

Introduction
Aortic valve (AV) sclerosis (AVS) is a form of AV disease affecting an estimated 1 in 4 people above the age of 65 in the United States.1 An aging population and more widespread use of noninvasive imaging are increasing the incidence of AVS. AVS is typically defined as calcification of the aortic leaflets without impairment in leaflet excursion or a significant transvalvular pressure gradient.2 It is characterized by a gradual progression beginning with calcium deposition that may ultimately transform to aortic stenosis (AS) with obstruction of outflow from the left ventricle. Severe AS eventually leads to ventricular remodeling and hemodynamic compromise with a high morbidity and mortality if not treated. Long considered an incidental age‐related degenerative process as a result of progressive wear and tear, there is substantial emerging evidence related to AVS that challenges this assumption.
Recent observations have shown that the development of AVS and AS may involve chronic inflammatory infiltrates, deposition of atherosclerotic lipoproteins, and calcification, akin to coronary artery disease (CAD). However, AVS has unique features, including a calcium predominance on histology, gradual progression, and location at a site of high pressure that serves as a gateway from the heart to the systemic circulation. Some investigators have reported the frequent coexistence of either AVS or AS in patients with underlying CAD.3 Several studies have demonstrated that independent risk factors in the progression of CAD, such as dyslipidemia, hypertension, and male sex, may also affect development of AVS and its progression to AS.4–6 These observations not only highlight the many shared characteristics of CAD and AVS but have also prompted investigators to test the efficacy of medical interventions that may have salutary effects on both conditions.
Heart disease is the leading cause of death in the United States. The majority of these deaths are attributed to CAD.7 Improvements in treatment for CAD, such as statins, angiotensin inhibitors (ACEI), and revascularization, have resulted in a larger proportion of the population living with CAD.8 Realizing the potential similarities between the underlying pathophysiology for AVS and CAD, many clinically relevant questions remain unanswered.9–10 For instance, does the presence of AVS suggest existence or progression of underlying CAD? Should AVS be considered a novel risk factor for the development of CAD? Does the finding of AVS warrant the initiation and careful titration of medications with lifestyle changes analogous to current strategies used in treating patients with diabetes mellitus? In this review, we discuss the shared pathophysiological aspects of AVS and CAD, summarize the present literature on mechanisms that lead to disease progression, and provide insights for future research to identify novel therapeutic targets.
Pathophysiology of CAD and AVS
Mechanical Forces
The AV is equipped with the capacity for dynamic movement in a high‐pressure setting. Endothelial injury from this stressful environment is thought to be the inciting factor for AVS. Lesions frequently occur on the aortic side of the leaflets, an area of high turbulent flow and tensile stress with low shear stress.11 The center of the valve cusp has the greatest mechanical stress and is more frequently involved than the commissures. Furthermore, there is a predilection for involvement of the noncoronary cusp, likely secondary to the lower shear stress given the lack of diastolic flow over this cusp.2 In contrast, the coronary arteries passively fill during diastole in a lower pressure environment. However, like AVS, coronary atherosclerotic lesions more commonly occur at sites with the highest oscillatory shear stress, such as coronary branch vessel bifurcations.10
Bicuspid AV is the most common congenital heart condition due to failure of leaflets to fuse during development, occurring in about 1% of the population.12 The AV is derived from mesenchymal cells from the neural crest as well as the endocardium.13 Genetic and molecular factors are thought to cause 2 of the leaflets to fuse, leaving a remnant ridge called a raphe. The genetics of bicuspid AV pathology have not been well defined but, at least in some cases, appear to be inherited in an autosomal dominant fashion with incomplete penetrance. First‐degree relatives of those with a bicuspid AV had a 9.1% chance of having a bicuspid AV in 1 investigation.14 The bicuspid AV is subjected to altered mechanical forces that incite AVS and progression to AS earlier than normal trileaflet AVs. In 1 study of patients undergoing AV replacement, those with bicuspid AVs were on average 7 years younger than their trileaflet AV counterparts.15 Using simulations of postmortem AVs, a study showed that bicuspid AVs typically have restricted motion that does not allow them to fully open to the size of the aorta.16 Furthermore, there is folding and redundancy of the valves with each cardiac cycle, thereby subjecting them to increased stress and asymmetrical turbulence, which likely explains their earlier compromise.
Early Lesions
The initial lesions in both AVS and CAD involve lipid deposition and focal sclerosis (Figure 1).9,17–20 The AV leaflets are composed of 3 layers—the ventricularis (on ventricular aspect of the leaflet containing elastin), spongiosa (consisting of loose connective tissue in the basal third of the valve), and fibrosa (composed of collagen core).21 The deposition of lipids typically occurs on the aortic side of the leaflets, given the aforementioned mechanical forces. Akin to arteries, endothelial cells line the surface of valve leaflets, maintaining homeostasis via the trafficking of mechanical and biochemical signals. In contrast to vascular endothelial cells, which assemble parallel to the direction of flow, valvular endothelium aligns perpendicular to flow.22 The difference in arrangement is accompanied by reorganization of focal adhesion complexes at the ends of long axis of cells, which likely contributes to focal thickening of leaflets at the sites with the highest turbulent flow. The endothelium in these high‐impact areas responds by increasing adhesion molecules and inducing inflammatory genes. Inflammatory cells then infiltrate and promote lipid deposition with disruption of the basement membrane. In both disease states, macrophages, T lymphocytes, and intracellular and extracellular lipids are present.11,23–25
Figure 1.

Pathophysiology of aortic sclerosis, aortic stenosis, and coronary artery disease. The evolution from normal vasculature to aortic stenosis and coronary artery disease shares several important cellular mechanisms including lipid deposition, inflammatory cell infiltration, cytokine release, and calcification. While a smooth muscle cap overlying a lipid core develops in coronary artery disease, aortic stenosis is characterized by thick calcifications. Pathology of human aortic valves and coronary arteries illustrates this transition at the tissue level. ACE indicates angiotensin‐converting enzyme; Ca, calcium; LDL, low‐density lipoprotein; M‐CSF, macrophage colony stimulating factor; MMP, matrix metalloproteinase; NO, nitric oxide; PDGF, platelet‐derived growth factor; TGF‐β, transforming growth factor β; TNF‐α, tumor necrosis factor α. Cynthia S. Gordon © 2014 MedAnimations.com.
Progression of Lesions
Local endothelial damage occurs as a result of insults from mechanical, genetic, and inflammatory cell–mediated factors in both CAD and AVS. Subsequent inflammation has been proposed as the hallmark of CAD and AVS pathogenesis. Several mediators are released, including tumor necrosis factor‐α, transforming growth factor‐β, and macrophage colony stimulating factor.9,17,26 Macrophage colony stimulating factor induces monocyte maturation into macrophages, which then take up low‐density lipoproteins to form foam cells. Toll‐like receptors are upregulated and serve to further activate macrophages, producing more cytokines and free radicals.27 While atherosclerosis is a known inflammatory disease, recent observations have suggested that AVS progression also involves an inflammatory pathway in individuals with certain common risk factors to initiate, propagate, and activate sclerotic lesions in the AV. In fact, histology of human stenotic and sclerotic AVs and atheromas has demonstrated active inflammatory cells.17,23,28
Although AVS and CAD have similar histology at their onset, more mature lesions exhibit structural differences. At the cellular level, an atheroma is characterized by a smooth muscle cap, which is not present in AVS.29 Instead, myofibroblasts encompass the predominant mesenchymal cell in AVS and release cytokines as well as increase the expression of matrix metalloproteinases and bone morphogenetic proteins, which promotes calcium deposition.30 In vitro, matrix metalloproteinases appear to work at least partially via stimulation of bone‐specific alkaline phosphatase.31 Myofibroblasts are thought to be stimulated by low nitric oxide and growth factors such as transforming growth factor‐β and platelet‐derived growth factor.32 Angiotensin‐converting enzyme is present in both AVS and CAD lesions, leading to increased angiotensin II.33 Interestingly, this enzyme is often coupled with apolipoprotein B particles, thereby implicating low‐density lipoprotein in the inflammatory process.33
Heterotopic calcium deposition is common to both diseases, although it is much more integral to the pathology of AVS. Examinations of valves with AS have demonstrated increased expression of osteocalcin, bone‐specific alkaline phosphatase, CBFA‐1 (Core‐binding factor α1), receptor activator of nuclear‐κB (RANK) ligand, osteopontin, and osteonectin, all of which are implicated in the pathogenesis of calcification.34 Notch1 is a transmembrane receptor that regulates bony differentiation in embryonic development and is involved in AV calcification in animal models and in vitro studies.35 When bound by ligand, its intracellular domain cleaves and translocates into the nucleus, eventually inhibiting Runx2.36–37 Inhibition of this transcriptional regulator of osteoblast fate leads to decreased calcification. Therefore, mutations in the NOTCH1 gene may ultimately cause disinhibition of calcium deposition, which can result in progressive AS. Moreover, NOTCH1 haploinsufficiency has been well described in some families with bicuspid AVs.13 Various growth and transcription factors such as transforming growth factor‐β, vascular endothelial growth factor, ErbB, Wnt, and GATA families have also been implicated in the pathogenesis of bicuspid AVs, although their roles are less defined.13 These data highlight the importance of NOTCH1 in the normal development of the AV and in its role in preventing AV calcification in adult‐onset disease.
