

Abstract
Background
Endoplasmic reticulum (ER) resident protein 44 (ERp44) is a member of the protein disulfide isomerase family, is induced during ER stress, and may be involved in regulating Ca2+ homeostasis. However, the role of ERp44 in cardiac development and function is unknown. The aim of this study was to investigate the role of ERp44 in cardiac development and function in mice, zebrafish, and embryonic stem cell (ESC)‐derived cardiomyocytes to determine the underlying role of ERp44.
Methods and Results
We generated and characterized ERp44−/− mice, ERp44 morphant zebrafish embryos, and ERp44−/− ESC‐derived cardiomyocytes. Deletion of ERp44 in mouse and zebrafish caused significant embryonic lethality, abnormal heart development, altered Ca2+ dynamics, reactive oxygen species generation, activated ER stress gene profiles, and apoptotic cell death. We also determined the cardiac phenotype in pressure overloaded, aortic‐banded ERp44+/− mice: enhanced ER stress activation and increased mortality, as well as diastolic cardiac dysfunction with a significantly lower fractional shortening. Confocal and LacZ histochemical staining showed a significant transmural gradient for ERp44 in the adult heart, in which high expression of ERp44 was observed in the outer subepicardial region of the myocardium.
Conclusions
ERp44 plays a critical role in embryonic heart development and is crucial in regulating cardiac cell Ca2+ signaling, ER stress, ROS‐induced oxidative stress, and activation of the intrinsic mitochondrial apoptosis pathway.
Keywords: apoptosis, Ca2+, ERp44−/−, ESC‐derived cardiomyocytes, heart development and cardiomyopathy
Introduction
The endoplasmic reticulum (ER) is an important organelle within the cell that has various functions, primarily involving synthesis and folding of proteins, as well as Ca2+ storage and signaling. Normal ER function is essential for heart development, proper oxidative folding, quality control of membrane‐bound and secretory proteins, and in regulating stress responses.1 Changes in intracellular Ca2+, molecular chaperone activities, the protein glycosylation machinery, or redox status in the ER lumen will induce unfolded protein response (UPR) that is a compensatory mechanism to manage these insults in the ER. The UPR process includes inhibition of protein synthesis, protein refolding, and degradation of misfolded proteins.2 Prolonged ER stress in cardiac muscle can induce cell death, cardiomyopathy, and heart failure (HF).3–5.
The ER resident protein 44 (ERp44; originally referred to as Txnd4) is a UPR‐induced ER protein of the protein disulphide isomerase (PDI) family.6 ERp44 may regulate oxidative protein folding and client protein homeostasis through thiol‐mediated protein retention in the ER.6–8 ERp44 has been shown to inhibit Ca2+ release through binding and inactivating inositol trisphosphate receptor 1 (IP3R1).8 ERp44 binds to the third luminal loop of the IP3R1, which is the home to structural differences between IP3R subtypes, suggesting that the interaction of ERp44 might be specific for type 1 IP3R.8–9 ERp44 binding with IP3R increases with H+ concentration; however, at neutral pH, oxidized ERp44 dissociates from the IP3R, whereas the reduced form of ERp44 can still bind with IP3R.8–9 Altogether, ERp44 binding appears to confer a sensitivity to Ca2+, pH, and redox state upon the IP3R.8–9 Furthermore, Mikoshiba showed that agonist‐induced Ca2+ release was inhibited by overexpression of ERp44 and enhanced by siRNA‐mediated knockdown of endogenous ERp44 in HeLa cells.8–9 In a “normal” ER environment, little ERp44 binds IP3R, which allows for normal Ca2+ release function; but, during early ER stress, ERp44 levels becomes elevated, binds with IP3R and inactivates IP3R‐stimulated Ca2+ release to maintain the Ca2+ store in the ER. As the redox state of the ER influences ERp44's binding to IP3R, the prolonged ER stress induction of ER oxidase 1 alpha (Ero1α) downstream of CCAAT‐enhancer‐binding protein homology protein (CHOP) activation, leading to hyperoxidation of the ER lumen and causing the oxidized ERp44 to dissociate from the IP3R. Moreover, the up‐regulated Ero1α competes with ERp44 binding to IP3R and can lead to the potentiation of the Ca2+ flux through the IP3R channel and trigger excessive Ca2+ signaling events, leading to cell death.10 However, despite this knowledge, the physiological role of ERp44 in the heart is still unclear.
To gain insight into the physiological role of ERp44 in the heart in vivo and in vitro, we generated multiple model systems, including an ERp44 knockout (KO)/lacZ knock‐in (KI) mouse, zebrafish morphants, and ERp44−/− embryonic stem cell (ESC)‐derived cardiomyocytes (ESCCs), and show that ERp44 plays a crucial role in heart development and function.
Methods
Generation and Genotyping of ERp44 KO Mice
The University of Toronto Animal Care and Use Committee approved all experiments (Toronto, Ontario, Canada). Gene‐trapped ESC line XB599 (MMRRC Stock No. 015890‐UCD; Bay Genomics, San Francisco, CA) was utilized to generate ERp44‐null mutant mice. By analysis of the 5′ tag ERp44‐XB599 sequence, we determined the gene trap vector inserted into ERp44 genomic intron 1 using a forward primer (F147 5′CCGTCGTTACCATGAATCCT3′) designed to recognize ERp44 exon 1 and a reverse primer designed to recognize the lacZ reporter gene (lacZR2 5′GACAGTATC GGCCTCAGGAAGATCG3′). A mutant allele resulted in an ERp44 exon1‐lacZ fusion product of 294 bp by reverse‐transcriptase polymerase chain reaction (RT‐PCR). Sequence analysis of the RT‐PCR product and genomic DNA PCR confirmed the gene‐trapping vector insertion into ERp44 intron 1. Southern blot hybridization was performed with a lacZ probe to detect the gene insertion event.
XB599 ESCs were aggregated with diploid embryos at The Toronto Center for Phenogenomics to obtain germ‐line transmission. Viable and fertile chimeras with the mutant allele were crossed to 129SvEv mice and backcrossed to C57BL/6 for a minimum of 7 generations. For genotyping, wild‐type (WT) allele primers (ERp44‐in) were designed against lacZ insertion sites in intron 1, and a reverse primer was designed downstream of lacZ knockin, producing a 993‐bp WT DNA fragment. A second set of WT primers were designed to produce a small fragment (ERp44 WT1–F131/mERp44‐R611). The WT allele gave a product of 481 bp. A final set of primers (ERp44 WT2 mERp44–In1F446/ERp44In1R612) produced a WT allele product of 167 bp. LacZ primers were designed against upstream lacZ cDNA sequences where lacZF270/lacZR630 primers against the mutant allele resulted in a product of 361 bp. All primers sequences are shown in Table 1.
Table 1.
Primer Sequences Used in Genotyping PCR, RT‐PCR, and Real‐Time RT‐PCR
| Primer Sequences (Mouse) 5′—3′ | Product Size, bp | |
|---|---|---|
| Mouse sequence | ||
| ERp44‐in | CCTTAAGCCTGCGGTCCACA GAAGACATATGTGGCCTCGTT |
993 |
| lacZ | CTGGCGTAATAGCGAAGAGG GTTGCACCACAGATGAAACG |
361 |
| ERp44 WT1 |
TACCTTCACTTTGGCCGTTC TGCTTCTAGGGTGCAGAGGT |
481 |
| ERp44 WT2 |
AAGCCCTCTGTTGTGGCTTA TGCTTCTAGGGTGCAGAGGT |
167 |
| ERp44‐1 | GAAGTTGTCCCGGGTGTTC TGAATCAAGACTTGCTATTTCAGC |
252 |
| ERp44‐2 | GAAGTTGTCCCGGGTGTTC GATGCAACATCTGGCTGAAA |
340 |
| ERp44‐3 | GAAGTTGTCCCGGGTGTTC CGCCAGACATGGTGTACTTG |
796 |
| ERp44‐4 | GAAGTTGTCCCGGGTGTTC CTTATCAACTGCCGGGCTAC |
1011 |
| XB599 Erp44‐lacZ |
CCGTCGTTACCATGA ATC CT GACAGTATCGGCCTCAGGAAGATCG |
294 |
| α‐MHC | GGGACAGTGGTAAAAGCAA TCCCTGCGTTCCACTATCTT |
542 |
| NK2.5 | CAAGTGCTCTCCTGCTTTCC CTGTCGCTTGCACTTGTAGC |
349 |
| ANP | CCTGTGTACAGTGCGGTGTC AGCCCTCAGTTTGCTTTTCA |
273 |
| BNP | GCCAGTCTCCAGAGCAATTC AAGAGACCCAGGCAGAGTCA |
322 |
| PDI | AACGGGAGAAGCCATTGTA AGGTGTCATCCGTCAGCTCT |
156 |
| XBP‐1 | GAACAAGGAGTTAAGAACACG AGGCAACAGTGTCAGGTCC |
205 |
| Ribophorin | GCCAGGAAGTGGTGTTTGTT CCAGAGGATTGGGTTCTTCA |
158 |
| ERp72 | ATACCTTCGCCATTGCTGAC CACCTTGACTGGTCCCTTGT |
245 |
| ERO1 | AACGACCATGTCCTTTCTGG TGGTCCTGCGAATCATCATA |
256 |
| CHOP | TATCTCATCCCCAGGAAACG CTGCTCCTTCTCCTTCATGC |
258 |
| ATF6 | GATGCAGCACATGAGGCTTA CAGGAACGTGCTGAGTTAA |
211 |
| BiP/GRp78 | GCTTCGTGTCTCCTCCTGAC GGAATAGGTGGTCCCCAAGT |
259 |
| GRp94 | CCAGTTTGGTGTCGGTTTTT GGGCTCCTCAACAGTCTCT |
298 |
| HSP40 | GCCATGGGCAAGGACTACTA TTCAGGCCTTCCTCTCCATA |
221 |
| HSP70 | CGTATGTGGCCTTCACTCCT TTTCTTCTGGGGCAAATGTC |
243 |
| GAPDH | CGTCCCGTAGACAAAATGGA TCTCCATGGTGGTGAAGACA |
328 |
| ß‐Actin | CCACCATGTACCCAGGCATT AGGGTGTAAAACGCAGCTCA |
253 |
| Zebrafish sequence | ||
| ERp44 MOs | Splice MO ACACAAACAAACAACTCACTGAGGA ATG MO GATGTTGTTGAATCTTGCCAGTCTC |
25 |
| ERp44‐1 | ATCGCAACCTCAACCAACAC TTTGACGCCTCCTCAAAGAT |
247 |
| ERp44‐2 | GAAGGAGGTGGTGTCGAGAG TTCCCCTCGTCCTGGATTAT |
158 |
| ERp44‐3 | GGAGGTGGTGTCGAGAGACT TTGACTAGAGCCACACCTGCT |
220 |
| XBP‐1 | GTTCAGGTACTGGAGTCCGC CTCAGAGTCTGCAGGGCCAG |
140 |
| GAPDH | ACTTTGTCATCGTTGAAGGT TGTCAGATCCACAACAGAGA |
239 |
ANP indicates atrial natriuretic peptide; BNP, B‐type natriuretic peptide; ERp44 indicates endoplasmic reticulum resident protein 44; α‐MHC, alpha myosin heavy chain; PCR, polymerase chain reaction; PDI, protein disulfide isomerase; RT‐PCR, reverse‐transcriptase polymerase chain reaction; WT, wild type.
