 (1209) |
| Completeness (%) | 99.0 (98.2) |
| Rmerge (within I+/I−)b | 0.01496 (0.03459) |
| Multiplicity | 2.0 (1.8) |
| Mean(I)/σ(I) | 58.93 (23.39) |
| Model refinement | |
| Rwork/Rfree (%)c,d | 15.12/19.56 (12.91/19.38) |
| Wilson B (Å2) | 10.0 |
| Overall average B (Å2) | 11.0 |
| n protein residues (atoms) | 160 (1286) |
| n ligands (atoms) | 3 Ca2+, 1 glycerol (9) |
| n waters (atoms) | 147 (147) |
| Ramachandran plot analysis | |
| Favored regions | 97% |
| Allowed regions | 3% |
| Outliers | 0% |
| Rms deviations | |
| Bond lengths (Å) | 0.007 |
| Bond angles (Å) | 1.004 |
aValues in parentheses refer to the highest resolution shell.
b, where Ihkl,i is the intensity of the ith measurement of reflection hkl and 〈Ihkl〉 is the mean value of Ihkl,i for all i measurements.
c, where Fobs is the observed structure factor, and Fcalc is the calculated structure factor.
dRfree is the same as Rwork except calculated with a subset, 5%, of data that are excluded from the refinement calculations. The PDB accession code is 4V11.
Figure 5.

Crystal structure of Thr84 phosphorylated Cluster-2 peptide bound to synaptotagmin-1 C2B domain. A, A ribbon diagram of the synaptotagmin-1 C2B domain (residues 270–422) bound to a peptide encompassing the Cluster-2 region of SV2A (residues 81–90) that is phosphorylated at Thr84 (PDB entry 4v11). The synaptotagmin-1 C2B domain is shown with β strands colored in cyan, helices in magenta, and loops in gray. The backbone of the SV2A peptide, binding at the polybasic region, is colored in yellow. The side chains of residues of the polybasic region interacting with the SV2A peptide are shown as stick models. The two Ca2+ ions, binding between loops 1 and 3, are shown as green spheres. B, Electrostatic surface representation of synaptotagmin-1 with the bound SV2A peptide. The negatively charged phosphate group of the SV2A peptide is binding to the positively charged surface area at the polybasic region of synaptotagmin-1. Negative charge (acidic) is shown in red and positive charge (basic) in blue. The electrostatic map is contoured from −5 to 5 kTe. C, Stereo-image and close-up analysis of the pocket on the C2B domain that interacts with Thr84 phosphorylated residue. The three highly conserved surface-exposed Lys residues on synaptotagmin-1 (Lys314, Lys326, and Lys328) interact with Thr84 of SV2A, and distances between the ε-amino groups of the lysine residues and the phosphate group of phosphorylated Thr84 are shown in angstroms. These Lys residues are distributed on anti-parallel β sheets 3 and 4, and their side chains form a specific binding pocket that precisely accommodates the phosphorylated Thr84 residue. The 2mFo-dFc map, before manual building of the peptide model and refinement, is shown as gray mesh and was contoured at 1σ within 2 Å of the SV2A peptide. D, Fluorescence polarization analysis of WT and indicated Lys substitution mutants (K314A, K326A, K328A, and K326A/K328A) of synaptotagmin-1 C2B domain and peptide encompassing Cluster-2 region of SV2A (residues 76–90), which was either phosphorylated at Thr84 or unphosphorylated as a negative control. Data points and error bars represent mean ± SD (n = 3), and similar results were obtained in at least two separate experiments for all data shown in this figure.
Initially, we also crystallized the triply phosphorylated Ser80, Ser81, and Thr84 Cluster-2 peptide (residues 76–90) with the C2B domain (residues 270–421) of synaptotagmin-1 because this peptide represented the phosphorylation status seen in the previous mass spectrometry experiments. Similarly, as seen with the peptide only phosphorylated at Thr84, electron density of residues 82–90 was clearly resolved. However, no density was observed for the Ser80 and Ser81 phosphorylated residues, also indicating that these residues may be disordered in this structure (data not shown).
Phosphorylation of SV2A at Cluster-2 does not affect its trafficking
We next addressed whether Cluster-2 phosphorylation of SV2A has a direct effect on its trafficking during action potential-evoked SV recycling. We generated an SV2A–pHluorin fusion protein by inserting the pH-sensitive GFP pHluorin into the intravesicular loop between the first and second transmembrane domains. This manipulation has no detrimental effect on evoked SV recycling (Kwon and Chapman, 2011). At neutral pH, the GFP pHluorin moiety is highly fluorescent, but its fluorescence is quenched in an acidic environment. Therefore, SV2A–pHluorin has little or no fluorescence when inside SVs (which have a pH of ∼5.5). The fusion of SV2A–pHluorin-expressing SVs can be viewed as an increase in fluorescence and retrieval of SV2A–pHluorin from the plasma membrane as a subsequent decrease (because SV endocytosis is the rate-limiting step rather than SV acidification; Sankaranarayanan and Ryan, 2000).
