

Abstract
Recent studies have found that those who suffer from posttraumatic stress disorder (PTSD) are more likely to experience dementia as they age, most often Alzheimer's disease (AD). These findings suggest that the symptoms of PTSD might have an exacerbating effect on AD progression. AD and PTSD might also share common susceptibility factors such that those who experience trauma-induced disease were already more likely to succumb to dementia with age. Here, we explored these two hypotheses using a mouse model of PTSD in wild-type and AD model animals. We found that expression of human familial AD mutations in amyloid precursor protein and presenilin 1 leads to sensitivity to trauma-induced PTSD-like changes in behavioral and endocrine stress responses. PTSD-like induction, in turn, chronically elevates levels of CSF β-amyloid (Aβ), exacerbating ongoing AD pathogenesis. We show that PTSD-like induction and Aβ elevation are dependent on corticotropin-releasing factor (CRF) receptor 1 signaling and an intact hypothalamic–pituitary–adrenal axis. Furthermore, we show that Aβ species can hyperexcite CRF neurons, providing a mechanism by which Aβ influences stress-related symptoms and PTSD-like phenotypes. Consistent with Aβ causing excitability of the stress circuitry, we attenuate PTSD-like phenotypes in vivo by lowering Aβ levels during PTSD-like trauma exposure. Together, these data demonstrate that exposure to PTSD-like trauma can drive AD pathogenesis, which directly perturbs CRF signaling, thereby enhancing chronic PTSD symptoms while increasing risk for AD-related dementia.
Keywords: Alzheimer's disease, crf, crfr1, crh, PTSD, stress
Introduction
Posttraumatic stress disorder (PTSD) is an affective disorder that results from exposure to life-threatening trauma, in many cases in the context of war. The diagnosis of PTSD involves the coincidence of intrusive thoughts and nightmares, avoidance of situations reminiscent of the trauma, decreased cognitive performance and mood, and changes in arousal (American Psychiatric Association, 2013). This confluence of symptoms suggests altered function of stress response pathways in PTSD, including changes in the activity of the hypothalamic–pituitary–adrenal (HPA) axis, which controls the release of cortisol (Cort) from the adrenal glands in response to corticotropin-releasing factor (CRF) release from the hypothalamus and subsequent ACTH release from the pituitary into the bloodstream (Vale et al., 1981; Yehuda et al., 1991). How changes in the activity of central neural circuits that respond to stress and control HPA axis activity produce features of PTSD remains unclear. However, chronic changes in HPA axis activity in PTSD patients can affect not only the neuropsychiatric features associated with PTSD but can also lead to susceptibility to other diseases, including cancer, cardiovascular disease, and neurodegenerative disorders (Reiche et al., 2004; Gold et al., 2005; Bao et al., 2008; Stuller et al., 2012; Volden and Conzen, 2013).
Veterans who suffer from PTSD are nearly twice as likely to be diagnosed with dementia as they age (Qureshi et al., 2010; Yaffe et al., 2010). This is likely attributable to chronically elevated stress, because multiple stressors have been shown to accelerate amyloid plaque formation (Dong et al., 2004; Ni et al., 2006; Lee et al., 2009; Devi et al., 2010), β-amyloid (Aβ) release (Kang et al., 2007; Rothman et al., 2012), tau hyperphosphorylation (Rissman et al., 2007; Carroll et al., 2011; Rissman et al., 2012), and neurodegeneration (Carroll et al., 2011) in model animals. Stress exacerbation of AD pathogenesis is attributable in part to elevated Cort levels, because glucocorticoids have been shown to have similar effects (Green et al., 2006; Sotiropoulos et al., 2008; Li et al., 2010; Joshi et al., 2012). However, many of these effects are directly dependent on CRF signaling and not exclusively dependent on Cort (Kang et al., 2007; Rissman et al., 2007; Rissman et al., 2012). AD pathogenesis-related protein regulation, such as levels of Aβ production and clearance, might also affect stress-responsive neural and endocrine pathways to exacerbate PTSD. Indeed, higher levels of circulating Cort have been reported in patients with mild cognitive impairment (MCI; Davis et al., 1986; Lind et al., 2007; Huang et al., 2009). This activation of the HPA axis correlates with accelerated conversion to AD (Lupien et al., 1998; Csernansky et al., 2006). Recently, multiple animal studies have demonstrated that an activation of the HPA axis occurs in AD model animals well before plaque formation occurs (Dong et al., 2008; Guo et al., 2012; Brureau et al., 2013; Hebda-Bauer et al., 2013). In this study, we tested the hypothesis that progressing AD pathogenesis activates CRF neurons to sensitize mice to PTSD-like induction and assayed the influence of PTSD-like induction on measures of AD progression.
Materials and Methods
Animal care and genetic crosses
Mice were housed up to five per cage with ad libitum access to food and water in a room with a 12 h light/dark cycle in a pathogen-free mouse facility. All procedures were performed in accordance with National Institutes of Health guidelines and with the approval of the Baylor College of Medicine and University of Texas, Houston Institutional Animal Care and Use Committees. Amyloid precursor protein (APP) knock-in and presenilin 1 (PS1) knock-in alleles were generously donated by their respective research groups (Flood et al., 2002; Köhler et al., 2005). Animals carrying homozygous APPhAβ/SL and homozygous PS1M146V (APP/hAβ/PS1 double knock-ins) and homozygous wild-type animals were separated from initial intercrosses and maintained as separate colonies (as described by Guo et al., 2012). Therefore, wild-type animals are derived from littermates of double knock-in animals (APP/hAβ/PS1) and are on the identical background as double knock-in animals (“wt for APP/hAβ/PS1” mice). To generate APP/hAβ/PS1 animals homozygous mutant for CRF receptor 1 (Crfr1), APP/hAβ/PS1 animals were crossed to a Crfr1 mutant allele (Smith et al., 1998) to obtain APP/hAβ/PS1;Crfr1+/− animals, which were then intercrossed to generate APP/hAβ/PS1;Crfr1−/− animals. Similarly wt for APP/hAβ/PS1 mice were outcrossed to Crfr1+/− mice and then intercrossed to generate wt for APP/hAβ/PS1;Crfr1−/− animals. Only males were used for experiments.
PTSD-like induction procedure
Animals were designated as control (no stress) or PTSD (stress). Control and PTSD animal groups were moved to the behavioral testing facility. In a separate room, animals in the PTSD group were immobilized on plastic cutting boards with four pieces of autoclave tape, one piece per arm or leg. If, during the stress, the animal got free from one of the restraints, a new piece of tape was used to retape that appendage. The area in which the animals remained during immobilization was brightly lit (>700 lux). After 120 min, both groups of animals were returned to their home cages and placed back on the housing rack. For the trigger, all animals again were moved to the behavioral testing area. The PTSD group was affixed to cutting boards with autoclave tape. After 15 min, they were returned to their home cages and placed back in the housing room, along with control animals. Cages were marked not to be changed for the remainder of the experiment and were left undisturbed for the next 7 d. For semagacestat (SEMA) experiments, SEMA (Med Chem Express) was dissolved at 25 mg/ml in ethanol and then diluted at 10% by volume in 10% Cremophor solution. Animals received a 100 mg/kg subcutaneous injection of SEMA 1 d before the procedure and on the stress and reminder days of PTSD-like induction.
Behavioral testing
After PTSD-like induction, animals were returned to their home housing room and left undisturbed for 7 d. On day 1 of behavioral testing, all animals were tested in the light–dark transfer (LDT) test. On day 2, animals were tested in the acoustic startle box and on day 3 in the open-field assay (OFA).
LDT test.
LDT testing was performed in activity monitoring frames fitted with Plexiglas partitions that divided a square open field into a light field (half of the field) and a dark field (the other half of the field), separated by a small opening. Animals were placed in the light field and allowed to move freely between the light and dark areas for 10 min, while movement was recorded by computer via horizontal beam breaks. Time in each partition (light and dark) was recorded, as well as the number of transfers between the two areas. After testing, animals were grouped by genotype and manipulation (PTSD or control), and an average score for each genotype and manipulation was determined.
Acoustic startle testing.
Animals were placed in a Plexiglas restraining chamber within a startle testing box (SR Labs). After 5 min of exposure to background white noise (60 dB), animals received a series of startle stimuli presented randomly. Each stimulus was presented six times. Stimuli included pure startle (60, 74, 78, 82, and 120 dB) and prepulse inhibition trials (74–120, 78–120, and 82–120). Amplitude of startle was measured for 500 ms after presentation of the stimulus, and the peak magnitude during that 500 ms window was measured as the startle magnitude (at a sampling frequency of 1 kHz). After testing, startle magnitude of each animal during presentation of a given stimulus was averaged between the six trials. Animals were grouped by genotype and manipulation, and group averages were computed for each stimulus parameter.
OFA.
Animals were placed in a 36 cm on each side square open field where their position in the field was tracked based on a photobeam-based system (Kinder Scientific) and were allowed to move freely for 30 min. Total distance, horizontal activity, and vertical activity were quantified, as well as time, distance, and horizontal activity within a surround zone comprising the edge portion of the field (9 cm bordering the edge of the field on all sides) and the center zone of the field (center 18 cm square field). In addition, the center to total distance and time were also calculated as a correlate of anxiety-related behavior. Animals were tested blind to genotype and manipulation [the experimenter that was performing the test did not know the genotype or manipulation (control vs stress)], identified, grouped, and averaged.
