

Abstract
Aβ accumulation plays a central role in the pathogenesis of Alzheimer's disease (AD). Recent studies suggest that process of Aβ nucleated polymerization is essential for Aβ fibril formation, pathology spreading and toxicity. Therefore, targeting this process represent an effective therapeutic strategy to slow or block disease progression. To discover compounds that might interfere with the Aβ seeding capacity, toxicity and pathology spreading, we screened a focused library of FDA-approved drugs in vitro using a seeding polymerization assay and identified small molecule inhibitors that specifically interfered with Aβ seeding-mediated fibril growth and toxicity. Mitoxantrone, bithionol and hexachlorophene were found to be the strongest inhibitors of fibril growth and protected primary cortical neuronal cultures against Aβ-induced toxicity. Next, we assessed the effects of these three inhibitors in vivo in the mThy1-APPtg mouse model of AD (8-month-old mice). We found that mitoxantrone and bithionol, but not hexachlorophene, stabilized diffuse amyloid plaques, reduced the levels of Aβ42 oligomers and ameliorated synapse loss, neuronal damage and astrogliosis. Together, our findings suggest that targeting fibril growth and Aβ seeding capacity constitutes a viable and effective strategy for protecting against neurodegeneration and disease progression in AD.
Keywords: Alzheimer's Disease, Aβ seeding-mediated aggregation, Aβ-propagation, amyloid protein, drug discovery, inhibitors
Introduction
Evidence from genetics, neuropathology, biochemistry and animal model studies continues to suggest that amyloid β protein (Aβ) aggregation and amyloid formation play central roles in the initiation and progression of neurodegeneration in Alzheimer's disease (AD) (Hartley et al., 1999 and McLean et al., 1999). However, the mechanisms by which these processes contribute to the pathogenesis of AD and the nature of the toxic species remain subjects of active investigation and debate. Genetic mutations (Goate et al., 1991, Lemere et al., 1996 and Nilsberth et al., 2001) or changes in the neurons that result in the increased production of Aβ or enhance its fibril formation have been linked to early onset forms of AD (McLean et al., 1999, Kumar-Singh et al., 2006).
Aβ is produced as a result of sequential proteolytic cleavage of the amyloid precursor proteins by β- and γ-secretase (Selkoe, 2012) resulting in the generation of Aβ peptides of variable lengths, with Aβ40 being the predominate species and Aβ42 being the most amyloidogenic and toxic form (Duyckaerts et al., 2009 and Mucke et al., 2000). Aβ Fibril formation occurs via multiple mechanisms involving primary and secondary nucleation events and involves the formation of on or off-pathway oligomers.
The nucleated polymerization mechanism (Fig. 1B) is characterized by a nucleation phase associated with the formation of assembly competent oligomers followed by a cooperative oligomer growth and fibril formation by monomer addition (Harper and Lansbury, 1997). Several oligomeric intermediates of different morphologies, including spherical, chain-like, and annular oligomers have been observed during Aβ fibril formation in vitro (Jan et al., 2010), (Fig. 1B) and similar structures were identified during post-mortem biochemical analysis of AD brains (Lemere et al., 1996; Lesne et al., 2006).
Figure 1. The molecular basis of the nucleated polymerization high-throughput assay to identify inhibitors of fibril growth and seeding capacity.
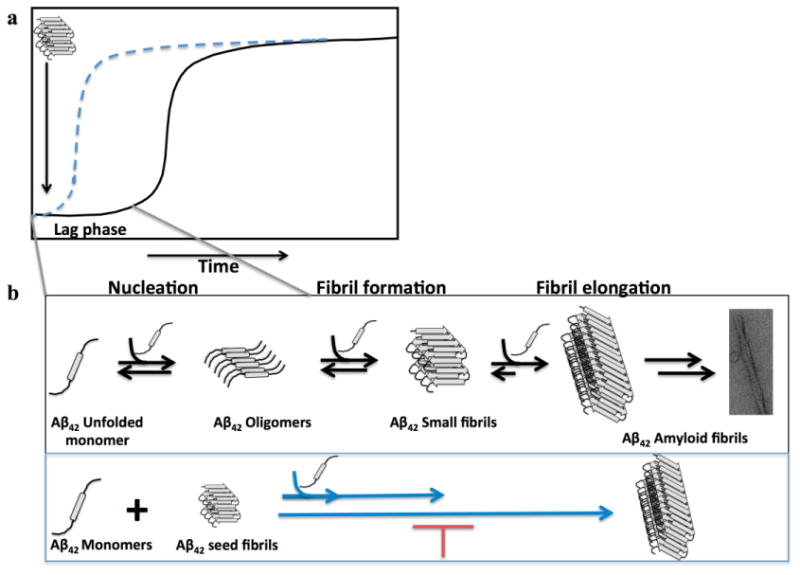
Fibril formation by Aβ follows a nucleated polymerization mechanism that is characterized by a nucleation phase associated with the formation of assembly competent oligomers followed by cooperative oligomer growth and fibril formation by monomer addition (a)16. This process can be seeded and accelerated by the addition of preformed fibrils (b) and is thought to serve as the underling mechanism for the spreading of Aβ pathology in the brain. The seeding polymerization assay we developed is based on adding a small amount of preformed fibrils to monomeric Aβ solutions and monitoring fibril growth in the presence or absence of small molecules. a) A schematic depiction of the kinetics of amyloid formation via a nucleated polymerization mechanism in the absence (in black) and presence (in blue) of preformed fibrillar seeds. b) The general mechanism of amyloid formation in vitro. The schematic depiction below illustrates the basis of our seeding polymerization assay to identify small molecules that interfere with fibril growth. The addition of small fibrils (seeds) eliminates the lag phase and accelerates amyloid formation in vitro (in blue).
This process Aβ fibril formation can be seeded and accelerated by the addition of preformed fibrils (Fig. 1B). The addition of a small amount of preformed fibrillar aggregates (seeds) eliminates the lag phase of Aβ aggregation and accelerates the fibrillization of monomeric Aβ in vitro and in vivo (Harper and Lansubry, 1997). The pathological relevance and consequences of this process remain subjects of intense debate and active investigation. We have recently shown that the toxicity of oligomeric Aβ preparations is enhanced by the addition of monomeric Aβ. These studies demonstrated a direct correlation between the ability of Aβ oligomers convert to fibrils and increased Aβ toxicity, supporting the hypothesis that the process of fibril formation and growth is also a key mediator of Aβ-induced toxicity. Increasing evidence from in vivo studies also suggests that the seeding-mediated aggregation of Aβ proteins is essential for the formation of amyloid plaques (Walker et al., 2013), amyloid propagation and spreading via a prion-like mechanism (Frost and Diamond, 2010).
Previous in vivo studies have shown that inoculation of brain homogenates from the brain of AD patient or aged βAPP transgenic mice into βAPP transgenic mice accelerates Aβ deposition via a seeding mechanism (Kane et al., 2000, Meyer-Luehmann et al., 2006 and Eisele et al., 2009). The amyloid-inducing activity of brain extracts was completely abolished by the Aβ-immunodepletion, by passive immunization of the βAPPtg mouse host with antibodies or by treatments of the extracts with formic acid, demonstrating that Aβ is a pre-requisite for the in vivo seeding process (Kane et al., 2000 and Meyer-Luehmann et al., 2006). Previous studies in different brain areas have also shown that Aβ aggregates spread from the site of the injection to more distal regions, and the spreading is consistent with normal age-related deposition in APPtg mouse hosts (Eisele et al., 2009). Recently, Prusiner and collaborators have shown that synthetic Aβ aggregates inoculated into young APPtg mice form Aβ deposits similar to the deposits induced by brain-derived Aβ aggregates (Sthor et al., 2012).
Together, these data suggest that seeding-mediated Aβ fibril formation deposition in vivo might be a key mediator in AD progression, and acting on this process could represent an effective therapeutic strategy to slow or block disease progression. Therefore, we sought to discover compounds that might interfere with seeding-mediated aggregation and toxicity. Towards this goal, we screened an FDA-approved library of bioactive compounds, and sixteen molecules were identified as strong inhibitors of Aβ42 seeding-mediated aggregation. Three of these inhibitors exhibited the strongest inhibition on seeding-mediated aggregation and were shown to protect against Aβ-induced neuronal toxicity; these inhibitors were selected for validation in an AD animal model. Here, we show that the administration of two of these compounds two months after the initiation of Aβ deposition reduced Aβ accumulation and oligomer formation and protected against Aβ-induced synapse loss and neuronal damage.
Materials and Methods
Preparation of the working compounds
A library containing 1040 small chemical compounds consisting of FDA-approved drugs was purchased from MicroSource Discovery System (Gaylordsville CT, USA). N-[N-(3,5-difluorophenylacetyl)-L-alanyl-]-(S)-phenylglycine-t-butyl ester (DAPT), egg yolk 1,2-Diacyl-sn-glycero-3-phosphocholine (PC) and 1,2-Diacyl-sn-glycero-3-phosphoethanolamine (PE) were purchased from Sigma-Aldrich (Steinheim, Germany).
Sample preparations for fibrillization studies and toxicity assays
Preparation and characterization of Aβ42 low molecular weight (LMW) and protofibrils (PF)- Aβ42
Aβ42 was synthesized and purified by Dr. James I. Elliot at Yale University (New Haven, CT, USA). To prepare the monomeric Aβ42 stock solution (90% of monomers and LMW in equilibrium), the Aβ42 was dissolved in 6 M guanidine-HCl at concentration of 1 mg/ml and subsequently centrifuged at 6,000 rcf for 5 minutes. To prepare the low molecular weight (LMW) and protofibril (PF) Aβ42 stock solution (250μM, 1mg/mL), the lyophilized Aβ42 was dissolved in 5% DMSO and 2 M Tris base, pH 7.6; it was then subjected to low speed centrifugation at 3000 rcf for 5 minutes. LMW Aβ42 and PF stock solution was loaded onto a gel filtration column (Superdex 75 HR 10/30 Amersham) previously equilibrated with 10 mM Tris buffer and then the fraction were separated and eluted in 10 mM Tris buffer. The concentration of Aβ42 (LMW and PF) was calculated using the theoretical molar extinction coefficient at 280 nm (1490 M-1cm-1). To perform aggregation studies, Aβ42 was incubated at 37°C in 1.5 mL polypropylene sterile tubes, with and without different concentrations of inhibitors, at molar ratios of Aβ42: inhibitor of 1:0.5 and 1:2 for 48 hours.
Preparation of Aβ42 seeds
The Aβ42 seeds were prepared by incubation of the Aβ42 solution (5% DMSO, 2 M Tris buffer, pH 7.6) at 37°C under agitation for 72 hours. After three days of incubation, the fibrils were mechanically fragmented to yield a narrow distribution of smaller fibrillar structures (100-300-nm long) by ultra-sonication on ice using a SONICS Vibra Cell™ equipped with a fine tip (20×5 second pulses, amplitude 40, output watts 6). The sonicated fibrils were diluted in 10 mM Tris buffer pH 7.6. Seeds and monomeric Aβ42 were incubated for 2-3 hours at 37°C with continuous shaking, with and without inhibitors, in black polystyrene 384-well plates (Nunc, Thermo Scientific, USA). The concentration of the seeds and fibrils were calculated on the basis of monomer concentration (250μM)
Characterization of the Aβ42 species
All the Aβ42 species generated using the protocols above (monomers, protofibrils, fibrils and fibril seeds) were characterized by size exclusion chromatography, ThT, and TEM as previously described in (Jan et al. 2010).