As a result of the durable calcium cap, it takes many years to progress from AVS to severe AS, which may manifest as angina, syncope, and heart failure. In contrast, an atheroma is often more fragile and susceptible to rupture, causing a clot and acute ischemia. Thus, CAD may be a gradual process accented by periods of rapid progression to ischemia, heart failure, arrhythmias, and death.
Clinical Insights Into AVS and CAD
Clinical research has enhanced the understanding of the relationship between AVS and CAD as well as specific patient populations that are affected by this association (Tables 1 and 2).4–6,4–44 Importantly, research to date has not been able to prove causality despite the frequent coexistence of these entities. In one investigation, symptomatic patients with stable angina (without prior cardiac history) who had AVS on a transthoracic echocardiogram had a higher rate of significant CAD compared with those without AVS (75% versus 47%, P<0.001).44 After multivariate adjustment for traditional CAD risk factors, those with AVS had an 8.6‐fold greater likelihood of having significant CAD, defined as >70% obstruction of a major epicardial artery, as opposed to those without AVS (P<0.01). Similar results have been reported in patients without known cardiac disease presenting to the hospital with chest pain. AVS was found to be an independent predictor of obstructive CAD (odds ratio [OR] 3.73, 95% CI 1.33 to 10.45).40 However, when stratified by age, the association only remained statistically significant for those <60 years old. In the younger group, 71% with AVS had significant CAD versus 24% without AVS (P=0.041). This suggests that the finding of AVS in a younger person (<60 years) may be an early marker suggestive of a systemic atherosclerotic process, as opposed to a degenerative condition. Renal failure is considered a risk factor for CAD.
Table 1.
Retrospective Studies of AVS and CAD
| Retrospective Study | Year | N | Patient Population | Risk of CAD (AVS vs Non‐AVS) |
|---|---|---|---|---|
| Soydinc et al38 | 2006 | 160 | Suspected CAD without significant valvular disease | 1‐ and 2‐vessel CAD: nonsignificant 3‐vessel CAD (40% vs 13.6%; P<0.001) Gensini score: 18±16.4 vs 40±38.05 (P<0.001) |
| Sui et al39 | 2006 | 138 | Known or suspected CAD | 63.8% in AVS vs 28.8% in non‐AVS (P<0.05) |
| Conte et al40 | 2007 | 93 | Patients without known heart disease hospitalized for chest pain | OR 3.73 (95% CI 1.33 to 10.45) |
| Roy et al41 | 2012 | 140 | Known or suspected CAD | AVS was independent predictor of CAD (P=0.018) |
AVS indicates aortic valve sclerosis; CAD, coronary artery disease; OR, odds ratio.
Table 2.
Prospective Studies of AVS and CAD
| Prospective Study | Year | N | Patient Population | Mean Follow‐up (y) | Main Outcome(s) (AVS vs Non‐AVS) |
|---|---|---|---|---|---|
| Aronow et al42 | 1999 | 1980 | Elderly without AS | 3.8 | MI or sudden cardiac death (RR 1.758, 95% CI 1.521 to 2.031) |
| Otto et al6 | 1999 | 4073 | No known CAD, population study | 5 | MI (RR 1.40, 95% CI 1.07 to 1.83) CHF (RR 1.28, 95% CI 1.01 to 1.63); cardiovascular mortality (RR 1.52, 95% CI 1.12 to 2.05) All‐cause mortality (RR 1.35, 95% CI 1.12 to 1.61) |
| Chandra et al4 | 2004 | 415 | Patients in emergency department with chest pain | 1 | All‐cause mortality: 18.7% vs 2.4% (P<0.0001) Cardiovascular mortality: 14.7% vs 1.4% (P<0.0001) No significant difference in cardiac death or MI after adjustment for risk factors, CAD, and CRP |
| Shah et al43 | 2007 | 814 | Outpatients with known CAD without AS | 4 | MI (HR 1.8, 95% CI 1.1 to 3.1) Statin use attenuated this risk. |
| Kim et al44 | 2009 | 165 | Outpatients with angina and inconclusive treadmill stress test | 0.9 | No significant difference in cardiac events Risk of CAD (OR 8.58, 95% CI 3.74 to 19.67) |
| Owens et al5 | 2012 | 6685 | Population‐based without known heart disease | 5.8 | Major cardiovascular event (HR 1.50, 95% CI 1.10 to 2.03) Major coronary event (HR 1.72, 95% CI 1.19 to 2.49) |
All studies underwent multivariate adjustment for cardiac risk factors. AVS indicates aortic valve sclerosis; AS, aortic stenosis; CAD, coronary artery disease; ; MI, myocardial infarction; RR, relative risk; CHF, congestive heart failure; CRP, C‐reactive protein; HR, hazard ratio.
Studies have shown an increased prevalence of AVS and AS in patients on dialysis, especially as the time on dialysis increases.45–46 This may be secondary to concomitant cardiac risk factors and alterations in calcium and phosphorus homeostasis. Furthermore, several investigations have found statistically significant correlations between decreased glomerular filtration rate and the presence of AVS, thereby suggesting that even mild renal insufficiency predisposes individuals to the development of AVS.5–6,47 However, whether renal insufficiency is an independent predictor for AVS remains an area of uncertainty and subject to active research.
Importantly, when examining whether AVS is associated with adverse cardiovascular events, a prospective study found a higher incidence of cardiovascular events (16.8% versus 7.1%, P=0.002) and worse event‐free survival at 1 year between AVS and non‐AVS groups.4 However, after adjustment for confounders (such as baseline CAD and C‐reactive protein), no statistically significant differences were found. Of note, the highest rate of cardiac events was in patients with AVS and the highest quartile C‐reactive protein. Taken together, these findings suggest that AVS is more likely to be a marker of CAD or inflammation than a direct cause of mortality or cardiovascular events.
AV calcification has also been used to improve prognostication in patients with no known cardiovascular disease for primary prevention. The Multi‐Ethnic Study of Atherosclerosis (MESA) followed 6685 participants prospectively for the development of cardiovascular events, including myocardial infarction, stroke, cardiac arrest, and cardiac death.5 All subjects aged 45 to 84 received a computed tomography coronary calcium score, which was used to assess the extent of coronary artery and AV calcification. Approximately 87% of those with AV calcium had coronary artery calcification as opposed to 45.1% without calcified valves (P<0.0001). After adjustment for traditional risk factors, the presence of AV calcification increased the chances of cardiovascular (hazard ratio 1.50, 95% CI 1.1 to 2.04) and coronary events (myocardial infarction 1.72, 95% CI 1.19 to 2.49) over a median follow‐up of 5.8 years. This is particularly interesting because AVS normally does not cause sufficient hemodynamic compromise to impact cardiac function. One limitation of this study is that the use of computed tomography calcium score in this investigation may have included subjects with subclinical AS because hemodynamics could not be assessed.
