

Abstract
Background
Increased concentrations of circulating fibroblast growth factor 23 (FGF‐23) have been associated with higher risk of cardiovascular disease. The association between FGF‐23 and the risk of atrial fibrillation (AF), a common arrhythmia, is less defined. Thus, we explored whether FGF‐23 concentration was associated with AF incidence in a large community‐based cohort.
Methods and Results
We studied 12 349 men and women enrolled in the Atherosclerosis Risk in Communities (ARIC) study, without prevalent AF at baseline in 1990–1992. Serum intact FGF‐23 concentration was measured with the Kainos 2‐site ELISA. Incident AF through 2010 was ascertained from study ECGs and hospital discharge codes. Cox proportional hazards models adjusted for potential confounding factors, including kidney function, were used to estimate the association between FGF‐23 and AF risk. We identified 1572 AF events during a mean follow‐up of 17 years. In multivariable analysis, a difference of 1 SD (16 pg/mL) in baseline FGF‐23 was not associated with the risk of AF (hazard ratio [HR], 1.04; 95% confidence interval [CI], 0.99, 1.09). Results were similar when FGF‐23 was modeled in quartiles (HR, 1.09; 95% CI, 0.94, 1.26, comparing extreme quartiles). Reduced kidney function was associated with increased AF risk across quartiles of FGF‐23 levels.
Conclusion
In this large community‐based cohort, baseline FGF‐23 levels were not associated with AF risk independently of kidney function. Our results do not support a major role for FGF‐23 as a risk factor for AF or as a mediator of the association between chronic kidney disease and AF.
Keywords: atrial fibrillation, epidemiology, kidney, risk factors
Introduction
Fibroblast growth factor 23 (FGF‐23) is a hormone involved in the regulation of phosphorus homeostasis, vitamin D metabolism, and bone mineralization. Secreted primarily by osteocytes in response to increased serum phosphorus levels, FGF‐23 reduces the expression and activity of Na/Pi cotransporters in the proximal tubules, lowers serum calcitriol levels, and suppresses synthesis of parathyroid hormone (PTH).1 In addition to being a potential marker of renal tubular dysfunction and of kidney disease risk,2–3 increased circulating levels of FGF‐23 have been associated with the incidence of heart failure (HF), coronary heart disease (CHD), and cardiovascular mortality (CVM),4–5 as well as with left ventricular hypertrophy (LVH).6–7 The same mechanisms underlying these associations could also be involved in the development of atrial fibrillation (AF), a common arrhythmia. Indeed, a recent publication from the Multi‐Ethnic Study of Atherosclerosis (MESA) and the Cardiovascular Health Study (CHS) cohorts, restricted to individuals free of cardiovascular disease (CVD), reported an increased risk of AF associated with higher levels of FGF‐23.8
Levels of FGF‐23 are elevated in individuals with chronic kidney disease (CKD), possibly in response to their reduced renal ability to eliminate phosphorus. Given the described higher risk of AF among individuals with CKD,9 exploring the association of FGF‐23 with AF incidence requires adequate adjustment for kidney function. In addition, an assessment of this association could illuminate the mechanisms linking CKD and AF, providing a better understanding of the interplay between kidney dysfunction, FGF‐23 levels, and AF risk. Different mechanisms, including development of hypertension and LVH, alterations in the renin‐angiotensin‐aldosterone system, and sympathetic activation, have been proposed to explain the elevated risk of AF among CKD patients.9 Exploring jointly the association of FGF‐23 and kidney function with AF risk may contribute to our understanding of the role of FGF‐23 as an alternative potential mechanism linking CKD with AF development.
We used data from the community‐based Atherosclerosis Risk in Communities (ARIC) Study to test the hypothesis that individuals with high FGF‐23 would have greater risk of developing AF independently of kidney function and other risk factors.
Methods
Study Design
The ARIC Study is a community‐based cohort investigating the determinants of atherosclerosis and CVD in the population. Its overall design and objectives have been previously published.10 In brief, 15 792 men and women residing in 4 communities in the United States (Forsyth County, NC; Jackson, MS; northwest suburbs of Minneapolis, MN; Washington County, MD) were recruited in 1987–1989 (visit 1). Study participants were examined again in 1990–1992 (visit 2), 1993–1995 (visit 3), 1996–1998 (visit 4), and 2011–2013 (visit 5). Only blacks were recruited in Jackson, while participants in the other field centers reflected the underlying population (mostly white in Minneapolis and Washington County, white and black in Forsyth County). At baseline and subsequent visits, participants provided written informed consent. The ARIC Study has been approved by institutional review boards at all participating institutions.
Measurement of FGF‐23
At each study visit, blood samples were collected, processed into serum, which was frozen within 2 hours, and stored at −70°C following standardized protocols. Circulating intact FGF‐23 was measured in singlicate at the Advanced Research and Diagnostic Laboratory, University of Minnesota (Minneapolis, MN) in 2012–2013 in serum samples collected during ARIC visit 2 (1990–1992) using a 2‐site ELISA (FGF‐23 ELISA Kit; Kainos Laboratories, Inc, Tokyo, Japan). Measurement coefficient of variation (CV) from split paired samples was 16.6%, whereas the CV from internal laboratory QC samples was 8.8% at 41.4 pg/mL. Measurements of FGF‐23 were available in 13 500 of 14 348 visit 2 participants (94%).