Fibrillization studies
Seeding polymerization assay
Polymerization of soluble Aβ42 with or without Aβ42 seeds was assayed as described in Di Giovanni et al. (Di Giovanni et al., 2010). Initial studies were performed by incubating a solution of monomers (M) and seeds (S) at molar ratios of 10:2 and 10:4. The inhibitory activity of the 1040 FDA-approved drugs using the seeding fibrillization assay was determined using a Zephyr® Compact Liquid Handling Workstation (Caliper Life Science). The assay was initiated by adding 64 μL of M and 8 μL of S (10:4 μM) to each well of black polystyrene 384-well plates (Nunc, USA). During the assay, the plates were kept at 4°C to avoid aggregation of the protein solutions. A total of 8 μL of the compound solutions dissolved in 1% DMSO was added to the reaction mixture so that the final concentration of the tested compounds was 10 μM. The plates were incubated at 37°C for 3 hours under agitation. The kinetics of the fibrillization were monitored using the standard Thioflavin T (ThT) binding assay as described below. The assay was performed by processing the microplate collection of each compound in duplicate. Curcumin was used as a positive control, and the results are reported as the percent inhibition compared with control wells lacking the test compounds.
Drug hit validation assays
The hits from the seeding polymerization assay were validated for their ability to inhibit the fibrillization of monomeric or protofibrillar Aβ42 in standard 1.5-mL tubes. The time course of Aβ42 fibrillization was measured using a ThT fluorescence assay and TEM.
ThT fluorescence assay
For the seeding polymerization assay described above, an aliquot of 80 μL of 10 μM Aβ42 solutions, previously incubated at 37°C in the absence or presence of compounds, was added to 10 μL of 100 μM ThT and 10 μL of 50 μM glycine buffer (pH 8.5). Fluorescence measurements were performed with a spectrofluorometer (Analyst™ AD 96-384, Bucher. Biotech. AG, Basel) at 25°C using black polystyrene 384-well plates. The excitation wavelength was set to 450 nm, and emission was monitored at 485 nm. All measurements were performed in triplicate.
TEM analysis
The samples for TEM analysis were prepared by placement on a Formvar-carbon copper grid for 1 min before removing the excess solution. The grid was washed with 2 drops of distilled water and 1 drop of uranyl acetate before staining with 1% of fresh uranyl acetate for 30 s. The grids were analyzed using a Zeiss OM 10 electron microscope.
Preparation of cell cultures and administration of Aβ42
Primary cortical neuronal cultures (P1)
Primary cortical neuronal cultures were prepared from the neocortices of 1-day-old rats as previously described (Hartley et al., 1999).
Cortical hemispheres were isolated in Hank's balanced salt solution (HBSS, Invitrogen) and incubated in 0.1% papain (Sigma-Aldrich). After 10 minutes, the papain was removed, and the reaction was blocked using 30% fetal calf serum (FCS, Invitrogen). Individual cells were obtained by trituration (8X); the cells were then resuspended in NeuroBasal medium 1X (Invitrogen) supplemented with 0.5 mM B27 (Invitrogen), 200 mM L-glutamine (Invitrogen), 25 μM Glutamax, 5% heat-inactivated FCS and 2% penicillin/streptomycin (Pen/Strep, Invitrogen). Cells were seeded at a density of 3.5×105 cells on a Petri dish (3×104 cells/cm2) in NeuroBasal medium supplemented with B27, 0.5 mM glutamine and 2% Pen/Strep. After 7 days in vitro (DIV), cortical neurons were exposed to an Aβ42 crude preparation, which contained a mixture of Aβ monomers, oligomers and protofibrils obtained by the incubation of ∼ 1 mg/mL Aβ42 solution [5% DMSO, 2 mM Tris buffer (pH 7.6)].
The neurons were exposed to a concentration of 40 μM for 24 hours or coincubated with the compounds at different concentrations as described in the results.
Cell viability analysis
MTT assay
The viability of the cortical neuron cultures was evaluated using a thiazolyl blue (MTT) assay. The cells were incubated for 15 minutes at 37°C; the dark crystals that formed after the incubation were dissolved in 0.1 M Tris-HCl buffer containing 5% Triton X-100, and the absorbance was read at 570 nm in a multiple spectrophotometric reader (Victor). All quantitative data are presented as the mean±SE.
Purification of γ-secretase and C100-Flag
γ-Secretase was purified from S-20 cells as described previously (Cacquevel et al., 2008 and Fraering et al., 2005). APP-C100Flag, the recombinant APP-based protein substrate of γ-secretase, was overexpressed in E. coli and purified using a Flag-specific M2 affinity resin (Sigma-Aldrich) (Cacquevel et al., 2008 and Fraering et al., 2005).
γ-Secretase activity assays
In vitro γ-secretase assays using the recombinant APP-C100-Flag substrate and purified γ-secretase were performed as previously described (Cacquevel et al., 2008 and Fraering et al., 2005). Briefly, γ-secretase solubilized in 0.2% (wt/vol) CHAPSO-HEPES (pH 7.5) was incubated at 37°C for 4 hours with 1 mM APP-C100-Flag substrate, 0.1% (wt/vol) PC, 0.025% PE and 10 mM of bithionol, hexachlorophene or mitoxantrone. All compounds were added to the reactions from DMSO stock solutions before the addition of APP-C100Flag. After 4 hours of incubation at 37°C, the reactions were halted by adding Laemmli sample buffer for western blot analyses. The resulting products, APP intracellular domain (AICD)-Flag and Aβ, were detected with a Flag-specific M2 antibody (1:1000; Sigma-Aldrich) and an Aβ–specific 6E10 antibody (1:1000; Covance), respectively (Cacquevel et al., 2008 and Fraering et al., 2005).
Pharmacokinetics and brain distribution
For this purpose, the compounds were submitted to Sai Life Sciences Limited (Pune, India) for determination of compound levels in mouse plasma and brain samples after a single administration of 10 mg/kg. C57/BL6 mice (n=3 per time point) were treated, and blood and brain samples were collected at 0, 0.08, 0.25, 0.5, 1, 2, 4, 8 and 24 hours. Compound levels were determined using LC-ms/ms pharmacokinetic parameters and calculated using a non-compartmental analysis tool of Phoenix WinNolin (vs. 6.3).
APP transgenic mouse model
To perform in vivo studies, we used the previously described transgenic mouse model of AD (line 41) expressing hAPP751 cDNA containing the London (V717l) and Swedish (K670M/N671l) mutations under the regulatory control of the murine (m) Thy-1 gene (m Thy1-hAPP751) (Rockenstein et al., 2001 and Rockenstein et al., 2002); female mice were used. The rationale behind using only female mice in our project is that the distribution of plaques in these animals correlates with that observed in cortical areas of female AD patient (Kraszpulski et al., 2001). Gender-dependent elevated plaque formation has been reported in several APP transgenic mouse model: APP23 (Sturchler-Pierrat et al.,2000), APP/Tau double transgenic mice (Lewis et al., 2001).
The female APPtg mice (8 months old) were treated with PBS (as the control) or bithionol, mitoxantrone or hexachlorophene. Bithionol and mitoxantrone, which are able to cross the brain-blood barrier (BBB), were administered to APPtg mice via a daily intraperitoneal injection for 1 month at different concentrations (bithionol, 10 mg/kg; mitoxantrone, 1 mg/kg). Hexachlorophene is unable to cross the BBB; thus, it was injected into the APPtg mouse brain via a cannula and an osmotic pump implanted into the right hemi ventricle (-1.0, 1.0, -3.0).
Brain tissue preparation and fractionation
The posterior mouse brain samples were homogenized in a sucrose-containing buffer (Buffer A) and separated into cytosolic and membrane fractions. The samples (0.1 g) were fractionated in 0.9 ml of Buffer A (containing PBS (pH 7.4), 0.32 M sucrose, 50 mM HEPES, 25 mM MgCl2, 0.5 mM DTT, 200 μg/ml PMSF, 2 μg/ml pepstatin A, 4 μg/ml leupeptin and 30 μg/ml benzamidine hydrochloride). To avoid protein degradation and dephosphorylation and preserve the mouse brain tissues, , reparation and fractionation of the brain tissues (Pham et al., 2010). preparation and fractionation of the brain tissues was performed in the presence of protease and phosphatase inhibitor cocktails (Calbiochem, San Diego, CA, USA) as previously described in (Pham et al., 2010). The samples were first homogenized and centrifuged at 3000 rcf for 5 minutes at 4°C. The supernatant was collected and re-centrifuged at 100,000×g for 1 hour at 4°C. After centrifugation, the supernatant (cytosolic fraction) was kept, and the pellet (membrane fraction) was re-suspended in 0.4 ml of Buffer A and re-homogenized. The membrane fraction was used to detect the Aβ oligomers. A BCA assay was used to determine the protein concentrations of the membrane fraction and cytosolic fraction samples.
Immunoblot analysis
Mouse proteins (25 or 10 μg) in the membrane fraction were separated on precast NuPage 4-12% Bis-Tris Acetate gels (Invitrogen, Life Technologies) and transferred to a nitrocellulose membrane (Whatman protein nitrocellulose transfer membrane, 0.2 μm, Millipore). The membrane was blocked using 5% bovine serum albumin (Sigma-Aldrich) dissolved in 0.1% PBS-Tween 20 and then incubated at room temperature for 1 hour with agitation and blotted with the primary antibody overnight at 4°C. The subsequent day, the membrane was incubated with a secondary antibody conjugated to horseradish peroxidase (HRP) and developed in a Versa Doc gel imaging system (Bio-Rad, Hercules, CA, USA). The Quantity One software package (Bio-Rad) was used for densitometry analysis. The protein concentrations were normalized to β-actin (1:1000).
Antibodies
We analyzed the levels of Aβ oligomers in mouse brain tissues using an immunoblotting assay performed with the membrane fraction and probed with anti-Aβ antibodies 82E1 (mouse IBL; 1:1000, Minneapolis, MN, USA) and 6E10 (mouse monoclonal; 1:1000, Signet). We simultaneously analyzed the levels of a synaptic vesicle protein, postsynaptic density-95 (mouse; 1:1000, PSD95), which has been shown to correlate with levels of oligomers in the brain, as well as the pre-synaptic protein synaptophysin using the anti-synaptophysin protein 38 (SY38) antibody (mouse monoclonal; 1:1000, Abcam).
To analyze the neuronal structure, we used MAP-2 (MAP378 mouse monoclonal; 1:250, Millipore) and NeuN (mouse monoclonal; 1:1000, Millipore); GFAP was used for astrogliosis analysis (rabbit polyclonal 1:500, Millipore). To normalize the data, we used β-actin (mouse monoclonal antibody; 1:1000, Millipore).
Determination of Aβ1-42 levels using ELISA
Quantitative evaluation of Aβ42 in mouse brains was performed using a solid phase sandwich enzyme-linked immunosorbent Assay (ELISA; Human Aβ42 kit, Invitrogen). The quantification of Aβ1-42 by the ELISA kit was performed on mouse membrane fraction samples or in brain tissues fractionated in guanidine-HCl, as previously reported (Masliah et al., 2000).