Both WT (SV2AWT–pHluorin) and a triple mutant with all Cluster-2 sites mutated to alanine (SV2AP-null–pHluorin) were distributed in a punctate manner when expressed in neuronal culture, indicating their localization to nerve terminals (Fig. 6A). When transfected neurons were challenged with a train of 300 action potentials (10 Hz), we observed no significant alteration in the extent of the SV2AP-null–pHluorin response compared with SV2AWT–pHluorin [maximal evoked ΔF, reflective of mobilization of the recycling SV pool normalized to the total SV pool (revealed by alkaline buffer), SV2AWT–pHluorin, 0.39 ± 0.05; SV2AP-null–pHluorin, 0.43 ± 0.04; n = 4 for each; p = 0.52, Student's t test]. This indicates that SV exocytosis was unlikely to be affected by expression of this reporter. There was also no significant difference in the loss of fluorescence after action potential stimulation (Fig. 6B), indicating that Cluster-2 phosphorylation has no direct effect on SV2A retrieval during SV endocytosis.
Figure 6.

Inhibition of SV2A binding accelerates Syt1–pHluorin retrieval during SV endocytosis. A, Hippocampal neurons were transfected with either WT or phospho-null (P-null) SV2A–pHluorin. Grayscale images show that SV2A–pHluorin was correctly targeted to nerve terminals. Scale bar, 1 μm. B, Neurons transfected with either WT (blue) or P-null (red) SV2A–pHluorin were stimulated with a train of 300 action potentials (10 Hz). Graph displays the mean ΔF/F0 time course for SV2A–pHluorin ± SEM normalized to the peak of stimulation (n = 5 WT, n = 4 P-null). C, Neurons were transfected with either WT or K326/328A Syt1–pHluorin. Grayscale images show that Syt1–pHluorin was correctly targeted to nerve terminals. Scale bar, 1 μm. Surface expression of either Syt1WT–pHluorin or Syt1[K326/328A]–pHluorin displayed as a percentage of total pHluorin ± SEM (**p = 0.004). D, Neurons transfected with either WT (blue) or K326/328A (red) Syt1–pHluorin were stimulated with a train of 300 action potentials (10 Hz). Graph displays the mean ΔF/F0 time course for Syt1–pHluorin ± SEM normalized to the peak of stimulation (significantly different between time points 128–140 s; p < 0.05; n = 8 for both WT and K326/328A). In both B and D, the bar indicates period of stimulation.
Ablation of SV2A binding accelerates synaptotagmin-1 retrieval during SV endocytosis
We next examined whether Cluster-2 phosphorylation controlled the trafficking of its phospho-dependent interaction partner synaptotagmin-1. To achieve this, we examined the trafficking of an Syt1–pHluorin reporter (Diril et al., 2006) that cannot bind SV2A (Syt1[K326A/K328A]–pHluorin). Both WT Syt1–pHluorin and Syt1[K326A/K328A]–pHluorin were targeted to nerve terminals (Fig. 6C) and displayed a similar evoked fluorescence increase when challenged with 300 action potentials (10 Hz), indicating no obvious role for the SV2A interaction in SV exocytosis (maximal evoked ΔF, Syt1WT–pHluorin, 0.49 ± 0.02; Syt1[K326A/K328A]–pHluorin, 0.44 ± 0.03; n = 6 WT, n = 8 [K326A/K328A]; p = 0.17 Student's t test). Syt1[K326A/K328A]–pHluorin displayed significant accumulation at the plasma membrane (percentage surface fraction, Syt1WT–pHluorin, 30.6 ± 1.9; Syt1[K326A/K328A]–pHluorin, 45.8 ± 4.5; n = 9 WT, n = 6 [K326A/K328A]; p = 0.004 Student's t test), a phenotype characteristic of dysfunctional SV cargo retrieval during SV endocytosis (Yao et al., 2010; Gordon et al., 2011; Koo et al., 2011; Kononenko et al., 2013). However, we observed a significant acceleration in activity-dependent Syt1[K326A/K328A]–pHluorin retrieval when compared with Syt1WT–pHluorin (Fig. 6D). This acceleration was not attributable to escape of Syt1[K326A/K328A]–pHluorin from the immediate vicinity of the nerve terminal, because identical kinetics were observed when a larger axonal area was sampled out with nerve terminals (data not shown).
Phosphorylation of T84 on SV2A modulates the kinetics of synaptotagmin-1 retrieval
We next explored whether loss of SV2A would also accelerate synaptotagmin-1 retrieval. We designed an shRNA vector that coexpressed both Syt1WT–pHluorin and an oligonucleotide sequence published to knockdown SV2A expression (Dong et al., 2006). When this shRNA vector was expressed in culture, endogenous SV2A levels were reduced by >75% when compared with empty vector (Fig. 7A; endogenous SV2A expression, 23.3 ± 2.2% of empty vector). SV2A knockdown had no significant effect on the size of the evoked Syt1WT–pHluorin response (maximal evoked ΔF, empty shRNA vector, 0.42 ± 0.04; SV2A shRNA1, 0.49 ± 0.05; n = 6 empty, n = 5 shRNA1; p = 0.33 Student's t test). In contrast, Syt1WT–pHluorin retrieval was accelerated in neurons expressing SV2A shRNA compared with those expressing empty shRNA vector (Fig. 7B), mimicking the trafficking of Syt1[K326A/K328A]–pHluorin.
Figure 7.