AF Ascertainment
In the ARIC cohort, AF has been ascertained using ECGs performed at study visits and diagnostic codes from hospitalization discharge summaries.11–12 At each study visit, a 12‐lead 10‐second ECG was done with the participant lying in a supine position. ECGs were transmitted electronically to a reading center (EpiCare, Wake Forest University, Winston‐Salem, NC), where they were reviewed for technical quality and electronically processed using the 2001 version of the GE Marquette 12‐SL program (GE Marquette, Milwaukee, WI). ECGs automatically coded as AF or atrial flutter were visually checked and confirmed by a cardiologist.13
Hospitalizations during follow‐up are ascertained through annual phone calls (>90% participation) and surveillance of local hospitals. Trained abstractors collect information from all participants' hospitalizations, including International Classification of Diseases, Ninth Revision, Clinical Modification (ICD‐9‐CM) codes for diagnoses and procedures associated with each hospitalization. We defined AF if ICD‐9‐CM codes 427.31 or 427.32 were present in any hospitalization. AF events associated with open cardiac surgery were not included. In a validation study conducted within ARIC, the positive predictive value of this case definition was ≈90% and the sensitivity was >80%.11 AF incidence date was defined as the earliest date in which an AF diagnosis was made during follow‐up. Individuals with a diagnosis of AF at or before ARIC visit 2 (when FGF‐23 measurements were made) were considered to have prevalent AF and therefore were excluded from this analysis.
Assessment of Other Covariates
At each study exam, participants provided self‐reported information on smoking status and use of medications. Weight and height were obtained with the participant wearing light clothes. Systolic and diastolic blood pressures (SBP/DBP) were defined as the average of the last 2 of 3 measurements. Diabetes status was defined as having a fasting blood glucose ≥126 mg/dL, nonfasting blood glucose >200 mg/dL, use of antidiabetic medications, or self‐report of a medical diagnosis of diabetes. Prevalent CHD and HF at visit 2 were defined as previously described.14–15
Blood phosphorus, calcium, creatinine, cystatin C, high‐sensitivity C‐reactive protein (hsCRP), N‐terminal prohormone of brain natriuretic peptide (NT‐proBNP), 25‐hydroxyvitamin D, and PTH were measured in stored samples collected during visit 2. Calcium and phosphorous were measured on a Roche Modular P Chemistry Analyzer (Roche Diagnostics, Indianapolis, IN) using colorimetric methods. Creatinine levels were measured in frozen serum samples, using methods based on modified kinetic Jaffe‐picric acid. Cystatin C was measured using the Gentian cystatin C reagent on the Roche Modular P Chemistry analyzer. hsCRP protein was measured using a latex‐particle enhanced immunoturbidimetric assay kit (Roche Diagnostics) and read on the Roche Modular P Chemistry analyzer, while NT‐proBNP was measured on a Cobas e411 analyzer using the Elecsys proBNP II immunoassay (Roche Diagnostics). Total 25‐hydroxyvitamin D (as the sum of 25‐hydroxyvitamin D2 and 25‐hydroxyvitaminD3) was measured using liquid chromatography‐tandem mass spectrometry. Finally, intact PTH was measured on a Roche Elecsys 2010 Analyzer (Roche Diagnostics Corporation) using a sandwich immunoassay method (Roche Diagnostics). Estimated glomerular filtration rate (eGFR) was calculated using the 2012 CKD‐EPI (Chronic Kidney Disease Epidemiology Collaboration) equation, which incorporates both cystatin C and creatinine.16
Statistical Analysis
For the current analysis, we included individuals attending visit 2, free of diagnosed AF, and with available baseline FGF‐23 measurements. We excluded those who were not white or black and nonwhites from the Minneapolis and Washington County field centers. In addition, participants with missing values in any of the covariates or missing ECG at baseline were also excluded, leaving 12 349 individuals for analysis (Figure 1). We used Cox proportional hazards model to assess the association between baseline FGF‐23 and AF incidence. Time of follow‐up was defined as the time between visit 2 (baseline for this analysis) and the incidence of AF, death, loss to follow‐up, or December 31, 2010, whichever occurred earlier. The dose‐response association was explored using restricted cubic splines models adjusted for age, sex, and race. Because the association was mostly linear, we modeled FGF‐23 as a continuous variable in the primary analysis, scaled as SD (1 SD=16 pg/mL). This model provided the best fit to the data. In additional analyses, FGF‐23 was modeled using quartiles of FGF‐23 and log2(FGF‐23) as a continuous variable. Results obtained from the later analysis can be interpreted as hazard ratios (HRs) associated with a doubling of FGF‐23 levels. The proportional hazards assumption was tested, including interaction terms between follow‐up time and FGF‐23 levels. None of these tests were statistically significant, supporting the adequateness of the assumption.
Figure 1.
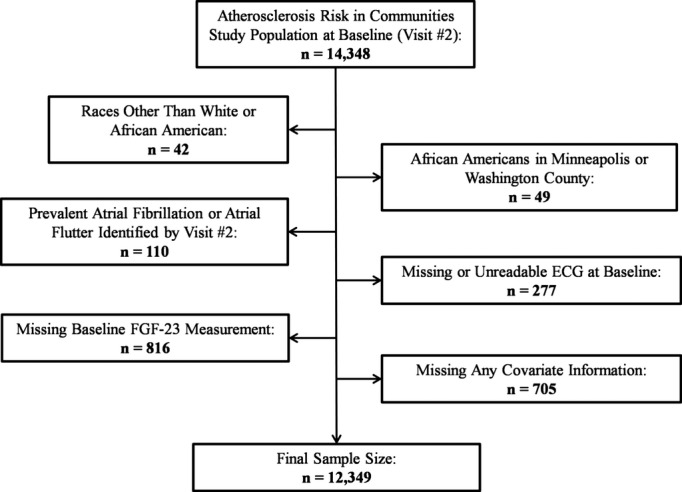
Inclusion flow chart, Atherosclerosis Risk in Communities Study, 1990–1992. FGF indicates fibroblast growth factor.