The large‐scale prospective Cardiovascular Health Study demonstrated similar findings. After 5 years of follow‐up, subjects with AVS and no known CAD had a statistically significantly higher risk of myocardial infarction (relative risk [RR] 1.4, 95% CI 1.07 to 1.83), cardiovascular mortality (RR 1.52, 95% CI 1.12 to 2.05), and all‐cause mortality (RR 1.35, 95% CI 1.12 to 1.61) compared with those without AVS, even after adjustment for traditional cardiac risk factors (but not CAD).6 In those with known CAD at the beginning of the study, AVS did not significantly have an impact on death. Hence, AVS may act as a marker of subclinical endothelial dysfunction and inflammation that amplifies mortality rates via effects on coronary arteries. Furthermore, in studies of elderly populations, AVS independently increased the risk of a major coronary event by 1.8‐fold.42,48 Retrospective studies of high‐risk patients also found AVS to be an independent predictor of CAD (P<0.05).39,41 The association of AVS with excess cardiovascular mortality warrants further research.
AVS has also demonstrated a correlation with the Gensini score, a qualitative and quantitative angiographic measure of overall burden of coronary atherosclerosis.38 In a high cardiovascular risk population with known or likely CAD, AVS was found to be a risk factor for significant 3‐vessel CAD but not for 1‐ or 2‐vessel disease. Other investigators have corroborated these findings, showing an association between severity of CAD and AVS.40
Flow‐mediated dilation is an ultrasound‐based measurement of endothelial function whereby arterial diameter is measured before and after exposure to increased shear stress.49 Normal vasculature undergoing shear stress will release vasoactive mediators that dilate the vessel; however, damaged endothelium will have an absent or a partial response. This technique has been proposed as a noninvasive metric for CAD risk but may have a relationship with AVS given similar pathophysiology. In a study of 102 hospitalized patients, those with AVS had lower flow‐mediated dilation than did subjects with a normal AV (2.2% versus 5.3%, P<0.01).50 This finding is corroborated in a recent publication of 107 hypertensive patients in whom flow‐mediated dilation was found to be an independent predictor of AVS after multivariate adjustment (OR 0.691, P=0.001).51 Furthermore, the presence of AVS had a 100% positive predictive value for endothelial dysfunction. Interestingly, another investigation showed that peripheral flow‐mediated dilation was impaired in those with bicuspid AVs compared with age‐matched controls, which could be related to their altered hemodynamics and premature calcification.52 Although research into this topic is sparse, these studies imply that systemic endothelial dysfunction is enmeshed with the pathogenesis of AVS.
Imaging Characteristics
Aortic sclerosis is an echocardiographic diagnosis based on presence of AV calcification without significant hemodynamic compromise—typically a peak velocity <2 m/s.53 In contrast, AS has a peak velocity >2.5 m/s. The traditional classification of AVS severity is based on the echocardiographer's assessment of the amount of calcification on the AV and thus is a subjective measurement. Comparing early stages of AVS may be difficult given that the rating is based on the reading cardiologist's discretion. Along with severity, the type and location of AV calcification may also be important for identifying the population with the greatest risk of CAD. Not all people with AVS progress to AS, yet it has been difficult to predict which group is at greatest risk for development of a hemodynamically significant stenosis. Prospective and retrospective studies have produced varied results but range from 9% progression of AVS to AS in 5 years to 33% progression over 3.7 years.54–55
Several studies have shown that more extensive calcifications in AVS, despite not having a hemodynamic effect, amplify the likelihood of CAD and cardiovascular events.5,40 Furthermore, in a study of 66 male veterans, diffuse and mixed patterns of AV calcification on echocardiography had a much stronger association with CAD (OR 3.7, 95% CI 1.2 to 11.1) than localized nodular or nonnodular types.56 This was replicated in another group of patients at high risk for CAD.41 These findings suggest that specific patterns of AV leaflet calcification (Figure 2) could identify a subpopulation of patients that should be evaluated more thoroughly for CAD.
Figure 2.
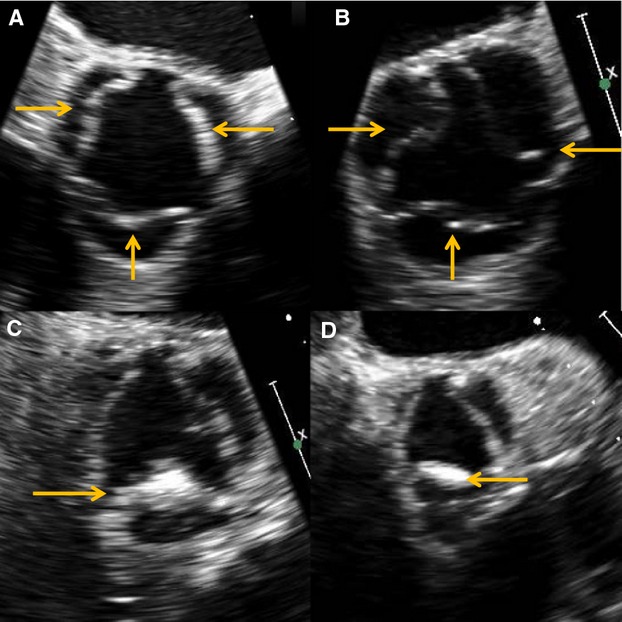
Patterns of aortic sclerosis seen on echocardiography. Diffuse (A) and mixed (B) types are associated with higher rates of coronary artery disease than are localized nodular (C) and localized nonnodular (D) forms. Arrows indicate areas of valvular sclerosis.
Potential Medical Therapies for AVS
Given the shared histological and clinical characteristics of AVS and CAD, many investigators have tested medical interventions such as lipid‐lowering agents and proremodeling agents (ie, ACEIs and angiotensin II receptor blockers [ARBs]) that may have salutary effects on both conditions.
Statins
A large pool of data, based on basic and clinical research, support the concept that dyslipidemia is often associated with AVS and AS.4–6,41,49 In addition to lowering lipids, statins have anti‐inflammatory effects and have proved to be beneficial in those with elevated C‐reactive protein but normal cholesterol.57 Thus, theoretically, statins could prevent the lipid deposition and inflammation in AVS. Whether there is causality for dyslipidemia and AVS remains undetermined.
Unfortunately, little research has been done on statin use in AVS, which may be a more appropriate target given that these lesions are characterized more by inflammation than their counterpart AS, which exhibits greater calcification and fibrosis. The early stages of AVS, when valvular stiffening and obstruction to flow have not yet developed, may provide a window of opportunity for statin treatment to potentially slow progression to overt AS. A retrospective study of 1689 patients with AVS found that statin use was associated with improved clinical outcomes, including decreased cardiovascular mortality (OR 0.73, 95% CI 0.56 to 0.98) and diminished risk of progression to AS (OR 0.64, 95% CI 0.42 to 0.97).58 Another investigation found an increased rate of myocardial infarction in those with AVS as opposed to normal valves, a difference that became insignificant after accounting for statin use.43 It is possible that AVS and AS could be a disease continuum where medical therapy may be more beneficial in the earlier stages.
Retrospective investigations into the impact of statin use on the progression of AS have produced conflicting results (Table 3). Several studies ranging from 65 to 211 subjects showed a statistically significant trend toward slower evolution of AS with administration of statins.47,59–63 However, a larger‐scale study of 1257 patients found no significant statin effect.64 In 1 analysis of people with existing AS, statins but not cholesterol levels affected the progression of AS.62 Thus, given the conflicting results and the inconsistencies in methodologies used in these studies, it is difficult to draw conclusions regarding the anti‐inflammatory, pleotrophic, or lipid‐lowering effects of statins on AS.
Table 3.