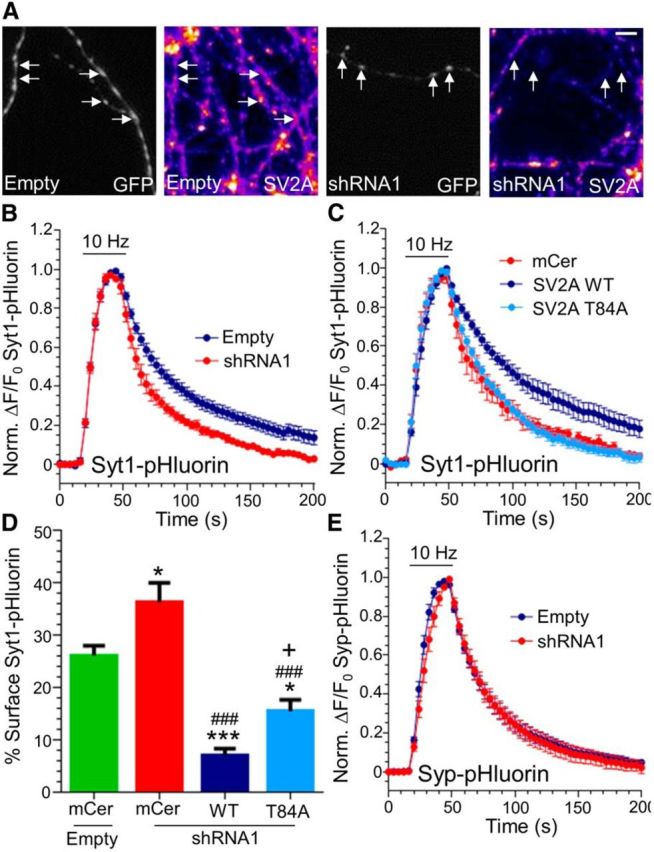
P-null SV2A does not rescue Syt1–pHluorin retrieval after shRNA knockdown. A, Images of cultures transfected with shRNA–Syt1–pHluorin vectors that are either empty or contain oligonucleotides against SV2A (shRNA1). Grayscale panels highlight transfected neurons (GFP), whereas false color panels display endogenous SV2A revealed by immunofluorescence staining. Arrows highlight nerve terminals. Scale bar, 1 μm. B, Neurons transfected with WT Syt1–pHluorin and either empty (blue) or SV2A shRNA (shRNA1, red) were stimulated with a train of 300 action potentials (10 Hz). Graph displays the mean ΔF/F0 time course for Syt1–pHluorin ± SEM normalized to the peak of stimulation (significantly different between time points 96–140 s; p < 0.05; n = 7 empty, n = 5 shRNA1). C, Neurons transfected with a pSUPER vector expressing both Syt1–pHluorin and shRNA1 were coexpressed with either mCer empty vector (mCer), WT mCer–SV2A, or T84A mCer–SV2A. Neurons were stimulated as in B, and the graph displays the mean ΔF/F0 time course for Syt1–pHluorin ± SEM normalized to the peak of stimulation (mCer and WT traces significantly different between 60 and 200 s; p < 0.05, T84A and WT traces significantly different from 68–200 s; p < 0.05; n = 5 mCer, n = 6 WT, n = 7 T84A). D, Surface expression of Syt1WT–pHluorin in neurons expressing a combination of empty or shRNA1 pSUPER vector and mCer, WT, or T84A mCer–SV2A displayed as a percentage of total pHluorin ± SEM (*p < 0.05, ***p < 0.001 to empty/mCer; ###p < 0.001 to shRNA1/mCer; +p < 0.05 to shRNA1/WT; n = 7 for all except n = 5 for shRNA1/T84A). E, Neurons transfected with WT synaptophysin–pHluorin (Syp–pHluorin) and either empty (blue) or SV2A shRNA (shRNA1, red) were stimulated as in B. Graph displays the mean ΔF/F0 time course for Syp–pHluorin ± SEM normalized to the peak of stimulation (n = 9 empty, n = 6 shRNA1, p > 0.05). In B, C, and E, the bar indicates the period of stimulation.
We next confirmed that overexpression of WT SV2A could rescue Syt1WT–pHluorin retrieval kinetics in neurons expressing SV2A shRNA. To achieve this, shRNA-resistant SV2A tagged with the fluorescent protein mCer (mCer–SV2A) was coexpressed with our modified shRNA vector expressing both SV2A oligonucleotides and Syt1WT–pHluorin. Coexpression of shRNA-resistant mCer–SV2A both restored SV2A expression (total SV2A expression, 231 ± 32% of empty vector control) and fully rescued Syt1WT–pHluorin retrieval kinetics (Fig. 7C). In contrast, mCer–SV2A lacking the critical Cluster-2 Thr84 phosphorylatable residue required for synpatotagmin-1 binding failed to rescue Syt1WT–pHluorin retrieval (Fig. 7C). The T84A mCer–SV2A mutant also failed to restore plasma membrane levels of Syt1WT–pHluorin to that observed with WT mCer–SV2A (Fig. 7D).
