

Abstract
Background
Antiphospholipid syndrome patients have antiphospholipid antibodies (aPLs) that promote thrombosis, and they have increased cardiovascular disease risk. Although the basis for the thrombosis has been well delineated, it is not known why antiphospholipid syndrome patients also have an increased prevalence of nonthrombotic vascular occlusion. The aims of this work were to determine if aPLs directly promote medial hypertrophy or neointima formation in mice and to identify the underlying mechanisms.
Methods and Results
Medial hypertrophy and neointima formation invoked by carotid artery endothelial denudation were evaluated in mice administered normal human IgG or aPLs. While aPLs had no effect on medial hypertrophy, they caused exaggerated neointima development. This was related to an aPL‐induced impairment in reendothelialization post denudation, and scratch assays in cell culture revealed that there are direct effects of aPLs on endothelium that retard cell migration. Further experiments showed that aPL antagonism of endothelial migration and repair is mediated by antibody recognition of β2‐glycoprotein I, apolipoprotein E receptor 2, and a decline in bioavailable NO. Consistent with these mechanisms, the adverse impacts of aPLs on reendothelialization and neointima formation were fully prevented by the NO donor molsidomine.
Conclusions
APLs blunt endothelial repair, and there is related aPL‐induced exaggeration in neointima formation after endothelial injury in mice. The initiating process entails NO deficiency mediated by β2‐glycoprotein I recognition by aPLs and apolipoprotein E receptor 2. The modulation of endothelial apolipoprotein E receptor 2 function or NO bioavailability may represent new interventions to prevent the nonthrombotic vascular occlusion and resulting cardiovascular disorders that afflict antiphospholipid syndrome patients.
Keywords: antiphospholipid syndrome, endothelium, neointima hyperplasia, nitric oxide
Introduction
The antiphospholipid syndrome (APS) is an autoimmune disorder characterized by increased risk of thrombosis and cardiovascular disease resulting from elevated levels of circulating antiphospholipid antibodies (aPLs).1–2 The cardiovascular disorders for which APS patients have greater predisposition include coronary artery disease, myocardial infarction, and stroke.3 In addition to the thrombotic diathesis, the processes contributing to greater cardiovascular disease risk in APS include an increased prevalence of nonthrombotic vascular occlusion. APS patients display greater carotid artery intima‐media thickness (IMT)4–8 and greater stenosis of the renal, intracranial, celiac, and mesenteric arteries than do non‐APS subjects.9–15 Why APS patients have increased nonthrombotic vascular occlusion is unknown. In particular, whether aPLs directly promote processes that lead to nonthrombotic vascular occlusion and the potential mechanisms underlying such effects of aPLs on the vascular wall are unknown.
Numerous studies in cell culture and in animal models have revealed that pathologically relevant aPLs are directed against the phospholipid‐binding protein β2 glycoprotein I (β2GPI).16–18 β2GPI is a plasma protein composed of 5 distinctive domains (domains I to V), and antibodies against domain I are an important disease‐promoting component of aPLs.19–20 We previously demonstrated in cultured endothelial cells and in mice that aPL binding to domain I of β2GPI and the resulting interaction of β2GPI with the low‐density lipoprotein receptor family member apolipoprotein E receptor 2 (ApoER2) in endothelium attenuate endothelial NO synthase (eNOS) activation, thereby promoting leukocyte–endothelial cell adhesion and thrombosis.21
In the current work, we designed experiments to test the hypothesis that aPLs promote medial hypertrophy and/or neointima formation. We used a mouse model that entails the induction of both of these processes after endothelial denudation in the carotid artery. The selection of such a model is based on recognition that an impairment in endothelial cell monolayer integrity most often underlies the initiation of medial hypertrophy or neointima formation.22–26 The results obtained reveal for the first time that although the development of medial hypertrophy is unaffected by aPLs, aPLs cause exaggerated neointima formation. Additional studies showed that this is related to a marked blunting of endothelial repair by aPLs involving direct actions on endothelial cells and that the impairment in reparative capacity is mediated by aPL recognition of domain I of β2GPI and ApoER2 and a decline in bioavailable NO. In contrast, aPLs have no direct effect on vascular smooth muscle cell (VSMC) proliferation or migration. We further demonstrated that aPL‐induced attenuation of reendothelialization and the resulting exaggeration of neointima formation in vivo are fully prevented by the NO donor molsidomine. Recognizing that NO is also antithrombotic,27–29 strategies to enhance bioavailable NO abundance or action may provide new mechanism‐based means to combat both the thrombotic and the nonthrombotic vascular complications of APS.
Materials and Methods
Animal Models
In vivo studies were performed in male C57BL/6 mice, in male ApoER2+/+ and ApoER2−/− littermates on an identical 129SvEv×C57BL/6J background, and in male FVB mice.21,30 All animal experiments were approved by the Institutional Animal Care and Utilization Committees at the University of Texas Southwestern Medical Center and the University of Cincinnati.