Retrospective Studies on the Impact of Statins on AVS and AS
| Retrospective Study | Year | N | Patient Characteristics | Impact of Statin Use |
|---|---|---|---|---|
| Pohle et al47 | 2001 | 104 | Patients with coronary and AV calcification | Lower LDL associated with slower progression of AV calcification |
| Aronow et al59 | 2001 | 180 | Patients with mild AS and 2 echocardiograms >2 years apart | Slower progression of AS |
| Novaro et al60 | 2001 | 174 | Patients with mild‐to‐moderate AS and 2 echocardiograms >12 months apart | Slower progression of AS |
| Shavelle et al61 | 2002 | 65 | Patients with AV calcification and 2 electron beam tomography scans >6 months apart | Slower progression of AV calcification |
| Bellamy et al62 | 2002 | 156 | Patients with AS, mean transvalvular gradient 10 mm Hg and AVA 2.0 cm2 | Slower progression of AS |
| Rosenhek et al63 | 2004 | 211 | Patients with aortic jet velocity >2.5 m/s and normal left ventricular ejection fraction | Slower AS progression, independent of LDL level |
| Antonini‐Canterin et al64 | 2005 | 1257 | Patients with AVS, mild or moderate AS | Overall, no significant difference in progression of AV pathology, but in subset with AVS, the rate of change in velocity was lower. |
| Ardehali et al58 | 2012 | 1689 | Patients with AVS | Reduced cardiovascular mortality |
AVS indicates aortic valve sclerosis; AS, aortic stenosis; AV, aortic valve; LDL, low‐density lipoprotein; AVA, aortic valve area.
Although there are inconsistent retrospective reports on the use of statin therapy, recent prospective studies have demonstrated failure to delay the progression of AS. The largest randomized controlled trial to date (1873 participants with mild to moderate AS) with the longest follow‐up (4.4 years) revealed that simvastatin in conjunction with ezetimibe did not reduce a variety of major cardiovascular outcomes, including those attributed to valvular dysfunction.65 In fact, rates of AV replacement were the same (myocardial infarction 1.00, 95% CI 0.84 to 1.18). Similar results were observed in studies of rosuvastatin and atorvastatin (Table 4).66–69 Additionally, several meta‐analyses found no significant differences between subjects treated with and without statins in terms of major echocardiographic findings (mean AV pressure gradient and AV area) or overall clinical outcomes.70–72 These collective findings suggest that (1) statins are unlikely to significantly affect the course of AS, (2) AVS may be a more appropriate target for statins but further research is needed given the scarcity of data, and (3) statins have multifactorial effects that may not be fully characterized yet.
Table 4.
Prospective Studies on the Impact of Statins on AVS and AS
| Prospective Study | Year | Study Design | N | Follow‐up (y) | Patient Characteristics | Impact of Statin Use |
|---|---|---|---|---|---|---|
| Cowell et al (SALTIRE)66 | 2005 | Double‐blind RCT | 151 | 2.1 | AS with aortic jet velocity >2.5 m/s with no statin indication | No difference in AS progression after treatment with atorvastatin |
| Moura et al (RAAVE)67 | 2007 | Open‐label, cohort | 121 | 1.5 | Moderate to severe AS with AVA 1.0 to 1.5 cm2, treated with statin only if indicated by guidelines | Slower progression of AS and lower serum LDL with rosuvastastin |
| Rossebø et al (SEAS)65 | 2008 | Double‐blind RCT | 1873 | 4.4 | Mild‐to‐moderate asymptomatic AS with aortic jet velocity of 2.5 to 4.0 m/s | No difference in AS‐related cardiovascular outcomes with simvastatin and ezetimibe treatment |
| Chan et al (ASTRONOMER)68 | 2010 | Double‐blind RCT | 269 | 3.5 | Mild‐to‐moderate AS with aortic jet velocity 2.5 to 4.0 m/s | No difference in AS progression after treatment with rosuvastatin |
| Panahi et al69 | 2013 | Double‐blind RCT | 75 | 1 | Mild‐to‐moderate AS | Lower mean and peak gradient in atorvastatin group but otherwise no difference in AS progression |
AVS indicates aortic valve sclerosis; AS, aortic stenosis; RCT, randomized controlled trial; AVA, aortic valve area; LDL, low‐density lipoprotein.
ACEIs and ARBs
As previously illustrated, the angiotensin pathway has been implicated in the process of AV calcification. Endothelial injury and lipid deposition during the early stages of AVS stimulate local formation and action of angiotensin II, which enhances collagen synthesis and attracts monocytes to accelerate disease progression.73 Therefore, this pathway has been proposed as a target for prevention of AVS and AS. In terms of patients with AVS, only 1 retrospective study has been performed, which found no benefit to ACEIs or ARBs in preventing AVS progression or mortality.58 ACEIs and ARBs have been retrospectively studied in AS with mixed results (Table 5).58,63,74–77 They pose particular difficulty in their evaluation given their hemodynamic effects, including afterload reduction (which may cause worsening symptoms of AS) and cardiac remodeling (which can prolong life). Prospective trials are warranted to evaluate whether ACEIs and ARBs are able to retard the progression of AVS and AS.
Table 5.
Effect of Angiotensin Pathway Inhibition on AVS and AS
| Retrospective Study | Year | N | Patient Characteristics | Impact of ACEI/ARB |
|---|---|---|---|---|
| Rosenhek et al63 | 2004 | 211 | Patients with aortic jet velocity >2.5 m/s and normal left ventricular ejection fraction | No effect on progression of AS |
| Sverdlov et al74 | 2004 | 212 | Randomly selected patients, measured AV backscatter over 4 years | Slowed progression of AV backscatter, a marker of calcification/stenosis |
| O'Brien et al75 | 2005 | 123 | Patients with AV calcification and 2 electron beam tomography scans | Decreased AV calcification |
| Nadir et al76 | 2011 | 2117 | AS detected on echocardiography | Lower all‐cause mortality and cardiovascular events |
| Wakabayashi et al77 | 2011 | 194 | AS detected on echocardiography | Slower progression of AS according to peak velocity |
| Ardehali et al58 | 2012 | 1689 | Patients with AVS | Reduction in admissions for ischemic heart disease and CHF; no impact on mortality or progression to AS |
AS indicates aortic stenosis; ACEI, angiotensin‐converting enzyme inhibitor; ARB, angiotensin II receptor blocker; AV, aortic valve; AVS, aortic valve sclerosis; CHF, congestive heart failure.
Other Agents
Nonsteroidal anti‐inflammatory drugs (NSAIDs) have a complex relationship with cardiovascular health, as is exemplified by the removal of many selective cyclooxygenase‐2 inhibitors from the market after increased risks of heart attack and stroke were noted. The American Heart Association recommends against nonaspirin NSAID use in patients with cardiovascular disease based on clinical data confirming increased mortality and cardiac events.78 However, given the inflammatory nature of AS and AVS, one may postulate that NSAIDs could offer a potential benefit. A large‐scale study of 2 cohorts produced divergent results, showing a slightly increased risk of worsening AV calcification in patients using aspirin in the American cohort (RR 1.60, 95% CI 1.19 to 2.15), while the German group experienced no difference (RR 1.06, 95% CI 0.87 to 1.28).79 Nonaspirin NSAIDs had no significant effects on either population. These results should be interpreted with caution given the lack of randomization, as well as other risk factors not accounted for in the group taking aspirin. The cardioprotective antiplatelet effect of aspirin likely outweighs any potential effect on AV calcification.