Immunohistochemistry analysis
Immunohistochemistry analyses were performed as described previously (Masliah et al., 2001). Vibratome sections were immunolabeled using a mouse monoclonal antibody against Aβ (clone 82E1, 1:500), anti-MAP2, anti-synaptophysin (SYN, clone SY38, 1:20; Chemicon), anti-NeuN, or anti-GFAP (a marker for astroglial cells) and detected using fluorescein isothiocynate-conjugated secondary antibodies (1:75; Vector Laboratories, Burlingame, CA, USA). The sections were processed simultaneously under the same conditions, and the experiments were performed twice to assess reproducibility. The FITC-labeled immunofluorescent sections were analyzed with the LSCM system (BioRad, Hercules, CA, USA), and the serial optical z-sections (0.2-μm thick) were imaged with a Zeiss 63X (N.A. 1.4) objective on an Axiovert 35 microscope (Zeiss, Thornwood, NY, USA).
Immunocytochemical staining
Neurons were fixed with paraformaldehyde (4% diluted in sodium phosphate buffer 1X, pH 7.4) for 20 minutes. The sections were incubated with primary antibody (anti-MAP2; 1:500) in primary antibody solution (5%BSA in PBS-Tween 0.01%) overnight at 4°C, and the cells were then washed and incubated with secondary antibody conjugated to Alexa Fluor 488 for 1 hour at room temperature. The coverslips were then washed in fresh antibody solution. The neurons on coverslips were imaged with a Zeiss 63X (N.A. 1.4) objective on an Axiovert 35 microscope (Zeiss, Thornwood, NY, USA).
Electron microscopy analysis of fibril formation in brain tissues
The mouse brain tissues were also analyzed through TEM. Vibratome sections were postfixed in 1% glutaraldehyde, treated with osmium tetraoxide, embedded in epon araldite and sectioned with an ultramicrotome (Leica, Nussloch, Germany). The grids were analyzed with a Zeiss OM 10 electron microscope.
Statistical analyses
Statistical analyses were performed using the SPSS 12.0 software package (SPSS Inc., Chicago, IL, USA). The Kolmogorov-Smirnov Test was used to check for normal distribution of the data, and t-tests were applied to identify significant (p<0.05) group differences. For multiple comparisons, a one-way ANOVA with post-hoc Fisher tests was used. All data proved to be normally distributed, and each experiment had a sample size of N=5. For the IC50 calculation, we used the dose response curves-inhibitors with variable slope and used [log] of the inhibitor (the concentration of the inhibitors was in a range from 0.001μM to 20μM) and the data was analyzed by Prism.
Results
To identify molecules that specifically interfere with and block the in vitro seeding mechanism (Fig. 1), we developed a robust seeding fibrillization assay (Z'=0.87) that is amenable to high-throughput screening, as shown in Fig. 2a. To avoid artifacts due to the rapid aggregation of Aβ42, sample evaporation and inhomogeneous precipitation of the protein, we optimized the aggregation conditions, and the fibrillization of Aβ42 monomers was complete within 2-3 hours at low concentrations (5-10 μM) in the presence of a small amount of preformed Aβ42 fibrils (seeds, 1-2 μM). Fibrillar seeds of Aβ42 were prepared by fragmenting preformed and purified fibrils by sonication (Di Giovanni et al., 2010). The extent of the Aβ42 fibril formation was then assessed using the Thioflavin T (ThT) fluorescence assay. The ThT signal of solutions containing only monomers did not change over the 4-hour incubation, whereas samples containing 2 μM of preformed Aβ fibrils showed a marked increase in ThT fluorescence after a 2-hour incubation and reached a maximum value within 4 hours of incubation at 37°C. The z factor value was 0.87, thereby confirming the robustness of our assay.
Figure 2. Identification of new inhibitors of the Aβ42 seeding mechanism by high-throughput assay validation of inhibitory mechanisms on the seeding assay and on the LMW and PF fibril elongation.
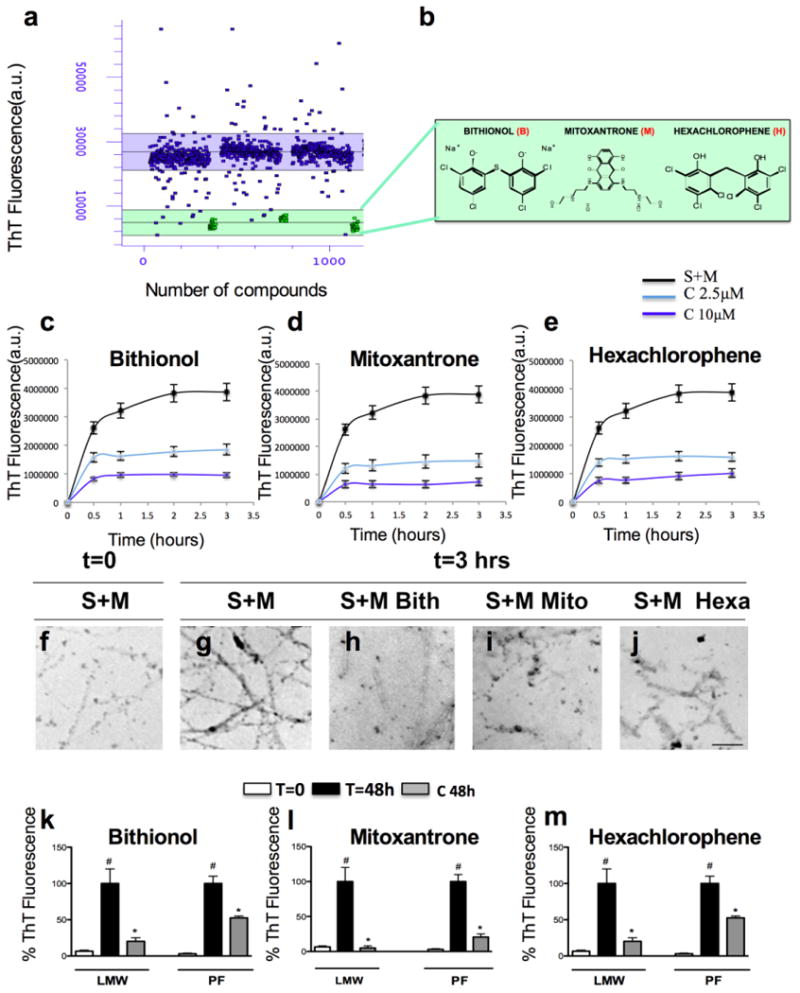
a) Using a high-throughput assay, we screened an FDA-approved library (NINDS Custom Collection II) composed of 1,040 bioactive compounds containing a diverse set of drugs, 85% of which are marketed drugs with a wide range of therapeutic usages including anti-inflammatory and analgesia use. The Aβ42 seeding polymerization assay was developed and optimized in 384-well plates, and a Z value of 0.869 was obtained by performing a ThT fluorescence assay using curcumin as a positive control. b) Identification of three strong inhibitors of the Aβ42 seeding mechanism: bithionol (B), mitoxantrone (M) and hexachlorophene (H) and the relative chemical structures (b). (c-e) In vitro analysis of the inhibitory effects of bithionol (c), mitoxantrone (d) and hexachlorophene (e) on the seeding mechanism evaluated by ThT. The Aβ42 peptide was incubated with the three compounds at a molar ratio of Aβ42:inhibitor (1:0.25 and 1:1), and the seeding reaction was monitored for 3 hours using the ThT assay. (f-j) In vitro analysis of the inhibitory effects of bithionol (c), mitoxantrone (d) and hexachlorophene (e) on the seeding mechanism evaluated using a transmission electron microscope (TEM). k-m) In vitro analysis of the inhibition of the bithionol (k), mitoxantrone (l) and hexachlorophene (m) on LMW and PF aggregation. The recombinant Aβ42 peptide was incubated with the compounds at molar ratios Aβ42: inhibitor (1:0.5 and 1:1) at 37°C for 48 hours, and the aggregation state was evaluated using a ThT assay. i-m) Structural analysis of the species stabilized after the inhibition has been performed by TEM after 3 hours of the incubation of Seeds (S) and Monomers (M) (j), or with co-treatments of the compounds bithionol (k), mitoxantrone (l) and hexachlorophene (m) at a molar ratio Aβ42:inhibitor (1:1).
Using this microplate-based Aβ42 seeding and fibril growth assay, we screened a library of 1040 compounds (80% of which are FDA-approved) and identified 17 inhibitors of Aβ42 fibril growth and seeding capacity (Fig. 1a). These small molecules comprised three classes. The first class consisted of aromatic phenolic or polyphenolic compounds, the second class comprised heteroaromatic amines, and the third hybrid contained structures with both phenolic and amine character compounds. We then performed predicted drugability profiles of these compounds based on the Lipinki rules, which included molecular mass, lipophilicity (clogP) and topological polar surface areas (tpsa), and computed the predicted blood-brain barrier values (logBB). On the basis of these analyses and after eliminating compounds with potential toxiphoric groups, we selected 3 inhibitors (bithionol, mitoxantrone and hexachlorophene; Fig. 2b) for validation in vitro and in an AD animal model.
Validation and characterization of the hit compounds
To validate the inhibitory effect on the seeding mechanism, different concentrations of the compounds were added to a freshly prepared solution of Aβ42 obtained using Guanidine HCl 6M. Fibrillar seeds at a final concentration of 2 μM were then added, and the kinetics of fibrillization were monitored through ThT fluorescence (Fig. 2c-e) and TEM (Fig. 2f-j). In the absence of compounds, fibrillization proceeded immediately to yield a dense network of amyloid fibrils, and the fibrillization reaction was complete within 2 hours (Fig. 2c-e). With 10 μM mitoxantrone (Fig. 2d), the seeding capacity of short fibrils was abolished by more than 80-90%. Bithionol and hexachlorophene at the same concentration showed an intermediate inhibitory effect of seeded fibril growth of approximately 60-80% (Fig. 2c&e). Next, we assessed the inhibitory effect of each compound on the seeded Aβ42 fibril growth at different concentrations, and the following IC50 values were obtained: mitoxantrone, IC50=1.7 μM; bithionol, IC50=5.33 μM; and hexachlorophene, IC50=5.9 μM.
The ability of these compounds to inhibit Aβ42 growth and seeding was also directly evaluated by TEM (Fig. 2f-j). Consistent with the ThT data, 3 hours of incubation with seeds and monomers resulted in the formation of mature fibrils (Fig. 2g), whereas co-incubation with bithionol (Fig. 2h), mitoxantrone (Fig. 2i) and hexachlorophene (Fig. 2j) resulted in significant inhibition of fibril growth and a reduction in the number of amyloid fibrils observed by TEM (Fig. 2c-e). Although these findings suggest that these compounds are strong inhibitors of Aβ42 seeding capacity acting at a low molar ratio protein:inhibitor, they do not rule out the possibility that they also influence the aggregation of Aβ42 by other mechanisms, notably, by stabilizing intermediates that precede the formation of mature fibrils or by altering the aggregation pathway in favor of non-fibrillar or ThT-negative Aβ42 aggregates.