Initial models were adjusted for age, sex, and race. In a second model, we adjusted additionally for study site, body mass index (BMI; continuous), smoking (never, past, or current), education (<high school, completed high school, or at least some college), height (continuous), diabetes (yes, no), SBP and DBP (continuous), use of antihypertensive medications (yes, no), prevalent CHD (yes, no), prevalent HF (yes, no), ECG‐based LVH (yes, no, missing), NT‐proBNP (continuous, log‐transformed), hsCRP (continuous, log‐transformed), and eGFR (categorized as <60, 60 to 89, 90, or higher mL/min per 1.73 m2). In a final model, we adjusted for serum calcium, phosphorus, PTH, and 25‐hydroxyvitamin D (all continuous). Interactions between FGF‐23 and age, sex, race, ECG‐derived LVH, and kidney function were tested, including multiplicative terms in the models. Three sensitivity analyses were conducted, first excluding individuals with prevalent CVD at baseline, as was done in the analysis from the MESA and CHS cohorts; second, restricting the follow‐up to the initial 10 years, to determine whether any association between FGF‐23 and AF risk was stronger earlier in the follow‐up; and third, conducting separate analyses by method of AF ascertainment (ECG only or hospitalization discharge code only).
Because of the observed association of CKD with higher AF risk, and the relationship between kidney function and FGF‐23 levels, we categorized individuals by eGFR categories (<60, 60 to 89, and ≥90 mL/min per 1.73 m2) and FGF‐23 quartiles and determined HRs of AF across the resulting 12 categories using those in the bottom FGF‐23 quartile and with eGFR≥90 mL/min per 1.73 m2 as the reference group.
Finally, we performed a meta‐analysis of the results obtained from the ARIC cohort with those from the MESA and CHS cohorts.8 Specifically, we pooled the HRs (95% confidence intervals [CIs]) obtained from modeling log2(FGF‐23) after multivariable adjustment. A random‐effects models was used given the presence of significant heterogeneity, as determined by the Cochran's Q test and the I2 statistic (P=0.04, I2=68.1%).17–18
Results
Among 12 349 individuals included in the analysis, 1572 participants were newly diagnosed with AF during a mean follow‐up of 17 years (incidence rate: 7.7 cases per 1000 person‐years). Mean (SD) and median (25th to 75th percentiles) of baseline FGF‐23 were 44.2 (16.0) and 41.8 (33.9 to 51.6) pg/mL, respectively. Table 1 presents study participants' characteristics at visit 2 by quartiles of FGF‐23. Individuals with higher levels of FGF‐23 were older, more likely to be male, with higher BMI, had higher prevalence of diabetes, CHD, and HF, and had lower eGFR and higher serum phosphorus and PTH levels.
Table 1.
Baseline Characteristics by Quartile of Serum FGF‐23, Atherosclerosis Risk in Communities Study, 1990–1992
| Q1 | Q2 | Q3 | Q4 | |
|---|---|---|---|---|
| FGF‐23, pg/mL | ||||
| Median | 28.8 | 37.8 | 46.2 | 60.5 |
| Range | 2.86 to 33.89 | >33.89 to 41.83 | 41.84 to 51.60 | 51.61 to 242.69 |
| N | 3088 | 3086 | 3088 | 3087 |
| Age, y | 56.4 (5.7) | 56.6 (5.7) | 57.0 (5.6) | 57.5 (5.7) |
| Female, % | 60.0 | 56.6 | 55.0 | 55.9 |
| African Americans, % | 23.5 | 21.5 | 23.0 | 25.8 |
| Body mass index, kg/m2 | 27.0 (5.3) | 27.8 (5.2) | 28.1 (5.2) | 28.9 (5.7) |
| Current smoker, % | 25.5 | 21.7 | 21.3 | 18.4 |
| Diabetes, % | 13.2 | 12.2 | 14.3 | 17.4 |
| Height, cm | 167.5 (9.1) | 168.3 (9.3) | 168.7 (9.3) | 168.6 (9.3) |
| High school graduate, % | 43.9 | 42.1 | 41.6 | 40.8 |
| Systolic BP, mm Hg | 119.7 (18.0) | 120.5 (18.2) | 121.3 (18.5) | 123.4 (19.6) |
| Diastolic BP, mm Hg | 71.3 (10.0) | 72.0 (10.1) | 72.4 (10.0) | 72.7 (10.8) |
| Antihypertensive medications, % | 25.7 | 27.3 | 32.6 | 42.4 |
| Prevalent CHD, % | 4.4 | 5.1 | 5.7 | 6.8 |
| Prevalent HF, % | 3.1 | 3.8 | 4.3 | 6.4 |
| LVH, % | 1.9 | 1.6 | 2.7 | 2.9 |
| hsCRP, mg/L* | 2.1 (3.4) | 2.1 (3.5) | 2.3 (3.8) | 2.7 (4.2) |
| NT‐proBNP, pg/mL* | 51.3 (65.4) | 48.7 (64.2) | 47.0 (68.2) | 51.6 (71.2) |
| PTH, pg/mL | 40.3 (15.2) | 41.5 (15.6) | 42.2 (16.0) | 44.1 (20.1) |
| Serum calcium, mg/dL | 9.3 (0.4) | 9.3 (0.4) | 9.4 (0.4) | 9.4 (0.5) |
| Serum phosphorous, mg/dL | 3.5 (0.5) | 3.5 (0.5) | 3.6 (0.5) | 3.6 (0.5) |
| Serum 25‐(OH)vitamin D, ng/mL | 23.5 (8.3) | 24.6 (8.4) | 24.7 (8.5) | 24.9 (8.7) |
| eGFR, mL/min per 1.73 m2 | 100.0 (15.2) | 97.3 (14.9) | 94.9 (15.6) | 89.1 (18.8) |
| 90+ | 75.8 | 71.1 | 64.0 | 51.8 |
| 60 to 89 | 23.5 | 28.0 | 34.4 | 41.1 |
| 15 to 59 | 0.7 | 1.0 | 1.7 | 7.2 |
Data shown as percent or mean (SD) except *=geometric mean (interquartile range). BP indicates blood pressure; CHD, coronary heart disease; eGFR, estimated glomerular filtration rate; FGF‐23, fibroblast growth factor 23; HF, heart failure; hsCRP, high‐sensitivity C‐reactive protein; LVH, left ventricular hypertrophy; NT‐proBNP, N‐terminal prohormone of brain natriuretic peptide; PTH, parathyroid hormone.