The osteogenic properties of AV lesions, which are also observed in atheromas, may be another therapeutic target. In an investigation of 55 patients with AS, those undergoing osteoporosis therapy (bisphosphonates, calcitonin, or estrogen receptor modulators) had a slower rate of AS progression on echocardiography.80 The mechanism of this intriguing result is not well delineated but may involve the RANK pathway. The RANK receptor is present on precursor osteoclasts and binds RANK ligand to promote maturation into active osteoclasts, which promote bone resorption and remodeling. In 1 study, RANK ligand was present in higher levels in AS than in controls.81 Despite the theoretical advantages of bisphosphonates, a larger retrospective study has not shown any benefit in slowing the progression of disease.82 Denosumab, a monoclonal antibody used in osteoporosis, inhibits RANK ligand and thus may be a potential therapeutic target for AVS. No research to date has been done on AV disease, although animal and human studies into thoracic aorta calcification have produced mixed results regarding the use of denosumab in preventing vascular calcification.83–84
Because calcium deposition is integral to the development of both CAD and AVS, some have hypothesized that higher levels of calcium and vitamin D may promote calcification. The relationship between CAD and vitamin D appears complex given that vitamin D increases coronary calcification in animal models.85 However, in humans, it appears to be protective against atherosclerosis.86 One potential explanation is that vitamin D has a therapeutic window with both very low and very high levels promoting calcium deposition. In terms of AV disease, information comes solely from animal research. One study found that vitamin D supplementation promoted AVS in rabbits when given alone and induced AS when given in conjunction with a high‐cholesterol diet.87 Yet, another study showed no difference in the development of AVS between rabbits treated with cholesterol and vitamin D versus those without either of these supplements.88 In contrast, a recent report demonstrated increased calcification in the aortic root of vitamin D receptor knockout mice, suggesting vitamin D deficiency may stimulate osteogenic factors involved in vascular calcification.89 Given the evidence that has emerged regarding the complex relationship between calcium and vitamin D supplementation and CAD, this topic should be investigated further with respect to AV disease. Paucity of appropriate animal models for AVS is a major limitation to pursue research in this area.
Antioxidants may also decrease inflammation and progression of AV disease. An in vitro study comparing normal AV, AVS, and AS discovered that there were lower levels of antioxidants in diseased valves.90 After exposure to reactive oxygen species, Runx2 levels were tripled, DNA repair was hindered, and calcification was increased in AS and AVS compared with controls. This suggests that calcified valves are more susceptible to osteogenic factors when exposed to reactive oxygen species. This effect was partially reversed when antioxidants were administered. Although these are exciting initial findings, they need to be replicated in human and animal models. Other targets may include matrix metalloproteinase inhibitors, Notch pathway augmentation, and enhancement of the nitric oxide signaling pathway, which require further research.74
Clinical Applicability
Results of the clinical studies reviewed here suggest that AVS may have a role in the evaluation of a patient with potential CAD. Combining echocardiography with an exercise treadmill test may increase the sensitivity and specificity of diagnosing CAD and avoid unnecessary coronary catheterizations.45 The presence of AVS in younger people should be of particular concern because it more likely represents an inflammatory as opposed to a degenerative process. However, because younger patients with AVS are normally asymptomatic, it is difficult to screen these individuals without an underlying clinical suspicion. Younger individuals with a family history of early AS, CAD, bicuspid valves, traditional risk factors such as diabetes mellitus, or chronic inflammatory diseases would be reasonable initial targets. For instance, patients with systemic lupus erythematosus have a higher risk of CAD compared with age‐matched controls according to a recent systematic review of 28 studies.91 The development of AVS on screening echocardiography could signify underlying CAD and prompt more aggressive management of risk factors.
The association of CAD with AVS presents an opportunity for screening. If early detection of AVS accelerated the diagnosis and treatment of CAD, this could theoretically decrease morbidity and mortality. However, currently there is no confirmatory evidence to support routine AV evaluation to screen patients for CAD. Understanding the mechanisms underlying the development of AVS and unraveling its association with CAD will undoubtedly generate potential areas for therapeutic interventions. It is possible that after further rigorous research, AVS could be used in concert with other factors such as family history, comorbidities, and C‐reactive protein, to help risk‐stratify patients and delineate how aggressive lifestyle and medical interventions should be pursued. This could be especially useful in the case of borderline patients without known CAD.
Future Directions
For many years, AV disease has been considered a progressive obstructive lesion that would ultimately require a mechanical approach to ameliorate symptoms and provide survival benefit. Surgical and recently transcatheter AV replacements with concomitant revascularization have been the standard management strategy for patients with severe AS and CAD.
AVS begins and progresses in a setting of complex interactions between mechanical forces and a dynamically changing tissue milieu that has both similarities and differences with CAD. The rapidly expanding body of knowledge regarding regulation and disruption of homeostasis in the AV and coronary vasculature will help with the discovery of common molecular pathways and therapeutic targets for clinical application. However, we should be cautious that treatment of surrogate risk factors, such as inflammation and calcification, may not provide functional benefit but allow us to identify patients at risk.
Finally, imaging methods, including molecular imaging that could identify sites of inflammation and calcification, with subsequent targeted therapy will be valuable. Future discovery of medical therapies to treat or slow progression of AVS will be challenging but necessary given the imminent aging population and significant financial costs.
Disclosures
None.
Acknowledgments
We wish to thank Dr Michael Fishbein for providing pathology images of AV disease and CAD and Dr Anthony Koppula for assistance in obtaining echocardiography images.
References
- 1.Stewart BF, Siscovick D, Lind BK, Gardin JM, Gottdiener JS, Smith VE, Kitzman DW, Otto CM. Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study. J Am Coll Cardiol. 1997; 29:630-634. [DOI] [PubMed] [Google Scholar]
- 2.Freeman RV, Otto CM. Spectrum of calcific aortic valve disease: pathogenesis, disease progression, and treatment strategies. Circulation. 2005; 111:3316-3326. [DOI] [PubMed] [Google Scholar]
- 3.Losi MA, Brevetti G, Schiano V, Barbati G, Parisi V, Contaldi C, Chiacchio E, Cavallaro M, Carpinella G, Fundaliotis A, Betocchi S, Brevetti L, Chiariello M. Aortic valve sclerosis in patients with peripheral and/or coronary arterial disease. Echocardiography. 2010; 27:608-612. [DOI] [PubMed] [Google Scholar]
- 4.Chandra HR, Goldstein JA, Choudhary N, O'Neill CS, George PB, Gangasani SR, Cronin L, Marcovitz PA, Hauser AM, O'Neill WW. Adverse outcome in aortic sclerosis is associated with coronary artery disease and inflammation. J Am Coll Cardiol. 2004; 43:169-175. [DOI] [PubMed] [Google Scholar]
- 5.Owens DS, Budoff MJ, Katz R, Takasu J, Shavelle DM, Carr JJ, Heckbert SR, Otto CM, Probstfield JL, Kronmal RA, O'Brien KD. Aortic valve calcium independently predicts coronary and cardiovascular events in a primary prevention population. JACC Cardiovasc Imaging. 2012; 5:619-625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Otto CM, Lind BK, Kitzman DW, Gersh BJ, Siscovick DS. Association of aortic‐valve sclerosis with cardiovascular mortality and morbidity in the elderly. N Engl J Med. 1999; 341:142-147. [DOI] [PubMed] [Google Scholar]
- 7.Centers for Disease Control and Prevention. Leading causes of death. 2010. Available at: http://www.cdc.gov/nchs/fastats/lcod.htm. Accessed January 12, 2014.