To test these possibilities and determine the mode of action of the three compounds, we investigated their capacity to inhibit the fibrillization of monomeric and protofibrillar Aβ42, which were freshly prepared by size exclusion chromatography (SEC) as described previously (Di Giovanni et al., 2010). Fresh monomeric Aβ42 solutions were incubated (37°C) with and without compounds at different molar ratios. The compounds were co-incubated with monomeric or protofibrillar Aβ42 at two different molar ratios (Aβ42 :compound, 1:0.5 and 1:2) for 48 hours (Fig. 2k-m), and the extent of fibril formation was assessed through ThT. Mitoxantrone was found to be the most active compound, showing a greater than 80% inhibition of Aβ42 fibrillization at 5-20 μM (Fig. 2l), whereas bithionol and hexachlorophene exhibited inhibition in the range of 80% (Fig. 2k&m), even at higher molar ratios (1:2, Aβ42:compound). Next, we assessed the inhibitory effect of each compound at different molar ratios of Aβ42:inhibitor (range, 1:0.01-1:2), and the following IC50 values were obtained: mitoxantrone, IC50=2.5 μM; bithionol, IC50=5.1 μM; and hexachlorophene, IC50=5 μM.
To determine whether these compounds act by targeting aggregation intermediates on the amyloid pathway, we investigated their ability to inhibit the conversion of protofibrils into mature amyloid fibrils. Mitoxantrone showed the strongest inhibition of protofibril to fibril conversion (>80%, Fig. 2l), whereas bithionol and hexachlorophene showed inhibition in the range of 50% (Fig. 2k&m). Given that these compounds were identified as inhibitors of fibril growth, it is not surprising that they were less effective at inhibiting protofibril growth. Protofibrils are heterogeneous mixtures of Aβ oligomers of different sizes and morphology that are in dynamic equilibrium with the monomers. Protofibrils lack the cross-β sheet structure that is characteristic of amyloid fibrils and grow very slowly in the absence of excess monomers. The strong inhibitory effects of the compounds observed in the case of monomeric Aβ could still be mediated by the inhibition of the growth and seeding capacity during the early stages of fibril formation.
Protection against Aβ42-induced toxicity in primary neuronal cultures
To determine whether the inhibition of Aβ growth and seeding capacity was sufficient to protect against Aβ42-induced toxicity, we investigated the neuroprotective properties of mitoxantrone, bithionol and hexachlorophene against Aβ42-induced toxicity on newborn (P1) mice cortical neurons (CNs) at 7 DIV as previously described39. First, we analyzed the effects of the three compounds on the primary neurons. We found that bithionol was not toxic to neurons (Fig. 3a). Mitoxantrone and hexachlorophene showed a significant reduction of the cellular viability only at high concentrations (20 and 50 μM; Fig. 3b-c). The primary neurons were treated for 24 hours with crude preparations of Aβ42 (40 μM), which contained predominantly monomeric and protofibrillar Aβ42 species in the absence or presence of 0.5, 5 or 10 μM of inhibitors (Di Giovanni et al., 2010). Exposure of the neurons to 40 μM of crude Aβ42 preparation reduced cellular viability by approximately 50-60% compared with untreated CNs (Fig. 3d-f). However, Aβ42 cytotoxicity was significantly reversed in the presence of low concentrations (5 and 10 μM) of mitoxantrone or bithionol (Fig. 3d-e) in CNs. The substoichiometric inhibition of Aβ toxicity is consistent with the proposed mode of actions of these compounds, i.e., that they target fibril growth rather than stabilizing monomeric Aβ42. Despite the fact that hexachlorophene was a stronger inhibitor of protofibrils to fibril conversion and exhibited similar inhibitory effects on Aβ42 monomer fibrillization, fibril growth and seeding capacity as bithionol, it showed only modest protection against Aβ-induced toxicity in CNs (Fig. 3f).
Figure 3. Neuroprotection of the compounds against the Aβ42-induced toxicity in cortical neurons (CN).
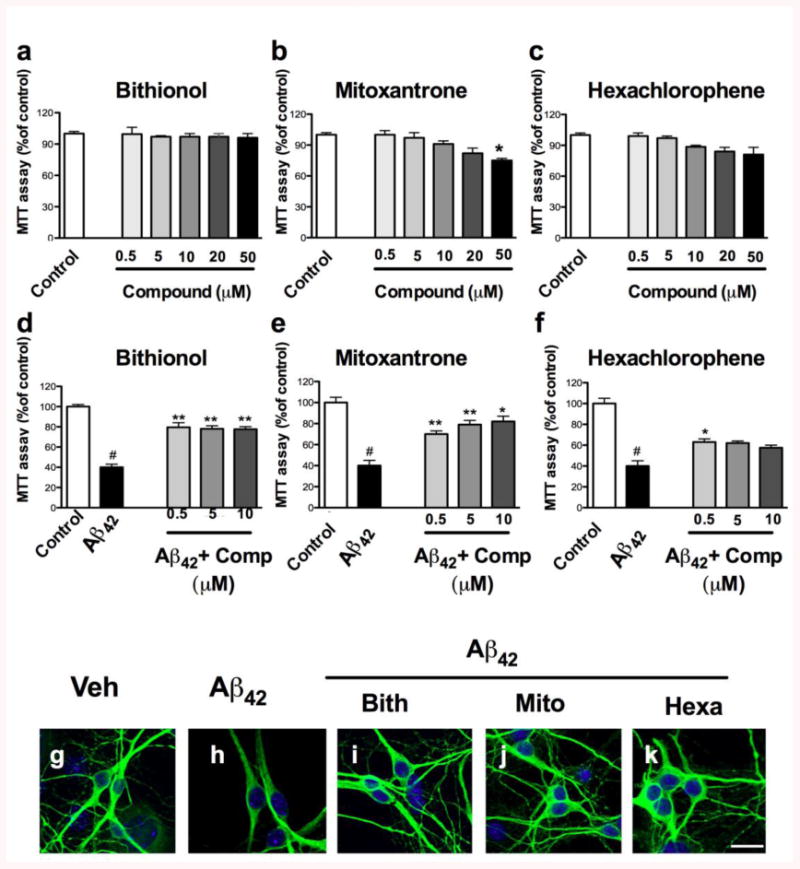
a-b) Cortical neurons were treated with different concentrations of the compounds (0.5, 5, 10, 20 and 50 μM) for 24 hours. d-f) The neurons were treated with 40 μM of the crude preparation of Aβ42 or co-treated with the crude preparation and the compounds at different concentrations (0.5, 5 and 10 μM) at 7 DIV. The effects of the compounds and the neuroprotection against the Aβ42-induced toxicity were evaluated using an MTT assay, and the control treatment was set to 100%. The error bars represent the mean±S.E. We used # to indicate the data compared to the control and * compared to the treatments with crude Aβ42 preparation, *p<0.05, **p<0.01, g-k). The neuroprotection of the compounds against Aβ42-induced toxicity was evaluated by staining the neurons with MAP2 (green) by immunofluorescence, and the nuclei were stained with DAPI (blue).
To determine morphological changes in the CNs after the exposure to the Aβ42 preparation and the neuroprotection of the compounds, we performed immunolabeling for the neuronal cytoskeleton protein MAP-2. Significant differences in MAP-2 staining were found between control and Aβ42–exposed cells (Fig. 3g-k). We found dystrophic neurons with several abnormal morphological features, including a dramatic reduction in the neuronal network (Fig. 3h). The analysis of the MAP-2 immunoreactivity in neurons co-incubated with Aβ42 crude preparation and compounds showed a significant reversion of the Aβ42–induced toxicity and a reduction of dystrophic neurites (Fig. 3i-k).
Bithionol and mitoxantrone reverse Alzheimer's disease features in APPtg mice
Motivated by the strong anti-amyloidgenic and neuroprotective properties observed for bithionol, mitoxantrone and hexachlorophene, we sought to validate our findings in an APP transgenic mouse model (APPtg). We first analyzed the plasma and brain pharmacokinetic properties of the three compounds. Following a single IV administration of each compound at 10 mg/kg, plasma and brain levels were determined by LC-Ms/Ms. For bithionol, the last AUC was 0.20 (20% brain penetration) with a half-life of 7.62 hours in the plasma and 1.77 hours in the brain. For mitoxantrone, the last AUC was 1.22 (100% brain penetration); the half-lives for plasma and brain were not determined. For hexachlorophene, the last AUC was 0.50 (50% brain permeability), and the half-lives were 4.4 hours in the plasma and 5.64 hours in the brain. After the brain and plasma levels of the three compounds were established, we analyzed the effects of three inhibitors on the amyloid plaques features including Aβ42 deposits, Aβ42 oligomers levels, synapse loss, neuronal damage and astrogliosis, which together represent the neuropathological hallmarks of AD (7), in APPtg mice; the data were compared with the vehicle-treated APP transgenic (APP Aβtg Vehicle) mice and the vehicle non-tg control (Non-tg Vehicle). To perform the in vivo studies, we used a transgenic mouse model that is characterized by the accumulation of mature plaques in the frontal cortex after 3-4 months of age and extends, at 5-7 months of age, to the hippocampus, thalamus and olfactory region. Moreover, the mice also produced high levels of the Aβ42 and exhibited synaptic damage and performance deficits in the water maze (Rockenstein et al., 2001 and Rockenstein et al., 2002). Bithionol and mitoxantrone cross the BBB and were administered to APPtg mice with a daily intraperitoneal injection for 1 month at the respective concentrations of 10 and 1 mg/Kg. In contrast, hexachlorophene is unable to cross the BBB and was injected in the APPtg mouse brain via a cannula and osmotic pump implanted into the right hemi ventricle.
Diffuse-like plaque detection in APPtg brains treated with bithionol and mitoxantrone
First, using immunostaining, we assessed the extent of Aβ42 accumulation and deposition into plaques in both APPtg mice (APPtg vehicle) and APPtg mice treated with the compounds in the frontal cortex (Fig. 4a-e) and the hippocampus (Fig. 4f-j). Subsequently, we performed a quantitative analysis of the area of neuropils (%) covered by Aβ42 immunoreactivity in the frontal cortex (Fig. 4p) and the hippocampus (Fig. 4q). Amyloid plaques are commonly classified as dense plaques surrounded by dystrophic neurites, reactive astrocytes, activated microglial cells and synaptic loss, while diffuse plaques are usually non-neuritic and are not associated with synaptic loss(Mucke et al., 2000). In vehicle-treated APPtg mouse sections, we found plaques with a dense amyloid deposit morphology in both regions (Fig. 4b&g) and an increase of the Aβ42 immunoreactivity with respect to the control (Non-tg Vehicle); these findings were in agreement with a previous analysis (Kumar-Singh et al., 2006). In APPtg mice treated with bithionol (Fig. 4c&h) and mitoxantrone (Fig. 4d&i), we observed diffuse-like plaques in the frontal cortex and the hippocampus and observed a significant decrease in the Aβ42-immunoreactivity in the frontal cortex when compared with the APPtg mice for both compounds (Fig. 4p) and in hippocampus for bithionol (Fig. 4q). In the APPtg mice treated with hexachlorophene (Fig. 4e&j), we detected dense plaques and high Aβ42-immunoreactivity (Fig. 4p-q).