Modeling levels of FGF‐23 as restricted cubic spline demonstrated a mostly linear association with the incidence of AF and, therefore the main analysis was done with FGF‐23 modeled as a linear variable (Figure 2). In a model adjusted for age, sex, and race, a difference of 16 pg/mL in FGF‐23 (corresponding to 1 SD) was associated with a 14% higher rate of AF (HR, 1.14; 95% CI, 1.09, 1.19; Table 2; Model 1). Adjustment for other covariates eliminated the association between FGF‐23 levels and AF incidence (Table 2; Models 2 and 3). Results were similar when we used log2(FGF‐23) or FGF‐23 quartiles instead of FGF‐23 levels, with moderately strong associations in minimally adjusted models and no association after adjustment for confounders (Table 2). Similar lack of association was observed in analyses including cases identified from study ECGs or hospital discharge codes separately (Table 3). No significant multiplicative interactions were found between FGF‐23 and race, sex, age, ECG‐derived LVH, or eGFR.
Figure 2.
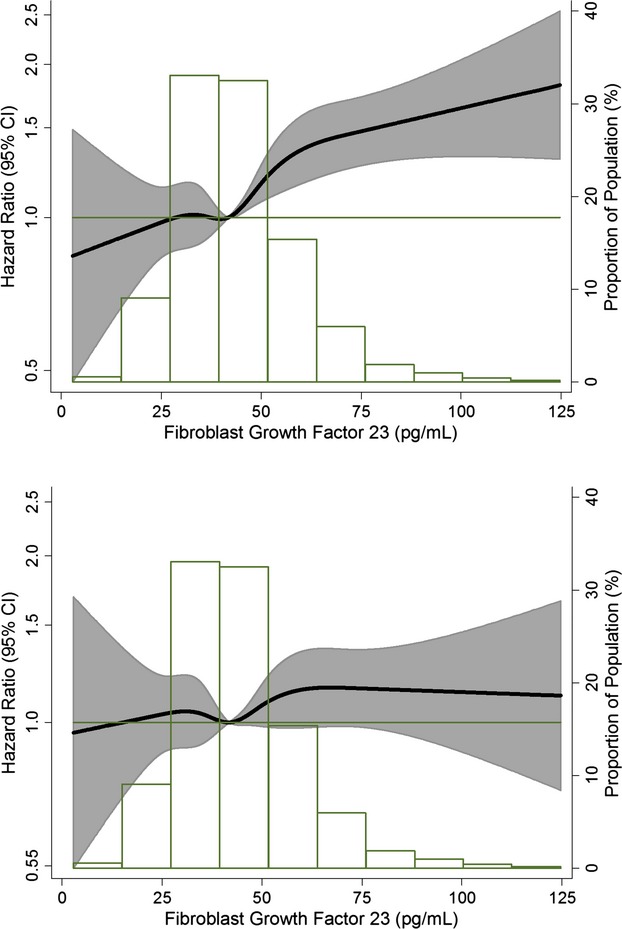
Association of circulating FGF‐23 concentrations with incidence of atrial fibrillation presented as hazard ratio (HR; solid line) and 95% confidence intervals (CI; shaded area). Results from Cox proportional hazards model with FGF‐23 modeled using restricted cubic splines, adjusted for age, sex, and race (top panel) and additionally adjusted for study site, body mass index, smoking, education, height, diabetes, systolic and diastolic blood pressure, use of antihypertensive medication, prevalent coronary heart disease, prevalent heart failure, ECG‐based left ventricular hypertrophy, NT‐proBNP, high‐sensitivity C‐reactive protein, and eGFR (bottom panel). Median value of FGF‐23 was considered the reference (HR=1). The histograms represent the frequency distribution of FGF‐23 in the study sample. Atherosclerosis Risk in Communities Study, 1990–2010. eGFR indicates estimated glomerular filtration rate; FGF, fibroblast growth factor; NT‐proBNP, N‐terminal prohormone of brain natriuretic peptide.
Table 2.
Hazard Ratios (HR) and 95% Confidence Intervals (CI) of Atrial Fibrillation (AF) Associated With FGF‐23 Concentrations, Atherosclerosis Risk in Communities Study, 1990–2010
| FGF‐23 (Linear)* | Log2(FGF‐23) | FGF‐23 (Quartiles) | ||||
|---|---|---|---|---|---|---|
| Q1 (2.9 to 33.8 pg/mL) | Q2 (33.9 to 41.8 pg/mL) | Q3 (41.9 to 51.6 pg/mL) | Q4 (51.7 to 242.7 pg/mL) | |||
| AF events | 1572 | 1572 | 340 | 366 | 382 | 484 |
| Model 1 | 1.14 (1.09, 1.19) | 1.31 (1.19, 1.45) | 1 (ref) | 1.03 (0.89, 1.20) | 1.06 (0.92, 1.23) | 1.39 (1.21, 1.60) |
| Model 2 | 1.04 (0.99, 1.09) | 1.07 (0.96, 1.18) | 1 (ref) | 0.99 (0.85, 1.15) | 0.95 (0.82, 1.10) | 1.09 (0.94, 1.26) |
| Model 3 | 1.04 (0.99, 1.09) | 1.07 (0.96, 1.19) | 1 (ref) | 1.00 (0.86, 1.16) | 0.95 (0.82, 1.11) | 1.10 (0.95, 1.27) |
Model 1: Cox proportional hazards model adjusted for age, race, sex. Model 2: As Model 1, additionally adjusted for study site, body mass index, smoking, education, height, diabetes, systolic and diastolic blood pressure, use of antihypertensive medication, prevalent coronary heart disease, prevalent heart failure, ECG‐based left ventricular hypertrophy, NT‐proBNP, high‐sensitivity C‐reactive protein, and eGFR. Model 3: As Model 2, additionally adjusted for serum calcium, phosphorus, parathyroid hormone, and 25‐hydroxyvitamin D. FGF indicates fibroblast growth factor; NT‐proBNP, N‐terminal prohormone of brain natriuretic peptide.