Corticosteroid measurement
To measure resting corticosteroid levels, single cages of mice were moved to a quiet, undisturbed room. After 2 h, an experimenter entered the room and performed retro-orbital eye bleeds on all of the mice in that room in <5 min. Approximately 150 μl of whole blood was taken from each animal. Resting bleeds were always performed at a similar time during the day (12:00 to 2:00 P.M.) to avoid differences attributable to diurnal cycling of Cort release. For peak corticosteroids, animals were moved to a quiet room and allowed to calm down for 30 min. They were then placed in a 50 ml conical tube with the pointed end removed for ventilation. After 20 min of restraint, animals were bled by retro-orbital eye bleed. Approximately 150 μl of whole blood was taken from each animal. For corticosteroid measurement during immobilization stress, animals were immobilized on boards and tail bled every 15–60 min. Approximately 30 μl of blood was sampled at each time point. For dexamethasone suppression, animals were given an injection of 30 μg/kg dexamethasone (Sigma-Aldrich) 2 h before blood samples were taken.
Blood samples were collected with 2 μl of 500 mm EDTA, pH 8.0, and centrifuged at 5000 × g for 10 min to remove blood cells. Plasma was removed from the pellet, placed in a fresh tube, and frozen until quantification. Cort was quantified using a Luminex platform and the Stress Hormone quantification assay (RSHMAG-69K; Millipore). Cort measurements for each animal were grouped by animal genotype and manipulation and averaged. In the case of resting Cort, a measurement was discarded if it was >100 ng/ml because this animal was most likely not at rest when sampled. Less than 2% of animals displayed >100 ng/ml resting corticosteroids, and these were not consistently from any genotype or manipulation group.
Immunohistochemistry
Immunohistochemistry was performed essentially as described previously (Justice et al., 2008). Briefly, mice were transcardially perfused with saline and then 4% paraformaldehyde, and the brain was removed, sucrose equilibrated, and sectioned on a frozen sliding microtome. Free-floating sections were washed in PBS and incubated in antibodies overnight. For mGluR5, we used rabbit anti-mGluR5 (AB5675; Millipore) at a concentration of 1:1000. After washing in PBS, sections were incubated in appropriate secondary antibodies for 2 h, washed, and mounted on gelatin-coated slides. Images were obtained on a Nikon A1 confocal microscope.
CSF collection
We performed CSF collection essentially as described by Liu and Duff (2008), with some minor modifications. Briefly, animals were anesthetized using isoflurane gas and placed in a stereotaxic frame. A sagittal cut was made in the scalp over the dorsal neck muscles. Using retractors, we separated the muscle to reveal the dura mater above the magna cisterna. A pulled capillary tube was inserted through the dura mater, and CSF flowed out into the capillary. CSF was expelled from the capillary into a storage tube and frozen until analysis. CSF analysis was performed using 3–5 μl of CSF on a Luminex platform, using the human neurodegenerative disease milliplex assay for human Aβ40 and human Aβ42 (HNDG4MAG-36K; Millipore), which is expressed in APP/hAβ/PS1 mice but not in wild-type mice.
Primary neuronal culture
Primary cultures were made from P0 mouse pups of the genotype CRF–cre;loxP–stop–loxP–tdTomato. Pups were decapitated, and the dorsal portion of the skull was removed. A cut was made with a razor blade at rostral (approximately the frontal cortex) and caudal (in front of the cerebellum) positions, and the center section of the brain was removed. From this section, the dorsal half containing the hippocampus and neocortex was removed. The remaining piece was washed in HBSS before being trypsinized for 10 min in 0.5% trypsin (Worthington Biochemical) and DNase. Trypsin inhibitor was added to stop the digestion, and the tissue was triturated to a single-cell suspension. Cells were centrifuged and resuspended in Neurobasal media supplemented with B27 (Invitrogen) before being counted and plated on poly-d-lysine-coated glass coverslips. Cultures were used for electrophysiological recordings at 10–14 d in culture.
Electrophysiological recording in culture
Coverslips with primary neuronal cultures were transferred to a chamber mounted on an Olympus upright microscope (BX50WI) and perfused with oxygenated artificial CSF (ACSF; in mm): 122 NaCl, 3 KCl, 1.2 NaH2PO4, 26 NaHCO3, 20 glucose, 2 CaCl2, and 1 MgCl2, pH 7.3 (305–310 mOsm). Borosilicate glass electrodes (Sutter Instruments) were used for whole-cell patch-clamp recordings. Electrodes were pulled with tip resistance between 4 and 7 MΩ and filled with internal solution (in mm): 120 K-gluconate, 5 KCl, 2 MgCl2, 0.05 EGTA, 10 HEPES, 2 Mg-ATP, 0.4 Mg-GTP, and 10 creatine phosphate, pH 7.3 (290–300 mOsm). Cells were visualized by both fluorescent and differential interference contrast imaging. Data were obtained via a Multiclamp 700B amplifier, low-pass Bessel filtered at 4 kHz, and digitized on computer disk (Clampex; Molecular Devices). No liquid-junction potential was corrected.
Slice electrophysiology
CRF–cre;loxP–stop–loxP–tdTomato mice (P15–P24) were anesthetized by isoflurane and decapitated. The brain was quickly excised and immersed into ice-cold cutting solution [in mm: 60 NaCl, 110 sucrose, 28 NaHCO3, 7 MgSO4, 3 KCl, 1.25 NaH2PO4, 0.5 CaCl2, and 0.6 Na-ascorbate (bubbled with 95% O2/5% CO2)] until the brain tissue was chilled completely. Subsequently, 250 μm coronal slices containing the paraventricular nucleus (PVN) were obtained using a Leica VT1200S vibratome. Slices were immediately transferred and incubated in normal ACSF [in mm: 125 NaCl, 26 NaHCO3, 1 MgSO4, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, and 10 glucose (bubbled with 95% O2/5% CO2)] at 35°C for at least 60 min before recording. Individual brain slices were placed in the recording chamber (Warner Instruments) and continuously perfused with either normal ACSF or modified ACSF supplemented with 20 μm CNQX, 30 μm d-AP-5, and 100 μm picrotoxin to block fast synaptic transmission mediated by AMPA, NMDA, and GABAA receptors, respectively. Control medium (CM), Aβ CM, Aβ CM + SEMA, and MPEP were all diluted in either normal or modified ACSF, as indicated in Results, and applied through whole chamber perfusion. Glass pipettes (Sutter Instrument) were pulled using a Narishige PC-10 puller. The pipette tip resistance was 3–5 MΩ when filled with the intracellular solution containing the following (in mm): 122 K-gluconate, 9 NaCl, 1.8 MgCl2, 0.9 EGTA, 9 HEPES, 14 Tris-creatine PO4, 4 Mg-ATP, and 0.3 Tris-GTP, with pH adjusted to 7.4 by KOH (osmolarity of 300 mOsm). CRF-positive PVN neurons were visualized using a 60× water-immersion objective lens and infrared differential interference contrast/fluorescence video microscopy (Olympus BX51WI with OLY-150IR video camera). Selected PVN neurons were voltage clamped at −70 mV before switching to current-clamp mode, and current was injected to allow action potential firing at low frequency. Cell firing was continuously monitored and recorded before and during drug administration. The temperature of the recording chamber was maintained at ∼32°C by passing the perfusion solution through an in-line heater (Warner Instruments) at 3 ml/min driven by a Rabbit peristaltic pump (Rainin Instrument). Data were acquired using an EPC10 amplifier operated by PatchMaster software (both from HEKA). Recordings were filtered at 3 kHz and digitized at 10 kHz. Firing frequency was calculated as spikes per second. All measurements were made offline using the analysis functions of the PatchMaster software. Representative traces in figures were reproduced in OriginPro (OriginLab).
CM collection
Untransfected CHO cells (control) or CHO cells stably transfected with human APP695 (SV695; kindly provided by S. Sisodia, University of Chicago, Chicago, IL) were plated at a density of 2 × 106 per 10 cm plate. Twenty-four hours later, cell culture media were removed, and cells were washed once with PBS and then incubated in ACSF buffer (see above, Slice electrophysiology); the same buffer that is used for electrophysiological recordings. After 24 h, media was removed from the cells and sterile filtered. Media were stored at 4°C.
Statistics
Most statistical comparisons between genotypes for behavioral and endocrine measurements were performed using a two-tailed Student's t test. Concentrations of corticosterone were analyzed using the MIXED procedure of SAS for repeated measures (Littell et al., 1998). The model included genotype, day, and the genotype × day interaction. The most appropriate covariance structure for the analysis was chosen from unstructured, compound symmetric, spatial power, and ante-dependence structures using Akaike's information criterion and Schwarz's Bayesian criterion (Littell et al., 2000). Kenward–Rogers approximation was used for the calculation of the degrees of freedom of the pooled error term. The random effect of mouse within each genotype accounted for the correlations among repeated observations on the same mouse. If the genotype × day interaction was significant (p < 0.05), means separations were evaluated on each day using the PDIFF function of SAS. If the main effect of genotype was significant (p < 0.05), then means separation were wild type versus APP/hAβ/PS1. Fisher's exact test was used to compare the percentages of mice within genotypes that displayed the PTSD-like phenotype. The number of animals used for each experiment is listed in the figure legends. For slice electrophysiology, Student's t test was used to compare the mean values of two groups of measurements. Repeated one-way ANOVA analysis followed by Newman–Keuls post hoc tests were used to compare firing frequency changes for the same group of individual cells that have been sequentially exposed to three different drug treatments. Data were presented as means ± SEMs. A difference between values was determined to be statistically significant if p < 0.05. In all figures, *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
PTSD-like induction protocol
To model PTSD in the mouse, we subjected mice to a single intense trauma, followed by a trigger the next day (Fig. 1A). The trauma used is a 120 min immobilization stress in which the arms and legs of mice are secured by tape to a board. This method of PTSD-like induction has been shown to produce chronic PTSD-like changes in HPA axis activity and anxiety-related behavior (Marti et al., 2001; Armario et al., 2008; Yamamoto et al., 2009). The trigger is a brief (15 min) immobilization that serves as a reminder and has been shown to reinforce the experience of the original trauma (Lebow et al., 2012). One week after trauma exposure, animals are tested for PTSD-like behavioral changes (Fig. 1A). During the following 2 weeks, blood is sampled from the animals to measure resting and peak Cort levels (Fig. 1A). Some animals are aged to measure the long-term effects of PTSD-like induction. During the initial immobilization, we measured corticosteroid levels repeatedly during and after immobilization stress (Fig. 1B). Wild-type animals display a sharp rise in Cort levels at the onset of the stress within 15 min. Cort levels then continue to rise for the remainder of the stress and begin to fall after the animals are released from restraint (Fig. 1B).