- 8.Nabel EG, Braunwald E. A tale of coronary artery disease and myocardial infarction. N Engl J Med. 2012; 366:54-63. [DOI] [PubMed] [Google Scholar]
- 9.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005; 352:1685-1695. [DOI] [PubMed] [Google Scholar]
- 10.Mohler ER., III Mechanisms of aortic valve calcification. Am J Cardiol. 2004; 94:1396-1402. [DOI] [PubMed] [Google Scholar]
- 11.Goldbarg SH, Elmariah S, Miller MA, Fuster V. Insights into degenerative aortic valve disease. J Am Coll Cardiol. 2007; 50:1205-1213. [DOI] [PubMed] [Google Scholar]
- 12.Nistri S, Basso C, Marzari C, Mormino P, Thiene G. Frequency of bicuspid aortic valve in young male conscripts by echocardiogram. Am J Cardiol. 2005; 96:718-721. [DOI] [PubMed] [Google Scholar]
- 13.Laforest B, Nemer M. Genetic insights into bicuspid aortic valve formation. Cardiol Res Pract. 2012; 2012:180297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huntington K, Hunter AG, Chan KL. A prospective study to assess the frequency of familial clustering of congenital bicuspid aortic valve. J Am Coll Cardiol. 1997; 30:1809-1812. [DOI] [PubMed] [Google Scholar]
- 15.Roberts WC, Ko JM. Frequency by decades of unicuspid, bicuspid, and tricuspid aortic valves in adults having isolated aortic valve replacement for aortic stenosis, with or without associated aortic regurgitation. Circulation. 2005; 111:920-925. [DOI] [PubMed] [Google Scholar]
- 16.Robicsek F, Thubrikar MJ, Cook JW, Fowler B. The congenitally bicuspid aortic valve: how does it function? Why does it fail? Ann Thorac Surg. 2004; 77:177-185. [DOI] [PubMed] [Google Scholar]
- 17.Otto CM, Kuusisto J, Reichenbach DD, Gown AM, O'Brien KD. Characterization of the early lesion of ‘degenerative’ valvular aortic stenosis. Histological and immunohistochemical studies. Circulation. 1994; 90:844-853. [DOI] [PubMed] [Google Scholar]
- 18.Stary HC, Chandler AB, Glagov S, Guyton JR, Insull W, Jr, Rosenfeld ME, Schaffer SA, Schwartz CJ, Wagner WD, Wissler RW. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis: a report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1994; 89:2462-2478. [DOI] [PubMed] [Google Scholar]
- 19.Skålén K, Gustafsson M, Rydberg EK, Hultén LM, Wiklund O, Innerarity TL, Borén J. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. 2002; 417:750-754. [DOI] [PubMed] [Google Scholar]
- 20.Leitinger N. Oxidized phospholipids as modulators of inflammation in atherosclerosis. Curr Opin Lipidol. 2003; 14:421-430. [DOI] [PubMed] [Google Scholar]
- 21.Otto CM. Why is aortic sclerosis associated with adverse clinical outcomes? J Am Coll Cardiol. 2004; 43:176-178. [DOI] [PubMed] [Google Scholar]
- 22.Butcher JT, Penrod AM, García AJ, Nerem RM. Unique morphology and focal adhesion development of valvular endothelial cells in static and fluid flow environments. Arterioscler Thromb Vasc Biol. 2004; 24:1429-1434. [DOI] [PubMed] [Google Scholar]
- 23.Jonasson L, Holm J, Skalli O, Bondjers G, Hansson GK. Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis. 1986; 6:131-138. [DOI] [PubMed] [Google Scholar]
- 24.Hansson GK, Holm J, Jonasson L. Detection of activated T lymphocytes in the human atherosclerotic plaque. Am J Pathol. 1989; 135:169-175. [PMC free article] [PubMed] [Google Scholar]
- 25.Branch KR, O'Brien KD, Otto CM. Aortic valve sclerosis as a marker of active atherosclerosis. Curr Cardiol Rep. 2002; 4:111-117. [DOI] [PubMed] [Google Scholar]
- 26.Jian B, Narula N, Li QY, Mohler ER, III, Levy RJ. Progression of aortic valve stenosis: TGF‐beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg. 2003; 75:457-465. [DOI] [PubMed] [Google Scholar]
- 27.Seneviratne AN, Sivagurunathan B, Monaco C. Toll‐like receptors and macrophage activation in atherosclerosis. Clin Chim Acta. 2012; 413:3-14. [DOI] [PubMed] [Google Scholar]
- 28.van der Wal AC, Becker AE, van der Loos CM, Tigges AJ, Das PK. Fibrous and lipid‐rich atherosclerotic plaques are part of interchangeable morphologies related to inflammation: a concept. Coron Artery Dis. 1994; 5:463-469. [PubMed] [Google Scholar]
- 29.Ghattas A, Griffiths HR, Devitt A, Lip GY, Shantsila E. Monocytes in coronary artery disease and atherosclerosis: where are we now? J Am Coll Cardiol. 2013; 62:1541-1551. [DOI] [PubMed] [Google Scholar]
- 30.Kaden JJ, Dempfle CE, Grobholz R, Tran HT, Kiliç R, Sarikoç A, Brueckmann M, Vahl C, Hagl S, Haase KK, Borggrefe M. Interleukin‐1 beta promotes matrix metalloproteinase expression and cell proliferation in calcific aortic valve stenosis. Atherosclerosis. 2003; 170:205-211. [DOI] [PubMed] [Google Scholar]
- 31.Clark‐Greuel JN, Connolly JM, Sorichillo E, Narula NR, Rapoport HS, Mohler ER, III, Gorman JH, III, Gorman RC, Levy RJ. Transforming growth factor‐beta1 mechanisms in aortic valve calcification: increased alkaline phosphatase and related events. Ann Thorac Surg. 2007; 83:946-953. [DOI] [PubMed] [Google Scholar]
- 32.Walker GA, Masters KS, Shah DN, Anseth KS, Leinwand LA. Valvular myofibroblast activation by transforming growth factor‐beta: implications for pathological extracellular matrix remodeling in heart valve disease. Circ Res. 2004; 95:253-260. [DOI] [PubMed] [Google Scholar]
- 33.O'Brien KD, Shavelle DM, Caulfield MT, McDonald TO, Olin‐Lewis K, Otto CM, Probstfield JL. Association of angiotensin‐converting enzyme with low‐density lipoproteins in aortic valvular lesions and in human plasma. Circulation. 2002; 106:2224-2230. [DOI] [PubMed] [Google Scholar]
- 34.Shetty R, Pepin A, Charest A, Perron J, Doyle D, Voisine P, Dagenais F, Pibarot P, Mathieu P. Expression of bone‐regulatory proteins in human valve allografts. Heart. 2006; 92:1303-1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Acharya A, Hans CP, Koenig SN, Nichols HA, Galindo CL, Garner HR, Merrill WH, Hinton RB, Garg V. Inhibitory role of Notch1 in calcific aortic valve disease. PLoS One. 2011; 6:e27743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, Grossfeld PD, Srivastava D. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005; 437:270-274. [DOI] [PubMed] [Google Scholar]
- 37.Hilton MJ, Tu X, Wu X, Bai S, Zhao H, Kobayashi T, Kronenberg HM, Teitelbaum SL, Ross FP, Kopan R, Long F. Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation. Nat Med. 2008; 14:306-314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soydinc S, Davutoglu V, Dundar A, Aksoy M. Relationship between aortic valve sclerosis and the extent of coronary artery disease in patients undergoing diagnostic coronary angiography. Cardiology. 2006; 106:277-282. [DOI] [PubMed] [Google Scholar]
- 39.Sui SJ, Ren MY, Xu FY, Zhang Y. A high association of aortic valve sclerosis detected by transthoracic echocardiography with coronary arteriosclerosis. Cardiology. 2007; 108:322-330. [DOI] [PubMed] [Google Scholar]
- 40.Conte L, Rossi A, Cicoira M, Bonapace S, Amado EA, Golia G, Zardini P, Vassanelli C. Aortic valve sclerosis: a marker of significant obstructive coronary artery disease in patients with chest pain? J Am Soc Echocardiogr. 2007; 20:703-708. [DOI] [PubMed] [Google Scholar]