Aortic Banding Surgery Experiments
Aortic banding (AB) surgery was performed in 12‐week‐old male (25 to 27 g) ERp44+/− mice and WT littermates, as described previously.11 Animals were observed every 2 hours after AB surgery for 24 hours and followed for up to 8 weeks. Some mice were euthanized post‐AB surgery at 48 hours and 1 and 2 weeks (n=5 surviving animals per time point, per group). Hearts were harvested, rinsed with cold PBS, frozen, and stored at −80°C. Cardiac function by echocardiography was assessed on surviving mice at postoperative weeks 1 and 2 (n=10 for sham per group, n=7 for WT, and n=9 for ERp44+/− in AB group). Animals were euthanized, and hearts were perfusion‐fixed with 4% paraformaldehyde (PFA) and sectioned for pathological studies.
Immunoblotting, Immunoprecipitation, and Quantitative RT‐PCR
Samples were lysed in buffer containing 20 mmol/L of HEPES (pH 7.9), 50 mmol/L of NaCl, 0.5 mol/L of sucrose, 0.1 mmol/L of EDTA, 0.5% Triton X‐100, and protease inhibitors (F. Hoffmann‐La Roche AG, Basel, Swizterland) and blotted. In some cases, nuclear extracts were isolated according to the Nuclear Extraction Protocol from Life Technologies (Carlsbad, CA). Antibodies used were rabbit anti‐ERp44, IP3R, GRp78, GRp94, PDI, calnexin, calsequestrin, peIF2, cytochrome c, caspase 3, caspase 12, phospholamban (PLN), pPLN (Ser16,Thr17), pRyR S2808, pRyR S2814, and mouse monoclonal anti‐CHOP, caspase 9 (all from Cell Signaling Technology, Beverly, MA); mouse monoclonal antibodies from Abcam (Cambridge, MA) were anti‐ryanodine receptor 2 (RyR2), sarcoplasmic reticulum Ca2+ ATPase (SERCA2a), sodium‐calcium exchanger (NCX1), and dihydropyridine receptor (DHPR); and mouse monoclonal anti‐antibodies from Sigma‐Aldrich (St. Louis, MO) were nuclear factor of activated T‐cell transcription factor 2 (NFAT2), GAPDH, and β‐actin.
For immunoprecipitations (IPs), cardiac cells were isolated and treated with the membrane‐permeable cross‐linker, dithiobis[succinimidyl propionate (DSP, 2 mmol/L; Pierce Chemicals, Rockford, IL), also referred to as Lomant's reagent, in Krebs‐Ringer phosphate buffer (KRPB) for 30 minutes at room temperature (RT). After washing with KRPB, cells were lysed with 20 mmol/L of HEPES (pH 7.9), 50 mmol/L of NaCl, 0.5 mol/L of sucrose, 0.1 mmol/L of EDTA, 0.5% Triton X‐100, and protease inhibitors (Roche). Five hundred microliters of lysate were incubated with 10 μg of ERp44 antibody and Protein G Sepharose for 24 hours at 4°C. The beads were washed 5 times, and proteins were eluted at 65°C for 10 minutes in SDS‐PAGE sampling buffer. The lysate (input), IP samples, and controls were analyzed by immunolotting.
For quantitative (q)RT‐PCR, RNA was extracted with Trizol and processed as previously described.12 First‐strand cDNA was synthesized using RT with random hexamers from 0.5 μg of total RNA in a 20‐μL reaction volume according to the manufacturer's protocol (Qiagen, Hilden, Germany), then one tenth of the reverse‐transcribed product was applied to 20 μL of PCR solutions utilizing SYBR Green 2XPCR master mix (Applied Biosystems, Foster City, CA). Gene amplification was performed with a sequence detection system (Prism 7700; Applied Biosystems). To correct for variations in total RNA content and unequal RT efficiency, all gene expression quantities were normalized to the amount of β‐actin or GAPDH mRNA.
Histology, Immunochemistry, and X‐Gal Staining
Isolated mouse heart and tissue were perfused and fixed with 4% PFA/PBS. Hematoxylin‐eosin (H&E), Masson's trichrome, or wheat germ agglutinin (WGA) staining were performed at the Pathology Laboratory of Toronto General Hospital (Toronto, Ontario, Canada). For X‐gal staining, the mouse embryos and tissues were collected and fixed for 1 hour at RT in fixation solution (0.2% glutaraldehyde, 2 mmol/L of mMgCl2, and 5 mmol/L of EGTA in 0.1 mol/L of PBS; pH7.4). Tissues were washed 3 times (0.01% [v/v] sodium deoxycholate, 0.02% [V/V] nonidet P‐40, 2 mmol/L of MgCl2, and 5 mmol/L of EGTA in PBS) and then stained with lacZ X‐gal staining solution (5 mmol/L K3Fe[CN]6, 5 mmol/L K4Fe[CN]6, 1 mg/mL of 5‐bromo‐4‐chloro‐3‐indolyl‐2‐d‐galactopyranoside [X‐gal]) in washing buffer for 24 hours at 37°C in the dark. Tissues were washed 3 final times and then paraffin embedded, sectioned, and counterstained with nuclear faster red in the University Health Network Pathology Core Lab (Toronto, Ontario, Canada).
For immunofluorescence (IF) staining, cryostat sections (5 μm) were fixed (4% PFA/PBS) for 15 minutes. Sections were washed twice with ice‐cold PBS and permeabilized with ice‐cold methanol for 8 minutes. Tissues were then washed with PBS and incubated with blocking solution (4% FBS and 0.1% Triton X‐100 in PBS) for 40 minutes at RT. Primary antibody solution was added overnight at 4°C. Samples were washed 5 times with ice‐cold PBS, incubated in secondary antibody for 60 minutes at RT, washed 3 times with ice‐cold PBS, and visualized with a Zeiss spinning disk confocal microscope (Zeiss Observer Z1; Carl Zeiss AG, Jena, Germany). Antibodies to detect ERp44 (Cell Signaling Technologies), IP3R (Cell Signaling Technologies), voltage‐gated K+ channel KV4.2 (Millipore, Billerica, MA), RyR2 (Abcam), and mouse monoclonal anti‐lacZ (Developmental Studies Hybridoma Bank, Iowa City, IA) were used.
Transmission Electron Microscopy
Transmission electron microscopy (TEM) experiments were performed at the Pathology Laboratory of Mount Sinai Hospital (Toronto, Ontario, Canada). Heart tissue, neonatal cardiomyocytes from ERp44 deficient and WT mice, and whole zebrafish were fixed in 2% glutaraldehyde and 0.1 mol/L of sodium cacodylate for 3 hours at RT, rinsed in PBS buffer, and postfixed in 1% osmium tetroxide PBS buffer followed by ethanol dehydration series and propylene oxide, and finally embedded in Quetol‐Spurr resin. Sections (100 nm thick) were obtained using an RMC MT6000 ultramicrotome (Boeckeler Instruments, Inc., Tucson, AZ), followed by staining with uranyl acetate and lead citrate, and the electron microscopy (EM) image were obtained by an FEI Tecnai 20 TEM.
Mouse Neonatal and Adult Cardiomyocyte Isolation
Neonatal myocytes were isolated according to our previously published procedures.13 Briefly, each heart was isolated and cut into small pieces, washed, and then incubated with 1 mg/mL of collagenase type II (Worthington Biochemical Corporation, Lakewood Township, NJ) and 0.5 mg/mL of pancreatin (Sigma‐Aldrich) for 2 hours, deactivated with 1 mL of 10% FBS DMEM/F12 media, and then cells were isolated and preplated for 2 hours to remove the cardiac fibroblasts. Ventricular cells were plated, allowed to recover, and cultured for 5 days. Five‐day‐old cells were used for Ca2+ imaging and other cellular assays. Mouse adult cardiomyocytes were isolated as previously described.14–16
Preparation of ESCs and Generation of ERp44−/− ESCs and ESCCs
ESCs were cultured and maintained as previously described.17 Briefly, the XB599 ERp44+/− ESC line (E14Tg2a.4 129/Ola) was maintained on feeder layer from primary embryonic fibroblast in high‐glucose DMEM (Wisent, Inc., Saint‐Jean‐Baptiste, Quebec, Canada) supplemented with 2 mmol/L of l‐glutamine, 100 μg/mL of penicillin/streptomycin, 0.1 mmol/L of nonessential amino acids (Sigma‐Aldrich), 0.05 mmol/L of β‐mercaptoethanol, 15% FBS (Wisent), and 10 ng/mL of leukemia inhibitor factor (LIF). The ERp44 gene‐trapping XB599 ESC line was originally targeted at one locus with a neomycin‐targeting cassette. This cell line was successfully selected with high concentrations of G418 to obtain double KO ERp44−/− ESCs by selecting in 10 times as much G418 (3 mg/mL) as was used in the original selection at a concentration of 300 μg/mL.18 The undifferentiated XB599 ESC line was cultivated on primary culture of embryonic fibroblasts with changing G418 (3 mg/mL) DMEM medium every other day for 12 days. G418‐resistant embryonic stem clones were isolated and subjected to PCR genotyping, RT‐PCR, and Western blot to confirm the homozygous double ERp44−/− ESC. ESCs were differentiated into cardiomyocytes by a previously described method.19 Briefly, ESCs were placed on a gelatin‐coated dish and cultured in ESC culture medium without LIF for 48 hours, and then the ESC suspension was spotted in the form of a hanging drop (400 cell/20 μL) in the lid of a Petri dish for 2 days to allow further proliferation and increase their size. Embryoid bodies (EBs) were then collected and transferred into dishes with fresh media and left in suspension for 3 days. Finally, the EBs were plated onto gelatin‐coated tissue culture dishes.19–20 The medium was changed to 5% FBS DMEM, and culture was continued for 10 to 25 days (differentiation phase). The first beating clusters formed at day 7 of differentiation. Ca2+ imaging was performed on cells at 10 to 15 days of differentiation.
Lentivector Construction and Transduction of Mouse Neonatal and Adult Cardiomyocytes
Lentiviral ERp44 cDNA (Catalog No.: OHS‐1770‐9381769; Open Biosystems, Huntsville, AL) was constructed using the Gateway expression system (Invitrogen, Carlsbad, CA) and transfected into mouse neonatal and adult cardiomyocytes (MNCs), as described previously.12 Briefly, neonatal cardiomyocytes were incubated with lentiviral supernatant overnight, and fresh medium was replaced after 24 hours, followed by 2 μg/mL of puromycin‐containing media for 72 hours to select the puromycin‐resistant homogenous population of transduced cells.