Figure 1.

PTSD-like induction procedure and phenotype. A, Diagram of the PTSD-like induction and testing procedure. B, Blood from wild-type animals was sampled every 15–30 min during the induction procedure. Circulating Cort rises dramatically at the onset of stress and slowly increases during immobilization before falling after the animal is released (n = 10). C, Cort levels were measured at rest (left) and after 20 min of restraint (right; peak). Cort levels in both conditions were lower in animals that had been exposed to PTSD-like induction (control, n = 18; stress, n = 15). D, Animals were tested for anxiety-related behavior. In the LDT test (2 left graphs), time in the light side and transitions to the light side were reduced after PTSD-like induction. In the OFA (3rd graph), the ratio between distance traveled in the center versus total distance traveled was reduced. In startle testing (right graph), wild-type animals displayed a nonstatistically significant increase in startle amplitude after PTSD-like induction (control, n = 15; stress, n = 21).
Wild-type mice display a PTSD-like phenotype after single-session trauma exposure
To establish a baseline PTSD-like phenotype, we first assessed chronic endocrine and behavioral changes that occur after PTSD-like induction in wild-type animals. We saw a nonstatistically significant decrease in resting Cort (17.1 ± 3.3 vs 20.2 ± 2.6 ng/ml, t(30) = 0.73, p = 0.47) and a decrease in peak Cort (265.4 ± 7.5 vs 305.1 ± 23.4 ng/ml, t(12) = 2.01, p < 0.05) in PTSD-like induced animals (Fig. 1C), as expected (Marti et al., 2001; Armario et al., 2008; Yamamoto et al., 2009). Similar decreases in circulating corticosteroid levels have been reported in humans with PTSD, although whether this is a consistent symptom among all PTSD patients is not clear (Yehuda, 2009; Marin et al., 2011). We tested control and PTSD-induced wild-type animals for anxiety-related behavior using multiple assays. In the LDT test, PTSD-like induced animals made fewer transitions to the light area (24.8 ± 2.4 vs 34.3 ± 2.5, t(34) = 2.63, p < 0.01) and spent less time in the light area of the maze (224.9 ± 17.1 vs 277.5 ± 17.6, t(34) = 2.09, p < 0.05), indicating elevated anxiety levels (Fig. 1D). In the OFA, animals also displayed elevated anxiety, traveling less distance in the center versus the surround (21.7 ± 1.4 vs 28.6 ± 1.1%, t(49) = 3.69, p < 0.001), a standard measure of anxiety-related behavior (Fig. 1D). PTSD patients often display elevated acoustic startle (Butler et al., 1990; Morgan et al., 1996; Glover et al., 2011), so we measured startle amplitude in response to a 120 dB sound pulse. PTSD-like induced animals displayed slightly elevated startle amplitudes (1039.1 ± 153.0 vs 801.8 ± 104.4 a.u., t(28) = 1.36, p = 0.21), although this did not reach statistical significance (Fig. 1D). Together, these endocrine and behavioral phenotypes seen in mice after single-session acute trauma approximate the symptomatic profile of patients diagnosed with PTSD (Yamamoto et al., 2009; Glover et al., 2011).
APP/hAβ/PS1 AD model animals display a stronger PTSD-like phenotype after trauma exposure
PTSD diagnosis correlates with elevated AD risk (Qureshi et al., 2010; Yaffe et al., 2010), so we tested whether knock-in AD model animals display a similar PTSD-like endocrine and behavioral phenotype when exposed to PTSD-like induction. We used a familial AD (FAD) knock-in AD model mouse in which homozygous FAD mutations and a humanized Aβ region are present in the APP genomic locus (Hartmann et al., 2004; Köhler et al., 2005), and a homozygous FAD mutation is carried in the PS1 locus (APP/hAβ/PS1 mice; Flood et al., 2002). This model does not display amyloid plaque formation until an advanced age (18 months) and therefore best models an early pre-plaque phase of AD pathogenesis, without high levels of ectopic APP expression. We measured Cort levels during the PTSD-like induction procedure in APP/hAβ/PS1 animals. In contrast to wild-type animals in which Cort levels continue to rise for the duration of the stress (Fig. 1B), Cort levels in APP/hAβ/PS1 animals peak at 15 min and plateau (F(1,117) = 10.61, p < 0.01; Fig. 2A). Surprisingly, given that the system appears to be hyperactivated in APP/hAβ/PS1 animals (Guo et al., 2012), Cort levels were significantly lower in APP/hAβ/PS1 animals compared with wild-type animals at 45 min (t(17) = 2.01, p < 0.05), 90 min (t(18) = 2.53, p < 0.05), and 120 min (t(17) = 2.29, p < 0.05) of immobilization stress (Fig. 1B). When we measured resting and peak Cort levels 2 weeks after the PTSD-like induction, although resting Cort levels did not change dramatically (25.0 vs 26.1 ng/ml, t(55) = 0.28, p = 0.96; Fig. 2B), peak Cort levels increased in APP/hAβ/PS1 animals exposed to PTSD-like induction (357.5 ± 29.2 vs 274.4 ± 10.9 ng/ml, t(56) = 2.63, p < 0.01; Fig. 2B). In addition, stressed APP/hAβ/PS1 animals displayed an enhanced suppression of corticosteroid levels 120 min after injection with dexamethasone when compared with stressed wild-type animals (13.8 ± 2.18 vs 17.2 ± 0.14 ng/ml, t(9) = 2.22, p < 0.05; Fig. 2B). Therefore, in contrast to wild-type animals, we see blunted acute HPA axis responses to immobilization stress, chronically elevated peak corticosteroid levels, and chronically enhanced negative feedback on the HPA axis, in APP/hAβ/PS1 AD model animals in response to trauma exposure.
Figure 2.

APP/hAβ/PS1 PTSD-like phenotype. A, Corticosteroid levels during immobilization of wild-type animals (blue line) and APP/hAβ/PS1 animals (red). APP/hAβ/PS1 animals display a blunted corticosteroid release during PTSD-like induction (n = 10 per genotype). B, Resting and peak corticosteroid levels in control (light red) and PTSD-like induced (dark red) animals. Resting Cort levels fall in stress-exposed APP/hAβ/PS1 animals. Peak Cort levels are elevated in PTSD-like induced animals (n = 29 per condition). APP/hAβ/PS1 animals displayed exaggerated dexamethasone (DST) suppression compared with wild-type animals after being exposed to trauma (right; n = 6 per genotype). C, D, Behavioral assessment of PTSD-like phenotype in APP/hAβ/PS1 animals. APP/hAβ/PS1 animals have a baseline anxiety-related phenotype. Increased anxiety levels after PTSD-like induction are evident on the LDT test (light entries) and acoustic startle (control, n = 19; stress, n = 34). E, The percentage of animals that display a PTSD-like phenotype. The APP/hAβ/PS1 genotype is associated with a higher frequency of animals displaying PTSD-like behavior on all three behavioral assays. F, Aβ40 and Aβ42 levels in young (2–3 months, left bars; control, n = 8; stress, n = 10) and old (6–12 months, right bars; control, n = 8; stress, n = 5) APP/hAβ/PS1 animals. Control animals (light red bars) did not experience trauma, whereas PTSD animals experienced trauma at 8–12 weeks of age.
We measured chronic changes in anxiety-related behavior in APP/hAβ/PS1 animals using the LDT test, OFA, and startle. APP/hAβ/PS1 animals display a baseline anxiety phenotype (Guo et al., 2012), and PTSD-like induction did not increase many measures of anxiety-related behavior above those already elevated levels (Fig. 2C). However, when measured for acoustic startle to a 120 dB sound, PTSD-like induction caused a dramatic increase in startle amplitude in APP/hAβ/PS1 animals (1256.3 ± 115.7 vs 817.1 ± 134.2, t(53) = 2.4, p < 0.05; Fig. 2D). Furthermore, the percentage of APP/hAβ/PS1 animals that displayed a PTSD-like phenotype (designated as being above the mean in all three behavioral tests) was higher than observed in wild-type cohorts (19.3 vs 9.2%, p < 0.05; Fig. 2E). Therefore, the behavioral and endocrine PTSD-like responses were characteristically different and exaggerated in APP/hAβ/PS1 mice exposed to PTSD-like induction.
CSF Aβ increases chronically after PTSD-like induction
Stress has been shown to exacerbate ongoing AD pathogenesis in transgenic APP/AD model mice (Dong et al., 2004; Rissman et al., 2007; Carroll et al., 2011). We tested whether PTSD-like induction would have a similar effect by measuring Aβ levels in the CSF of APP/hAβ/PS1 mice. CSF was collected after initial behavioral and endocrine testing (3–4 months) or after aging the animals to 9–12 months old and measured for human Aβ40 and Aβ42, which are not present in wild-type control mice. In young animals, we saw elevated Aβ40 and Aβ42 in CSF samples after PTSD-like induction compared with unstressed controls (Aβ40, 2.83 ± 0.31 vs 1.95 ± 0.22 ng/ml, t(16) = 2.22, p < 0.05; Aβ42, 6.90 ± 0.70 vs 4.55 ± 0.46 ng/ml, t(16) = 2.65, p < 0.05; Fig. 2F). This elevation in CSF Aβ remained in PTSD-like induced aged mice, 6–9 months after the exposure to trauma (Aβ40, 3.44 ± 0.70 vs 1.96 ± 0.38 ng/ml, t(11) = 2.45, p < 0.05; Aβ42, 7.60 ± 1.34 vs 4.01 ± 0.70, t(11) = 2.98. p < 0.01; Fig. 2F). Therefore, stress exposure can result in persistent changes in the production and/or clearance of soluble Aβ peptide for extended periods, perhaps accelerating AD pathogenesis in the context of PTSD.