- 41.Roy GC, Rahman F, Hoque MH, Habib MA, Banerjee SK, Siddique MA, Barua UK, Hossain AS, Bhuiyan GR, Haider MS. Aortic valve sclerosis is an indicator of coronary artery diseases. Mymensingh Med J. 2012; 21:226-232. [PubMed] [Google Scholar]
- 42.Aronow WS, Ahn C, Shirani J, Kronzon I. Comparison of frequency of new coronary events in older subjects with and without valvular aortic sclerosis. Am J Cardiol. 1999; 83:599-600. [DOI] [PubMed] [Google Scholar]
- 43.Shah SJ, Ristow B, Ali S, Na BY, Schiller NB, Whooley MA. Acute myocardial infarction in patients with versus without aortic valve sclerosis and effect of statin therapy (from the Heart and Soul Study). Am J Cardiol. 2007; 99:1128-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim DB, Jung HO, Jeon DS, Park CS, Jang SW, Park HJ, Kim PJ, Baek SH, Seung KB, Rho TH, Kim JH, Choi KB. Aortic valve sclerosis on echocardiography is a good predictor of coronary artery disease in patients with an inconclusive treadmill exercise test. Korean Circ J. 2009; 39:275-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schönenberger A, Winkelspecht B, Köhler H, Girndt M. High prevalence of aortic valve alterations in haemodialysis patients is associated with signs of chronic inflammation. Nephron Clin Pract. 2004; 96:48-55. [DOI] [PubMed] [Google Scholar]
- 46.Straumann E, Meyer B, Misteli M, Blumberg A, Jenzer HR. Aortic and mitral valve disease in patients with end stage renal failure on long‐term haemodialysis. Br Heart J. 1992; 67:236-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pohle K, Mäffert R, Ropers D, Moshage W, Stilianakis N, Daniel WG, Achenbach S. Progression of aortic valve calcification: association with coronary atherosclerosis and cardiovascular risk factors. Circulation. 2001; 104:1927-1932. [DOI] [PubMed] [Google Scholar]
- 48.Aronow WS, Ahn C, Shirani J, Kronzon I. Comparison of frequency of new coronary events in older persons with mild, moderate, and severe valvular aortic stenosis with those without aortic stenosis. Am J Cardiol. 1998; 81:647-649. [DOI] [PubMed] [Google Scholar]
- 49.Raitakari OT, Celermajer DS. Flow‐mediated dilatation. Br J Clin Pharmacol. 2000; 50:397-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Poggianti E, Venneri L, Chubuchny V, Jambrik Z, Baroncini LA, Picano E. Aortic valve sclerosis is associated with systemic endothelial dysfunction. J Am Coll Cardiol. 2003; 41:136-141. [DOI] [PubMed] [Google Scholar]
- 51.Erdoğan T, Cetin M, Kocaman SA, Durakoğlugil ME, Ergül E, Canga A. Aortic valve sclerosis is a high predictive marker of systemic endothelial dysfunction in hypertensive patients. Herz. 2013; 38:915-921. [DOI] [PubMed] [Google Scholar]
- 52.Ali OA, Chapman M, Nguyen TH, Chirkov YY, Heresztyn T, Mundisugih J, Horowitz JD. Interactions between inflammatory activation and endothelial dysfunction selectively modulate valve disease progression in patients with bicuspid aortic valve. Heart. 2014; 100:800-805. [DOI] [PubMed] [Google Scholar]
- 53.American College of Cardiology; American Heart Association Task Force on Practice Guidelines (writing committee to revise the 1998 guidelines for the management of patients with valvular heart disease); Society of Cardiovascular Anesthesiologists. Bonow RO, Carabello BA, Chatterjee K, de Leon AC, Jr, Faxon DP, Freed MD, Gaasch WH, Lytle BW, Nishimura RA, O'Gara PT, O'Rourke RA, Otto CM, Shah PM, Shanewise JS, Smith SC, Jr, Jacobs AK, Adams CD, Anderson JL, Antman EM, Fuster V, Halperin JL, Hiratzka LF, Hunt SA, Lytle BW, Nishimura R, Page RL, Riegel B. ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing committee to revise the 1998 guidelines for the management of patients with valvular heart disease) developed in collaboration with the Society of Cardiovascular Anesthesiologists endorsed by the Society for Cardiovascular Angiography and Interventions and the Society of Thoracic Surgeons. J Am Coll Cardiol. 2006; 48:1-148. [DOI] [PubMed] [Google Scholar]
- 54.Novaro GM, Katz R, Aviles RJ, Gottdiener JS, Cushman M, Psaty BM, Otto CM, Griffin BP. Clinical factors, but not C‐reactive protein, predict progression of calcific aortic‐valve disease: the Cardiovascular Health Study. J Am Coll Cardiol. 2007; 50:1992-1998. [DOI] [PubMed] [Google Scholar]
- 55.Faggiano P, Aurigemma GP, Rusconi C, Gaasch WH. Progression of valvular aortic stenosis in adults: literature review and clinical implications. Am Heart J. 1996; 132:408-417. [DOI] [PubMed] [Google Scholar]
- 56.Tolstrup K, Crawford M, Roldan CA. Morphologic characteristics of aortic valve sclerosis by transesophageal echocardiography: importance for the prediction of coronary artery disease. Cardiology. 2002; 98:154-158. [DOI] [PubMed] [Google Scholar]
- 57.Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM, Jr, Kastelein JJ, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJJUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C‐reactive protein. N Engl J Med. 2008; 359:2195-2207. [DOI] [PubMed] [Google Scholar]
- 58.Ardehali R, Leeper NJ, Wilson AM, Heidenreich PA. The effect of angiotensin‐converting enzyme inhibitors and statins on the progression of aortic sclerosis and mortality. J Heart Valve Dis. 2012; 21:337-343. [PMC free article] [PubMed] [Google Scholar]
- 59.Aronow WS, Ahn C, Kronzon I, Goldman ME. Association of coronary risk factors and use of statins with progression of mild valvular aortic stenosis in older persons. Am J Cardiol. 2001; 88:693-695. [DOI] [PubMed] [Google Scholar]
- 60.Novaro GM, Tiong IY, Pearce GL, Lauer MS, Sprecher DL, Griffin BP. Effect of hydroxymethylglutaryl coenzyme a reductase inhibitors on the progression of calcific aortic stenosis. Circulation. 2001; 104:2205-2209. [DOI] [PubMed] [Google Scholar]
- 61.Shavelle DM, Takasu J, Budoff MJ, Mao S, Zhao XQ, O'Brien KD. HMG CoA reductase inhibitor (statin) and aortic valve calcium. Lancet. 2002; 359:1125-1126. [DOI] [PubMed] [Google Scholar]
- 62.Bellamy MF, Pellikka PA, Klarich KW, Tajik AJ, Enriquez‐Sarano M. Association of cholesterol levels, hydroxymethylglutaryl coenzyme‐A reductase inhibitor treatment, and progression of aortic stenosis in the community. J Am Coll Cardiol. 2002; 40:1723-1730. [DOI] [PubMed] [Google Scholar]
- 63.Rosenhek R, Rader F, Loho N, Gabriel H, Heger M, Klaar U, Schemper M, Binder T, Maurer G, Baumgartner H. Statins but not angiotensin‐converting enzyme inhibitors delay progression of aortic stenosis. Circulation. 2004; 110:1291-1295. [DOI] [PubMed] [Google Scholar]
- 64.Antonini‐Canterin F, Popescu BA, Huang G, Korcova‐Miertusova R, Rivaben D, Faggiano P, Pavan D, Piazza R, Bolis A, Ciavattone A, Ruggiero A, Nicolosi GL. Progression of aortic valve sclerosis and aortic valve stenosis: what is the role of statin treatment? Ital Heart J. 2005; 6:119-124. [PubMed] [Google Scholar]
- 65.Rossebø AB, Pedersen TR, Boman K, Brudi P, Chambers JB, Egstrup K, Gerdts E, Gohlke‐Bärwolf C, Holme I, Kesäniemi YA, Malbecq W, Nienaber CA, Ray S, Skjaerpe T, Wachtell K, Willenheimer RSEAS Investigators. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med. 2008; 359:1343-1356. [DOI] [PubMed] [Google Scholar]