HR (95% CI) corresponds to a difference of 16 pg/mL.
Table 3.
Hazard Ratios (HR) and 95% Confidence Intervals (CI) of Atrial Fibrillation (AF) Associated With FGF‐23 Concentrations by Method of AF Ascertainment, Atherosclerosis Risk in Communities Study, 1990–2010
| FGF‐23 (Linear)* | Log2(FGF‐23) | FGF‐23 (Quartiles) | ||||
|---|---|---|---|---|---|---|
| Q1 (2.9 to 33.8 pg/mL) | Q2 (33.9 to 41.8 pg/mL) | Q3 (41.9 to 51.6 pg/mL) | Q4 (51.7 to 242.7 pg/mL) | |||
| Events ascertained from hospital discharge codes | ||||||
| AF events | 1565 | 1565 | 339 | 365 | 380 | 481 |
| HR (95% CI) | 1.03 (0.98, 1.09) | 1.06 (0.96, 1.18) | 1 (ref) | 0.99 (0.85, 1.15) | 0.95 (0.82, 1.10) | 1.09 (0.94, 1.26) |
| Events ascertained from study ECG | ||||||
| AF events | 77 | 77 | 15 | 20 | 22 | 20 |
| HR (95% CI) | 0.91 (0.71, 1.17) | 0.91 (0.56, 1.48) | 1 (ref) | 1.20 (0.61, 2.37) | 1.10 (0.56, 2.15) | 0.84 (0.42, 1.72) |
Cox proportional hazards model adjusted for age, race, sex, study site, body mass index, smoking, education, height, diabetes, systolic and diastolic blood pressure, use of antihypertensive medication, prevalent coronary heart disease, prevalent heart failure, ECG‐based left ventricular hypertrophy, NT‐proBNP, high‐sensitivity C‐reactive protein, and eGFR. eGFR indicates estimated glomerular filtration rate; FGF, fibroblast growth factor; NT‐proBNP, N‐terminal prohormone of brain natriuretic peptide.
HR (95% CI) corresponds to a difference of 16 pg/mL.
We conducted 2 sensitivity analyses, first including only participants without prevalent CVD at baseline and, second, restricting the analysis to the initial 10 years of follow‐up. Results remained basically unchanged: HRs (95% CI) of AF for 1‐SD increment were 1.03 (0.98, 1.09) among the 11 103 ARIC participants without CVD at baseline (1267 AF events) and 1.01 (0.93, 1.09) during the first 10 years of follow‐up (590 AF events).
In an additional analysis, we categorized individuals by eGFR categories and FGF‐23 quartiles to explore in more detail the association of eGFR with AF risk across levels of FGF‐23. Overall, we found that impaired kidney function, reflected as eGFR<60 mL/min per 1.73 m2, was associated with increased risk of AF at all levels of FGF‐23 (Figure 3) and that FGF‐23 was not associated with AF risk across eGFR categories (Figure 4).
Figure 3.
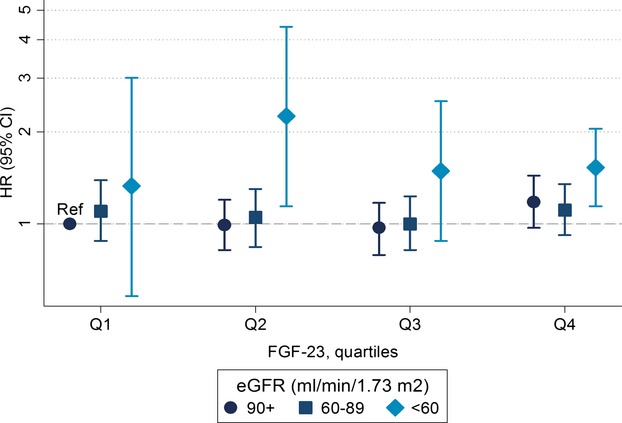
Hazard ratios (HR) and 95% confidence intervals (CI) of AF by categories of eGFR and FGF‐23 quartile, using participants with eGFR≥90 mL/min per 1.73 m2 and FGF‐23 in the first quartile as the reference. Models were adjusted for age, race, sex, study site, body mass index, smoking, education, height, diabetes, systolic and diastolic blood pressure, use of antihypertensive medication, prevalent coronary heart disease, prevalent heart failure, ECG‐based left ventricular hypertrophy, NT‐proBNP, and high‐sensitivity C‐reactive protein, Atherosclerosis Risk in Communities Study, 1990–2010. AF indicates atrial fibrillation; eGFR, estimated glomerular filtration rate; FGF, fibroblast growth factor; NT‐proBNP, N‐terminal prohormone of brain natriuretic peptide.
Figure 4.
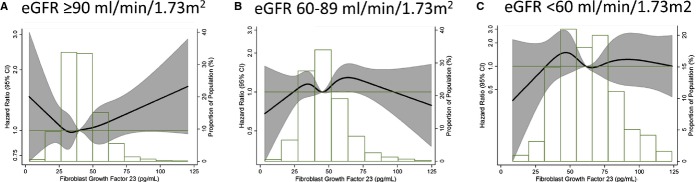
Association of circulating FGF‐23 concentrations with incidence of atrial fibrillation presented as hazard ratio (HR; solid line) and 95% confidence intervals (CI; shaded area) by categories of eGFR (<60 (panel C), 60 to 89 (panel B), and 90+ mL/min per 1.73 m2 (panel A)). Results from Cox proportional hazards model with FGF‐23 modeled using restricted cubic splines, adjusted for age, sex, race, study site, body mass index, smoking, education, height, diabetes, systolic and diastolic blood pressure, use of antihypertensive medication, prevalent coronary heart disease, prevalent heart failure, ECG‐based left ventricular hypertrophy, NT‐proBNP, high‐sensitivity C‐reactive protein, and eGFR. Median value of FGF‐23 was considered the reference (HR=1). The histograms represent the frequency distribution of FGF‐23 in each category of eGFR. Atherosclerosis Risk in Communities Study, 1990–2010. eGFR indicates estimated glomerular filtration rate; FGF, fibroblast growth factor; NT‐proBNP, N‐terminal prohormone of brain natriuretic peptide.