Biochemical Assays
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT; assays were carried out for detection of cell viability using the MTT Assay (Sigma‐Aldrich), according to the manufacturer's instructions. Cells were cultured and treated in 96‐well plates. Approximately, 0.5 mg/mL of MTT was added to each well, and the plate was placed in the incubator (37°C) for a period of 4 hours. Untransformed MTT was removed and formazan crystals were dissolved in dimethyl sulfoxide (DMSO; 150 μL/well). Formazan absorbance was measured at 570 nm. Mitochondrial membrane potential (MMP) was detected in cells using 50 nmol/L of vital mitochondrial dye JC‐1 (Molecular Probes, Eugene, OR), and apoptosis was measured by Annexin V and propidium iodide (PI) staining, fluorescence‐activated cell sorting (FACS) analysis, and by labeling of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)‐positive nuclei.13,21 Intracellular reactive oxygen species (ROS) of cultured cells were detected utilizing CellROX deep red reagent (Invitrogen), according to the manufacturer's instructions. Cells were plated into 96‐well plates (105/well) stressed with tunicamycin (Tu; 5 μg/mL) for 24 hours. The ROS fluorogenic probe (5 μmol/L) was then added. Fluorescence was measured at 665 nm with a PerkinElmer plate reader (PerkinElmer, Waltham, MA).
Ca2+ Imaging
Isolated and cultured day 5 MNCs from ERp44‐deficient and WT neonatal pups and different stage ESCCs were treated with or without different pharmacological stimulators, and then Ca2+ transients were measured utilizing the Ca2+ indicator, Fura‐2AM (Molecular Probes), as previously described.22 Briefly, cells were incubated for 30 minutes with 1 μmol/L of Fura‐2AM and then cells were washed with DMEM/F12 culture medium. Image capture and processing were carried out with a Ca2+ imaging system (Olympus, Tokyo, Japan). Ca2+ release amplitude was measured by normalized basal fluorescence to peak fluorescence intensity (FI). Rhod‐2AM (Invitrogen) was also used to visualize mitochondrial Ca2+ according to protocols resulting in the reduction of the ester by sodium borohydride that directs Rhod‐2 fluorescence to the mitochondria.23 Briefly, cells were incubated for 60 minutes with 5 μmol/L of Rhod‐2AM culture medium and then cells were washed with culture medium. Total fluorescence was measured at a 581‐nm wavelength with a PerkinElmer plate reader.
In Vivo Cardiac Function Measurements and Cardiac Morphometry
Cardiac function was monitored by noninvasive echocardiographic imaging. Echocardiography was performed under light anesthesia with isoflurane, as previously described.12,21 Morphometric analysis was performed on cardiac sections using a quantitative image digital analysis system. Relative ventricular diameters were determined as previously reported.21 For heart and body weight or tibia length measurements, each heart was weighted after being washed and blot‐dried before being snap‐frozen in liquid nitrogen. Measurements were expressed as heart weight to body weight (HW/BW) ratios in milligrams per gram and heart weight to tibia length (HW/TL) ratios in mg/mm.
Zebrafish Studies
Zebrafish were raised in a healthy aquatic circulating environment system at St. Michael's Hospital Zebrafish facility (Toronto, Ontario, Canada). The collection of fertilized eggs was obtained through pair‐wise breeding according to the standard method previously described.24–26 Morpholino oligonucleotides (MOs) against the zebrafish ERp44 transcript were custom‐synthesized by Gene Tools (Carvalis, OR), and their sequences are shown in Table 1. MOs for translation blockers (AMOs) were based on the ERp44 mRNA sequence near the ATG start site, and the spliced blockers splice morpholino (SMO) sequences were directed complementary to exon2 and intron 2 splice junction targets. For phenotype rescue assays, in vitro transcribed capped ERp44 RNA were generated utilizing human ERp44 cDNA in the Gateway Vector pDONR 223, obtained from Open Biosystems, as a CCSB Human ORFeome entry clone (Catalog No.: OHS‐1770‐9381769). In vitro synthesis reaction of capped RNA was performed using the Message Machine T7 kit (Ambion, Austin, TX). After RNA purification, 1 nL of RNA (35 ng/μL) and/or MOs (250 μmol/L) were injected into 1‐cell stage embryos.27–28 After microinjection, in vivo imaging of zebrafish embryogenesis was performed utilizing a dissecting fluorescent microscope (Olympus) or a spinning disk confocal microscope (Carl Zeiss AG). Zebrafish Ca2+ dynamics were performed on 3 to 7 day postfertilization (dpf) fish after MO microinjection. Fish were incubated with 30 μg/mL of Fura‐2AM (Invitrogen) containing 1% DMSO in double‐distilled (dd)H20 for 30 minutes at RT. Fish were then rinsed twice with ddH20 and placed on a custom‐made imaging plate. Fish were immobilized with 3% methylcellulose containing 50 μg/mL of tricaine (Sigma‐Aldrich)29 for 30 second. Ca2+ images were recording utilizing a dedicated Ca2+ image system with Rolera cameras (Olympus).
Statistical Analysis
All results are expressed as mean±SEM. Shapiro‐Wilk's tests were carried out in all the experiments in order to confirm homogeneity and normal distribution. Student t tests were utilized for comparisons between 2 groups. One‐way ANOVA was performed for testing differences between multiple groups. Two‐way ANOVA was performed to compare the effect of multiple levels of factors. Tukey's HSD (honest significant difference) post‐hoc tests were performed after the ANOVA analysis. Kaplan‐Meier's plots were utilized to generate the survival curves. The log‐rank test was used to analyze the survival curves. A P value <0.05 was considered significant.
Results
Generation and Characterization of ERp44 KO/LacZ KI Mice
We generated ERp44‐null mutant mice through ESC aggregation utilizing the gene‐trapped ES cell line, XB599 (IGTC). The ERp44 gene was disrupted by fusion of ERp44 with a lacZ neomycin cassette insertion into intron 1 (Figure 1A and 1B). This was confirmed using Southern blotting (Figure 1C) and genomic PCR (Figure 1D). ERp44 mRNA and protein levels were undetectable in the KO mouse monitored by RT‐PCR (Figure 1E) and immunoblot (Figure 1F). The KO mice showed significant embryonic and/or perinatal lethality, demonstrated retarded development, and possessed a very low survival rate into adulthood. The very few surviving KO mice have a shortened lifespan and died around 8 to 9 months of age (Figure 1G). The surviving KO mice have 40% to 50% smaller body size, compared to WT mice (Figure 1H and 1I). As shown in Table 2, the highest mortality of KO embryos was observed at E9.5 to E12.5. LacZ staining of tissue sections in a 6‐month‐old KO mouse showed that LacZ expression was driven in various tissues, including the kidney, spleen, gastrointestinal tract, liver, and heart (Figure 2).
Figure 1.

Generation and characterization of ERp44 knockout/lacZ knock‐in mice. A, ERp44 genomic DNA structure with a gene‐trapping vector containing splice acceptor (SA)‐lacZ neo inserted into the ERp44 gene intron 1. Small arrows represent PCR primers. B, Structure of gene‐trapping vector integration site in ERp44 locus. Gene‐trapping vector pGT1dTMpfs containing SA and lacZ/neomycin resistance gene was inserted into ERp44 genomic DNA locus. Forward primer “a” was designed against ERp44 exon 1 and reverse primer “c” against upstream lacZ cDNA. C, Southern blot analyses of genomic DNA digested with‐EcoRV with a lacZ probe. Analysis revealed a single 9.3‐kb band in lacZ+ mice. D, Genotyping by mouse genomic PCR. lacZ primers produced a 361‐bp DNA fragment. WT primers were designed outside lacZ insertion sites in intron 1 labeled “c” and “d” in (A) and produced a 995‐bp DNA fragment. E, RT‐PCR analysis for ERp44 and β‐actin. ERp44 primers produced a 251‐bp fragment. β‐actin primers produced a 325‐bp fragment. F, Immunoblots of mouse neonatal cardiomyocytes (MNCs) from WT (+/+) and KO (−/−) mice. G, Kaplan‐Meier's survival curves in ERp44+/+ (n=280), ERp44+/−(n=200), and ERp44−/− (n=10) mice. Log‐rank testing shows significantly decreased survival in both ERp44+/− and ERp44−/− mice, compared to ERp44+/+ mice (**P<0.01). H, Body weight of ERp44+/+, ERp44+/−, and ERRp44−/− littermates (n=13, 9, and 5 for 3 weeks, 3 months, and 6 months group, respectively). I, Micrograph of ERp44+/+ and ERRp44−/− mice. ERp44 indicates endoplasmic reticulum resident protein 44; KO, knockout; RT‐PCR, real time‐polymerase chain reaction; WT, wild type.
Table 2.
Genotyping PCR Results From ERp44+/− Breeder Offspring
| Age | Total (n) | WT (%) | H&E (%) | KO (%) | Dead KO (%) |
|---|---|---|---|---|---|
| E9.5 | 103 | 25.2 | 58.3 | 6.8 | 9.7 |
| E 10.5 | 131 | 27.5 | 58.8 | 3.8 | 9.9 |
| E12.5 | 139 | 27.3 | 57.6 | 3.6 | 11.5 |
| E13.5 | 154 | 29.9 | 64.9 | 3.9 | 1.3 |
| E19.5 | 246 | 29.7 | 64.2 | 4.9 | 1.2 |
| 3 weeks | 730 | 28.5 | 63.8 | 1.8 | 1.8 |
Embryo age, total number of dissected embryos, and the ERp44 genotyping results are shown as indicated. ERp44 indicates endoplasmic reticulum resident protein 44; H&E, hematoxylin‐eosin; KO, knockout; PCR, polymerase chain reaction; WT wild type.
Figure 2.
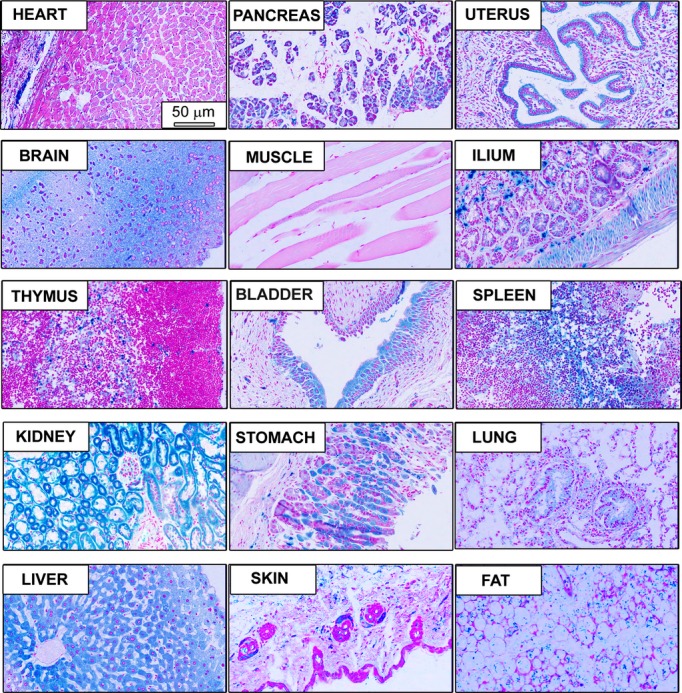
LacZ expression in tissues from ERp44 KO mice. Whole‐mount lacZ staining in fixed tissue sections obtained from 6‐month‐old ERp44 KO mice. Scale bar is 50 μm. ERp44 indicates endoplasmic reticulum resident protein 44; KO, knockout.