Figure 4. Immunocytochemical analysis of the Aβ immunoreactivity in the frontal cortex and the hippocampus of APPtg mice to evaluate the effect of the treatments with bithionol, mitoxantrone and hexachlorophene.
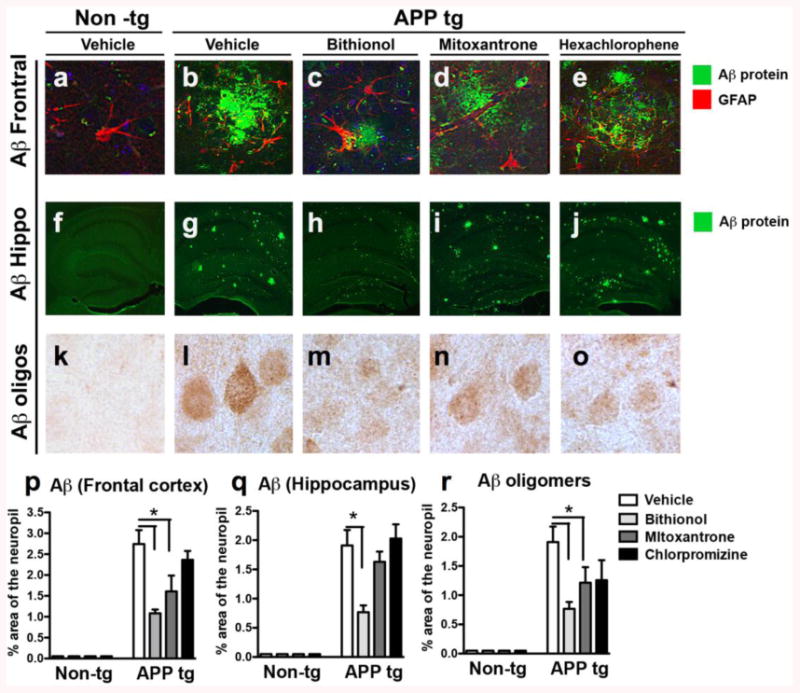
(a-j) Vibratome sections of the frontal cortex and the hippocampus were immunolabeled with thioflavine S, and plaques were analyzed in the frontal cortex and the hippocampus of the Non-tg mice. (a-f) The APPtg mice (b-g) were compared with the diffuse plaque-like structures found in APPtg mice treated with bithionol (c-h) and mitoxantrone (d-i) or in more dense structures in the presence of hexachlorophene (c-j). (k-o) Aβ deposits in the frontal cortex were also evaluated using the 6E10 antibody; in the graphs (p-q), the data are related to a computer-aided quantitative analysis of the area of the neuropils covered (%) by the anti-Aβ antibody in the frontal cortex and the hippocampus' Aβ oligomers in Non-tg and APPtg mice treated with the compounds. *p<0.05 compared to APPtg mice by ANOVA with post-hoc Dunnett's test.
Moreover, we analyzed the levels of Aβ plaques blotted with 6E10 antibody using DAB staining (Fig. 4k-o), thus confirming the ThS staining of the plaques.
Using electron microscopy, we also investigated the micrographs of neuropils and analyzed the status of synapses (SYN) and dendrites in control mice, in vehicle-treated APP mice and in APP mice after the administration of bithionol, mitoxantrone and hexachlorophene (Fig. 5a-o). The neuropil electron micrographs are depicted in Fig. 5a-e. In the non-tg mice, the synapses and dendrites demonstrated normal characteristics and were well preserved (Fig. 5a), whereas in the vehicle-treated APPtg mice, abundant dystrophic neurites (DN) with electrodense bodies typically associated with plaques were observed (Fig. 5b). In the bithionol- and mitoxantrone-treated APP mice (Fig. 5c-d), the axons were comparable to controls, while in the hexachlorophene-treated APP mice (Fig. 5e), abnormal dystrophic neurites were found. The neuropils with amyloid-containing areas were evaluated in Fig. 5f-o. No amyloid plaques were found in the non-tg mice (Fig. 5f&k). In the APP vehicle (Fig. 5g&l), there were abundant patches of amyloid fibrils (arrowheads) forming plaques. In the bithionol and mitoxantrone groups, only few fibrillar aggregates were found scattered in the neuropils (Fig. 5h&m and 5i&n). In contrast, hexachlorophene APP-tg mice showed abundant fibrils and plaques (Fig. 5e&o).
Figure 5. The micrographs of the neuropil by EM; an analysis of the synapses (SYN) and dendritic status.
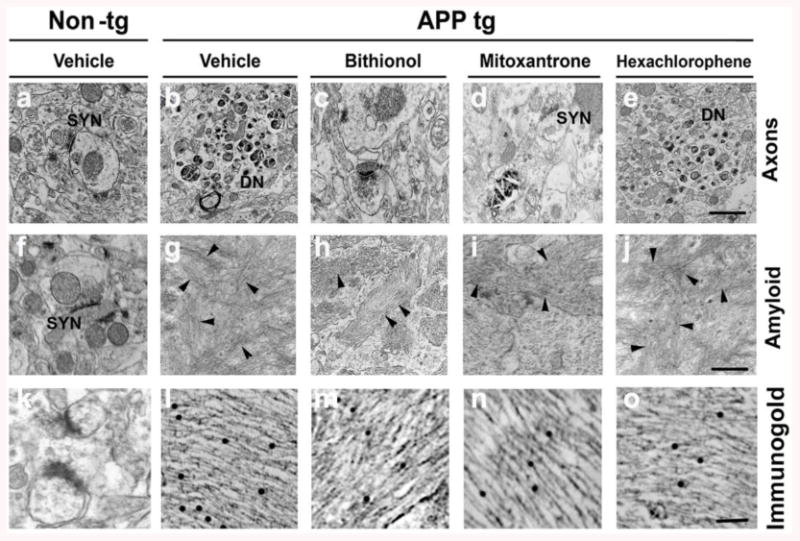
Analysis of the control in vehicle-treated APP mice and in APP mice after the administration of bithionol, mitoxantrone and hexachlorophene. Analysis of the electron micrographs of the neuropil (a-e) in the non-tg mice (a), in the vehicle-treated APPtg mice (b), in the bithionol-, mitoxantrone- and hexachlorophene-treated APP mice (c-e); analysis of the neuropil with the amyloid-containing areas (f-j) and of the amyloid-containing area by immunogold (k-o), in the non-tg mice (f-&k), in the APP vehicle (g&l) and in the bithionol (h&m), mitoxantrone (i&n) and hexachlorophene (j&o)-treated APP-tg mice.
Bithionol and mitoxantrone reduced the Aβ42 intermediate species
The posterior parts of the mouse brains were fractionated by ultracentrifugation, and the levels of Aβ42 monomers and oligomers in the membrane fraction were detected through immunoblotting with an Aβ42 antibody (82E1 clone). As shown in Fig. 6a, the immunoblot analysis revealed different bands corresponding to Aβ42 intermediate species. In agreement with previous studies (Pham et al., 2010), the analysis of the vehicle-treated APP transgenic brains displayed a significant increase in the levels of the bands corresponding to monomers and intermediate species (Fig. 6a) compared with the non-tg control. In contrast, the treatment of the APPtg mice with bithionol and mitoxantrone induced a significant reduction in the levels of Aβ42 monomers and intermediate species (Fig. 6a-b). In the APPtg mice treated with hexachlorophene, the Aβ42 level intermediate (Fig. 6a-b) were comparable to that observed in the vehicle-treated APPtg mice. To confirm the reduction of the Aβ42 levels in APPtg mice treated with bithionol and mitoxantrone, we quantified the Aβ42 oligomers present in the brain samples using ELISA; the Aβ42 has already been identified as the most abundant species in the APPtg (line 41). We detected the Aβ42 oligomers in membrane fractions (Fig. 6c) and simultaneously extracted the brain samples in guanidine-HCl to maximize the detection of total Aβ42 levels (Fig. 6d). The administration of bithionol and mitoxantrone induced a significant and dramatic reduction in Aβ42 levels compared with the vehicle-treated APPtg mice in both the membrane fractions and the guanidine-HCl brain extractions (Fig. 6c-d). In contrast, hexachlorophene treatment did not significantly affect Aβ42 levels. Together, the results obtained through immunoblot analysis (Fig. 6a-b) and ELISA (Fig. 6c-d) confirmed a strong effect of bithionol and mitoxantrone on the reduction of Aβ42 levels.
Figure 6. Immunoblot analysis of Aβ species, ELISA test and γ-secretase activity in APPtg mice treated with bithionol, mitoxantrone and hexachlorophene.
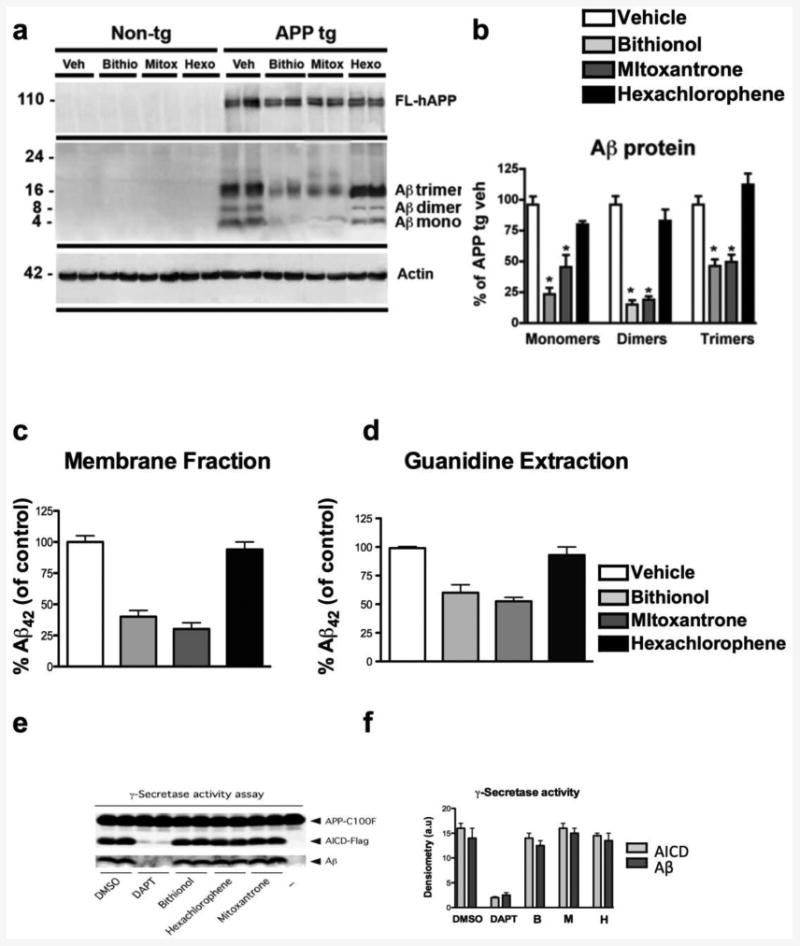
a)The mouse posterior brain tissues were homogenized in Buffer A, and the membrane fraction was probed with antibodies against Aβ (82E1) and β-actin. b) Semi-quantitative densitometric analyses of the bands representing Aβ monomers (4 kDa), dimers (8 kDa), and trimers (12 kDa). N=5 cases per group, *p<0.05 compared with APP/tg mice by ANOVA with post-hoc Dunnett's test. c) and d) quantitative analysis of Aβ42 levels in APPtg mice treated with bithionol, mitoxantrone and hexachlorophene by ELISA. The quantification of Aβ42 levels in the membrane fraction (c) and guanidine-HCl extracted samples (d) N=5 cases per group, female gender. *p<0.05 compared with APPtg mice by ANOVA with post-hoc Dunnett's test.
e-f) the compounds do not affect the processing of APP-C100-Flag and Aβ production by purified γ-secretase. γ-Secretase solubilized in 0.2% CHAPSO-HEPES, pH 7.5, was incubated at 37°C for 4 h with 1 μM C100-Flag substrate, 0.1% PC, 0.025% PE and 10 μM of bithionol, hexachlorophene and mitoxantrone. Activity assays included control reactions with 10 μM of the γ-secretase inhibitor DAPT, DMSO (1.6% w/v) or a reaction with omitted γ-secretase (-). The reactions were halted by adding 0.5% SDS, and the resulting products were detected with anti-Aβ and -AICD-Flag antibodies 6E10 and M2, respectively. The relative levels of AICD-Flag or Aβ were estimated by densitometry (mean±SD; n=2).