Loss of CRFR1 blocks PTSD-like induced changes in behavior and Aβ release
We previously showed that the CRF system is perturbed in APP/hAβ/PS1 animals (Guo et al., 2012). To determine whether this perturbed CRF circuitry might account for PTSD-like sensitivity in APP/hAβ/PS1 animals, we next asked whether Crfr1 mutations abrogate PTSD-like phenotypes. We crossed APP/hAβ/PS1 double knock-in animals with Crfr1−/− animals to generate APP/hAβ/PS1 animals lacking CRFR1. Crfr1 mutants display decreased anxiety-related behavior and a hypoactive and unresponsive HPA axis (Smith et al., 1998). We performed PTSD-like induction on animals mutant for Crfr1 and analyzed resulting behavioral phenotypes. The absence of CRFR1 caused PTSD-like induction to be ineffective in modifying behavior in APP/hAβ/PS1;Crfr1−/− animals and in Crfr1−/− mutants not carrying AD-causing mutations (Fig. 3A). In the LDT test and OFA, APP/hAβ/PS1 animals and control animals that lacked CRFR1 displayed equivalent numbers of light-side entries regardless of exposure to PTSD-like induction. PTSD-like induction also failed to increase startle amplitudes when Crfr1 is mutant, as it does in APP/hAβ/PS1 animals. These results indicate that CRF signaling and an intact HPA axis are required to express behavioral aspects of the PTSD-like phenotype.
Figure 3.
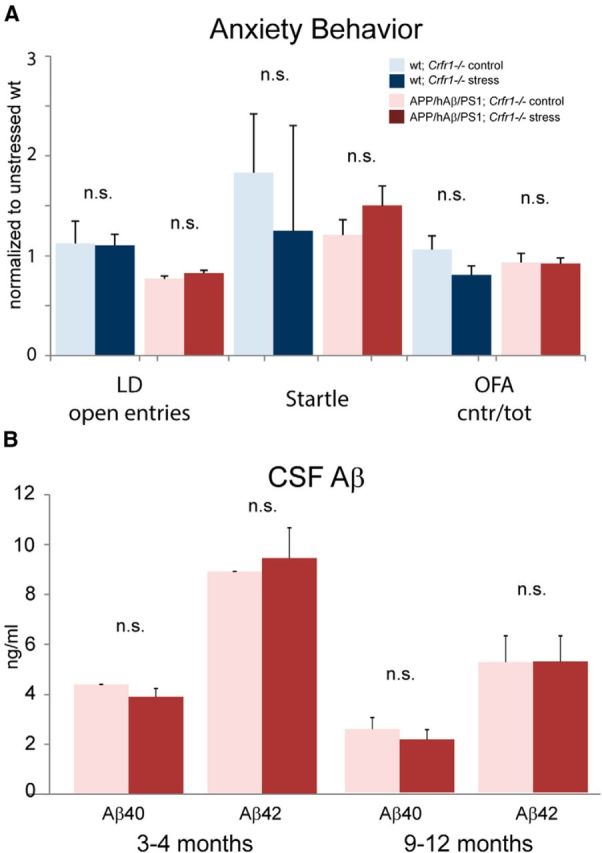
PTSD-like phenotype requires Crfr1. A, Wild-type (blue) and APP/hAβ/PS1 (red) animals were crossed into a Crfr1−/− background. In resulting mice, PTSD-like induction (darker bars) did not alter anxiety-related behavior or acoustic startle in either genotype. Data have been normalized to scores for wild-type, unstressed animals for each test (wild-type Crfr1−/− control, n = 6; wild-type Crfr1−/− stress, n = 5; APP/hAβ/PS1;Crfr1−/− control, n = 14; APP/hAβ/PS1;Crfr1−/− stress, n = 18). B, CSF Aβ levels were measured in APP/hAβ/PS1;Crfr1−/− animals at both a young (3–4 months) and old (6–12 months) time point. PTSD-like induction did not alter Aβ levels when animals lacked Crfr1 (young APP/hAβ/PS1;Crfr1−/−, n = 5 per condition; old APP/hAβ/PS1;Crfr1−/−, n = 5 control, n = 6 stress).
To examine the dependence of CSF Aβ elevations after PTSD-like induction on CRF signaling and the HPA axis, we measured CSF Aβ levels in APP/hAβ/PS1;Crfr1−/− PTSD-like induced mice at 3–4 months and 9–12 months of age. In contrast to APP/hAβ/PS1 animals, we saw no changes in Aβ40 or Aβ42 levels after PTSD-like induction at both the early and later time points (Fig. 3B), Interestingly, in 3- to 4-month-old APP/hAβ/PS1 animals, we saw consistently higher levels of Aβ40 and Aβ42 in APP/hAβ/PS1;Crfr1−/− animals (Fig. 3B compared with Fig. 2F). It remains possible that Crfr1 loss-of-function-caused increases in Aβ might mask Aβ changes that occur in response to PTSD-like induction. However, together with behavioral results, the lack of a change in Aβ levels is consistent with intact CRF signaling being critical to both the expression of the PTSD-like phenotype and the exacerbation of AD pathogenesis.
CRF neurons are hyperactivated by soluble Aβ species
AD-causing mutations in APP and PS1, which are present in the APP/hAβ/PS1 model animal, cause the overproduction of toxic Aβ species (Flood et al., 2002; Hartmann et al., 2004; Köhler et al., 2005). Given our observations that APP/hAβ/PS1 AD model animals are sensitized to PTSD-like induction, we asked whether Aβ might directly affect CRF neuron excitability and therefore PTSD-like sensitivity. We identified CRF neurons using a genetic strategy to fluorescently label neurons that express CRF, which is released in response to stress (Vale et al., 1981). Primary neuronal cultures were made from the ventral forebrain of P0 mouse pups containing a CRF–cre recombinase transgene (Crhtm1(cre)Zjh) and a floxed-stop tdTomato reporter gene [Ai9;B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J]. Neurons were cultured for 12–21 d, at which point they form synaptically integrated networks, of which a very small proportion (one to five per coverslip) express CRF and therefore tdTomato, causing them to be red fluorescent (Fig. 4B). Red fluorescent CRF-expressing cells were patched, and conditioned media were bath applied, diluted in ACSF recording solution. While recording whole-cell patch electrical activity in current-clamp mode, neurons were exposed to recording solution containing 10% conditioned media from control CHO cells (Fig. 4A), and baseline activity was measured as the rate of action potential firing (Fig. 4C). Maintaining the patch, we switched the bathing media to 10% conditioned media from SV695 cells that secrete Aβ into the culture medium because of a stable transgene expressing human APP695 (Hare, 2006). CRF neuron activity increased with the addition of conditioned media containing Aβ within 5 min (t(14) = 2.23, p < 0.05; Fig. 4C). To confirm that the effect is attributable to Aβ in the culture media, we performed the identical experiment but instead used conditioned media from SV695 cells cultured in 1 μm SEMA (Aβ + SEMA), a potent γ-secretase antagonist that blocks Aβ production, which contained no detectable Aβ (Fig. 4A). Aβ + SEMA conditioned media did not increase the firing rate of patched CRF neurons (t(4) = 0.16, p = 0.88; Fig. 4C,D).
Figure 4.

Aβ elevates CRF neuron activity in culture. A, Conditioned media from CHO cells (control CM) or CHO cells stably transfected with APP695 (SV695; Aβ CM) was measured for Aβ40 concentration. In Aβ CM, we detected significantly more Aβ than in control CM, in which Aβ levels were below the detection limit of the assay (dashed line). CHO-K1 or SV695 cells incubated with 1 μm SEMA, a γ-secretase inhibitor, also had undetectable Aβ levels. B, Neurons were prepared from P0 mouse pups of the genotype CRF–cre;lox–stop–lox tomato (CRF–tomato). CRF neurons from these pups are fluorescent in culture. C, Whole-cell patch of CRF neurons with the addition of 10% conditioned media. Aβ conditioned media increased the action potential firing rate of CRF neurons (n = 8 cells). Both CM from CHO-K1 or SV695 cells incubated with SEMA did not affect CRF neuron firing rate (n = 3 cells). D, Quantification of results in C, normalized to the average firing rate of CRF neurons in 10% control CM. Aβ CM induced cells to fire, on average, three to four times faster than control CM.
Next, we performed similar experiments in brain slices prepared from CRF–cre;lsl–tdTomato mice. We patched red fluorescent CRF neurons located in the PVN (Fig. 5A) and perfused the slices with ACSF containing conditioned media. Again, we observed increases in action potential firing frequency in CRF neurons during exposure to Aβ containing conditioned media but not in conditioned media that was collected in the presence of 1 μm SEMA (F(2,20) = 18.35, p < 0.01; Fig. 5B). CRF neuron excitation might be attributable to elevated synaptic network activity or to the cell-autonomous effects of Aβ on CRF neurons. To distinguish between these possibilities, we incubated the slices in APV, CNQX, and picrotoxin to block synaptic currents and perfused slices with 10% Aβ conditioned media. With synaptic activity blocked, we again saw an increase in action potential firing frequency (F(2,14) = 7.71, p < 0.05; Fig. 5C), indicating that Aβ directly affects CRF neuron excitability. Aβ oligomers have been found recently to selectively excite subpopulations of neurons (Busche et al., 2012; Um et al., 2013). This can occur through the action of Aβ bound to prion protein activating mGluR5, a metabotropic glutamate receptor (Um et al., 2013). To test whether mGluR5 on CRF neurons mediates Aβ sensitivity, we included 100 μm MPEP, an mGlur1/5 antagonist, in 10% Aβ conditioned media while recording CRF neuron activity by patch electrode in slice. MPEP blocked the effect of Aβ in the conditioned media (t(5) = 0.54, p = 0.61; Fig. 5D), suggesting that mGluR5 mediates the excitation of CRF neurons by Aβ. Consistent with this finding, we saw positive labeling for mGluR5 in CRF neurons in the PVN (Fig. 5A). Finally, to test whether the effect of Aβ conditioned media is dose dependent, we patched CRF neurons in the PVN in slice and perfused sections with different percentages of Aβ CM diluted in ACSF. We found that even 1% Aβ conditioned media elicited an increase in firing rate (Fig. 5E). Increasing the concentration of Aβ CM increased the firing frequency to a maximal response at 10% (F(4,14) = 5.95, p < 0.05; Fig. 5E). We recorded from CRF neurons in the central nucleus of the amygdala and bed nucleus of the stria terminalis (BST) and did not see an excitatory effect (data not shown), suggesting that Aβ selectively activates CRF neurons located in the PVN. These neurons project to the median eminence and release CRF to the pituitary, so Aβ perturbation could directly activate HPA axis activity and peripheral corticosteroid release.