We analyzed the cardiac phenotypes in surviving KO mice. ERp44 was detected in WT cardiac sections, but not in KO heart, using immunohistochemistry (IHC; Figure 3A). Heart sections stained for H&E revealed slightly abnormal cardiac morphology in KO mice, compared to WT, with evidence of mild fibrosis and increased cell size in KO mice (Figure 3B). Heart sections were stained with WGA (Figure 3C and 3D) to visualize cell dimension, which showed greater cardiac cell area in KO versus control (Figure 3E; n=150/genotype; P<0.05). The HW/BW and HW/TL ratio of the KO was significantly higher versus the WT (Figure 3F; n=5/genotype; P<0.05), suggesting cardiac hypertrophy in the KO mice. EM of cardiac sections revealed significant myocyte swelling, mitochondrial membrane rupture, and cardiac myofibril disorganization in KO mice (Figure 3G). M‐mode echocardiography (Figure 3H) showed cardiac dysfunction in the few surviving 6‐month‐old KO mice, with increased left ventricular end‐diastolic diameter (LVEDD) and reduced fractional shortening (FS; Figure 3I and 3J). Real‐time qRT‐PCR showed that hypertrophic markers atrial natriuretic factor (ANF) and B‐type natriuretic factor (BNF) were significantly increased in KO mouse hearts (Figure 3K).
Figure 3.

Cardiac phenotype in adult ERp44−/− mice. A, Immunohistochemistry for ERp44 in hearts of ERp44+/+ and ERp44−/− mice and (B) H&E‐stained heart sections. C and D, Wheat germ agglutinin (WGA) staining in ERp44+/+ and ERp44−/− hearts. E, Quantification of cell size using ImageJ (National Institutes of Health, Bethesda, MD) analysis of WGA‐stained hearts (n=150/genotype; P<0.05). F, Heart mass to tibia length (HW/TL) and body weight (HW/BW) (n=5/genotype; *P<0.05). G, EM analysis reveals swelling, structural disruption, and membrane rupture in the mitochondria (arrows) in late‐stage ERp44−/− mice. H, Cardiac function assessed in mice using M‐mode echocardiography. I, LV internal diameters in diastole (LVIDd) and (J) fractional shortening (FS) measurements. K, Expression of ANF and BNF in hearts were quantified by qRT‐PCR (n=3/genotype; *P<0.05). (L) Quantification of mitochondrial size using ImageJ analysis of EM heart images. Mean values±SEM were determined (n=5/genotype; *P<0.05). ANF indicates atrial natriuretic factor; BNF, B‐type natriuretic factor; EM, electron microscopy; ERp44, endoplasmic reticulum resident protein 44; LV, left ventricle; LVEDD, LV end‐diastolic diameter; qRT‐PCR, quantitative real‐time polymerase chain reaction.
ERp44‐lacZ Expression During Heart Development
The lacZ KI reporter gene allowed us to examine ERp44 expression patterns in the hearts of ERp44+/LacZ. We determined ERp44‐lacZ expression by lacZ staining of whole‐mount embryos and tissue sections from WT and ERp44+/LacZ mice. Whole‐mount staining showed lacZ expression in the earliest stages (7.5 days postconception [dpc]) of the developing embryo that remained high through 12.5 dpc (Figure 4A). Immunoblot analysis of isolated hearts from WT embryos showed ERp44 highly expressed in early development and levels slowly declining as the hearts developed (Figure 4B). LacZ staining showed high expression of lacZ in the E13 and P1 hearts, with expression into adulthood (Figure 4C). Sectioning of these hearts revealed that lacZ and endogenous ERp44 have a very specific expression pattern during maturation in the heart (Figure 4D). Double IF staining with anti‐lacZ/ERp44 antibodies reveals the higher ERp44/lacZ expression in epicardium, compared to myocardium, in P7 old mice (Figure 4E). In the 12‐week‐old mice, ERp44 fluorescence intensity was significantly higher in the epicardium, compared to myocardium (Figure 3F; n=3/genotype; P<0.05). ERp44 FI was significantly lower in the epicardium of 12‐week‐old ERp44 heterozygous mice, compared to WT mice (Figure 4F; n=3/genotype; P<0.05). Endogenous ERp44 in cardiac sections showed very strong IHC staining in E13 embryo (Figure 4G and 4H). After E13, high expression of ERp44 in the outer epicardial region and the epicardial/myocardial border was observed (Figure 4I). Potassium voltage‐gated channel subfamily D member 2 (KV4.2) has been shown to be highly expressed in the epicardial myocardial region of the adult heart; hence, we stained the adult heart with KV4.2 antibody to observe its pattern of expression, compared to ERp44. KV4.2 was expressed in the epicardium myocardium region of the adult heart, a very similar expression pattern to ERp44 in the adult heart (Figure 4J and 4K). Based on the known interaction between ERp44 and IP3R, adult hearts were stained with IP3R; and given the homology and functional overlap between IP3R and RyR2, we also assessed RyR2 confocal localization. The result shows that although IP3R staining, and RyR2 to a lesser extent, showed a very slight enhanced expression in the outer epicardial region, they did not have the same transmural expression gradient as ERp44 (Figure 4K). At 12.5 dpc, there were few dead KO embryos (3 of 5) that showed a phenotype similar to a condition called encephalocele (Figure 4L).
Figure 4.

ERp44 expression in the mouse embryo and developing heart. A, Whole‐mount lacZ staining of 7.5 to 12.5 dpc ERp44+/+ and ERp44+/− embryos. B, Immunoblot analysis of E9.5 to 12‐week‐old mouse heart tissue from ERp44+/+ and ERp44+/− mice using anti‐ERp44 or GAPDH antibody (n=3/genotype; *P<0.05). C, Whole‐mount lacZ staining of isolated hearts of ERp44+/− mice starting at E13 to 12 weeks of age. D, lacZ staining and ERp44 immunofluorescence (IF) staining of cryostat sections from ERp44+/− mouse heart at E13 to 12 weeks. E, lacZ and ERp44 double IF staining of hearts from ERp44+/− at postnatal day 7 (P7). F, Quantification of ERp44 IF intensity from epicardium and myocardium ERp44+/+ and ERp44+/− mouse heart at 12 weeks (n=3/genotype; **P<0.01). G, Immunohistochemistry staining of E13 (E and F) and 3‐week‐old (G) mouse heart using anti‐ERp44. H and I, Transmural expression of ERp44 in adult WT heart. J, IF confocal images of adult heart section stained with ERp44 (green), phalloidin (red; F‐actin dye), CD31 (white), and Hoechst (blue). K, Adult heart stained with ERp44 (red), KV4.2 (green), IP3R (green), and RyR2 (green), and Hoechst (blue). L, Whole‐mount lacZ staining 12.5‐dpc ERp44−/− embryo showing encephalocele. ERp44 indicates endoplasmic reticulum resident protein 44; IP3R, inositol trisphosphate receptor; KV4.2, potassium voltage‐gated channel subfamily D member 2; RyR2, ryanodine receptor 2; WT, wild type.
Ca2+ Homeostasis in ERp44‐Deficient Models
To explore the function of endogenous ERp44 in regulating Ca2+ handling and ER stress responses in cardiac cells, we characterized MNCs isolated from ERp44−/−, ERp44+/− and ERp44+/+ neonatal mice. ERp44 gene and protein expression reduced by ≈65% in the ERp44+/− and were completely absent in ERp44−/− myocytes (Figure 5A and 5B). Transfecting WT cardiomyocytes with lentivirus‐HIS‐tagged ERp44, followed by IP of ERp44 with an anti‐ERp44 antibody showed a clear coprecipitation of IP3R with ERp44 and was confirmed using the anti‐His antibody (results not shown). To investigate the in vivo ERp44 and IP3R interaction in cardiac cells, we isolated P1 and 3‐month‐old cardiomyocytes and treated them with the membrane‐permeable crosslinker, DSP (2 mmol/L), followed by an IP with anti‐ERp44 and control rabbit IgG, consistent with earlier conditions used by Mikoshiba's group in similar experiments using Hela cell lysates.8 In both the P1 neonatal and 3‐month‐old adult tissues, we were able to coprecipitate IP3R from ERp44‐conjugated beads (Figure 5C).
Figure 5.

Immunoblot analysis of ERp44‐deficient MNCs. A and B, qRT‐PCR and immunoblot analysis of ERp44 expression in ERp44+/+, ERp44+/−, and ERp44−/− MNCs (n=6/genotype). C, P1 and 3‐month cardiomyocytes were isolated and treated with 2 mmol/L of DPS in Krebs‐Ringer phosphate buffer (KRPB) for 30 minutes and washed 3 times with KRPB and subjected to IP with anti‐ERp44 and control set of beads using rabbit IgG with beads alone, and samples analyzed by immunoblot with the anti‐IP3R, and anti‐ERp44 antibodies. Input controls were included. D, Immunoblot analysis of ER proteins, Ca2+ channels handling proteins and p‐RyR S2808, p‐PLN (Ser16/Thr17) in the ERp44+/+, +/−, and −/− mouse neonatal P1 heart tissues. Nuclear NFAT2 levels were assessed using enriched nuclear proteins (n=3/genotype; *P<0.05). Experiments were repeated 3 times independently. DHPR indicates dihydropyridine receptor; DSP, dithiobis[succinimidyl propionate]; ERp44, endoplasmic reticulum resident protein 44; IP, immunoprecipitation; IP3R, inositol trisphosphate receptor; MNCs, mouse neonatal cardiomyocytes; NCX, sodium‐calcium exchanger; NFAT2, nuclear factor of activated T‐cell transcription factor 2; PDI, protein disulfide isomerase; PLN, phospholamban; qRT‐PCR, quantitative real‐time polymerase chain reaction; RyR, ryanodine receptor; SERCA2a, sarcoplasmic reticulum Ca2+ ATPase; WB, Western blot.