Finally, we performed a meta‐analysis of the results from the ARIC, MESA, and CHS cohorts. Specifically, we used HR and 95% CI associated with 1 unit increment in log2(FGF‐23), which corresponds to a doubling of FGF‐23 levels, in multivariable adjusted models. Cohort‐specific baseline characteristics are provided in Table S1. Because significant between‐study heterogeneity was present (P=0.04, I2=68.1%), a random‐effects model was used. The meta‐analysis showed a statistically significant association of moderate strength between FGF‐23 levels and AF risk (HR, 1.22; 95% CI, 1.02, 1.46; P=0.04, per doubling of FGF‐23 levels; Figure 5). Because of the small number of studies, however, this analysis provides a poor estimate of the width of the distribution of the associations between FGF‐23 and AF risk and, therefore results should be interpreted with caution.
Figure 5.
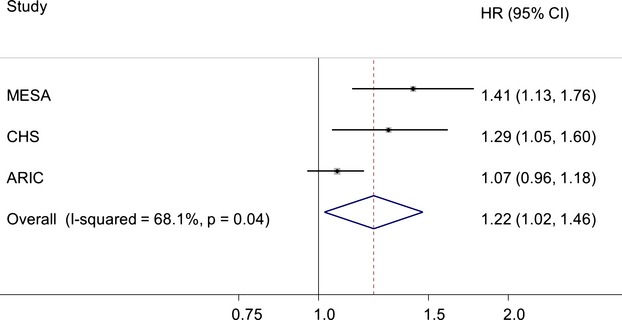
Random‐effects meta‐analysis of the association between fibroblast growth factor (FGF)‐23 concentrations and atrial fibrillation incidence in the Multi‐Ethnic Study of Atherosclerosis (MESA), Cardiovascular Health Study (CHS), and Atherosclerosis Risk in Communities (ARIC) cohorts. Study‐specific results correspond to multivariable hazard ratios (HR) and 95% confidence intervals (CI) for 1 unit increment in log2(FGF‐23) levels. Size of the study‐specific markers is proportional to the meta‐analysis weights.
Discussion
In this analysis of the ARIC study, we found that serum FGF‐23 was not associated with the incidence of AF independently of other risk factors. The lack of association was consistently observed in several sensitivity analyses. However, a meta‐analysis of our results with those previously reported in the MESA and CHS cohorts supported the role of FGF‐23 as a potential risk factor for AF. We also found that CKD was associated with increased risk of AF independently of serum FGF‐23 levels, suggesting that FGF‐23 is not a major mediator of the previously described association between CKD and AF.
The lack of association between serum FGF‐23 and AF risk in the ARIC study is in direct contrast with results recently reported from the MESA and CHS cohorts. In these studies, which included a total of 520 incident AF cases among 7748 participants, a doubling of FGF‐23 levels was associated with multivariable HRs (95% CI) of 1.41 (1.13, 1.76) and 1.29 (1.05, 1.60), respectively, compared with 1.07 (0.96, 1.18) in the ARIC study.8 All 3 cohorts adjusted for major potential confounders, including kidney function. However, differences in the study sample and analytical approach could explain the inconsistency, including the higher proportion of individuals with CKD and the exclusion of individuals with CVD in the MESA and CHS cohorts, and the shorter follow‐up in the MESA cohort. However, in sensitivity analyses in the ARIC cohort that applied similar exclusion criteria and follow‐up, our main results remained unaltered. Other methodological aspects, including ascertainment of AF and covariate measurement, were comparable across studies. Also, serum FGF‐23 measurement in the ARIC and MESA cohorts were performed using the same assays, which measures biologically intact FGF‐23. In CHS, a different assay measuring inactive C‐terminal FGF‐23 concentrations was used. Similarly, the current ARIC results contradict the observed associations of FGF‐23 with other outcomes in the general population, including CVM, HF, and kidney disease.3–5
Despite the lack of association between serum FGF‐23 and AF incidence in our study, we found that a combined estimate of the ARIC, MESA, and CHS cohorts suggested that higher levels of FGF‐23 may be associated with increased risk of AF. Several mechanisms can be hypothesized to explain this observation. Higher circulating levels of FGF‐23 have been associated with higher levels of inflammatory markers,19 LVH,6–7 and vascular dysfunction.20 In addition, FGF‐23 inhibits the synthesis of 1,25‐dihydroxyvitamin D. These mechanisms could predispose to AF directly or indirectly through an increased risk of HF or CHD.21–23 Additional research is needed to disentangle the role of these pathways in the etiology of AF.
A higher incidence of AF among individuals with CKD has been consistently reported in the literature.9,24–26 Given the increase in circulating FGF‐23 that accompanies worsening of kidney function, and the potential association between FGF‐23 and AF risk, we hypothesized that elevations in FGF‐23 could be partly responsible for the impact of CKD on AF risk. The previously described association between higher serum phosphorus levels and increased AF risk also supports this hypothesis.8,27 In the current analysis of the ARIC cohort, however, we found that CKD was a risk factor for AF independently of FGF‐23 levels. Our results suggest that other mechanisms, including oxidative stress,28 sympathetic activation,29 inflammation,30 or imbalances in the renin‐angiotensin‐aldosterone system,31 are probably responsible for the CKD‐AF link.
Our analysis has important strengths, such as the inclusion of a community‐based, large, and diverse cohort, the availability of potential confounders, including kidney function, the high number of events, and the excellent retention. An important limitation, however, is the method of AF ascertainment, with most events found through hospital discharge codes. Thus, individuals with asymptomatic AF or those managed in an outpatient setting not requiring hospital admission were more likely to remain unidentified. Nonetheless, the validity of hospital discharge codes for identifying incident AF in epidemiologic studies has previously been demonstrated.11,32 The lack of repeated measures of FGF‐23 is an additional weakness. Using a single assessment of FGF‐23 may be insufficient to characterize the long‐term effects of elevated circulating FGF‐23, potentially diluting the underlying associations.