Genomic deletion of the monomeric adaptor for synaptotagmin, stonin 2, accelerates both Syt1WT–pHluorin retrieval and SV endocytosis (Kononenko et al., 2013). Thus, ablation of the SV2A–synaptotagmin-1 interaction may also have a global effect on SV endocytosis. To test this, we determined the effect of SV2A knockdown on a distinct SV cargo, synaptophysin, because this SV protein is absent from SV2A protein complexes (Schivell et al., 1996). Knockdown of SV2A had no effect on either the peak fluorescent response (maximal evoked ΔF, empty shRNA vector, 0.36 ± 0.04; SV2A shRNA1, 0.32 ± 0.04; n = 7 empty, n = 5 shRNA1; p = 0.43, Student's t test) or the retrieval kinetics of synaptophysin–pHluorin (Fig. 7E). Thus, SV2A phosphorylation is specifically required for synaptotagmin-1 retrieval and not SV endocytosis in general.
Discussion
CK1 isoforms phosphorylate SV2A at highly conserved sites
We demonstrate in vitro that four CK1 and two TTBK isoforms phosphorylated the cytoplasmic N-terminal domain of SV2A[1–160] at two regions termed Cluster-1 (Ser42, Ser45, and Ser47) and Cluster-2 (Ser80, Ser81, and Thr84; Figs. 1, 2). Both clusters are highly conserved (Fig. 2E), indicating that the phosphorylation of these residues may play critical roles. In agreement, the key residue Thr84 is conserved in both SV2B and SV2C, as is Ser81. Because phosphorylation of substrates by CK1 kinases is facilitated by an acidic residue or priming phosphorylation at the −3 position (Flotow et al., 1990; Meggio et al., 1991), phosphorylation of Ser81 primes for phosphorylation of Thr84. Consistent with this, mutation of Ser80 and Ser81 prevented phosphorylation of Thr84 in SV2A overexpressed in HEK293 cells (Table 2). When we crystallized the C2B domain of synaptotagmin-1 with the Cluster-2 Ser80, Ser81, and Thr84 triply phosphorylated peptide, we observed no electron density for the phosphorylated Ser80 and Ser81, indicating that these residues may not be interacting specifically with the C2B domain. Consistent with this, the triply phosphorylated peptide did not interact with the synaptotagmin-1 C2B domain with higher affinity than the singly Thr84 phosphorylated peptide (Fig. 4). Therefore, it is likely that the principal role of Ser80 and Ser81 is to prime Thr84 for phosphorylation by CK1 family kinase(s) rather than to mediate interaction with synaptotagmin-1.
Which CK1 family member(s) phosphorylate SV2A at Cluster-1 and Cluster-2 sites in vivo? To address this question, systematic analysis of SV2A phosphorylation in CK1 isoform knockdown or knock-out systems may be required. This analysis may be complicated by the redundancy of multiple CK1 isoforms phosphorylating SV2A, as suggested by our in vitro experimentation (Fig. 1). None of the widely used pan CK1 inhibitors described to date (D4476, CKI-7, and IC261) are sufficiently potent (in vitro IC50 values >1 μm), or selective (see http://www.kinase-screen.mrc.ac.uk/kinase-inhibitors), to be used to determine which CK1 isoforms regulate SV2A phosphorylation in vivo. Although certain CK1 family kinases tested, such as TTBK1, TTBK2[1–316], and CK1δ, phosphorylated SV2A more efficiently in vitro than other family members (CK1α1 and CKγ1), it is not possible to rule out that this was a result of the recombinant kinases possessing low intrinsic activity because of their expression in E. coli and/or lack of factors required to promote phosphorylation of SV2A by CK1 kinase(s). The finding that phosphorylation of Ser80 and Ser81 promotes phosphorylation of Thr84 in vivo (Tables 1, 2) is consistent with CK1 family kinase(s) known to require a −3/−4 phosphorylation priming mechanism.
Mapping of the synaptotagmin-1–SV2A interaction site
The N terminus of SV2A interacts with synaptotagmin-1 (Schivell et al., 1996, 2005), and this interaction is enhanced by CK1 phosphorylation in vitro (Pyle et al., 2000). Our data establish that CK1 phosphorylation of Thr84 promotes the interaction of SV2A with the C2B domain of synaptotagmin-1 (Fig. 4). Crystallographic analysis reveals that the phosphorylated Thr84 residue interacts with a specific pocket on the surface of the synaptotagmin-1 C2B domain (Fig. 5). This lies within the “polybasic region” (Fernandez et al., 2001). This pocket is formed from three surface-exposed Lys residues (Lys314, Lys326, and Lys328) that make hydrogen bonds with the phosphate moiety of the phosphorylated Thr84 residue (Fig. 5). These Lys residues are conserved in all species of synaptotagmin-1 we have analyzed and in 13 of the 17 synaptotagmin isoforms present in humans, suggesting that these will all possess the ability to interact with CK1-phosphorylated SV2 isoforms. The Lys residues equivalent to Lys326 and Lys328 are absent in the C2A domain of synaptotagmin-1, likely accounting for its failure to bind the Cluster-2 Thr84 phosphorylated peptide.