Figure 5.
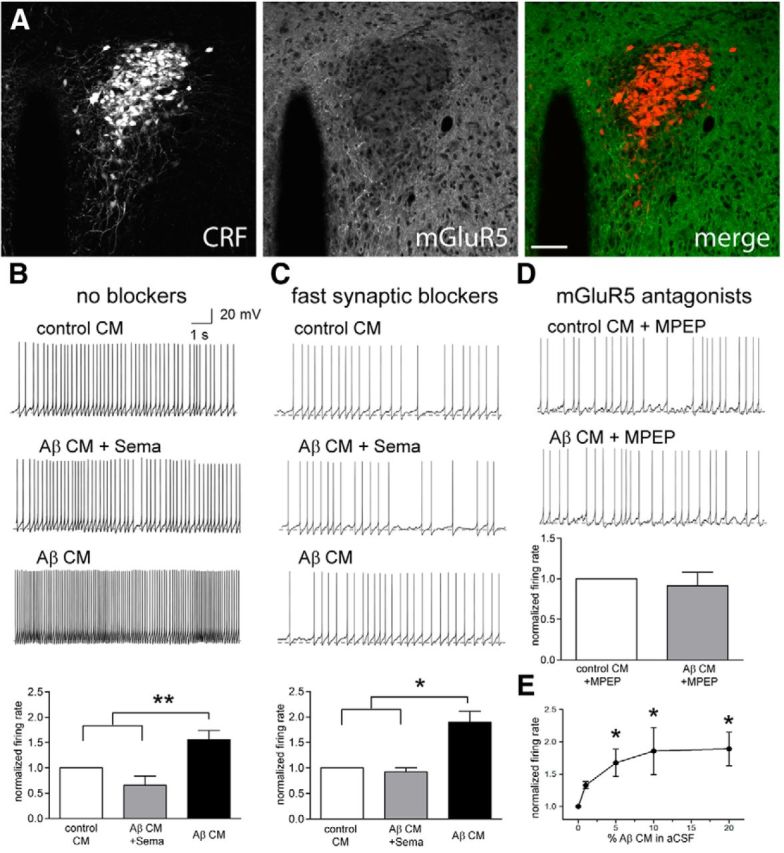
PVN CRF neurons are excited by Aβ conditioned media. Crf–cre;lsl–tdTomato mice were imaged for tomato (left) and stained for mGluR5 (middle). CRF neurons in the PVN express a moderate level of mGluR5. Scale bar, 100 μm. B, CRF neurons in the PVN were patched in slice and perfused with control CM (top trace), CM from APP-expressing cells incubated with 1 μm SEMA (Aβ CM + Sema; middle trace), and CM from APP cells (Aβ CM; bottom trace). PVN CRF neurons increase action potential firing when exposed to Aβ containing CM (quantified in the bottom graph; n = 7 cells). C, Slices were incubated in synaptic blockers to isolate individual neuron excitability. CRF neurons increase action potential firing rate in Aβ CM (bottom trace; n = 5 cells) but not in control CM (top trace) or Aβ CM collected in the presence of SEMA (middle trace). D, PVN CRF neuron excitation by Aβ in the conditioned media is blocked when coapplied with MPEP, an mGluR5 antagonist (n = 6 cells). E, Dose response of PVN CRF neurons to varying dilutions of CM in ACSF. One percent Aβ CM caused a small increase in action potential firing. Ten percent Aβ CM caused a maximal response equivalent to the response to 20% Aβ CM (n = 3 cells).
γ-Secretase inhibitors reverse PTSD-like behavioral phenotypes in APP/hAβ/PS1 mice
Given that local Aβ release can influence the excitability of CRF neurons, we wanted to test whether this might account for why APP/hAβ/PS1 animals are sensitive to PTSD-like induction. To test this in vivo, we injected mice with SEMA (100 mg/kg, s.c.), a γ-secretase inhibitor, to block production of Aβ and lower Aβ levels, 1 d before and on the stress and trigger days of the PTSD-like induction procedure. Behavioral testing of APP/hAβ/PS1 animals 2 h after injection with 100 mg/kg SEMA did not reveal decreases in anxiety-related behavior (data not shown). We tested SEMA- and vehicle-treated APP/hAβ/PS1 animals for behavioral and endocrine responses 7 d after trauma exposure, according to the same schedule used in previous experiments. We saw that APP/hAβ/PS1 animals that were injected with SEMA during trauma exposure now displayed an abrogated PTSD-like phenotype. This was not most evident on the LDT test, in which we saw an nonsignificant increase in light time in APP/hAβ/PS1 compared with stressed vehicle controls (257.1 ± 39.1 vs 217.6 ± 22.1, t(18) = 1.15, p = 0.27; Fig. 6A) and no change in the number of light entries (23.2 ± 3.7 vs 25.5 ± 5.5, t(11) = 0.36, p = 0.73; Fig. 6A). However, on the acoustic startle test, we saw a significant reduction of startle amplitude in APP/hAβPS1 animals given SEMA during trauma exposure compared with vehicle-treated stressed APP/hAβ/PS1 animals (825.0 ± 93.1 vs 1237.5 ± 89.8, t(24) = 3.13, p < 0.01; Fig. 6B). Similarly on the OFA, we saw an increase in center/total distance ratio in SEMA-treated animals compared with vehicle-treated animals (28.1 ± 2.0 vs 23.3 ± 2.7%, t(10) = 2.34, p < 0.05; Fig. 6C). SEMA during trauma also lowered peak corticosteroids chronically in stressed APP/hAβ/PS1 animals (256.8 ± 10.8 vs 282.4 ± 8.0 ng/ml; t(31) = 2.09, p < 0.05; Fig. 6D). Finally, we measured CSF Aβ levels in nonstressed SEMA-treated APP/hAβ/PS1 animals immediately after the third injection and found that this lowers CSF Aβ40 levels (1702.3 ± 385.8 vs 3307.8 ± 228.0 pg/ml, t(12) = 3.58, p < 0.01; Fig. 6E). These findings are consistent with SEMA decreasing Aβ levels and thereby relieving Aβ perturbation of CRF neurons during trauma exposure, which reduces chronic PTSD-like behavioral changes in APP/hAβ/PS1 animals.
Figure 6.

γ-Secretase inhibitors reverse PTSD-like phenotypes in APP/hAβ/PS1 animals. A, Treatment with SEMA during PTSD-like trauma exposure did not significantly alter behavior on the LDT test. B, SEMA decreased acoustic startle to a 120 dB sound. C, SEMA increased center/total distance traveled in the OFA. D, SEMA treatment decreased peak corticosteroid levels observed in trauma-exposed animals 2 weeks after the stress (for A–D, vehicle, n = 17; SEMA, n = 14). E, CSF sampled from unstressed APP/hAβ/PS1 animals after receiving 3 d of SEMA injections contained less Aβ40 compared with vehicle controls (n = 7 per treatment).
Discussion
Interactions between PTSD and early-stage AD-related pathogenesis
Human subjects with PTSD are more likely to suffer dementia as they age, with the majority of these cases having AD (Qureshi et al., 2010; Yaffe et al., 2010). This might be attributable to an exacerbating effect of changes in nervous system regulation that occur with PTSD, specifically alterations in the regulation of the HPA axis (Yehuda et al., 1991; Ehlert et al., 2001; Yehuda, 2009; Morris et al., 2012). In addition, there might be factors that cause susceptibility to both PTSD and AD, such that those who succumb to PTSD in the event of trauma exposure are predisposed to dementia with age. For example, smaller hippocampal volume and lower IQ have been linked to elevated incidence of both PTSD and AD (Gilbertson et al., 2002; Richards and Sacker, 2003; Csernansky et al., 2005; Koenen et al., 2007). To examine how these two mechanisms might account for increased dementia risk in PTSD patients, we used an animal model of PTSD in normal and knock-in AD model mice. We found that AD model mice have an exaggerated response, by both behavioral and endocrine measurements, to PTSD-like induction (Fig. 2). AD model mice also displayed a PTSD-like phenotype at a higher frequency after trauma exposure (Fig. 2). These findings support a scenario in which animals predisposed to AD-related pathology incur more severe PTSD-like symptoms after trauma exposure. In line with stress sensitivity being caused by early AD pathogenesis, patients at the earliest stages of AD diagnosis often have elevated corticosteroid levels (Davis et al., 1986; Lupien et al., 1998; Csernansky et al., 2006). In addition, late-life depression has been shown recently to elevate the risk for AD by twofold (Barnes et al., 2012), again suggesting a coincidence between abnormal activity of stress pathways associated with depression and AD progression. This effect was much larger in those who displayed late-life versus mid-life depression, consistent with stress axis disruption occurring with increasing levels of AD-related pathology (Barnes et al., 2012). Numerous studies have now shown real-time changes in soluble Aβ levels in the brain with varying levels of stress and mental status (Kang et al., 2007, 2009). Whether daily changes in Aβ levels might be in play at the relatively young ages that many PTSD patients experience the traumatic event associated with chronic PTSD remains unclear. However, we have shown that genetically induced early AD pathogenesis has the potential to disrupt stress responses and influence chronic PTSD-like acquisition.