ERp44 has been implicated in Ca2+ dynamics; hence, immunoblots were performed to determine the levels of Ca2+ handling proteins in ERp44+/+, ERp44+/−, and ERp44−/− neonatal mice and 6‐month‐old adult mice. These studies showed that the protein levels of the major cardiac Ca2+ handling proteins, namely, IP3R, RyR2, SERCA2, NCX, and PLN, were not significantly affected, whereas DHPR expression was increased in ERp44 KO neonatal mice (n=3/genotype; P<0.05). In the 6‐month‐old adult mice, RyR2 and SERCA2 protein levels were significantly decreased, whereas DHPR levels remained elevated (Figure 7B). Nuclear NFAT2 levels were elevated in ERp44 KO neonatal and 6‐month‐old adult mice (Figures 5D and 7B). Levels of phosphorylated PLN and RyR were also elevated in the mutant models (Figures 5D and 7B).
Figure 7.

Activation of cellular responses in the ERp44‐deficient models. A, qRT‐PCR detecting expression of ER stress genes in adult hearts of ERp44−/− and ERp44+/+ mice (n=3/genotype). B, Immunoblot analysis of ER proteins, Ca2+ handling proteins, apoptosis markers, cytochrome c, and nuclear NFAT2 in heart from ERp44+/+, +/−, and −/− mice (n=3/genotype). C, MTT viability assay (n=6/genotype). D, MMP assay (n=6/genotype) and (E) mkitochondrial Ca2+ levels (Rhod‐2AM) were detected in MNCs (n=6/genotype). F, ROS were assessed after treatment with Tu (5 μg/mL) for 24 hours using the ROS assay kit (n=6/genotype; *P<0.05; **P<0.01). Experiments were repeated 3 times independently. ATF indicates activating transcription factor; CHOP, CCAAT‐enhancer‐binding protein homology protein; DHPR, dihydropyridine receptor; ERo, endoplasmic reticulum membrane oxidoreductase; ERp44, endoplasmic reticulum resident protein 44; GRp, glucose‐regulated protein; HSP, heat shock protein; IP3R, inositol trisphosphate receptor; MMP, mitochondrial membrane potential; MNCs, mouse neonatal cardiomyocytes; MTT, 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide; NCX, sodium‐calcium exchanger; NFAT2, nuclear factor of activated T‐cell transcription factor 2; PDI, protein disulfide isomerase; PLN, phospholamban; qRT‐PCR, quantitative real‐time polymerase chain reaction; ROS, reactive oxygen species; RyR, ryanodine receptor; SERCA2a, sarcoplasmic reticulum Ca2+ ATPase; XBP, X‐box protein.
We next assessed Ca2+ transients in ERp44+/+, ERp44+/−, and ERp44−/− MNCs using the Ca2+ indicator, Fura‐2AM (1 μg/mL; Figure 6A). As shown in Figure 6A and 6B, decreased spontaneous Ca2+ frequency and enhanced Ca2+ transient amplitude were observed in the ERp44+/− and ERp44−/− MNCs, compared to the control cells (n=25/genotype; P<0.05), indicative of a greater Ca2+ store and/or enhanced Ca2+ release from ER/SR. Given previous reports that ERp44 binding to IP3R can inhibit IP3R‐dependent Ca2+ signaling, we treated cells with histamine (an IP3R agonist; 1 mmol/L) or 2‐aminoethoxydiphenyl borate (2‐APB; an IP3R inhibitor; 10 μmol/L; Figure 6A and 6B). The normalized FI of Fura2AM measured at 380 nm in the untreated ERp44+/+, ERp44+/−, and ERp44−/− MNCs were 267±16, 620±40, and 662±51 AU, respectively (n=25/genotype; P<0.01). The FI in the histamine‐treated ERp44+/+, ERp44+/−, and ERp44−/− MNCs were 430±18, 875±37, and 887±36 AU, respectively (Figure 6B). Thus, Ca2+ amplitude in the histamine‐treated ERp44+/− and ERp44−/− MNCs increased significantly, compared to the ERp44+/+ cells (n=25/genotype; P<0.05; Figure 6B). We further observed the FI in the 2‐APB‐treated ERp44+/+, ERp44+/−, and ERp44−/− MNCs were 60±5, 380±23, and 434±24 AU, respectively (Figure 6B). These results showed that the Ca2+ amplitude FI in the 2‐APB‐treated ERp44+/− and ERp44−/− cells was significantly higher than ERp44+/+ cells (Figure 6B; n=25/genotype; P<0.01). Two‐way ANOVA and Tukey's post‐hoc tests were performed (P<0.01). Finally, overexpressing ERp44 in ERp44‐deficient cells by infection of ERp44 lentivirus was able to restore the Ca2+ transient amplitude and frequencies back to WT levels (Figure 6C through 6E).
Figure 6.

Enhanced Ca2+ release in ERp44‐deficient MNCs. A, Ca2+ transients were obtained with the Ca2+ indicator, Fura‐2AM. B, Quantification of Ca2+ transient parameters: amplitude fluorescent intensity (Fi; n=25/genotype; 2‐way ANOVA analysis and Tukey's post‐hoc tests were performed; *P<0.05; **P<0.01 for comparing the ERp44+/+ vs ERp44+/−; #P<0.05; ##P<0.01 for comparing untreated, histamine treated, and 2‐APB‐treated groups. C, Immunoblots of ERp44 protein levels in control cells and after ERp44 lentiviral infection (n=6/group: control vs. lentiviral group experiments repeated 3 times; **P<0.01). D, Representative Ca2+ transient mock‐transduced cells and ERp44 viral‐transduced cells. E, Ca2+ amplitude measured in control and ERp44 viral‐infected MNCs (n=25/genotyping; *P<0.05). Experiments were repeated 6 times independently. 2‐APB indicates 2‐aminoethoxydiphenyl borate; ERp44, endoplasmic reticulum resident protein 44; MNCs, mouse neonatal cardiomyocytes.
ER Stress‐Induced Apoptosis in ERp44‐Deficient Models
Given the Ca2+ transient defects and that Ca2+ released from the ER has a profound effect on ER stress and cell viability,30 we determined ER stress levels, cell viability, mitochondrial health, and apoptosis in ERp44‐deficient cardiac cells. In ERp44−/− 6 months old hearts there was a significant increase in gene expression for nearly all the ER stress response members, including PDI, glucose‐regulated protein 78 kDa (GRP78), CHOP, ERp72, activating transcription factor 6 (ATF6), ER membrane oxidoreductase 1 (ERO1), heat shock protein 70 (HSP70), and X‐box‐binding protein 1 (XBP‐1), when compared to WT mice (Figure 7A; n=3/genotype; P<0.05). The ER stress response was also observed in the ERp44‐deficient mouse neonatal cardiac cells (Figure 5D). Moreover, the ER stress response proteins, GRp78, GRp94, peIF2, and CHOP, and apoptotic markers cytochrome c and caspase‐3 protein expression levels were all increased in hearts of 6‐month‐old ER44‐deficient mice (Figure 7B), whereas other ER Ca2+‐binding proteins calnexin and calsequestrin were not affected.
Cell viability measured by an MTT assay was decreased significantly in ERp44‐deficient MNCs (Figure 7C), compared to WT cells. MMP was assessed using MitoTracker and JC‐1 fluorescence assays and showed that ERp44‐deficient cells had significantly decreased fluorescence indicative of mitochondrial dysfunction (Figure 7D). Furthermore, Rhod‐2AM used to measure mitochondrial [Ca2+] showed an increased level of Rhod‐2AM fluorescence in ERp44‐deficient cells versus control (Figure 7E), indicating higher mitochondrial [Ca2+] in the ERp44‐deficient cells. Finally, we observed elevated ROS levels in ERp44‐deficient cells, compared to WT cells (Figure 7F).
ESCCs can mimic cardiac development and closely recapitulate embryonic development.31–32 To overcome the fact that KO mice have a very high embryonic lethality, we also studied the cardiac phenotype of ERp44‐deficient ESCCs. Using high G418 selection of the ERp44 gene‐targeted ES cell line,18 we established ERp44−/− ESCs as determined by genotyping PCR, RT‐PCR, Western blot, and lacZ/fluorescein di‐V‐galactoside FACS analysis (results not shown). We generated ESSCs from these cells and confirmed that these ESCCs expressed cardiac marker genes, including Nkx2.5, alpha‐myosin heavy chain (α‐MHC), and cardiac‐specific α‐actin by RT‐PCR on day 25 ESCCs, and most cells were cardiac troponin T positive (results not shown). ERp44‐null cells have lower levels of both α‐MHC and Nkx2.5, suggesting that alteration of cardiac cell development, cell viability was significantly decreased, and the MMP was decreased indicating mitochondrial dysfunction (results not shown). Similarly to MNCs, ERp44‐null ESCCs showed higher [Ca2+] transient amplitudes, elevated ROS, and showed enhanced sensitivity to Tu (5 μg/mL)‐induced ER stress apoptosis that could also be rescued by infection with lentiviral ERp44 expression in KO ESCCs (results not shown). Altogether, our ESCC model reproduced the mouse neonatal work with great fidelity.
Cardiac Phenotypes of the AB ERp44+/− Model
Adult ERp44+/− mice appeared generally normal, compared to ERp44−/− mice. We subjected these mice to transverse aortic constriction to induce left ventricular pressure overload and cardiac hypertrophy. Animals were banded and followed for up to 8 weeks postsurgery. As shown in Figure 8A, survival of WT and ERp44+/− mice was comparable in sham animals, but decreased survival was observed in ERp44+/− mice after banding. This was particularly evident within 24 hours after AB surgery. Survival curve analysis was performed by a log‐rank test showing significantly decreased survival in the ERp44+/− AB group, compared to ERp44+/+(P=0.0011). AB resulted in cardiac hypertrophy in both WT and ERp44+/− mice within 2 weeks postsurgery (Figure 8B through 8D). Of the surviving mice, qRT‐PCR showed that ANF was significantly up‐regulated in the ERp44+/− heart (Figure 8E). Histological analysis of heart sections along with Masson's trichrome staining, H&E staining, and WGA staining showed significant cardiac cell hypertrophy and increased collagen in ERp44+/− AB mice, compared to WT mice 2 weeks postsurgery (Figure 8F and 8G). EM examination revealed an aberrant myocardial ultrastructure in ERp44+/− myocardium after 2 weeks postsurgery, characterized by mitochondrial swelling, degeneration of the mitochondrial matrix, poorly developed cristae and electron‐dense matrix deposits, and crystalline and amorphous inclusions and vacuolization, together with cardiac myofibril disorganization, none of which were apparent in the myocardium of WT AB cardiomyocytes (Figure 8H). After 2 weeks, the thickness of the interventricular septum diameter (IVSd), LV posterior wall diameter (PWd), the LV end‐systolic diameter (LVESD), and LVEDD were all increased significantly in the ERp44+/− heart, whereas FS and HR were significantly decreased, compared to WT mice (P<0.05; Table 3). AB further led to substantially increased expression of ER stress response genes, including PDI, ERo1, CHOP, ATF6, GRp78, HSP70, and spliced XBP‐1 in ERp44+/− mice, compared to WT control, within 2 weeks postsurgery (results not shown). TUNEL staining of heart sections showed a significant increase of TUNEL‐positive cells in ERp44+/−, compared to WT mice (Figure 8I). Immunoblots for cleaved caspase‐12, ‐9, and ‐3 showed significantly increased levels in ERp44+/− banded hearts, compared to WT mice (Figure 8J).