The regions surrounding the phosphorylated Thr84 binding pocket on the synaptotagmin-1 C2B domain bind to other components, including phosphoinositides (Schiavo et al., 1996), adaptor protein 2 (AP-2; Zhang et al., 1994), calcium channels (Leveque et al., 1992), other synaptotagmins (Chapman et al., 1996), and target-soluble N-ethylmaleimide-sensitive factor attachment protein receptor (t-SNARE) dimers (Bhalla et al., 2006). NMR analysis (Joung et al., 2012) revealed that inositol hexaphosphate IP6 binds to a pocket that lies adjacent to that for the phosphorylated Thr84 binding pocket. The phosphate groups on IP6 form key interactions with Lys328, Thr330, and Lys332 residues in human synaptotagmin-1. Inspection of the structure of the C2B domain does not rule out the possibility that the C2B domain could bind simultaneously to the phosphorylated Cluster-2 SV2A peptide and IP6/phosphoinositide, with Lys328 functioning as a pivotal residue capable of interacting with both phosphorylated Thr84 and an inositol phosphate group. Phosphoinositide binding has been proposed to control the interaction of the C2B domain with the t-SNARE dimer (Rickman et al., 2004). Thus, Lys328 may act as a molecular switch, binding either phosphorylated SV2A for its retrieval or IP6 for t-SNARE-specific interactions. Other studies have suggested that the Lys327 and Lys328 residues (corresponding residues in human SYT1) control C2B dimerization and regulate interactions with both AP-2 and voltage-gated calcium channels (Chapman et al., 1998; Grass et al., 2004). Therefore, more work is required to study the interplay between the interaction of synaptotagmin-1 with CK1-phosphorylated SV2A and other binding components.
Phosphorylation of SV2A controls synaptotagmin-1 retrieval during SV endocytosis
We found that removal of the phosphorylation-dependent SV2A interaction by either mutagenesis of the C2B domain, shRNA knockdown of SV2A, or mutagenesis of T84 on SV2A all result in increased retrieval kinetics of Syt1–pHluorin. This was surprising, because disruption of endocytic sorting mechanisms by either knockdown of adaptor protein complexes or monomeric adaptors (Kim and Ryan, 2009; Willox and Royle, 2012; Kononenko et al., 2014) or mutagenesis of key endocytosis motifs (Voglmaier et al., 2006; Yao et al., 2010; Koo et al., 2011; Foss et al., 2013) usually result in retarded SV cargo retrieval. This acceleration was not attributable to defective retention of Syt1–pHluorin in nerve terminals, because retrieval kinetics were identical when axonal regions were analyzed. Interestingly, accelerated Syt1–pHluorin retrieval was also observed in neurons derived from stonin 2 knock-out mice (Kononenko et al., 2013). Stonin 2 is a synaptotagmin-specific adaptor molecule, and knock-out neurons displayed both accumulation of Syt1–pHluorin at the plasma membrane and enhanced SV endocytosis kinetics. We also observed increased plasma membrane accumulation of Syt1–pHluorin on perturbation of SV2A interactions, but we see no effect on global SV endocytosis. The most likely explanation for these results is that Syt1–pHluorin is being retrieved by a parallel endocytic mode with faster retrieval kinetics than classical clathrin-mediated endocytosis. It is possible that accumulation of Syt1–pHluorin at the plasma membrane may even trigger this mode. Thus, although there is an underlying defect in Syt1–pHluorin retrieval via clathrin-mediated endocytosis (reflected in increased surface accumulation in resting neurons), the retrieval of Syt1–pHluorin newly deposited on the cell surface by neuronal activity could be mediated by an alternate endocytosis mode.
SV2A is a phospho-dependent synaptotagmin-1 chaperone
We propose that SV2A could act as a phospho-dependent chaperone for the efficient post-exocytic trafficking of synaptotagmin-1, in addition to AP-2 and stonin 2. The requirement for a classical adaptor protein complex, a monomeric adaptor protein, and SV chaperone has close parallels with recent investigations of the retrieval mechanism of the essential vesicle v-SNARE synaptobrevin II. Synaptobrevin II has no canonical motif for AP-2 binding, so the monomeric adaptor AP180 clusters this SV cargo by simultaneously binding to AP-2 and the SNARE motif of synaptobrevin II (Koo et al., 2011). However, the SV protein synaptophysin is also essential for efficient synaptobrevin II retrieval (Gordon et al., 2011; Gordon and Cousin, 2013). Thus, interactions between SV cargo at the nerve terminal plasma membrane may serve to aid cargo clustering, present cargo in the correct conformation for adaptor proteins, or restrict entry into ectopic “dead-end” complexes (Gordon and Cousin, 2014). A recent quantitative study demonstrated a similar copy number for both SV2A and synaptotagmin-1 on the SV (Wilhelm et al., 2014), supporting a potential chaperone role for SV2A.
How might SV2A control synaptotagmin-1 retrieval in concert with other clathrin adaptor molecules? Stonin 2 primarily interacts with the C2A domain (Jung et al., 2007), meaning that both SV2A and stonin 2 could interact simultaneously with synaptotagmin-1 via different C2 domains. In addition, both stonin 2 and SV2A interact with AP-2 via canonical internalization motifs (Diril et al., 2006; Yao et al., 2010). Thus, after SV fusion, synaptotagmin-1 will be sequestered by both stonin 2 and SV2A for clustering with AP-2. In agreement, SV2A binding increases the AP-2 affinity for the synaptotagmin-1 C2B domain (Haucke and De Camilli, 1999; Haucke et al., 2000). Because the SV2A–synaptotagmin-1 interaction is phosphorylation dependent, this model predicts that SV2A is only phosphorylated after exocytosis is complete, allowing synaptotagmin-1 on the SV to participate in evoked neurotransmitter release. This suggests that the phosphorylation state of presynaptic chaperones may be a regulatory mechanism that contributes to the efficiency of SV cargo retrieval. Therefore, it will be of great future interest to determine where (on SVs and/or plasma membrane) and when (before, during, or after action potential stimulation) SV2A is phosphorylated by a CK1 family kinase.