We found that exposure to PTSD-like inducing trauma elevated CSF Aβ levels in both the short term (1–2 months) and chronically (6–12 months). This increase in Aβ levels might accelerate AD pathogenesis, leading to exacerbated amyloid plaque deposition, as has been seen in many AD models when exposed to acute stressors (Dong et al., 2004; Lee et al., 2009; Devi et al., 2010; Carroll et al., 2011). In addition, given findings presented here that Aβ can perturb the stress circuitry, this increase in Aβ production might also exacerbate chronic changes in behavior and corticosteroid regulation, resulting in a higher incidence of PTSD. Once chronic PTSD symptoms are established, chronically altered corticosteroid levels, which have been shown to exacerbate AD pathogenesis (Green et al., 2006; Catania et al., 2009; Li et al., 2010; Joshi et al., 2012; discussed below), as well as aberrant activity of stress-circuits centrally (Kang et al., 2007; Rissman et al., 2007), might combine to cause an accelerated loss of cognitive function, and the diagnosis of dementia at an earlier age. With both stress sensitivity caused by trauma-induced overproduction of Aβ and subsequent stress exacerbation of later AD pathogenesis, these two mechanisms potentially function in a vicious cycle to drive neuropathological progression of AD in the context of PTSD.
Early-stage AD pathogenesis alters stress circuit and HPA axis function
One feature that unites many of the transgenic and knock-in AD model mice is elevations in stress-related behavior, as well as changes in activity of the HPA axis and elevated circulating corticosteroid levels, often preceding any pathological signs of disease, such as amyloid plaque formation (Dong et al., 2008; Guo et al., 2012; Rothman et al., 2012; Hebda-Bauer et al., 2013). Interestingly, early-onset changes in circulating corticosteroid levels are also common in humans at the MCI stage of AD (Davis et al., 1986; Lupien et al., 1998; Csernansky et al., 2006). Pre-AD incidence of neuropsychiatric symptoms has also been reported. Carriers of FAD mutations in PS1 or APP have been reported to have a higher incidence of depression and stress-related symptoms (Kennedy et al., 1993; Harvey et al., 1998; Kasuga et al., 2009). Depression is also associated with a higher incidence of AD (Barnes et al., 2012). An Alzheimer's Disease Neuroimaging Initiative study showed that those who presented with both depression and MCI had significantly more loss in brain scanning density than those without depression when scanned by MRI 2 years later (Lee et al., 2012). This multifaceted evidence from both model mice and humans strongly suggests that events that occur before overt cognitive loss that leads to the MCI diagnosis correspond to disease-related stress axis disruption. Many direct and indirect mechanisms may contribute to the activation of the HPA axis and resulting behavioral and endocrine changes that occur with AD. One proposed mechanism for these changes is a corollary of the glucocorticoid cascade hypothesis (Sapolsky et al., 1986). In this hypothesis, loss of hippocampal function, in this case attributable to AD pathogenesis, would lead to decreased inhibitory control of the HPA axis, most likely through a neural circuit that includes the BST and the PVN, resulting in higher Cort levels. Another indirect hypothesis is that elevated inflammation caused by AD pathogenesis activates central autonomic pathways that activate the HPA axis via the nucleus of the solitary tract (Serrats and Sawchenko, 2006; Serrats et al., 2010). Although these indirect mechanisms may account for alterations in the HPA axis with AD, we chose to test direct mechanisms of HPA axis alteration by looking at activation of CRF neurons, which control HPA axis output, by elevated levels of toxic species being generated in AD.
CRF neuron activation by Aβ signals progressing disease
Soluble Aβ oligomers have been shown to have a wide range of toxic effects on neurons, including synaptic destabilization, dendritic spine loss, and reduced LTP (Walsh et al., 2002; Shankar et al., 2007, 2008; Zempel et al., 2010, 2013; Mairet-Coello et al., 2013). Recently, Aβ has been shown to have an acute excitotoxic capacity, exciting ∼25% of hippocampal neurons during exogenous application of Aβ (Busche et al., 2012). This could be attributable to the interaction of Aβ with a number of reported Aβ receptors (Mezler et al., 2012; Kim et al., 2013; Um et al., 2013). Here, we show that the excitatory drive of Aβ has a functional effect by activating CRF neurons that control HPA axis activity. The HPA axis is activated in response to a wide range of stimuli and is thought to be a generalized signal of distress or movement away from physiological homeostasis (Selye, 1948, 1950). In this case, activation of CRF neurons by proteotoxic species such as Aβ might indicate the inability of the brain to maintain proteostasis, activating corticosteroid release. The action of corticosteroids might function to sensitize the brain to additional insults of advancing pathogenesis (Sapolsky, 1999) or to increase Aβ clearance to reduce toxicity (discussed below). However, the clinical result of an activated stress axis is the production of neuropsychiatric symptoms, such as depression, insomnia, aggression, and anxiety, that can often be present with AD (Tune, 1998; Braun and Kunik, 2004; Barnes et al., 2012).
Cort might be both beneficial and detrimental in AD
Much emphasis has been placed on the detrimental effects of Cort on the progression of AD (Green et al., 2006; Sotiropoulos et al., 2008; Huang et al., 2009; Joshi et al., 2012) because of the known effect of elevated Cort on neuronal health (Sapolsky, 1996). However, our results suggest that Cort can also lower Aβ levels. In AD model animals that lack Crfr1 (APP/hAβ/PS1;Crfr1−/−), Aβ levels are elevated dramatically in young animals (Fig. 3B). Although it is formally possible that this is attributable to loss of CRF signaling centrally, we favor the hypothesis that normal Cort levels are required to prevent excess Aβ buildup, by either limiting production or increasing clearance of the peptide. Therefore, maintaining appropriate levels of circulating Cort is important to limiting AD progression. In many PTSD patients, there are chronically lower levels of circulating corticosteroids (Yehuda et al., 1991; Yehuda, 2009). Either depressed or elevated corticosteroid levels could alter Aβ production or clearance to accelerate AD pathogenesis, contributing to more frequent diagnosis of AD in PTSD patients.
Footnotes
This work was supported by National Institutes of Health Grants K01 AG036738 (N.J.J.), R01 NS078294 (B.R.A.), and R01 AG020670 and R01 NS076117 (H.Z.). We thank Rebecca Berdeaux and Qingchun Tong for critical reading of this manuscript. We thank the Baylor College of Medicine Intellectual and Developmental Disabilities Research Center behavioral core and Corinne Spencer for help with setting up behavioral experiments.
The authors declare no competing financial interests.
References
- American Psychiatric Association. The diagnostic and statistical manual of mental disorders DSM-5. Ed 5. Arlington, VA: American Psychiatric Association; 2013. [Google Scholar]
- Armario A, Escorihuela RM, Nadal R. Long-term neuroendocrine and behavioural effects of a single exposure to stress in adult animals. Neurosci Biobehav Rev. 2008;32:1121–1135. doi: 10.1016/j.neubiorev.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Bao AM, Meynen G, Swaab DF. The stress system in depression and neurodegeneration: focus on the human hypothalamus. Brain Res Rev. 2008;57:531–553. doi: 10.1016/j.brainresrev.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Barnes DE, Yaffe K, Byers AL, McCormick M, Schaefer C, Whitmer RA. Midlife vs late-life depressive symptoms and risk of dementia: differential effects for Alzheimer disease and vascular dementia. Arch Gen Psychiatry. 2012;69:493–498. doi: 10.1001/archgenpsychiatry.2011.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun UK, Kunik ME. Behavioral disturbances in dementia: finding the cause(s) Geriatrics. 2004;59:32, 34. [PubMed] [Google Scholar]
- Brureau A, Zussy C, Delair B, Ogier C, Ixart G, Maurice T, Givalois L. Deregulation of hypothalamic-pituitary-adrenal axis functions in an Alzheimer's disease rat model. Neurobiol Aging. 2013;34:1426–1439. doi: 10.1016/j.neurobiolaging.2012.11.015. [DOI] [PubMed] [Google Scholar]
- Busche MA, Chen X, Henning HA, Reichwald J, Staufenbiel M, Sakmann B, Konnerth A. Critical role of soluble amyloid-beta for early hippocampal hyperactivity in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2012;109:8740–8745. doi: 10.1073/pnas.1206171109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler RW, Braff DL, Rausch JL, Jenkins MA, Sprock J, Geyer MA. Physiological evidence of exaggerated startle response in a subgroup of Vietnam veterans with combat-related PTSD. Am J Psychiatry. 1990;147:1308–1312. doi: 10.1176/ajp.147.10.1308. [DOI] [PubMed] [Google Scholar]