Figure 8.

Cardiac phenotypes of ERp44+/− AB mice. A, Kaplan‐Meier's survival curves analysis for sham and banded ERp44+/+ and ERp44+/− mice. Log‐rank tests show that survival levels in aortic‐banded ERp44+/+ were significantly higher than ERp44+/− mice (**P<0.01). Survival level in both sham groups were significantly lower than both aortic banded groups (##P<0.01). B, Heart weight to body weight (HW/BW). C, Heart weight to tibia length (HW/TL) of ERp44+/− (n=7) and control (n=10; **P<0.01) for comparing the ERp44+/+ and ERp44+/− groups (#P<0.05; ##P<0.01 for comparing the sham and AB groups by 2‐way ANOVA analysis and Tukey's post‐hoc test. D, Cardiac cell area was determined using ImageJ (National Institutes of Health, Bethesda, MD) analysis (n=100/genotype). E, Expression of ANF in hearts were quantified by qRT‐PCR for sham and banded ERp44+/− and +/+ mice. (n=3/genotype; #P<0.05, for comparing the sham and AB groups by 2‐way ANOVA analysis and Tukey's post‐hoc test; **P<0.01). F, Cardiac cell size and collagen expression was measured in ERp44+/− and WT AB mice 2 weeks after AB by Masson's trichrome and (G) WGA staining (n=3/genotype). H, EM analysis of ERp44+/+ and ERp44+/− hearts 2 week after AB surgery. I, TUNEL staining of heart tissue 1 week after AB in ERp44+/− and control mice. J, Immunoblots for the apoptotic markers: Cleaved caspase‐12, ‐9, and ‐3 are shown. Experiments were repeated 3 times independently (n=3/genotype; *P<0.05; **P<0.01). AB indicates aortic banding; ANF, atrial natriuretic factor; EM, electron microscopy; ERp44, endoplasmic reticulum resident protein 44; qRT‐PCR, quantitative real‐time polymerase chain reaction; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling; WGA, wheat germ agglutinin; WT, wild‐type.
Table 3.
Echocardiographic Parameters in WT and ERp44+/− Mice After 2‐Week Aortic Banding
| LVEDD (mm) | LVESD (mm) | FS (%) | IVS (mm) | PW (mm) | HR (min) | |
|---|---|---|---|---|---|---|
| +/+ (Baseline) (n=10) | 3.33±0.02 | 1.62±0.02 | 51.4±0.5 | 0.62±0.01 | 0.61±0.01 | 608.1±9.9 |
| +/− (Baseline) (n=10) | 3.31±0.03 | 1.57±0.02* | 52.4±0.4 | 0.62±0.01 | 0.61±0.01 | 622.2±6.7* |
| +/+ (Sham) (n=7) | 3.31±0.03 | 1.59±0.02 | 51.8±0.4 | 0.62±0.01 | 0.62±0.13 | 614.9±11.2 |
| +/− (Sham) (n=7) | 3.45±0.03** | 1.79±0.02** | 48.5±2.6 | 0.66±0.02** | 0.67±0.02^ | 588.2±9.0** |
| +/+ (AB) (n=9) | 4.01±0.13 | 2.95±0.16 | 32.0±2.2 | 0.76±0.02 | 0.75±0.01 | 569.9±11.2 |
| +/− (AB) (n=9) | 4.36±0.10# | 3.27±0.12# | 24.9±1.3# | 0.79±0.01# | 0.78±0.01# | 534.4±6.7# |
Noninvasive echocardiography assessment was performed on anesthetized mice utilizing isoflurane. Data displayed as mean±SE. One‐way ANOVA analysis was performed followed by Tukey's honest significant difference test. bpm indicates heart beat per minute; FS, fractional shortening, calculated as (LVEDD−LVESD)/LVEDD×100; HR, heart rate; IVSTD, interventricular septal thickness in diastole; LVEDD, left ventricular end‐diastolic dimension; LVESD, left ventricular‐systolic dimension; PW, posterior wall thickness (left ventricular); WT, wild type.
*P<0.05 baseline group ERp44+/+ versus ERp44+/−; **P<0.05 sham group ERp44+/+ versus ERp44+/−; #P<0.05 AB group ERp44+/+ versus ERp44+/−; ^P<0.05 sham group ERp44+/+ versus ERp44+/−.
Cardiac and Embryo Development Phenotypes of Zebrafish zERp44 Morphant
The zebrafish ERp44 (zERp44) protein has high homology (78%) to the human ERp44 protein sequence and is abundantly expressed during zebrafish embryonic development, as demonstrated by RNA in situ hybridization studies (http://zfin.org). We observed that zERp44 is abundantly expressed during embryonic development by RT‐PCR (Figure 9A) and immunoblotting (Figure 9B), although expression is reduced in the more mature fish (1 month old). We knocked down (KD) zERp44 expression by designing gene‐specific SMO and AMO (Figure 9C and 9D). After MO injection, 4‐dpf embryos were collected and immunoblot analyses showed significantly reduced (70% to 80%) zERp44 levels in the zERp44 SMO morphant, compared to control MO (CMO). The zERp44 AMO morphant showed near complete disruption of ERp44 expression (Figure 9C). Consistent with the severe embryonic lethality observed in ERp44 KO mice, we observed that the vast majority of AMO zebrafish embryos died within 1 to 3 days after injection. We thus focused our studies using the SMO morphant. In the embryos microinjected with SMO, there was a marked decrease in thickness of the ventricular and atrial wall in morphants, in particular the atrial wall, which is almost made out of a single layer of cardiomyocytes with fewer cells in the endocardium cushions area (Figure 9E). We also observed consistently enlarged heart chambers in hearts isolated from SMO‐injected embryos at 7 dpf, compared to CMO‐injected fish (Figure 9F). Ninety‐five percent of the embryos showed consistently retarded embryonic development with a “curved” body at 24 hours postfertilization (hpf), 48 hpf, and 4 dpf, compared to CMO‐injected embryos (Figure 9I). In hearts of SMO‐injected cmlc2:EGFP33 embryos, we observed an enlarged heart atrial chamber and abnormal looping at 48 and 96 hpf (Figure 9J). Pericardial edema, slowed blood circulation, and blood retention was frequently observed in SMO‐injected embryos (Figure 9I). Phenotype‐rescue experiments by coinjecting ERp44 mRNA together with the SMO resulted in the rescue of the heart and body developmental phenotypes, as well as Ca2+ transient (Figure 9O and 9P) and ultrastructural defects in zERp44 morphants (Figure 9G).
Figure 9.

Phenotype of zebrafish zERp44 morphants. A, RT‐PCR for ERp44 and (B) ERp44 immunoblot analysis were performed on zebrafish embryos from 0 hours postfertilization (hpf) to 2‐month‐old zebrafish. C, Immunoblots of ERp44, IP3R, and β‐actin protein levels at 96 hpf. D, Strategy for zebrafish zERp44 knockdown. ATG blocking morpholino (AMO) was designed complementary to the translation start site, and splice blocking morpholino (SMO) was complementary to exon 2 and intron 2 splice junction targets. E, Toluidine blue staining in the SMO and CMO heart of 96‐hpf zebrafish showing ventricular (V) and atrial (A) walls in the SMO morphants. F, Fluorescent image (red) of isolated hearts from 7 days postfetilization (dpf) MO‐injected embryos. G, Electron microscopy of hearts from 4‐dpf zERp44 SMO morphants, compared to CMO, and RNA+SMO‐injected zebrafish. H, Cardiomyocytes were isolated from SMO‐ and CMO‐injected 3‐dpf zebrafish and treated with 5 μg/mL of Tu for 4 hours, then stained with Annexin V FITC/propidium iodide (PI) to assess the number of apoptotic cells. The results revealed a ≈2‐fold increase in the number of apoptotic cells (Annexin V+/PI+) in cardiomyocytes from KD zebrafish, compared to control zebrafish (12.7±2.6% vs. 6.1±1.2%, respectively; n=6; P<0.05). Results shown are representative of more than 3 independent experiments. I, Whole‐body images showing body size and tail morphology in morphant fish at 24 to 96 hpf. J, Fluorescent image (green) of MOs injected cmcl2:EGFP embryos, compared to CMO controls. K, qRT‐PCR of ER stress marker splice XBP‐1 transcript (n=6/group). L, Representative tracings of Ca2+ transients from hearts of MO zebrafish with/without histamine (2 mmol/L) or 2‐APB (20 μmol/L; n=30/group). M and N, Quantification of Ca2+ transient data with or without histamine or 2‐APB treated (n=30/group). Two‐way ANOVAs were performed and Tukey's post‐hoc tests were used (**P<0.01 for comparing the CMO vs. SMO groups; ##P<0.01 for comparing the untreated and treated groups). O, Representative tracings of Ca2+ transients from hearts of 4‐dpf KD zebrafish with or without injected ERp44RNA1 (35 ng/mL) or RNA2 (70 ng/mL). P, Quantification of Ca2+ release amplitude in fish with or without comicroinjection of human ERp44mRNA with MOs (n=30/group; **P<0.01). All experiments were repeated at least 3 times independently. 2‐APB indicates 2‐aminoethoxydiphenyl borate; CMO, control MO; EGFP, enhanced green fluorescent protein; ERp44, endoplasmic reticulum resident protein 44; FITC, fluorescein isothiocyanate; IP3R, inositol trisphosphate receptor; KD, knocked down; MOs, morpholino oligonucleotides; qRT‐PCR, quantitative real‐time polymerase chain reaction; Tu, tunicamycin; zERp44, zebrafish ERp44.
Discussion
ERp44 Plays a Crucial Role in Cardiac and Embryonic Development
Disruption of the mouse ERp44 gene results in embryonic lethality as early as embryonic day (E)9.5. ERp44 KO embryos were smaller and had clear neural tube defects at E12.5. KD of ERp44 in the zebrafish also led to embryonic lethality within 1 to 3 dpc. ERp44 KD in the zebrafish revealed various cardiac defects, including failure of looping, thin ventricular wall, decreased endocardial cushion, and abnormal atrioventricular canal. These abnormalities suggest that ERp44 is critical for embryogenesis and is essential for normal heart development. ERp44 is expressed throughout embryo development with high expression in the developing heart. Increased expression was observed in the outflow tract, atrioventricular cushion, developing valves, and upper part of the interventricular septum in the E13 mouse embryo.