Bithionol and mitoxantrone neuroprotection against synapse loss and neuronal damage
To evaluate the neuroprotective effects of bithionol and mitoxantrone against synapse loss in APPtg mice, we analyzed the levels of two proteins: synaptophysin, a major protein constituent of the membranes of pre-synaptic vesicles, and postsynaptic density-95 protein (PSD95). PSD95 levels have previously been correlated with the level of Aβ oligomers in the brain, and reduced PSD95 protein levels have also been observed in AD patients and APPtg mice (Pham et al., 2010). We analyzed the levels of synaptophysin and PSD95 in vehicle-treated APPtg mice and in APPtg mice after the administration of bithionol, mitoxantrone and hexachlorophene through immunoblot analysis (Fig. 7a). In addition, we performed a semi-quantitative analysis of the bands through densitometry (Fig. 7b). The mouse brains were fractionated, and the membrane fractions were probed with anti-PSD95 and anti-SY38 antibodies. In agreement with previous studies in APPtg mice, we found a significant reduction of PSD95 and synaptophysin levels compared with the control and a significant reversion of the damage in the presence of the three compounds (Fig. 7a-b).
Figure 7. Immunoblot analysis of the synaptic proteins (PSD95 and synaptophysin) and an analysis of the synaptophysin immunoreactivity in the frontal cortex and the hippocampus of APPtg mice treated with bithionol, mitoxantrone and hexachlorophene.
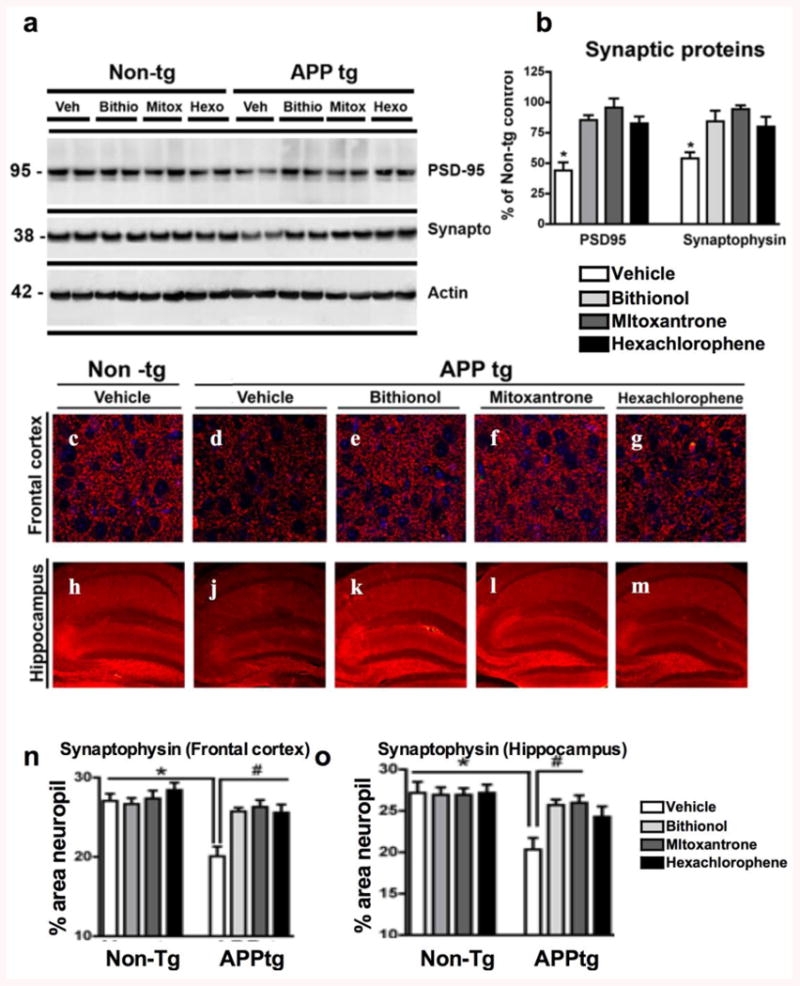
a) Posterior brain tissues were homogenized in Buffer A, and the membrane fraction was probed with antibodies against PSD95 and synaptophysin (SY38). b) Semi-quantitative densitometric analyses of the bands for PSD95 and synaptophysin, N=5 cases per group, *p<0.05 compared with APP/tg mice by ANOVA with post-hoc Dunnett's test; (c-m) Sections of the frontal cortex (c-g) and the hippocampus (h-m) were immunolabeled with an anti-SYN antibody, a presynaptic terminal marker, and the nuclei are stained blue with DAPI; the brain sections were analyzed with the laser scanning confocal microscope to evaluate the protective effects of the compounds towards the synapsis damage. (n-o) Computer-aided quantitative analysis of % area of neuropil covered by SYN-immunoreactive terminals in the frontal cortex and the hippocampus of Non-tg and APPtg mice treated with the compounds. *p<0.05 compared with APPtg mice by ANOVA with post-hoc Dunnett's test.
Moreover, to further investigate the neuroprotection of bithionol and mitoxantrone against synapse loss, we analyzed the levels of synaptophysin immunoreactivity in the frontal cortex (Fig. 7c-g) and hippocampus (Fig. 7h-m) of vehicle-treated APPtg mice (Fig. 7d&j) and mice treated with bithionol (Fig. 7e&k), mitoxantrone (Fig. 7f&l) and hexachlorophene (Fig. 7g&m). The Non-tg mice were used as the control (Fig. 7c&h). In the frontal cortex and hippocampus of vehicle-treated APPtg mice, there was a progressive loss of SYN-IR (Fig. 7d&j) compared with the control. In contrast, APPtg mice treated with the compounds showed increased SYN-IR levels in both regions. We analyzed the neuroprotection in the brain samples treated with the compounds using a computer-aided quantitative analysis of the area (%) of neuropils covered by SYN-immunoreactivity terminals in the frontal cortex (Fig. 7n) and in the hippocampus (Fig. 7o). We found a significant reduction of synaptophysin immunoreactivity in APPtg mice treated with PBS compared with the controls in the frontal cortex (Fig. 7d and 7n) and in the hippocampus (Fig. 7j and Fig. 7o). The synapse loss in these two regions was significantly reversed in the presence of bithionol (Fig. 7e&k and 7n-o), mitoxantrone (Fig. 7f&l and 7n-o) and hexachlorophene (Fig. 7g&m and 7n-o).
Moreover, we analyzed the effects of the compounds on the neuronal structure and integrity in the frontal cortex and hippocampus by immunostaining the mouse brain tissues for microtubule-associated protein 2 (MAP2), which is a specific mature neuronal marker (Fig. 8a-j); we then quantified MAP2 immunoreactivity (Fig. 8k-l). In vehicle-treated APPtg mice, we found a loss of microtubule structures in the frontal cortex and the hippocampus (Fig. 8b&g) compared with the control (Fig. 8a&f), reflecting neuronal damage in these structures. This reduction was confirmed by quantifying the MAP2-IR levels (Fig. 8k-l). In APPtg mice treated with bithionol, mitoxantrone and hexachlorophene, we found a reversion of the damage in the frontal cortex for the three compounds (Fig. 8c-e and 8k) as well as in the hippocampus when treated with bithionol and mitoxantrone (Fig. 8h-i and 8l). Finally, we studied the effects of the compounds on neuronal loss by evaluating NeuN immunoreactivity, a nuclear neuronal marker in the frontal cortex of APPtg mice (Fig. 8m-q and 8w) (McLean et al., 1999). In the brain sections of vehicle-treated APPtg mice, we found reduced NeuN immunoreactivity (Fig. 8n), which was confirmed by cell counting (Fig. 8w compared with control Fig. 8m). An increase in NeuN immunoreactivity was found in APPtg mice treated with bithionol (Fig. 8o&8w), mitoxantrone (Fig. 8p&8w) and hexachlorophene (Fig. 8q&8w).
Figure 8. Immunolabeling analysis for microtubule-associated protein 2 (MAP2), neuronal density and astrogliosis in the frontal cortex of APPtg mice treated with bithionol, mitoxantrone and hexachlorophene.