Footnotes
This work was supported by Medical Research Council (MRC) Grant G1002117 (M.A.C.) and MRC core support (D.R.A.), Wellcome Trust Senior Fellowship Grant 087590 (D.M.F.v.A.), Wellcome Trust Equipment Award Grant 094090, and Astra Zeneca, Boehringer-Ingelheim, GlaxoSmithKline, Merck, Janssen Pharmaceutica, and Pfizer, who supported the Division of Signal Transduction Therapy Unit. We thank Paul Davies for aid with the isothermal calorimetric-binding analysis and for the excellent technical support of the MRC Protein Phosphorylation and Ubiquitylation Unit (PPU) DNA Sequencing Service (coordinated by Nicholas Helps), the MRC–PPU tissue culture team (coordinated by Kirsten Airey and Janis Stark), the Division of Signal Transduction Therapy protein production and antibody purification teams (coordinated by Hilary McLauchlan and James Hastie), and the Centre for Advanced Scientific Technologies X-Ray Crystallography Facility (coordinated by Dr. Paul Fyfe).
The authors declare no competing financial interests.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anggono V, Smillie KJ, Graham ME, Valova VA, Cousin MA, Robinson PJ. Syndapin I is the phosphorylation-regulated dynamin I partner in synaptic vesicle endocytosis. Nat Neurosci. 2006;9:752–760. doi: 10.1038/nn1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajjalieh SM, Frantz GD, Weimann JM, McConnell SK, Scheller RH. Differential expression of synaptic vesicle protein 2 (SV2) isoforms. J Neurosci. 1994;14:5223–5235. doi: 10.1523/JNEUROSCI.14-09-05223.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla A, Chicka MC, Tucker WC, Chapman ER. Ca(2+)-synaptotagmin directly regulates t-SNARE function during reconstituted membrane fusion. Nat Struct Mol Biol. 2006;13:323–330. doi: 10.1038/nsmb1076. [DOI] [PubMed] [Google Scholar]
- Brose N, Petrenko AG, Südhof TC, Jahn R. Synaptotagmin—a calcium sensor on the synaptic vesicle surface. Science. 1992;256:1021–1025. doi: 10.1126/science.1589771. [DOI] [PubMed] [Google Scholar]
- Buckley K, Kelly RB. Identification of a transmembrane glycoprotein specific for secretory vesicles of neural and endocrine cells. J Cell Biol. 1985;100:1284–1294. doi: 10.1083/jcb.100.4.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DG, Morrice NA. Identification of protein phosphorylation sites by a combination of mass spectrometry and solid phase Edman sequencing. J Biomol Tech. 2002;13:119–130. [PMC free article] [PubMed] [Google Scholar]
- Chapman ER, An S, Edwardson JM, Jahn R. A novel function for the second C2 domain of synaptotagmin. Ca2+-triggered dimerization. J Biol Chem. 1996;271:5844–5849. doi: 10.1074/jbc.271.10.5844. [DOI] [PubMed] [Google Scholar]
- Chapman ER, Desai RC, Davis AF, Tornehl CK. Delineation of the oligomerization, AP-2 binding, and synprint binding region of the C2B domain of synaptotagmin. J Biol Chem. 1998;273:32966–32972. doi: 10.1074/jbc.273.49.32966. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Sequeira SM, Malinina L, Tereshko V, Söllner TH, Patel DJ. Crystallographic identification of Ca2+ and Sr2+ coordination sites in synaptotagmin I C2B domain. Protein Sci. 2004;13:2665–2672. doi: 10.1110/ps.04832604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton EL, Sue N, Smillie KJ, O'Leary T, Bache N, Cheung G, Cole AR, Wyllie DJ, Sutherland C, Robinson PJ, Cousin MA. Dynamin I phosphorylation by GSK3 controls activity-dependent bulk endocytosis of synaptic vesicles. Nat Neurosci. 2010;13:845–851. doi: 10.1038/nn.2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowder KM, Gunther JM, Jones TA, Hale BD, Zhang HZ, Peterson MR, Scheller RH, Chavkin C, Bajjalieh SM. Abnormal neurotransmission in mice lacking synaptic vesicle protein 2A (SV2A) Proc Natl Acad Sci U S A. 1999;96:15268–15273. doi: 10.1073/pnas.96.26.15268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diril MK, Wienisch M, Jung N, Klingauf J, Haucke V. Stonin 2 is an AP-2-dependent endocytic sorting adaptor for synaptotagmin internalization and recycling. Dev Cell. 2006;10:233–244. doi: 10.1016/j.devcel.2005.12.011. [DOI] [PubMed] [Google Scholar]
- Dong M, Yeh F, Tepp WH, Dean C, Johnson EA, Janz R, Chapman ER. SV2 is the protein receptor for botulinum neurotoxin A. Science. 2006;312:592–596. doi: 10.1126/science.1123654. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr. 2013;69:1204–1214. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez I, Araç D, Ubach J, Gerber SH, Shin O, Gao Y, Anderson RG, Südhof TC, Rizo J. Three-dimensional structure of the synaptotagmin 1 C2B-domain: synaptotagmin 1 as a phospholipid binding machine. Neuron. 2001;32:1057–1069. doi: 10.1016/S0896-6273(01)00548-7. [DOI] [PubMed] [Google Scholar]