- 66.Cowell SJ, Newby DE, Prescott RJ, Bloomfield P, Reid J, Northridge DB, Boon NAScottish Aortic Stenosis and Lipid Lowering Trial, Impact on Regression (SALTIRE) Investigators. A randomized trial of intensive lipid‐lowering therapy in calcific aortic stenosis. N Engl J Med. 2005; 352:2389-2397. [DOI] [PubMed] [Google Scholar]
- 67.Moura LM, Ramos SF, Zamorano JL, Barros IM, Azevedo LF, Rocha‐Gonçalves F, Rajamannan NM. Rosuvastatin affecting aortic valve endothelium to slow the progression of aortic stenosis. J Am Coll Cardiol. 2007; 49:554-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chan KL, Teo K, Dumesnil JG, Ni A, Tam JASTRONOMER Investigators. Effect of lipid lowering with rosuvastatin on progression of aortic stenosis: results of the aortic stenosis progression observation: measuring effects of rosuvastatin (ASTRONOMER) trial. Circulation. 2010; 121:306-314. [DOI] [PubMed] [Google Scholar]
- 69.Panahi Y, Sahebkar A, Taghipour HR, Dadjou Y, Pishgoo B, Rakhshankhah AS. Atorvastatin therapy is not associated with slowing the progression of aortic stenosis: findings of a randomized controlled trial. Clin Lab. 2013; 59:299-305. [DOI] [PubMed] [Google Scholar]
- 70.De Vecchis R, Di Biase G, Esposito C, Ciccarelli A, Cioppa C, Giasi A, Ariano C, Cantatrione S. Statin use for nonrheumatic calcific aortic valve stenosis: a review with meta‐analysis. J Cardiovasc Med. 2013; 14:559-567. [DOI] [PubMed] [Google Scholar]
- 71.Teo KK, Corsi DJ, Tam JW, Dumesnil JG, Chan KL. Lipid lowering on progression of mild to moderate aortic stenosis: meta‐analysis of the randomized placebo‐controlled clinical trials on 2344 patients. Can J Cardiol. 2011; 27:800-808. [DOI] [PubMed] [Google Scholar]
- 72.Parolari A, Tremoli E, Cavallotti L, Trezzi M, Kassem S, Loardi C, Veglia F, Ferrari G, Pacini D, Alamanni F. Do statins improve outcomes and delay the progression of non‐rheumatic calcific aortic stenosis? Heart. 2011; 97:523-529. [DOI] [PubMed] [Google Scholar]
- 73.Helske S, Lindstedt KA, Laine M, Mäyränpää M, Werkkala K, Lommi J, Turto H, Kupari M, Kovanen PT. Induction of local angiotensin II‐producing systems in stenotic aortic valves. J Am Coll Cardiol. 2004; 44:1859-1866. [DOI] [PubMed] [Google Scholar]
- 74.Sverdlov AL, Ngo DT, Chan WP, Chirkov YY, Gersh BJ, McNeil JJ, Horowitz JD. Determinants of aortic sclerosis progression: implications regarding impairment of nitric oxide signalling and potential therapeutics. Eur Heart J. 2012; 33:2419-2425. [DOI] [PubMed] [Google Scholar]
- 75.O'Brien KD, Probstfield JL, Caulfield MT, Nasir K, Takasu J, Shavelle DM, Wu AH, Zhao XQ, Budoff MJ. Angiotensin‐converting enzyme inhibitors and change in aortic valve calcium. Arch Intern Med. 2005; 165:858-862. [DOI] [PubMed] [Google Scholar]
- 76.Nadir MA, Wei L, Elder DH, Libianto R, Lim TK, Pauriah M, Pringle SD, Doney AD, Choy AM, Struthers AD, Lang CC. Impact of renin‐angiotensin system blockade therapy on outcome in aortic stenosis. J Am Coll Cardiol. 2011; 58:570-576. [DOI] [PubMed] [Google Scholar]
- 77.Wakabayashi K, Tsujino T, Naito Y, Ezumi A, Lee‐Kawabata M, Nakao S, Goda A, Sakata Y, Yamamoto K, Daimon T, Masuyama T. Administration of angiotensin‐converting enzyme inhibitors is associated with slow progression of mild aortic stenosis in Japanese patients. Heart Vessels. 2011; 26:252-257. [DOI] [PubMed] [Google Scholar]
- 78.Antman EM, Bennett JS, Daugherty A, Furberg C, Roberts H, Taubert KAAmerican Heart Association. Use of nonsteroidal antiinflammatory drugs: an update for clinicians: a scientific statement from the American Heart Association. Circulation. 2007; 115:1634-1642. [DOI] [PubMed] [Google Scholar]
- 79.Delaney JA, Lehmann N, Jöckel KH, Elmariah S, Psaty BM, Mahabadi AA, Budoff M, Kronmal RA, Nasir K, O'Brien KD, Möhlenkamp S, Moebus S, Dragano N, Winterstein AG, Erbel R, Kälsch H. Associations between aspirin and other non‐steroidal anti‐inflammatory drugs and aortic valve or coronary artery calcification: the Multi‐Ethnic Study of Atherosclerosis and the Heinz Nixdorf Recall Study. Atherosclerosis. 2013; 229:310-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Skolnick AH, Osranek M, Formica P, Kronzon I. Osteoporosis treatment and progression of aortic stenosis. Am J Cardiol. 2009; 104:122-124. [DOI] [PubMed] [Google Scholar]
- 81.Kaden JJ, Bickelhaupt S, Grobholz R, Haase KK, Sarikoç A, Kiliç R, Brueckmann M, Lang S, Zahn I, Vahl C, Hagl S, Dempfle CE, Borggrefe M. Receptor activator of nuclear factor kappaB ligand and osteoprotegerin regulate aortic valve calcification. J Mol Cell Cardiol. 2004; 36:57-66. [DOI] [PubMed] [Google Scholar]
- 82.Aksoy O, Cam A, Goel SS, Houghtaling PL, Williams S, Ruiz‐Rodriguez E, Menon V, Kapadia SR, Tuzcu EM, Blackstone EH, Griffin BP. Do bisphosphonates slow the progression of aortic stenosis? J Am Coll Cardiol. 2012; 59:1452-1459. [DOI] [PubMed] [Google Scholar]
- 83.Samelson EJ, Miller PD, Christiansen C, Daizadeh NS, Grazette L, Anthony MS, Egbuna O, Wang A, Siddhanti SR, Cheung AM, Franchimont N, Kiel DP. RANKL inhibition with denosumab does not influence 3‐year progression of aortic calcification or incidence of adverse cardiovascular events in postmenopausal women with osteoporosis and high cardiovascular risk. J Bone Miner Res. 2014; 29:450-457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Helas S, Goettsch C, Schoppet M, Zeitz U, Hempel U, Morawietz H, Kostenuik PJ, Erben RG, Hofbauer LC. Inhibition of receptor activator of NF‐kappaB ligand by denosumab attenuates vascular calcium deposition in mice. Am J Pathol. 2009; 175:473-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hsu JJ, Tintut Y, Demer LL. Vitamin D and osteogenic differentiation in the artery wall. Clin J Am Soc Nephrol. 2008; 3:1542-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Watson KE, Abrolat ML, Malone LL, Hoeg JM, Doherty T, Detrano R, Demer LL. Active serum vitamin D levels are inversely correlated with coronary calcification. Circulation. 1997; 96:1755-1760. [DOI] [PubMed] [Google Scholar]
- 87.Drolet MC, Couet J, Arsenault M. Development of aortic valve sclerosis or stenosis in rabbits: role of cholesterol and calcium. J Heart Valve Dis. 2008; 17:381-387. [PubMed] [Google Scholar]
- 88.Hekimian G, Passefort S, Louedec L, Houard X, Jacob MP, Vahanian A, Michel JB, Messika‐Zeitoun D. High‐cholesterol + vitamin D2 regimen: a questionable in‐vivo experimental model of aortic valve stenosis. J Heart Valve Dis. 2009; 18:152-158. [PubMed] [Google Scholar]
- 89.Schmidt N, Brandsch C, Kühne H, Thiele A, Hirche F, Stangl GI. Vitamin D receptor deficiency and low vitamin D diet stimulate aortic calcification and osteogenic key factor expression in mice. PLoS One. 2012; 7:e35316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Branchetti E, Sainger R, Poggio P, Grau JB, Patterson‐Fortin J, Bavaria JE, Chorny M, Lai E, Gorman RC, Levy RJ, Ferrari G. Antioxidant enzymes reduce DNA damage and early activation of valvular interstitial cells in aortic valve sclerosis. Arterioscler Thromb Vasc Biol. 2013; 33:66-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schoenfeld SR, Kasturi S, Costenbader KH. The epidemiology of atherosclerotic cardiovascular disease among patients with SLE: a systematic review. Semin Arthritis Rheum. 2013; 43:77-95. [DOI] [PubMed] [Google Scholar]