The embryonic lethality observed in the ERp44‐deficient mouse and zebrafish model may be a result of the interaction between IP3R and ERp44. ERp44 has been shown to inhibit Ca2+ release through binding and inactivating IP3R1.8 IP3Rs play a key role in early embryonic development, such as early body‐axis formation, cell‐cycle progression, and Ca2+ signaling in cardiac development and mediate responses to cellular stress.34–35 IP3Rs are expressed in the early stage embryos at E5.5 and are clearly evident within the developing heart tube at E8.5, which reveals that the functional IP3‐gated Ca2+ release channels periodic Ca2+.36 The highest embryonic lethality in the ERp44 KO mouse model was observed at E12.5. The lethality at this stage is most likely a result of an inadequate conduction system; hence, lack of ERp44 to regulate IP3R during cardiogenesis could have led to Ca2+ dysregulation and caused embryonic lethality.37 Interestingly, the IP3R1 and IP3R2 double KO mouse model showed embryonic lethality at E11.5 with cardiac defects evident at E9.75.38 The cardiac defect in this KO model was similar to what we observed in the zERp44 SMO morphants at 4 dpf, which included thinning of the ventricular wall.
Other ER proteins have also been shown to play an important role in embryogenesis. For instance, calreticulin (CRT) KO mice are embryonic lethal with severe defects in heart development and function resulting in death by E15.39 CRT regulates Ca2+ homeostasis and plays an important part in cardiac development as a component of the Ca2+/calcineurin/NFAT/GATA‐4 transcription pathway.39 Similar to ERp44, CRT is also highly expressed in the developing heart and similarly expression declines in the mature heart.39
We also observed ≈60% of the KO mice at E12.5 displayed encephalocele, a condition caused by improper neural tube closure. As observed in the embryo whole‐mount staining, ERp44 is highly expressed in the developing brain and seems to be highly expressed during neurulation, a complex process involving interactions between neuroepithelial cells and the mesenchyme. Defects in neural tube closure have been observed in various gene KOs, in which the protein plays a role in cell migration, and Ca2+ signaling.40 Interestingly, the brain and the heart develop concurrently in the fetus. The development of both organs intersect at several levels, such as stem cell and progenitor proliferation, tissue maturation, as well as left/right and dorsal/ventral developmental patterning.41 Many of these morphological events in brain and heart development share genetic regulation of the same genes, such as, Notch, Jagged, Nkx2.5, and Nodal.41
ERp44+/lacZ protein also presented a transmural gradient expression pattern as the heart developed. In the adult heart, ERp44 distribution showed very prominent enrichment in the outer walls of the heart, with high expression in the epicardium myocardium and epicardial layers. In fact, the staining pattern for ERp44 shows striking similarities to the transient cardiac outward potassium current (Ito) consisting of Kv4.2 (KCND2) and/or Kv4.3 (KCND3) subunits. Recent work has shown that KV4.2 is enriched in the general epicardium region of the heart,42 along with KV channel‐interacting protein 2. Besides the outward potassium current, SERCA2, PLN, and connexin 43 (CX43) have also been shown to play a part in transmural disparities and are important for normal excitation‐contraction coupling (ECC). Studies have shown that disruption and/or amplification in transmural expression may occur in ischemia, infarction, or arrhythmia.43 For instance, increased expression of CX43 in the endocardial region led to late contraction and caused arrhythmia.44 The transmural gradient ERp44/lacZ expression pattern in the adult heart suggests that ERp44 is probably involved in the functional remodeling of ECC and Ca2+ handling in pathophysiological condition by producing a myocardial electrical gradient through regulation of IP3R1 or some other ion channels.
IP3R, RyR2, and Ca2+ Dynamics in ERp44‐Deficient Models
Experiments on zERp44 morphants, the ERp44‐deficient MNCs, and ESCCs showed consistent results of enhanced Ca2+ release amplitude in MNCs, zebrafish morphants, and ESCCs. These effects could be reversed by either expression of lentiviral ERp44 in ERp44 deficient MNCs by comicroinjection of ERp44 mRNA and MOs into zebrafish. RyR2 and IP3Rs are 2 major intracellular Ca2+ release channels located in the cardiomyocyte sarcoplasmic reticulum (SR). RyR2 plays a major role in controlling Ca2+ release in excitation contraction in the heart. IP3Rs are also able to release Ca2+ from the SR, but because they represent only a small fraction (≈1%) of the total Ca2+ release channels. In the failing heart, IP3R1 expression is increased significantly, whereas levels of RyR2 are down‐regulated.45 Dysregulation of IP3R can lead to pathological changes in Ca2+ signaling, signal initiation, amplitude and frequency of Ca2+ signals, duration of Ca2+ elevation, and abnormal growth and apoptosis of cells.9 Complex regulation of Ca2+ signaling is required for cells to live and function, and this task can only be managed when the IP3R properly binds with its numerous binding partners.46 ERp44 is an inhibitor of IP3R18,47; therefore, Ca2+ dysregulation in ERp44‐deficient cardiac cells is likely through an IP3R1 Ca2+ release pathway. Our results indicated that dysfunctional ERp44‐enhanced Ca2+ release amplitude can be further amplified by stimulation with 1 mmol/L of histamine (IP3R agonist), which supports the idea that ERp44 inhibits IP3R1 in cardiomyocytes in vitro and in vivo, at least under normal cellular environments. Without ERp44 inhibition, uncontrolled IP3R1 led to the increase of Ca2+ release to the cytosol from the ER in ERp44‐deficient models. The increase of Ca2+ release from the IP3R1 could also cause an activation in RyR2 through Ca2+‐induced Ca2+ release and cause a release of RyR2 Ca2+ sparks. Hence, Ca2+ from the IP3R through the RyR2‐mediated Ca2+ sparks may increase amplitude of electrically evoked Ca2 transients. ERp44 could also be interacting with RyR2, given that RyR2 is structurally similar to IP3R and contains homologous loop domains. We explored the possibility of ERp44 interacting with RyR2 and observed that ERp44 binds, to a lesser extent, the RyR2 M5‐M10 domain (data not show). Although the Ca2+ dynamic of ERp44‐deficient models can be inhibited by treating with 2‐APB (an IP3Rs inhibitor), we still observed ERp44‐deficient attenuated cellular responses to 2‐APB, where Ca2+ transients showed increases in both Ca2+ release amplitude and Ca2+ transient frequency, compared to WT. Thus, these results suggest that ERp44 likely also interacts with other receptors besides IP3R.
ER Stress and Apoptosis in ERp44‐Deficient Models
ER stress and apoptosis are detected in ERp44 KO mice and zebrafish morphants. Significant increase of ER stress genes, such as PDI, CHOP, and XBP‐1, is observed in ERp44‐deficient cardiac tissue. Decreased cell viability, MMP, and increase of Ca2+ concentration in the mitochondria and ROS detection after Tu treatment is also observed in ERp44‐deficient MNCs, ESCCs, and cardiomyocytes of zebrafish morphant cardiomyocytes. Interestingly, the ERp44+/− AB mice also showed greater increase of ER stress genes in 2 weeks postsurgery, compared to WT AB mice.
ERp44 has been shown to be involved in formation of mixed disulfide bonds with various proteins to regulate oxidative protein folding and client protein homeostasis.6 ERp44 is also active in the Cis‐Golgi compartment, where it retrieves unfolded and/or misfolded proteins back to the ER for proper folding to take place.48 Hence, deficiency of ERp44 can result in the accumulation of unfolded and misfolded proteins and potential cellular dysfunction and pathological consequences of ER stress. Lack of ERp44 can also lead to dysregulation of IP3R, which causes significant increase of Ca2+ release to the cytosol and also in the mitochondria,8 events that could trigger caspase‐9‐ and ‐3‐induced apoptosis. IP3R1 and ERp44 colocalized to the ER and nucleus membranes, and overexpression of ERp44 can result in reduced ATP‐induced nuclear Ca2+ release through IP3R1.8
Altogether, our data suggest that ERp44 deficiency can contribute to disruption in Ca2+ homeostasis, causing ER stress and eventually apoptosis in cardiac development and cardiomyopathy.
Sources of Funding
This project was funded by the Heart and Stroke Foundation of Ontario (T‐6281 and NS‐6636) to Gramolini, Canadian Institute of Health Research to Gramolini (MOP‐106538 and GPG‐102166), the Heart and Stroke Richard Lewar Center of Cardiovascular Excellence, and the Ontario Research Fund—Global Leadership Round in Genomics and Life Sciences to Gramolini and Liu (GL2‐01012). Gramolini is a Canada Research Chair in Cardiovascular Proteomics and Molecular Therapeutics.
Disclosures
None.
Acknowledgments
The authors thank Shunichi Yokoe, Aaron Wilson, Spencer Tong, and Wenping Li for excellent technical support.