- Flotow H, Graves PR, Wang AQ, Fiol CJ, Roeske RW, Roach PJ. Phosphate groups as substrate determinants for casein kinase I action. J Biol Chem. 1990;265:14264–14269. [PubMed] [Google Scholar]
- Foss SM, Li H, Santos MS, Edwards RH, Voglmaier SM. Multiple dileucine-like motifs direct VGLUT1 trafficking. J Neurosci. 2013;33:10647–10660. doi: 10.1523/JNEUROSCI.5662-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E, Hoogland C, Gattiker A, Duvaud Se, Wilkins M, Appel R, Bairoch A. Protein identification and analysis tools on the ExPASy server. In: Walker J, editor. The proteomics protocols handbook. New York: Humana; 2005. pp. 571–607. [Google Scholar]
- Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Südhof TC. Synaptotagmin-I—a major Ca2+ sensor for transmitter release at a central synapse. Cell. 1994;79:717–727. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- Gordon SL, Cousin MA. X-linked intellectual disability-associated mutations in synaptophysin disrupt synaptobrevin II retrieval. J Neurosci. 2013;33:13695–13700. doi: 10.1523/JNEUROSCI.0636-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon SL, Cousin MA. The Sybtraps: control of synaptobrevin traffic by synaptophysin, alpha-synuclein and AP-180. Traffic. 2014;15:245–254. doi: 10.1111/tra.12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon SL, Leube RE, Cousin MA. Synaptophysin is required for synaptobrevin retrieval during synaptic vesicle endocytosis. J Neurosci. 2011;31:14032–14036. doi: 10.1523/JNEUROSCI.3162-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grass I, Thiel S, Höning S, Haucke V. Recognition of a basic AP-2 binding motif within the C2B domain of synaptotagmin is dependent on multimerization. J Biol Chem. 2004;279:54872–54880. doi: 10.1074/jbc.M409995200. [DOI] [PubMed] [Google Scholar]
- Griffin BA, Adams SR, Tsien RY. Specific covalent labeling of recombinant protein molecules inside live cells. Science. 1998;281:269–272. doi: 10.1126/science.281.5374.269. [DOI] [PubMed] [Google Scholar]
- Haucke V, De Camilli P. AP-2 recruitment to synaptotagmin stimulated by tyrosine-based endocytic motifs. Science. 1999;285:1268–1271. doi: 10.1126/science.285.5431.1268. [DOI] [PubMed] [Google Scholar]
- Haucke V, Wenk MR, Chapman ER, Farsad K, De Camilli P. Dual interaction of synaptotagmin with mu2- and alpha-adaptin facilitates clathrin-coated pit nucleation. EMBO J. 2000;19:6011–6019. doi: 10.1093/emboj/19.22.6011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honigmann A, van den Bogaart G, Iraheta E, Risselada HJ, Milovanovic D, Müller V, Mullar S, Diederichsen U, Fasshauer D, Grubmüller H, Hell SW, Eggeling C, Kühnel K, Jahn R. Phosphatidylinositol 4,5-bisphosphate clusters act as molecular beacons for vesicle recruitment. Nat Struct Mol Biol. 2013;20:679–686. doi: 10.1038/nsmb.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung MJ, Mohan SK, Yu C. Molecular level interaction of inositol hexaphosphate with the C2B domain of human synaptotagmin I. Biochemistry. 2012;51:3675–3683. doi: 10.1021/bi300005w. [DOI] [PubMed] [Google Scholar]
- Jung N, Wienisch M, Gu M, Rand JB, Müller SL, Krause G, Jorgensen EM, Klingauf J, Haucke V. Molecular basis of synaptic vesicle cargo recognition by the endocytic sorting adaptor stonin 2. J Cell Biol. 2007;179:1497–1510. doi: 10.1083/jcb.200708107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Ryan TA. Synaptic vesicle recycling at CNS snapses without AP-2. J Neurosci. 2009;29:3865–3874. doi: 10.1523/JNEUROSCI.5639-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kononenko NL, Diril MK, Puchkov D, Kintscher M, Koo SJ, Pfuhl G, Winter Y, Wienisch M, Klingauf J, Breustedt J, Schmitz D, Maritzen T, Haucke V. Compromised fidelity of endocytic synaptic vesicle protein sorting in the absence of stonin 2. Proc Natl Acad Sci U S A. 2013;110:E526–E535. doi: 10.1073/pnas.1218432110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kononenko NL, Puchkov D, Classen GA, Walter AM, Pechstein A, Sawade L, Kaempf N, Trimbuch T, Lorenz D, Rosenmund C, Maritzen T, Haucke V. Clathrin/AP-2 mediate synaptic vesicle reformation from endosome-like vacuoles but are not essential for membrane retrieval at central synapses. Neuron. 2014;82:981–988. doi: 10.1016/j.neuron.2014.05.007. [DOI] [PubMed] [Google Scholar]