- Carroll JC, Iba M, Bangasser DA, Valentino RJ, James MJ, Brunden KR, Lee VM, Trojanowski JQ. Chronic stress exacerbates tau pathology, neurodegeneration, and cognitive performance through a corticotropin-releasing factor receptor-dependent mechanism in a transgenic mouse model of tauopathy. J Neurosci. 2011;31:14436–14449. doi: 10.1523/JNEUROSCI.3836-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catania C, Sotiropoulos I, Silva R, Onofri C, Breen KC, Sousa N, Almeida OF. The amyloidogenic potential and behavioral correlates of stress. Mol Psychiatry. 2009;14:95–105. doi: 10.1038/sj.mp.4002101. [DOI] [PubMed] [Google Scholar]
- Csernansky JG, Wang L, Swank J, Miller JP, Gado M, McKeel D, Miller MI, Morris JC. Preclinical detection of Alzheimer's disease: hippocampal shape and volume predict dementia onset in the elderly. Neuroimage. 2005;25:783–792. doi: 10.1016/j.neuroimage.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Csernansky JG, Dong H, Fagan AM, Wang L, Xiong C, Holtzman DM, Morris JC. Plasma cortisol and progression of dementia in subjects with Alzheimer-type dementia. Am J Psychiatry. 2006;163:2164–2169. doi: 10.1176/appi.ajp.163.12.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis KL, Davis BM, Greenwald BS, Mohs RC, Mathé AA, Johns CA, Horvath TB. Cortisol and Alzheimer's disease, I: Basal studies. Am J Psychiatry. 1986;143:300–305. doi: 10.1176/ajp.143.3.300. [DOI] [PubMed] [Google Scholar]
- Devi L, Alldred MJ, Ginsberg SD, Ohno M. Sex- and brain region-specific acceleration of beta-amyloidogenesis following behavioral stress in a mouse model of Alzheimer's disease. Mol Brain. 2010;3:34. doi: 10.1186/1756-6606-3-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Goico B, Martin M, Csernansky CA, Bertchume A, Csernansky JG. Modulation of hippocampal cell proliferation, memory, and amyloid plaque deposition in APPsw (Tg2576) mutant mice by isolation stress. Neuroscience. 2004;127:601–609. doi: 10.1016/j.neuroscience.2004.05.040. [DOI] [PubMed] [Google Scholar]
- Dong H, Yuede CM, Yoo HS, Martin MV, Deal C, Mace AG, Csernansky JG. Corticosterone and related receptor expression are associated with increased beta-amyloid plaques in isolated Tg2576 mice. Neuroscience. 2008;155:154–163. doi: 10.1016/j.neuroscience.2008.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlert U, Gaab J, Heinrichs M. Psychoneuroendocrinological contributions to the etiology of depression, posttraumatic stress disorder, and stress-related bodily disorders: the role of the hypothalamus-pituitary-adrenal axis. Biol Psychol. 2001;57:141–152. doi: 10.1016/S0301-0511(01)00092-8. [DOI] [PubMed] [Google Scholar]
- Flood DG, Reaume AG, Dorfman KS, Lin YG, Lang DM, Trusko SP, Savage MJ, Annaert WG, De Strooper B, Siman R, Scott RW. FAD mutant PS-1 gene-targeted mice: increased A beta 42 and A beta deposition without APP overproduction. Neurobiol Aging. 2002;23:335–348. doi: 10.1016/S0197-4580(01)00330-X. [DOI] [PubMed] [Google Scholar]
- Gilbertson MW, Shenton ME, Ciszewski A, Kasai K, Lasko NB, Orr SP, Pitman RK. Smaller hippocampal volume predicts pathologic vulnerability to psychological trauma. Nat Neurosci. 2002;5:1242–1247. doi: 10.1038/nn958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover EM, Phifer JE, Crain DF, Norrholm SD, Davis M, Bradley B, Ressler KJ, Jovanovic T. Tools for translational neuroscience: PTSD is associated with heightened fear responses using acoustic startle but not skin conductance measures. Depress Anxiety. 2011;28:1058–1066. doi: 10.1002/da.20880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold SM, Mohr DC, Huitinga I, Flachenecker P, Sternberg EM, Heesen C. The role of stress-response systems for the pathogenesis and progression of MS. Trends Immunol. 2005;26:644–652. doi: 10.1016/j.it.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Green KN, Billings LM, Roozendaal B, McGaugh JL, LaFerla FM. Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer's disease. J Neurosci. 2006;26:9047–9056. doi: 10.1523/JNEUROSCI.2797-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Zheng H, Justice NJ. Central CRF system perturbation in an Alzheimer's disease knockin mouse model. Neurobiol Aging. 2012;33:2678–2691. doi: 10.1016/j.neurobiolaging.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare JF. Intracellular pathways of folded and misfolded amyloid precursor protein degradation. Arch Biochem Biophys. 2006;451:79–90. doi: 10.1016/j.abb.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Hartmann J, Erb C, Ebert U, Baumann KH, Popp A, König G, Klein J. Central cholinergic functions in human amyloid precursor protein knock-in/presenilin-1 transgenic mice. Neuroscience. 2004;125:1009–1017. doi: 10.1016/j.neuroscience.2004.02.038. [DOI] [PubMed] [Google Scholar]
- Harvey RJ, Ellison D, Hardy J, Hutton M, Roques PK, Collinge J, Fox NC, Rossor MN. Chromosome 14 familial Alzheimer's disease: the clinical and neuropathological characteristics of a family with a leucine→serine (L250S) substitution at codon 250 of the presenilin 1 gene. J Neurol Neurosurg Psychiatry. 1998;64:44–49. doi: 10.1136/jnnp.64.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebda-Bauer EK, Simmons TA, Sugg A, Ural E, Stewart JA, Beals JL, Wei Q, Watson SJ, Akil H. 3xTg-AD mice exhibit an activated central stress axis during early-stage pathology. J Alzheimers Dis. 2013;33:407–422. doi: 10.3233/JAD-2012-121438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CW, Lui CC, Chang WN, Lu CH, Wang YL, Chang CC. Elevated basal cortisol level predicts lower hippocampal volume and cognitive decline in Alzheimer's disease. J Clin Neurosci. 2009;16:1283–1286. doi: 10.1016/j.jocn.2008.12.026. [DOI] [PubMed] [Google Scholar]
- Joshi YB, Chu J, Praticò D. Stress hormone leads to memory deficits and altered tau phosphorylation in a model of Alzheimer's disease. J Alzheimers Dis. 2012;31:167–176. doi: 10.3233/JAD-2012-120328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice NJ, Yuan ZF, Sawchenko PE, Vale W. Type 1 corticotropin-releasing factor receptor expression reported in BAC transgenic mice: implications for reconciling ligand-receptor mismatch in the central corticotropin-releasing factor system. J Comp Neurol. 2008;511:479–496. doi: 10.1002/cne.21848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JE, Cirrito JR, Dong H, Csernansky JG, Holtzman DM. Acute stress increases interstitial fluid amyloid-beta via corticotropin-releasing factor and neuronal activity. Proc Natl Acad Sci U S A. 2007;104:10673–10678. doi: 10.1073/pnas.0700148104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, Fujiki N, Nishino S, Holtzman DM. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326:1005–1007. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasuga K, Ohno T, Ishihara T, Miyashita A, Kuwano R, Onodera O, Nishizawa M, Ikeuchi T. Depression and psychiatric symptoms preceding onset of dementia in a family with early-onset Alzheimer disease with a novel PSEN1 mutation. J Neurol. 2009;256:1351–1353. doi: 10.1007/s00415-009-5096-4. [DOI] [PubMed] [Google Scholar]
- Kennedy AM, Newman S, McCaddon A, Ball J, Roques P, Mullan M, Hardy J, Chartier-Harlin MC, Frackowiak RS, Warrington EK, Rossor MN. Familial Alzheimer's disease. A pedigree with a mis-sense mutation in the amyloid precursor protein gene (amyloid precursor protein 717 valine→glycine) Brain. 1993;116:309–324. doi: 10.1093/brain/116.2.309. [DOI] [PubMed] [Google Scholar]
- Kim T, Vidal GS, Djurisic M, William CM, Birnbaum ME, Garcia KC, Hyman BT, Shatz CJ. Human LilrB2 is a beta-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer's model. Science. 2013;341:1399–1404. doi: 10.1126/science.1242077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenen KC, Moffitt TE, Poulton R, Martin J, Caspi A. Early childhood factors associated with the development of post-traumatic stress disorder: results from a longitudinal birth cohort. Psychol Med. 2007;37:181–192. doi: 10.1017/S0033291706009019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler C, Ebert U, Baumann K, Schröder H. Alzheimer's disease-like neuropathology of gene-targeted APP-SLxPS1mut mice expressing the amyloid precursor protein at endogenous levels. Neurobiol Dis. 2005;20:528–540. doi: 10.1016/j.nbd.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Lebow M, Neufeld-Cohen A, Kuperman Y, Tsoory M, Gil S, Chen A. Susceptibility to PTSD-like behavior is mediated by corticotropin-releasing factor receptor type 2 levels in the bed nucleus of the stria terminalis. J Neurosci. 2012;32:6906–6916. doi: 10.1523/JNEUROSCI.4012-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GJ, Lu PH, Hua X, Lee S, Wu S, Nguyen K, Teng E, Leow AD, Jack CR, Jr, Toga AW, Weiner MW, Bartzokis G, Thompson PM. Depressive symptoms in mild cognitive impairment predict greater atrophy in Alzheimer's disease-related regions. Biol Psychiatry. 2012;71:814–821. doi: 10.1016/j.biopsych.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Kim JB, Seo JS, Kim TK, Im JY, Baek IS, Kim KS, Lee JK, Han PL. Behavioral stress accelerates plaque pathogenesis in the brain of Tg2576 mice via generation of metabolic oxidative stress. J Neurochem. 2009;108:165–175. doi: 10.1111/j.1471-4159.2008.05769.x. [DOI] [PubMed] [Google Scholar]