References
- 1.Prins D, Michalak M. Endoplasmic reticulum proteins in cardiac development and dysfunction. Can J Physiol Pharmacol. 2009; 87:419-425. [DOI] [PubMed] [Google Scholar]
- 2.Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012; 197:857-867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hamada H, Suzuki M, Yuasa S, Mimura N, Shinozuka N, Takada Y, Suzuki M, Nishino T, Nakaya H, Koseki H, Aoe T. Dilated cardiomyopathy caused by aberrant endoplasmic reticulum quality control in mutant KDEL receptor transgenic mice. Mol Cell Biol. 2004; 24:8007-8017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, Hirata A, Fujita M, Nagamachi Y, Nakatani T, Yutani C, Ozawa K, Ogawa S, Tomoike H, Hori M, Kitakaze M. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004; 110:705-712. [DOI] [PubMed] [Google Scholar]
- 5.Xu C, Bailly‐Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005; 115:2656-2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anelli T, Alessio M, Mezghrani A, Simmen T, Talamo F, Bachi A, Sitia R. ERp44, a novel endoplasmic reticulum folding assistant of the thioredoxin family. EMBO J. 2002; 21:835-844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anelli T, Alessio M, Bachi A, Bergamelli L, Bertoli G, Camerini S, Mezghrani A, Ruffato E, Simmen T, Sitia R. Thiol‐mediated protein retention in the endoplasmic reticulum: the role of ERp44. EMBO J. 2003; 22:5015-5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Higo T, Hattori M, Nakamura T, Natsume T, Michikawa T, Mikoshiba K. Subtype‐specific and ER lumenal environment‐dependent regulation of inositol 1,4,5‐trisphosphate receptor type 1 by ERp44. Cell. 2005; 120:85-98. [DOI] [PubMed] [Google Scholar]
- 9.Choe C‐U, Ehrlich BE. The inositol 1,4,5‐trisphosphate receptor (IP3R) and its regulators: sometimes good and sometimes bad teamwork. Sci STKE. 2006; 2006:re15. [DOI] [PubMed] [Google Scholar]
- 10.Anelli T, Bergamelli L, Margittai E, Rimessi A, Fagioli C, Malgaroli A, Pinton P, Ripamonti M, Rizzuto R, Sitia R. Ero1alpha regulates Ca(2+) fluxes at the endoplasmic reticulum‐mitochondria interface (MAM). Antioxid Redox Signal. 2012; 16:1077-1087. [DOI] [PubMed] [Google Scholar]
- 11.Sun M, Chen M, Dawood F, Zurawska U, Li JY, Parker T, Kassiri Z, Kirshenbaum LA, Arnold M, Khokha R, Liu PP. Tumor necrosis factor‐alpha mediates cardiac remodeling and ventricular dysfunction after pressure overload state. Circulation. 2007; 115:1398-1407. [DOI] [PubMed] [Google Scholar]
- 12.Bousette N, Chugh S, Fong V, Isserlin R, Kim KH, Volchuk A, Backx PH, Liu P, Kislinger T, MacLennan DH, Emili A, Gramolini AO. Constitutively active calcineurin induces cardiac endoplasmic reticulum stress and protects against apoptosis that is mediated by alpha‐crystallin‐B. Proc Natl Acad Sci USA. 2010; 107:18481-18486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chis R, Sharma P, Bousette N, Miyake T, Wilson A, Backx PH, Gramolini AO. Alpha‐crystallin B prevents apoptosis after H2O2 exposure in mouse neonatal cardiomyocytes. Am J Physiol Heart Circ Physiol. 2012; 303:H967-H978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Asahi M, Otsu K, Nakayama H, Hikoso S, Takeda T, Gramolini AO, Trivieri MG, Oudit GY, Morita T, Kusakari Y, Hirano S, Hongo K, Hirotani S, Yamaguchi O, Peterson A, Backx PH, Kurihara S, Hori M, MacLennan DH. Cardiac‐specific overexpression of sarcolipin inhibits sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA2a) activity and impairs cardiac function in mice. Proc Natl Acad Sci USA. 2004; 101:9199-9204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gramolini AO, Trivieri MG, Oudit GY, Kislinger T, Li W, Patel MM, Emili A, Kranias EG, Backx PH, Maclennan DH. Cardiac‐specific overexpression of sarcolipin in phospholamban null mice impairs myocyte function that is restored by phosphorylation. Proc Natl Acad Sci USA. 2006; 103:2446-2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wickenden AD, Kaprielian R, Parker TG, Jones OT, Backx PH. Effects of development and thyroid hormone on K+ currents and K+ channel gene expression in rat ventricle. J Physiol. 1997; 504:271-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang D, Stewart AK, Zhuang L, Zhu Y, Wang Y, Shi C, Keating A, Slutsky A, Zhang H, Wen XY. Enhanced adaptive immunity in mice lacking the immunoinhibitory adaptor Hacs1. FASEB J. 2010; 24:947-956. [DOI] [PubMed] [Google Scholar]
- 18.Lefebvre L, Dionne N, Karaskova J, Squire JA, Nagy A. Selection for transgene homozygosity in embryonic stem cells results in extensive loss of heterozygosity. Nat Genet. 2001; 27:257-258. [DOI] [PubMed] [Google Scholar]
- 19.Gissel C, Doss MX, Hippler‐Altenburg R, Hescheler J, Sachinidis A. Generation and characterization of cardiomyocytes under serum‐free conditions. Methods Mol Biol. 2006; 330:191-219. [DOI] [PubMed] [Google Scholar]
- 20.Kadir SHSA, Ali NN, Mioulane M, Brito‐Martins M, Abu‐Hayyeh S, Foldes G, Moshkov AV, Williamson C, Harding SE, Gorelik J. Embryonic stem cell‐derived cardiomyocytes as a model to study fetal arrhythmia related to maternal disease. J Cell Mol Med. 2009; 13:3730-3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gramolini AO, Kislinger T, Alikhani‐Koopaei R, Fong V, Thompson NJ, Isserlin R, Sharma P, Oudit GY, Trivieri MG, Fagan A, Kannan A, Higgins DG, Huedig H, Hess G, Arab S, Seidman JG, Seidman CE, Frey B, Perry M, Backx PH, Liu PP, MacLennan DH, Emili A. Comparative proteomics profiling of a phospholamban mutant mouse model of dilated cardiomyopathy reveals progressive intracellular stress responses. Mol Cell Proteomics. 2008; 7:519-533. [DOI] [PubMed] [Google Scholar]
- 22.Ryan T, Sharma P, Ignatchenko A, MacLennan DH, Kislinger T, Gramolini AO. Identification of novel ryanodine receptor 1 (RyR1) protein interaction with calcium homeostasis endoplasmic reticulum protein (CHERP). J Biol Chem. 2011; 286:17060-17068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trollinger DR, Cascio WE, Lemasters JJ. Selective loading of Rhod 2 into mitochondria shows mitochondrial Ca2+ transients during the contractile cycle in adult rabbit cardiac myocytes. Biochem Biophys Res Commun. 1997; 236:738-742. [DOI] [PubMed] [Google Scholar]
- 24.Nusslein‐Volhard C, Dahm R. Zebrafish. 2002New York: Oxford University Press [Google Scholar]
- 25.Avdesh A, Chen M, Martin‐Iverson MT, Mondal A, Ong D, Rainey‐Smith S, Taddei K, Lardelli M, Groth DM, Verdile G, Martins RN. Regular care and maintenance of a zebrafish (Danio rerio) laboratory: an introduction. J Vis Exp. 2012; 69:e419610.3791/4196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Westerfield M. The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio rerio). 20004th edEugene: University of Oregon Press [Google Scholar]
- 27.Yuan S, Sun Z. Microinjection of mRNA and morpholino antisense oligonucleotides in zebrafish embryos. J Vis Exp. 2009; 710.3791/1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nasevicius A, Ekker SC. The zebrafish as a novel system for functional genomics and therapeutic development applications. Curr Opin Mol Ther. 2001; 3:224-228. [PubMed] [Google Scholar]
- 29.Muntean BS, Horvat CM, Behler JH, Aboualaiwi WA, Nauli AM, Williams FE, Nauli SM. A comparative study of embedded and anesthetized zebrafish in vivo on myocardiac calcium oscillation and heart muscle contraction. Front Pharmacol. 2010; 1:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joseph SK, Hajnoczky G. IP3 receptors in cell survival and apoptosis: Ca2+ release and beyond. Apoptosis. 2007; 12:951-968. [DOI] [PubMed] [Google Scholar]
- 31.Keller G. Embryonic stem cell differentiation: emergence of a new era in biology and medicine. Genes Dev. 2005; 19:1129-1155. [DOI] [PubMed] [Google Scholar]
- 32.Van Vliet P, Wu SM, Zaffran S, Puceat M. Early cardiac development: a view from stem cells to embryos. Cardiovasc Res. 2012; 96:352-362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang CJ, Tu CT, Hsiao CD, Hsieh FJ, Tsai HJ. Germ‐line transmission of a myocardium‐specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Dev Dyn. 2003; 228:30-40. [DOI] [PubMed] [Google Scholar]
- 34.Gorza L, Schiaffino S, Volpe P. Inositol 1,4,5‐trisphosphate receptor in heart: evidence for its concentration in purkinje myocytes of the conduction system. J Cell Biol. 1993; 121:345-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kockskamper J, Zima AV, Roderick HL, Pieske B, Blatter LA, Bootman MD. Emerging roles of inositol 1,4,5‐trisphosphate signaling in cardiac myocytes. J Mol Cell Cardiol. 2008; 45:128-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosemblit N, Moschella MC, Ondriasa E, Gutstein DE, Ondrias K, Marks AR. Intracellular calcium release channel expression during embryogenesis. Dev Biol. 1999; 206:163-177. [DOI] [PubMed] [Google Scholar]
- 37.Savolainen SM, Foley JF, Elmore SA. Histology atlas of the developing mouse heart with emphasis on E11.5 to E18.5. Toxicol Pathol. 2009; 37:395-414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uchida K, Aramaki M, Nakazawa M, Yamagishi C, Makino S, Fukuda K, Nakamura T, Takahashi T, Mikoshiba K, Yamagishi H. Gene knock‐outs of inositol 1,4,5‐trisphosphate receptors types 1 and 2 result in perturbation of cardiogenesis. PLoS One. 2010; 510.1371/journal.pone.0012500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mesaeli N, Nakamura K, Zvaritch E, Dickie P, Dziak E, Krause KH, Opas M, MacLennan DH, Michalak M. Calreticulin is essential for cardiac development. J Cell Biol. 1999; 144:857-868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith A, Robinson V, Patel K, Wilkinson DG. The EphA4 and EphB1 receptor tyrosine kinases and ephrin‐B2 ligand regulate targeted migration of branchial neural crest cells. Curr Biol. 1997; 7:561-570. [DOI] [PubMed] [Google Scholar]
- 41.McQuillen PS, Goff DA, Licht DJ. Effects of congenital heart disease on brain development. Prog Pediatr Cardiol. 2010; 29:79-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Costantini DL, Arruda EP, Agarwal P, Kim KH, Zhu Y, Zhu W, Lebel M, Cheng CW, Park CY, Pierce SA, Guerchicoff A, Pollevick GD, Chan TY, Kabir MG, Cheng SH, Husain M, Antzelevitch C, Srivastava D, Gross GJ, Hui CC, Backx PH, Bruneau BG. The homeodomain transcription factor Irx5 establishes the mouse cardiac ventricular repolarization gradient. Cell. 2005; 123:347-358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soltysinska E, Olesen SP, Christ T, Wettwer E, Varro A, Grunnet M, Jespersen T. Transmural expression of ion channels and transporters in human nondiseased and end‐stage failing hearts. Pflugers Arch. 2009; 459:11-23. [DOI] [PubMed] [Google Scholar]
- 44.Yamada KA, Kanter EM, Green KG, Saffitz JE. Transmural distribution of connexins in rodent hearts. J Cardiovasc Electrophysiol. 2004; 15:710-715. [DOI] [PubMed] [Google Scholar]
- 45.Gutstein DE, Marks AR. Role of inositol 1,4,5‐trisphosphate receptors in regulating apoptotic signaling and heart failure. Heart Vessels. 1997; suppl 12:53-57. [PubMed] [Google Scholar]
- 46.Pan C, Zheng J, Wu Y, Chen Y, Wang L, Zhou Z, Yin W, Ji G. ERp44 C160S/C212S mutants regulate IP3R1 channel activity. Protein Cell. 2011; 2:990-996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li G, Mongillo M, Chin KT, Harding H, Ron D, Marks AR, Tabas I. Role of ERO1‐alpha‐mediated stimulation of inositol 1,4,5‐triphosphate receptor activity in endoplasmic reticulum stress‐induced apoptosis. J Cell Biol. 2009; 186:783-792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vavassori S, Cortini M, Masui S, Sannino S, Anelli T, Caserta IR, Fagioli C, Mossuto MF, Fornili A, van Anken E, Degano M, Inaba K, Sitia R. A pH‐regulated quality control cycle for surveillance of secretory protein assembly. Mol Cell. 2013; 50:783-792. [DOI] [PMC free article] [PubMed] [Google Scholar]