- Koo SJ, Markovic S, Puchkov D, Mahrenholz CC, Beceren-Braun F, Maritzen T, Dernedde J, Volkmer R, Oschkinat H, Haucke V. SNARE motif-mediated sorting of synaptobrevin by the endocytic adaptors clathrin assembly lymphoid myeloid leukemia (CALM) and AP180 at synapses. Proc Natl Acad Sci U S A. 2011;108:13540–13545. doi: 10.1073/pnas.1107067108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon SE, Chapman ER. Synaptophysin regulates the kinetics of synaptic vesicle endocytosis in central neurons. Neuron. 2011;70:847–854. doi: 10.1016/j.neuron.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzell DR, Belizaire R, Thakur P, Sherry DM, Janz R. SV2B regulates synaptotagmin 1 by direct interaction. J Biol Chem. 2004;279:52124–52131. doi: 10.1074/jbc.M407502200. [DOI] [PubMed] [Google Scholar]
- Leveque C, Hoshino T, David P, Shoji-Kasai Y, Leys K, Omori A, Lang B, el Far O, Sato K, Martin-Moutot N, Newsom-Davis J, Takahashi M, Seagar MJ. The synaptic vesicle protein synaptotagmin associates with calcium channels and is a putative Lambert-Eaton myasthenic syndrome antigen. Proc Natl Acad Sci U S A. 1992;89:3625–3629. doi: 10.1073/pnas.89.8.3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meggio F, Perich JW, Reynolds EC, Pinna LA. A synthetic beta-casein phosphopeptide and analogues as model substrates for casein kinase-1, a ubiquitous, phosphate directed protein kinase. FEBS Lett. 1991;283:303–306. doi: 10.1016/0014-5793(91)80614-9. [DOI] [PubMed] [Google Scholar]
- Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY, Halim VA, Bagshaw RD, Hubner NC, Al-Hakim A, Bouchard A, Faubert D, Fermin D, Dunham WH, Goudreault M, Lin ZY, et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat Methods. 2013;10:730–736. doi: 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza-Torreblanca JG, Vanoye-Carlo A, Phillips-Farfán BV, Carmona-Aparicio L, Gómez-Lira G. Synaptic vesicle protein 2A: basic facts and role in synaptic function. Eur J Neurosci. 2013;38:3529–3539. doi: 10.1111/ejn.12360. [DOI] [PubMed] [Google Scholar]
- Pyle RA, Schivell AE, Hidaka H, Bajjalieh SM. Phosphorylation of synaptic vesicle protein 2 modulates binding to synaptotagmin. J Biol Chem. 2000;275:17195–17200. doi: 10.1074/jbc.M000674200. [DOI] [PubMed] [Google Scholar]
- Rickman C, Archer DA, Meunier FA, Craxton M, Fukuda M, Burgoyne RD, Davletov B. Synaptotagmin interaction with the syntaxin/SNAP-25 dimer is mediated by an evolutionarily conserved motif and is sensitive to inositol hexakisphosphate. J Biol Chem. 2004;279:12574–12579. doi: 10.1074/jbc.M310710200. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, Ryan TA. Real-time measurements of vesicle-SNARE recycling in synapses of the central nervous system. Nat Cell Biol. 2000;2:197–204. doi: 10.1038/35008615. [DOI] [PubMed] [Google Scholar]
- Schiavo G, Gu QM, Prestwich GD, Söllner TH, Rothman JE. Calcium-dependent switching of the specificity of phosphoinositide binding to synaptotagmin. Proc Natl Acad Sci U S A. 1996;93:13327–13332. doi: 10.1073/pnas.93.23.13327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schivell AE, Batchelor RH, Bajjalieh SM. Isoform-specific, calcium-regulated interaction of the synaptic vesicle proteins SV2 and synaptotagmin. J Biol Chem. 1996;271:27770–27775. doi: 10.1074/jbc.271.44.27770. [DOI] [PubMed] [Google Scholar]
- Schivell AE, Mochida S, Kensel-Hammes P, Custer KL, Bajjalieh SM. SV2A and SV2C contain a unique synaptotagmin-binding site. Mol Cell Neurosci. 2005;29:56–64. doi: 10.1016/j.mcn.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Studier FW, Moffatt BA. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- Voglmaier SM, Kam K, Yang H, Fortin DL, Hua Z, Nicoll RA, Edwards RH. Distinct endocytic pathways control the rate and extent of synaptic vesicle protein recycling. Neuron. 2006;51:71–84. doi: 10.1016/j.neuron.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Wilhelm BG, Mandad S, Truckenbrodt S, Kröhnert K, Schäfer C, Rammner B, Koo SJ, Claßen GA, Krauss M, Haucke V, Urlaub H, Rizzoli SO. Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science. 2014;344:1023–1028. doi: 10.1126/science.1252884. [DOI] [PubMed] [Google Scholar]
- Willox AK, Royle SJ. Stonin 2 is a major adaptor protein for clathrin-mediated synaptic vesicle retrieval. Curr Biol. 2012;22:1435–1439. doi: 10.1016/j.cub.2012.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Nowack A, Kensel-Hammes P, Gardner RG, Bajjalieh SM. Cotrafficking of SV2 and synaptotagmin at the synapse. J Neurosci. 2010;30:5569–5578. doi: 10.1523/JNEUROSCI.4781-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JZ, Davletov BA, Südhof TC, Anderson RG. Synaptotagmin I is a high affinity receptor for clathrin AP-2: implications for membrane recycling. Cell. 1994;78:751–760. doi: 10.1016/S0092-8674(94)90442-1. [DOI] [PubMed] [Google Scholar]