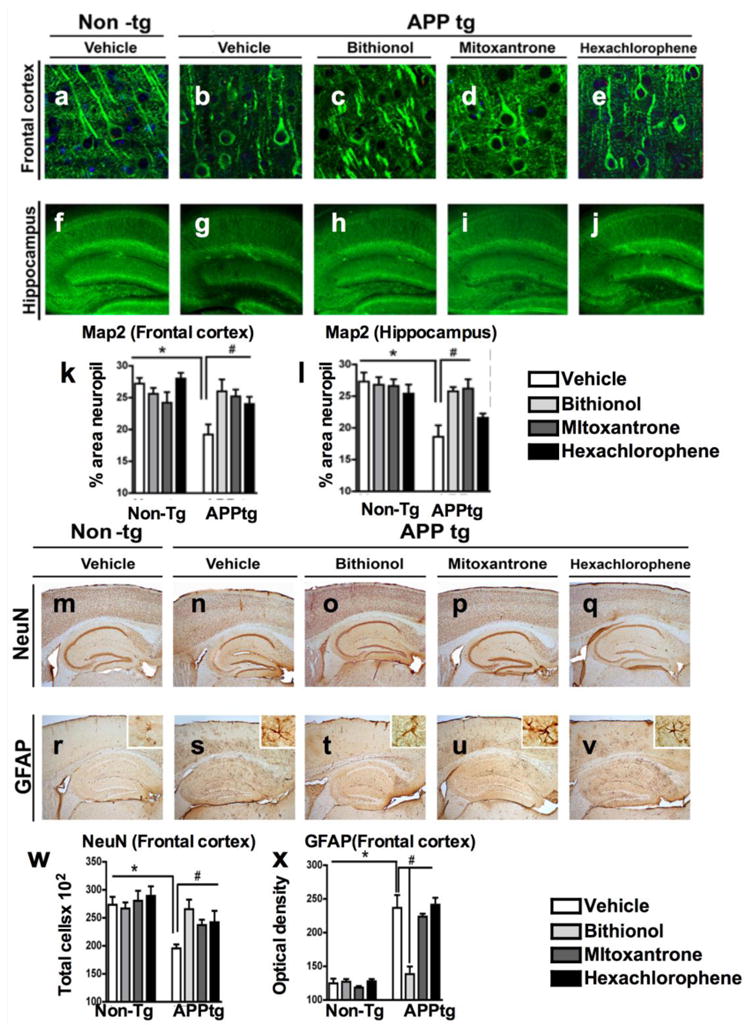
Vibratome sections of the frontal cortex (a-e) and the hippocampus (f-j) were immunolabeled with anti-MAP2 antibody, a neuronal-specific protein localized to the cytoskeleton, and the nuclei were stained by DAPI. The sections were visualized with the laser scanning confocal microscope (k-l). Computer-aided quantitative analysis of the area of neuropils (%) covered by MAP2 immunoreactivity. The effect of the compounds was also examined for neuronal density and astrogliosis using an antibody against the neuronal marker NeuN (m-q), and against the astrocytic marker GFAP (r-v). In the graphs (w-x), the analysis of the frontal cortex NeuN immunoreactivity and GFAP immunoreactivity across the experimental groups. *p<0.05 compared with APPtg
Bithionol and mitoxantrone protected neurons against astroglial reactivity
Reactive astrogliosis is commonly associated with dense core plaques, indicating that amyloid-β is a major trigger of this glial response. To study the effects of the compounds against astroglial reactivity, we analyzed the immunoreactivity of a glial marker, glial fibrillar acidic protein (GFAP), in the frontal cortex (Fig. 8f-x). In control mouse sections, astrocytes were not reactive (Fig. 8r), and GFAP immunoreactivity was very low (Fig. 8x). In vehicle-treated APPtg mice, the astrocytes exhibited an increase in the number of their intermediate filaments, changing their phenotypic appearance (Fig. 8s); an increase in the GFAP immunoreactivity was also observed (Fig. 8x). The treatment of APPtg mice with bithionol induced a strong and significant reduction of GFAP immunoreactivity (Fig. 8x). A significant effect on astroglial reactivity was also observed in animals treated with mitoxantrone (Fig. 8u&x) but not with hexachlorophene (Fig. 9r&x).
Effects of the compounds on γ-secretase activity
To determine whether the three selected compounds might influence Aβ production by altering the levels of secretases or by acting as direct γ-secretase inhibitors, we investigated the ability of these compounds to inhibit γ-secretase activity in a cell-free γ-secretase assay performed with highly purified γ-secretase and APP-C100-Flag, a recombinant APP-based substrate (Cacquevel et al., 2008 and Fraering et al., 2005). We did not observe any significant changes in secretase levels (Fig. 6e-f). Furthermore, and in contrast to the control γ-secretase inhibitor DAPT, none of the compounds tested at a final concentration of 10 μM inhibited the γ-secretase-dependent processing of APP-C100-Flag or the production of both AICD and Aβ (Fig. 9). Together, our results demonstrate that bithionol, hexachlorophene and mitoxantrone do not target the γ-secretase-dependent processing of APP-C99 and Aβ production.
Discussion
Recently, we and others have shown that nucleated polymerization and seeding-mediated aggregation are essential for amyloid toxicity (Jan et al., 2010), progressive neurodegeneration and pathology spreading by several amyloid forming proteins (Jucker and Walker, 2011) including Aβ (Aguzzi et al., 2007, Langer and al., 2011 and Lee et al., 2010), α-synuclein (Luk et al., 2012 and Volpicelli-Daley et al., 2011) and IAPP (Gurlo et al., 2010). These findings have significant implications for the development of effective treatments to slow AD progression, especially because a significant amount of amyloid plaques are present in the brains of AD patients at the time of diagnosis. Amyloid formation, a nucleated polymerization process (Supplemental Fig. 1), is significantly accelerated by the presence or addition of preformed fibrils (Harper and Lansbury, 1997). Therefore, once significant amyloids have formed, preventing further aggregation and the spreading of the pathology becomes more challenging because the energetic barriers associated with the nucleation of aggregation are significantly lower, and further aggregation can occur below the critical concentration required for aggregation in the absence of a seed.
We hypothesized that inhibiting oligomer and seeding-mediated aggregation constitutes a viable and effective strategy for protecting against neurodegeneration and disease progression in AD. Unlike monomeric Aβ, which is unstructured and difficult to target using small molecules, Aβ oligomers and fibrils in particular are highly structured and possess hydrophobic pockets that can be targeted by small molecules (Autiero et al., 2013).
To test this hypothesis, we utilized an in vitro, cell-free high-throughput screening method to identify novel inhibitors of the Aβ42 seeding capacity and fibril growth. Specifically, bithionol, mitoxantrone and hexachlorophene were selected as potent inhibitors of the in vitro seeding-mediated aggregation. Of the three compounds, bithionol and mitoxantrone were the most effective at blocking Aβ pathology in APPtg mice, even when administered to an AD animal model 2-3 months after amyloid deposition. In addition, we found that these compounds were able to block the conversion of the monomers and protofibrils into mature fibrils by stabilizing oligomeric intermediates in the amyloid pathway. Stabilization of intermediate species with bithionol, mitoxantrone and hexachlorophene also reduced Aβ42-induced toxicity in primary cortical neurons.
After demonstrating that these compounds can prevent seeding-mediated fibril growth and protect neurons from Aβ-induced toxicity, we tested the inhibitors in vivo in APPtg mice. We found that bithionol and mitoxantrone but not hexachlorophene significantly reduced Aβ accumulation and neurodegenerative pathology in APPtg mice by stabilizing diffuse-like plaques and reducing the levels of Aβ intermediates and higher order species. Moreover, the levels of amyloid deposition and the neuro-inflammatory response, as exemplified by the levels of astrogliosis, were significantly reduced.
Another interesting finding of our study was that although the three compounds showed similar effects in vitro, the most significant effects on reducing Aβ-related pathology in vivo were observed with mitoxantrone and bithionol. Mitoxantrone is an anthracenedione-derived antineoplastic agent that has also been used for the treatment of multiple sclerosis. Mitoxantrone has a MW of 444, a clogP of 2.29 and a logBB of -1.9; it has been shown in vitro to inhibit B-cell, T-cell and macrophage proliferation and impair antigen presentation as well as interfere with the secretion of interferon gamma (IN-γ), TNFα and IL-2. Mitoxantrone, a DNA-reactive agent that intercalates into DNA through hydrogen bonding, causing crosslinks and strand breaks, also interferes with RNA; it is a potent inhibitor of topoisomerase II, an enzyme responsible for uncoiling and repairing damaged DNA.
In contrast, bithionol is a halogenated bisphenol with a MW of 370, a logP of 5.72 and a logBB of +0.34 that has been used as local anti-infective and anti-platyhelmintic agent. The mechanisms for its anti-infective actions are not well understood. Hexachlorophene is similar to bithionol and is a chlorinated version of a bisphenol with an antiseptic and bacteriostatic action against Gram-positive organisms, but is substantially less effective against Gram-negative organisms. The antiseptic mechanism of action occurs by inhibiting the membrane-bound portion of the electron transport chain, respiratory D-lactate dehydrogenase, thus blocking cellular respiration. Hexachlorophene has a MW of 371, a clogP of 6.39 and a logBB of +0.50. Bithionol has not been tested in AD models; however, recent studies have shown that this compound is a robust activator of Slack channels. The Slack (Sequence like a calcium-activated K channel) (Slo2.2) gene is abundantly expressed in the mammalian brain and encodes a sodium-activated K+ (KNa) channel. Although the specific roles of Slack channel subunits in neurons remain to be identified, they may play a role in the adaptation of the firing rate and in protection against ischemic injury (Yang et al., 2006).
Although hexachlorophene has better parameters for trafficking into the CNS, both mitoxantrone and biothinol displayed more robust effects with respect to reducing pathology in APPtg mice, suggesting that the differences in activity for these compounds are unrelated to CNS exposure. Hexachlorophene has been shown to be neurotoxic in rats by inhibiting brain succinate dehydrogenase (SDH) activity, thus inducing spongiosis and brain edema (Kinosita et al., 2000). Hexachlorophene has not been tested before in models of neurodegenerative disorders. In contrast, mitoxantrone has been used in multiple sclerosis and has been shown to modulate the expression of Tau by interfering with the mRNA stem loop structure (Liu et al., 2009). Our results are consistent with previous studies showing that mitoxantrone can block Aβ oligomerization and fibril formation (Colombo et al., 2009). These compounds represent a proof of concept that inhibiting the seeding aggregation pathway might be a potential therapeutical value and provide a starting point for the development of promising anti-amyloidgenic drugs for the treatment of AD. However the compounds need to be further refined and developed to reduce the reportd toxic effects.
Anatomical analysis of the pathological Aβ deposition in AD brains showed that Aβ aggregate deposition followed a hierarchical distribution (Thal et al., 2002), suggesting spreading of the aggregates during disease progression. AD brain and aged APPtg mouse brain extracts injected in young APPtg mice induced β-amyloidosis not only at the injection site but also in other brains areas. Spreading of the Aβ deposits was observed from the hippocampus region, i.e., the injection site, progressively to the dorsal lateral geniculate nucleus, the corpus callosum, the entorhinal cortex and in vasculatures of the thalamus and the pia mater (Eisele et al., 2009); this effect followed a template prion-like spreading mechanism (Aguzzi et al, 2007). Recent studies have shown that amyloid proteins can also spread in PD (Luk et al., 2012, Volpicelli-Daley et al., 2011 and Lee et al., 2011) and Huntington's disease (Lee et al., 2011). Together, these data showed an important role for the spreading in the progression of neurodegenerative disease, and suggest blocking the spreading mechanism in AD, as in other neurodegenerative disorders, could represent a new therapeutic avenue (Lee et al., 2011).
In summary, utilizing novel in vitro assays, we identified novel compounds (mitoxantrone and bithionol) that inhibit Aβ seeding-mediated aggregation in vitro and demonstrated that these compounds block the Aβ-associated pathology in APPtg mice. Our in vitro studies suggest that the neuroprotective effects of these compounds are not mediated by their effects on APP processing or Aβ production, but instead by their ability to act at multiple steps on the amyloid cascade pathway, including the inhibition of Aβ seeding-mediated aggregation and aggregation-mediated toxicity. It is likely that interfering with fibril growth and seeding-mediated aggregation will also enhance the clearing of Aβ aggregates via other physiological clearance mechanisms. Together, our work suggests that targeting seeding-mediated aggregation has great potential for developing novel small molecule drugs to prevent Aβ pathology formation and/or inhibit pathology spreading and neurodegeneration at different stages of AD progression.
Supplementary Material
Highlights.