In conclusion, levels of circulating FGF‐23 were not associated with AF risk in a large community‐based cohort once potential confounders, including kidney function, were taken into account. Still, the existing published evidence supports a possible role of FGF‐23 as a risk factor for AF. In addition, our results indicate that the higher risk of AF among individuals with CKD is unlikely to be mediated by elevations in FGF‐23.
Supplementary Material
Appendix Supplemental Table S1. Baseline characteristics of participants in the ARIC, MESA and CHS cohorts
Sources of Funding
The Atherosclerosis Risk in Communities Study is carried out as a collaborative study supported by National Heart, Lung and Blood Institute (NHLBI) contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, and HHSN268201100012C). Additional support was provided by grant R01 HL103706 from the NHLBI to Dr Lutsey, grants RC1 HL099452 from the NHLBI and 09SDG2280087 from the American Heart Association to Dr Alonso, and grant R01 DK089174 from the National Institute of Diabetes and Digestive and Kidney Diseases to Dr Selvin. Roche provided material support (reagents) for the C‐reactive protein and NT‐proBNP assays.
Disclosures
None.
Acknowledgments
The authors thank the staff and participants of the ARIC study for their important contributions.
References
- 1.Heine GH, Seiler S, Fliser D. FGF‐23: the rise of a novel cardiovascular risk marker in CKD. Nephrol Dial Transplant. 2012; 27:3072-3081. [DOI] [PubMed] [Google Scholar]
- 2.Dominguez JR, Shlipak MG, Whooley MA, Ix JH. Fractional excretion of phosphorus modifies the association between fibroblast growth factor‐23 and outcomes. J Am Soc Nephrol. 2013; 24:647-654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown JR, Katz R, Ix JH, de Boer IH, Siscovick DS, Grams ME, Shlipak M, Sarnak MJ. Fibroblast growth factor‐23 and the long‐term risk of hospital‐associated AKI among community‐dwelling older individuals. Clin J Am Soc Nephrol. 2014; 9:239-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ix JH, Katz R, Kestenbaum BR, de Boer IH, Chonchol M, Mukamal KJ, Rifkin D, Siscovick DS, Sarnak MJ, Shlipak MG. Fibroblast growth factor‐23 and death, heart failure, and cardiovascular events in community‐living individuals: CHS (Cardiovascular Health Study). J Am Coll Cardiol. 2012; 60:200-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lutsey PL, Alonso A, Selvin E, Pankow JS, Michos ED, Agarwal SK, Loehr LR, Eckfeldt JH, Coresh J. Fibroblast growth factor‐23 and incident coronary heart disease, heart failure and cardiovascular mortality: the Atherosclerosis Risk in Communities (ARIC) Study. J Am Heart Assoc. 2014; 3:e00093610.1161/JAHA.114.000936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Faul C, Amaral AP, Oskouei B, Hu M‐C, Sloan A, Isakova T, Gutiérrez OM, Aguillon‐Prada R, Lincoln J, Hare JM, Mundel P, Morales A, Scialla J, Fischer M, Soliman EZ, Chen J, Go AS, Rosas SE, Nessel L, Townsend RR, Feldman HI, St. John Sutton M, Ojo A, Gadegbeku C, Di Marco GS, Reuter S, Kentrup D, Tiemann K, Brand M, Hill JA, Moe OW, Kuro‐o M, Kusek JW, Keane MG, Wolf M. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011; 121:4393-4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seiler S, Cremers B, Rebling NM, Fornof F, Jeken J, Kersting S, Steimle C, Ege P, Fehrenz M, Rogacev KS, Scheller B, Bohm M, Fliser D, Heine GH. The phosphatonin fibroblast growth factor 23 links calcium‐phosphate metabolism with left ventricular dysfunction and atrial fibrillation. Eur Heart J. 2011; 32:2688-2696. [DOI] [PubMed] [Google Scholar]
- 8.Mathew JS, Sachs MC, Patton KK, Heckbert SR, Hoofnagle AN, Alonso A, Chonchol M, Deo R, Ix JH, Siscovick DS, Kestenbaum BR, De Boer IH. Fibroblast growth factor‐23 and incident atrial fibrillation: the Multi‐Ethnic Study of Atherosclerosis (MESA) and the Cardiovascular Health Study (CHS). Circulation. 2014; 130:298-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alonso A, Lopez FL, Matsushita K, Loehr LR, Agarwal SK, Chen LY, Soliman EZ, Astor BC, Coresh J. Chronic kidney disease is associated with the incidence of atrial fibrillation: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2011; 123:2946-2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.The ARIC Investigators. The Atherosclerosis Risk in Communities (ARIC) study: design and objectives. Am J Epidemiol. 1989; 129:687-702. [PubMed] [Google Scholar]
- 11.Alonso A, Agarwal SK, Soliman EZ, Ambrose M, Chamberlain AM, Prineas RJ, Folsom AR. Incidence of atrial fibrillation in whites and African‐Americans: the Atherosclerosis Risk in Communities (ARIC) study. Am Heart J. 2009; 158:111-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chamberlain AM, Agarwal SK, Ambrose M, Folsom AR, Soliman EZ, Alonso A. Metabolic syndrome and incidence of atrial fibrillation among blacks and whites in the Atherosclerosis Risk in Communities (ARIC) Study. Am Heart J. 2010; 159:850-856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soliman EZ, Prineas RJ, Case D, Zhang Z‐M, Goff DC., Jr Ethnic distribution of electrocardiographic predictors of atrial fibrillation and its impact on understanding the ethnic distribution of ischemic stroke in the Atherosclerosis Risk in Communities Study (ARIC). Stroke. 2009; 40:1204-1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.White AD, Folsom AR, Chambless LE, Sharret AR, Yang K, Conwill D, Higgins M, Williams OD, Tyroler HAThe ARIC Investigators. Community surveillance of coronary heart disease in the Atherosclerosis Risk in Communities (ARIC) Study: methods and initial two years' experience. J Clin Epidemiol. 1996; 49:223-233. [DOI] [PubMed] [Google Scholar]