- Li WZ, Li WP, Yao YY, Zhang W, Yin YY, Wu GC, Gong HL. Glucocorticoids increase impairments in learning and memory due to elevated amyloid precursor protein expression and neuronal apoptosis in 12-month old mice. Eur J Pharmacol. 2010;628:108–115. doi: 10.1016/j.ejphar.2009.11.045. [DOI] [PubMed] [Google Scholar]
- Lind K, Edman A, Nordlund A, Olsson T, Wallin A. Increased saliva cortisol awakening response in patients with mild cognitive impairment. Dement Geriatr Cogn Disord. 2007;24:389–395. doi: 10.1159/000109938. [DOI] [PubMed] [Google Scholar]
- Littell RC, Henry PR, Ammerman CB. Statistical analysis of repeated measures data using SAS procedures. J Anim Sci. 1998;76:1216–1231. doi: 10.2527/1998.7641216x. [DOI] [PubMed] [Google Scholar]
- Littell RC, Pendergast J, Natarajan R. Modelling covariance structure in the analysis of repeated measures data. Stat Med. 2000;19:1793–1819. doi: 10.1002/1097-0258(20000715)19:13<1793::AID-SIM482>3.0.CO%3B2-Q. [DOI] [PubMed] [Google Scholar]
- Liu L, Duff K. A technique for serial collection of cerebrospinal fluid from the cisterna magna in mouse. J Vis Exp. 2008;pii:960. doi: 10.3791/960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupien SJ, de Leon M, de Santi S, Convit A, Tarshish C, Nair NP, Thakur M, McEwen BS, Hauger RL, Meaney MJ. Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nat Neurosci. 1998;1:69–73. doi: 10.1038/271. [DOI] [PubMed] [Google Scholar]
- Mairet-Coello G, Courchet J, Pieraut S, Courchet V, Maximov A, Polleux F. The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Abeta oligomers through Tau phosphorylation. Neuron. 2013;78:94–108. doi: 10.1016/j.neuron.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin MF, Lord C, Andrews J, Juster RP, Sindi S, Arsenault-Lapierre G, Fiocco AJ, Lupien SJ. Chronic stress, cognitive functioning and mental health. Neurobiol Learn Mem. 2011;96:583–595. doi: 10.1016/j.nlm.2011.02.016. [DOI] [PubMed] [Google Scholar]
- Martí O, García A, Vallès A, Harbuz MS, Armario A, Vellès A. Evidence that a single exposure to aversive stimuli triggers long-lasting effects in the hypothalamus-pituitary-adrenal axis that consolidate with time. Eur J Neurosci. 2001;13:129–136. doi: 10.1046/j.1460-9568.2001.01355.x. [DOI] [PubMed] [Google Scholar]
- Mezler M, Barghorn S, Schoemaker H, Gross G, Nimmrich V. A beta-amyloid oligomer directly modulates P/Q-type calcium currents in Xenopus oocytes. Br J Pharmacol. 2012;165:1572–1583. doi: 10.1111/j.1476-5381.2011.01646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan CA, 3rd, Grillon C, Southwick SM, Davis M, Charney DS. Exaggerated acoustic startle reflex in Gulf War veterans with posttraumatic stress disorder. Am J Psychiatry. 1996;153:64–68. doi: 10.1176/ajp.153.1.64. [DOI] [PubMed] [Google Scholar]
- Morris MC, Compas BE, Garber J. Relations among posttraumatic stress disorder, comorbid major depression, and HPA function: a systematic review and meta-analysis. Clin Psychol Rev. 2012;32:301–315. doi: 10.1016/j.cpr.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Y, Zhao X, Bao G, Zou L, Teng L, Wang Z, Song M, Xiong J, Bai Y, Pei G. Activation of beta2-adrenergic receptor stimulates gamma-secretase activity and accelerates amyloid plaque formation. Nat Med. 2006;12:1390–1396. doi: 10.1038/nm1485. [DOI] [PubMed] [Google Scholar]
- Qureshi SU, Kimbrell T, Pyne JM, Magruder KM, Hudson TJ, Petersen NJ, Yu HJ, Schulz PE, Kunik ME. Greater prevalence and incidence of dementia in older veterans with posttraumatic stress disorder. J Am Geriatr Soc. 2010;58:1627–1633. doi: 10.1111/j.1532-5415.2010.02977.x. [DOI] [PubMed] [Google Scholar]
- Reiche EM, Nunes SO, Morimoto HK. Stress, depression, the immune system, and cancer. Lancet Oncol. 2004;5:617–625. doi: 10.1016/S1470-2045(04)01597-9. [DOI] [PubMed] [Google Scholar]
- Richards M, Sacker A. Lifetime antecedents of cognitive reserve. J Clin Exp Neuropsychol. 2003;25:614–624. doi: 10.1076/jcen.25.5.614.14581. [DOI] [PubMed] [Google Scholar]
- Rissman RA, Lee KF, Vale W, Sawchenko PE. Corticotropin-releasing factor receptors differentially regulate stress-induced tau phosphorylation. J Neurosci. 2007;27:6552–6562. doi: 10.1523/JNEUROSCI.5173-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissman RA, Staup MA, Lee AR, Justice NJ, Rice KC, Vale W, Sawchenko PE. Corticotropin-releasing factor receptor-dependent effects of repeated stress on tau phosphorylation, solubility, and aggregation. Proc Natl Acad Sci U S A. 2012;109:6277–6282. doi: 10.1073/pnas.1203140109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman SM, Herdener N, Camandola S, Texel SJ, Mughal MR, Cong WN, Martin B, Mattson MP. 3xTgAD mice exhibit altered behavior and elevated Abeta after chronic mild social stress. Neurobiol Aging. 2012;33:830.e1–830.e12. doi: 10.1016/j.neurobiolaging.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapolsky RM. Stress, glucocorticoids, and damage to the nervous system: the current state of confusion. Stress. 1996;1:1–19. doi: 10.3109/10253899609001092. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM. Glucocorticoids, stress, and their adverse neurological effects: relevance to aging. Exp Gerontol. 1999;34:721–732. doi: 10.1016/S0531-5565(99)00047-9. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Krey LC, McEwen BS. The neuroendocrinology of stress and aging: the glucocorticoid cascade hypothesis. Endocr Rev. 1986;7:284–301. doi: 10.1210/edrv-7-3-284. [DOI] [PubMed] [Google Scholar]
- Selye H. Factors influencing the corticotropin production of the anterior pituitary. J Clin Endocrinol Metab. 1948;8:588. [PubMed] [Google Scholar]
- Selye H. Stress and the general adaptation syndrome. Br Med J. 1950;1:1383–1392. doi: 10.1136/bmj.1.4667.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrats J, Sawchenko PE. CNS activational responses to staphylococcal enterotoxin B: T-lymphocyte-dependent immune challenge effects on stress-related circuitry. J Comp Neurol. 2006;495:236–254. doi: 10.1002/cne.20872. [DOI] [PubMed] [Google Scholar]
- Serrats J, Schiltz JC, García-Bueno B, van Rooijen N, Reyes TM, Sawchenko PE. Dual roles for perivascular macrophages in immune-to-brain signaling. Neuron. 2010;65:94–106. doi: 10.1016/j.neuron.2009.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GW, Aubry JM, Dellu F, Contarino A, Bilezikjian LM, Gold LH, Chen R, Marchuk Y, Hauser C, Bentley CA, Sawchenko PE, Koob GF, Vale W, Lee KF. Corticotropin releasing factor receptor 1-deficient mice display decreased anxiety, impaired stress response, and aberrant neuroendocrine development. Neuron. 1998;20:1093–1102. doi: 10.1016/S0896-6273(00)80491-2. [DOI] [PubMed] [Google Scholar]
- Sotiropoulos I, Catania C, Riedemann T, Fry JP, Breen KC, Michaelidis TM, Almeida OF. Glucocorticoids trigger Alzheimer disease-like pathobiochemistry in rat neuronal cells expressing human tau. J Neurochem. 2008;107:385–397. doi: 10.1111/j.1471-4159.2008.05613.x. [DOI] [PubMed] [Google Scholar]
- Stuller KA, Jarrett B, DeVries AC. Stress and social isolation increase vulnerability to stroke. Exp Neurol. 2012;233:33–39. doi: 10.1016/j.expneurol.2011.01.016. [DOI] [PubMed] [Google Scholar]
- Tune LE. Depression and Alzheimer's disease. Depress Anxiety. 1998;8(Suppl 1):91–95. [PubMed] [Google Scholar]
- Um JW, Kaufman AC, Kostylev M, Heiss JK, Stagi M, Takahashi H, Kerrisk ME, Vortmeyer A, Wisniewski T, Koleske AJ, Gunther EC, Nygaard HB, Strittmatter SM. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron. 2013;79:887–902. doi: 10.1016/j.neuron.2013.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science. 1981;213:1394–1397. doi: 10.1126/science.6267699. [DOI] [PubMed] [Google Scholar]
- Volden PA, Conzen SD. The influence of glucocorticoid signaling on tumor progression. Brain Behav Immun [Suppl] 2013;30:S26–S31. doi: 10.1016/j.bbi.2012.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Yaffe K, Vittinghoff E, Lindquist K, Barnes D, Covinsky KE, Neylan T, Kluse M, Marmar C. Posttraumatic stress disorder and risk of dementia among US veterans. Arch Gen Psychiatry. 2010;67:608–613. doi: 10.1001/archgenpsychiatry.2010.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Morinobu S, Takei S, Fuchikami M, Matsuki A, Yamawaki S, Liberzon I. Single prolonged stress: toward an animal model of posttraumatic stress disorder. Depress Anxiety. 2009;26:1110–1117. doi: 10.1002/da.20629. [DOI] [PubMed] [Google Scholar]
- Yehuda R. Status of glucocorticoid alterations in post-traumatic stress disorder. Ann N Y Acad Sci. 2009;1179:56–69. doi: 10.1111/j.1749-6632.2009.04979.x. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Giller EL, Southwick SM, Lowy MT, Mason JW. Hypothalamic-pituitary-adrenal dysfunction in posttraumatic stress disorder. Biol Psychiatry. 1991;30:1031–1048. doi: 10.1016/0006-3223(91)90123-4. [DOI] [PubMed] [Google Scholar]
- Zempel H, Thies E, Mandelkow E, Mandelkow EM. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010;30:11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zempel H, Luedtke J, Kumar Y, Biernat J, Dawson H, Mandelkow E, Mandelkow EM. Amyloid-beta oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. EMBO J. 2013;32:2920–2937. doi: 10.1038/emboj.2013.207. [DOI] [PMC free article] [PubMed] [Google Scholar]