Hightrougthput screening in vitro of a library of FDA approved drugs
Identification new inhibitors of Aβ seeding mediated aggregation: Mitoxantrone and Bithionol
Mitoxantrone and Bithionol protected primary cortical neurons from Aβ-induced toxicity
Administration of the compounds in APPtg mice stabilize diffuse plaques
Compounds in vivo administration ameliorated synapse loss, neuronal damage and astrogliosis
Acknowledgments
This work is supported by the Swiss Federal Institute of Technology, Lausanne (HAL), the Strauss Foundation (HAL, SE, SD) and NIH grants AG18440, AG022074, AG031097, AG010435 (to EM). We would like to express our gratitude to Nathalie Jordan, John Perrin and Amy Paulino for their outstanding technical support and Abid Oueslati for assistance in preparing Supplemental Figure S1.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aguzzi A, Heikenwalder M, Polymenidou M. Insight into prions strains and neurotoxicity. Nat Rev Mol Cell Biol. 2007;8(7):552–61. doi: 10.1038/nrm2204. Review. [DOI] [PubMed] [Google Scholar]
- 2.Autiero I, Saviano M, Langella E. In silico investigation and targeting of amyloid β oligomers of different size Mol. Biosyst. 2013;9(8):2118–24. doi: 10.1039/c3mb70086k. [DOI] [PubMed] [Google Scholar]
- 3.Cacquevel M, Aeschbach L, Osenkowski P, Li D, Ye W, Wolfe MS, Li H, Selkoe DJ, Fraering PC. Rapid purification of active gamma-secretase, an intermembrane protease implicated in Alzheimer's disease. J Neurochem. 2008;104:210–220. doi: 10.1111/j.1471-4159.2007.05041.x. [DOI] [PubMed] [Google Scholar]
- 4.Colombo R, Carotti A, Catto M, Racchi M, Lanni C, Verga L, Caccialanza G, De Lorenzi E. CE can identify small molecules that selectively target soluble oligomers of amyloid beta protein and display antifibrillogenic activity. Electrophoresis. 2009;30:1418–29. doi: 10.1002/elps.200800377. [DOI] [PubMed] [Google Scholar]
- 5.Di Giovanni S, Eleuteri S, Paleologou KE, Yin G, Zweckstetter M, Carrupt PA, Lashuel HA. Entacapone and tolcapone, two catechol O-methyltransferase inhibitors, block fibril formation of alpha-synuclein and beta-amyloid and protect against amyloid-induced toxicity. J Biol Chem. 2010;285:14941–54. doi: 10.1074/jbc.M109.080390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duyckaerts C, Delatour B, Potier MC. Classification and basic pathology of Alzheimer's disease. Acta Neuropathology. 2009;118:5–36. doi: 10.1007/s00401-009-0532-1. [DOI] [PubMed] [Google Scholar]
- 7.Eisele YS, Bolmont T, Heikenwalder M, Langer F, Jacobson LH, Yan ZX, Roth K, Aguzzi A, Staufenbiel M, Walker LC, Jucker M. Induction of cerebral beta-amyloidosis: intracerebral versus systemic Abeta inoculation. PNAS. 2009;106(31):12926–31. doi: 10.1073/pnas.0903200106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fraering PC, Ye W, LaVoie MJ, Ostaszewski BL, Selkoe DJ, Wolfe MS. gamma-secretase substrate selectively can be modulated directly via interaction with a nucleotide-binding site. J Biol Chem. 2005;280:41987–41996. doi: 10.1074/jbc.M501368200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frost B, Diamond MI. Prion-like mechanism in neurodegenerative diseases. Nature Rev Neuroscinece. 2010;11(3):155–9. doi: 10.1038/nrn2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, Mant R, Newton P, Rooke K, Roques P, Talbot C, Pericak-Vance M, Roses A, Williamson R, Rossor M, Owen M, Hardy J. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 11.Gurlo T, Ryazantsev S, Huang CJ, Yeh MW, Reber HA, Hines OJ, O'Brien TD, Glade CG, Butler PC. Evidence for proteotoxicity in beta cells type 2 diabetes: toxic islet amyloid polypeptide oligomers form intracellulary in the secretory pathway. Am J Pathol. 2010;176:861–869. doi: 10.2353/ajpath.2010.090532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harper JD, Lansbury PT., Jr Model of amyloid seeding in Alzheimer's disease and scrapie mechanistic truths and physiological consequences of the time-dependent solubility and amyloid proteins. Ann Rev Biochem. 1997;66:385–407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- 13.Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19:8876–888. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jan A, Hartley DM, Lashuel HA. Preparation and characterization of toxic Abeta aggregates for structural and functional studies in Alzheimer's disease research. Nat Protoc. 2010;5(6):1186–1209. doi: 10.1038/nprot.2010.72. [DOI] [PubMed] [Google Scholar]
- 15.Jucker M, Walker LC. Pathogenic protein seeding in Alzheimer's disease and other neurodegenerative disorders. Ann Neurol. 2011;70:532–40. doi: 10.1002/ana.22615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kane MD, Lipinski WJ, Callahan MJ, Bian F, Durham RA, Schwarz RD, Roher AE, Walker LC. Evidence for seeding of beta -amyloid by intracerebral infusion of Alzheimer brain extracts in beta -amyloid precursor protein-transgenic mice. J Neurosci. 2000;20(10):3606–11. doi: 10.1523/JNEUROSCI.20-10-03606.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kinosita Y, Matsumura H, Yokota A. Hexachlorophene-induced brain edema in rat observed by proton magnetic resonance. Brain Res. 2000;873:127–30. doi: 10.1016/s0006-8993(00)02523-3. [DOI] [PubMed] [Google Scholar]
- 18.Kraszpulski M, Soininen H, Helisalmi S, Alafuzzoff I. The load and distribution of beta-amyloid in brain tissue of patients with Alzheimer's disease. Acta Neurol Scand. 2001;103:88–92. doi: 10.1034/j.1600-0404.2001.103002088.x. [DOI] [PubMed] [Google Scholar]
- 19.Kumar-Singh S, Theuns J, Van Broeck B, Pirici D, Vennekens K, Corsomit E, Cruts M, Dermaut B, Wang R, Van Broeckhoven C. Mean age-of-onset of familial Alzheimer's disease caused by presenilin mutations correlates with both increased Aβ42and decreased Aβ40. Hum Mutat. 2006;27:686–695. doi: 10.1002/humu.20336. [DOI] [PubMed] [Google Scholar]
- 20.Langer F, Eisele YS, Fritschi SK, Staufenbiel M, Walker LC, Junker M. Soluble Aβ are potent inducers of cerebral β-amyloid. J Neurosci. 2011;31:14488–95. doi: 10.1523/JNEUROSCI.3088-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee SJ, Desplats P, Sigurolson C, Tsigelny I, Masliah E. Cell-to-cell transmission of non prion protein aggregates. Nat Rev Neur. 2010;6:702–6. doi: 10.1038/nrneurol.2010.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee SJ, Lim HS, Masliah E, Lee HJ. Protein aggregate spreading in neurodegenerative diseases: Problems and prospectives. Neuroscience Research. 2011;70:339–348. doi: 10.1016/j.neures.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lemere CA, Blusztajin JK, Yamaguchi H, Wisniewski T, Saido TC, Selkoe DJ. Sequence of deposition of heterogeneous amyloid beta peptides and APOE in Down syndrome; implications for initial events in amyloid plaque formation. Neurob Dis. 1996;3:79–84. doi: 10.1006/nbdi.1996.0003. [DOI] [PubMed] [Google Scholar]
- 24.Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 25.Lewis J, Dickson DW, Lin WL, Chisholm L, Chisholmx A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 26.Liu Y, Peacey E, Dickson J, Donahue CP, Zheng S, Varani G, Wolfe MS. Mitoxantrone analogues as ligands for a stem-loop structure of tau pre-mRNA. J Med Chem. 2009;52:6523–6. doi: 10.1021/jm9013407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luk KC, Kehm VM, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegeneration α-synucleinopathy in mice. J Exp Med. 2012;209:975–986. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hasimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 29.Masliah E, Rockenstein E, Veinbergs I, Sagara Y, Mallory M, Hashimoto M, Mucke L. β –amyloid peptides enhance α-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer's disease and Parkinson's disease. PNAS. 2001;98:12245–12250. doi: 10.1073/pnas.211412398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McLean CA, Cherny RA, Fraser FW. Soluble pool of A-beta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 31.Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Walsh DM, Mathews PM, Ghiso J, Staufenbiel M, Walker LC, Jucker M. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313(5794):1781–4. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- 32.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholondeko D, Jhonson-Wood K, McConlouge L. High-level neuronal expression of A beta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nilsberth C, Westlind-Daniellsson A, Eckman CB, Condron MM, Axelman K, Forsell C, Stenh C, Luthman J, Teplow DB, Younkin SG, Naulund J, Lannfelt L. The Arctic APP mutation (E693G) causes Alzheimer's disease by enhanced Aβ protofibril formation. Nature Nuroscience. 2001;4:887–893. doi: 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- 34.Pham E, Crews L, Ubhi K, Hansen L, Adame A, Cartier A, Salmon D, Galasko D, Michael S, Savas JN, Yates JR, Glabe C, Masliah E. Progressive accumulation of amyloid-b oligomers in Alzheimer's disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. FEBS Journal. 2010;227:3051–3067. doi: 10.1111/j.1742-4658.2010.07719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rockenstein E, Mallory M, Mante M, Sisk A, Masliah E. Early formation of mature amyloid-β protein deposits in a mutant APP transgenic model depends on levels of Aβ1-42. J Neurosci Res. 2001;66:573–582. doi: 10.1002/jnr.1247. [DOI] [PubMed] [Google Scholar]
- 36.Rockenstein E, Mallory M, Alfrod M, Windisch M, Moessler H, Masliah E. Effects of cerebrolysin on amyloid-beta deposition in a transgenic model of Alzheimer's disease. J Neural Transm Suppl. 2002;62:327–36. [PubMed] [Google Scholar]
- 37.Selkoe DJ. Alzheimer's Disease: genes, proteins and Therapy. Physiol Rev. 2012;81:741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 38.Sthor J, Watts J, Mensiger ZL, Oehler A, Grillo SK, Dearmond SJ, Prusiner SB, Giles K. Purified and synthetic Alzheimer's amyloid beta prions. PNAS. 2012;109(27):11025–30. doi: 10.1073/pnas.1206555109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sturchler-Pierrat C, Staufenbiel M. Phatogenic mechanisms of Alzheimer's disease analyzed in the APP23 transgenic mouse model. Ann N Y Acad Sci. 2000;920:134–139. doi: 10.1111/j.1749-6632.2000.tb06915.x. [DOI] [PubMed] [Google Scholar]
- 40.Thal DR, Rub U, Orantes M, Braak H. Phases of amyloid-β-deposition in the human brain and its revelance for the development of AD. Neurology. 2002;58(12):1791–800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 41.Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–67. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walker LC, Diamond MI, Duff KE, Hyman BT. Mechanism of protein seeding in neurodegenerative disease. JAMA Neurol. 2013;70(3):304–10. doi: 10.1001/jamaneurol.2013.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wougulis M, Wright S, Cunningham D, Chilcote T, Powell K, Rydel RE. Nucleation-dependent polymerization is an essential component of amyloid-mediated neuronal cell death. J Neuroscience. 2005;25(5):1071–81. doi: 10.1523/JNEUROSCI.2381-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang B, Gribkoff VK, Pan J, Damagnez V, Dworetzky SI, Boissard CG, Bhattacharjee A, Yan Y, Sigworth FJ, Kaczmarek LK. Pharmacological activation and inhibition of Slack (Slo2.2) channels. Neuropharmacology. 2006;51:896–906. doi: 10.1016/j.neuropharm.2006.06.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.