- 15.Loehr LR, Rosamond WD, Chang PP, Folsom AR, Chambless LE. Heart failure incidence and survival (from the Atherosclerosis Risk in Communities Study). Am J Cardiol. 2008; 101:1016-1022. [DOI] [PubMed] [Google Scholar]
- 16.Inker LA, Schmid CH, Tighiouart H, Eckfeldt JH, Feldman HI, Greene T, Kusek JW, Manzi J, Van Lente F, Zhang YL, Coresh J, Levey AS. Estimating glomerular filtration rate from serum creatinine and cystatin C. N Engl J Med. 2012; 367:20-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DerSimonian R, Laird N. Meta‐analysis in clinical trials. Control Clin Trials. 1986; 7:177-188. [DOI] [PubMed] [Google Scholar]
- 18.Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ. 2003; 327:557-560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munoz Mendoza J, Isakova T, Ricardo AC, Xie H, Navaneethan SD, Anderson AH, Bazzano LA, Xie D, Kretzler M, Nessel L, Hamm LL, Negrea L, Leonard MB, Raj D, Wolf Mfor the Chronic Renal Insufficiency Cohort. Fibroblast growth factor 23 and inflammation in CKD. Clin J Am Soc Nephrol. 2012; 7:1155-1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mirza MAI, Larsson A, Lind L, Larsson TE. Circulating fibroblast growth factor‐23 is associated with vascular dysfunction in the community. Atherosclerosis. 2009; 205:385-390. [DOI] [PubMed] [Google Scholar]
- 21.Guo Y, Lip GYH, Apostolakis S. Inflammation in atrial fibrillation. J Am Coll Cardiol. 2012; 60:2263-2270. [DOI] [PubMed] [Google Scholar]
- 22.Alonso A, Krijthe BP, Aspelund T, Stepas KA, Pencina MJ, Moser CB, Sinner MF, Sotoodehnia N, Fontes JoD, Janssens ACJW, Kronmal RA, Magnani JW, Witteman JC, Chamberlain AM, Lubitz SA, Schnabel RB, Agarwal SK, McManus DD, Ellinor PT, Larson MG, Burke GL, Launer LJ, Hofman A, Levy D, Gottdiener JS, Kääb S, Couper D, Harris TB, Soliman EZ, Stricker BHC, Gudnason V, Heckbert SR, Benjamin EJ. Simple risk model predicts incidence of atrial fibrillation in a racially and geographically diverse population: the CHARGE‐AF consortium. J Am Heart Assoc. 2013; 2:e00010210.1161/JAHA.112.000102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robinson‐Cohen C, Hoofnagle AN, Ix JH, Sachs MC, Tracy RP, Siscovick DS, Kestenbaum BR, de Boer IH. Racial differences in the association of serum 25‐hydroxyvitamin D concentration with coronary heart disease events. JAMA. 2013; 310:179-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nelson SE, Shroff GR, Li S, Herzog CA. Impact of chronic kidney disease on risk of incident atrial fibrillation and subsequent survival in Medicare patients. J Am Heart Assoc. 2012; 1:e00209710.1161/JAHA.112.002097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watanabe H, Watanabe T, Sasaki S, Nagai K, Roden DM, Aizawa Y. Close bidirectional relationship between chronic kidney disease and atrial fibrillation: the Niigata preventive medicine study. Am Heart J. 2009; 158:629-636. [DOI] [PubMed] [Google Scholar]
- 26.Horio T, Iwashima Y, Kamide K, Tokudome T, Yoshihara F, Nakamura S, Kawano Y. Chronic kidney disease as an independent risk factor for new‐onset atrial fibrillation in hypertensive patients. J Hypertens. 2010; 28:1738-1744. [DOI] [PubMed] [Google Scholar]
- 27.Lopez FL, Agarwal SK, Grams ME, Loehr LR, Soliman EZ, Lutsey PL, Chen LY, Huxley RR, Alonso A. Relation of serum phosphorus levels to the incidence of atrial fibrillation (from the Atherosclerosis Risk In Communities [ARIC] Study). Am J Cardiol. 2013; 111:857-862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fukunaga N, Takahashi N, Hagiwara S, Kume O, Fukui A, Teshima Y, Shinohara T, Nawata T, Hara M, Noguchi T, Saikawa T. Establishment of a model of atrial fibrillation associated with chronic kidney disease in rats and the role of oxidative stress. Heart Rhythm. 2012; 9:2023-2031. [DOI] [PubMed] [Google Scholar]
- 29.Schlaich MP, Socratous F, Hennebry S, Eikelis N, Lambert EA, Straznicky N, Esler MD, Lambert GW. Sympathetic activation in chronic renal failure. J Am Soc Nephrol. 2009; 20:933-939. [DOI] [PubMed] [Google Scholar]
- 30.Shlipak MG, Fried LF, Crump C, Bleyer AJ, Manolio TA, Tracy RP, Furberg CD, Psaty BM. Elevations of inflammatory and procoagulant biomarkers in elderly persons with renal insufficiency. Circulation. 2003; 107:87-92. [DOI] [PubMed] [Google Scholar]
- 31.Siragy HM, Carey RM. Role of the intrarenal renin‐angiotensis‐aldosteron system in chronic kidney disease. Am J Nephrol. 2010; 31:541-550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jensen PN, Johnson K, Floyd J, Heckbert SR, Carnahan R, Dublin S. A systematic review of validated methods for identifying atrial fibrillation using administrative data. Pharmacoepidemiol Drug Saf. 2012; 21suppl 1:141-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix Supplemental Table S1. Baseline characteristics of participants in the ARIC, MESA and CHS cohorts