Antibody Preparation
Polyclonal aPLs were isolated from patients with APS, which were characterized as having high‐titer aPL levels (>140 IgG phospholipid units GPL U), thromboses, and/or pregnancy losses.21 The relevant clinical and laboratory features of the patients who provided aPLs are given in Table. Individuals provided informed consent before participating in these studies, and all protocols were approved by the institutional review boards of the Hospital for Special Surgery and the University of Texas Southwestern Medical Center. Human IgG from healthy nonautoimmune individuals (NHIgG) was purified by using an identical method. All IgG samples were treated to deplete endotoxin as previously described.21 Mouse monoclonal antibodies directed to domain I of β2GPI (3F8) and the control mouse monoclonal antibody (BBG) were prepared as previously reported.21,31
Table 1.
Clinical and Laboratory Features of the Patients
| Patient | Age | Sex | aCL (>140 GPLU) | Anti‐β2GPI | Clinical Features |
|---|---|---|---|---|---|
| 1 | 50 | F | + | + | Arterial thrombosis, pregnancy losses, catastrophic APS, myocardial infarction |
| 2 | 57 | M | + | + | Arterial thrombosis, recurrent pulmonary hemorrhage, catastrophic APS |
| 3 | 31 | M | + | + | Adrenal hemorrhage, skin necrosis, nemolytic anemia, multiple pulmonary emboli, catastrophic APS |
aCL indicates anti‐cardiolipin antibodies; GPLU, IgG phospholipid units; β2GPI, β2‐glycoprotein I; APS, antiphospholipid antibody.
Carotid Artery Reendothelialization
Carotid artery reendothelialization was evaluated after perivascular electric injury in 12‐ to 16‐week‐old male mice as previously described.32–33 Mice were anesthetized via intraperitoneal administration of avertin, and the left common carotid artery was exposed through an anterior incision of the neck. Electric current of 4 W was applied through 2‐mm forceps (2 W/mm) for 3 sec in microbipolar mode (Force 2 Electrosurgical Generator; Valleylab). The mice were allowed to recover, and analgesia was provided via intraperitoneal administration of buprenorphine immediately after surgery and 24 hours later. At 72 hours after injury, animals were injected intravenously with 5% Evans blue dye (Sigma‐Aldrich), the carotid arteries were harvested, and the area of denudation (which incorporates the dye) was quantified in a blinded manner through image analysis by using Scion Image (free software from National Institutes of Health). NHIgG or aPLs (100 μg per mouse) were injected intraperitoneally 24 hours before injury, on the day of injury, and 24 hours after injury. For evaluation of the effect of molsidomine on reendothelialization, mice were injected intraperitoneally with molsidomine (Sigma‐Aldrich, 5 mg/kg per day) or vehicle (saline) daily starting 72 hours before injury and ending 48 hours after injury.
Carotid Artery Neointima Formation
The procedures used to induce endothelial denudation in mouse carotid arteries, tissue preparation, and the evaluation of neointima formation were performed as described previously.30,34 Studies were performed in 8‐week‐old male FVB mice injected intraperitoneally with NHIgG (100 μg/mouse) or aPLs (100 μg/mouse) 24 hours before and every 48 hours after the endothelial injury for 14 days. Briefly, to denude the carotid artery endothelium, mice were anesthetized with a mixture of ketamine (80 mg/kg; Fort Dodge Laboratories) and xylazine (16 mg/kg; The Butler Co). The left external carotid artery was looped proximally and ligated distally with 7‐0 silk sutures (Ethicon). An epon‐resin probe was produced by forming a bead slightly larger than the diameter of the carotid artery (≈0.45 mm) on a 3‐0 nylon suture. A transverse arteriotomy was made between the 7‐0 sutures, the resin probe was inserted, and the probe was advanced toward the aortic arch and withdrawn 5 times. The probe was then removed and the proximal 7‐0 suture was ligated. Mice were killed 1 hour or 14 days after injury by perfusion fixation with 10% phosphate‐buffered formalin (pH 7.0) for 15 min at physiological pressure. The entire block of neck tissue containing both uninjured right and injured left common carotid arteries was harvested, fixed with the same solution for an additional 48 hours, and decalcified before being embedded in paraffin. Ten identical 5‐μm cross sections at 500‐μm intervals were made from the distal aspect of the neck beginning at the location of the proximally ligated 7‐0 suture. Parallel sections were subjected to routine hematoxylin‐and‐eosin staining and to Verhoeff–Van Gieson staining of elastic lamina. For each animal, 4 whole‐neck cross sections were digitized, and the vessels were quantified with Scion Image analysis software (Scion). Measurements of luminal area, area inside the internal elastic lamina, and the area encircled by external elastic lamina were made on each section. Medial area was calculated by subtracting internal elastic lamina from external elastic lamina area, and intimal area was calculated by subtracting luminal area from internal elastic lamina. To evaluate the effect of molsidomine on neointima formation, mice were injected intraperitoneally with molsidomine (5 mg/kg per day) or saline vehicle daily from the time of injury to tissue harvest 14 days later.
Cell Culture and RNA Interference
Primary bovine aortic endothelial cells (BAECs) were isolated by using previously described methods,21,33 cultured in EGM2 media (Lonza), and used within 3 to 7 passages. In small interfering RNA (siRNA) experiments, BAECs were transfected with siRNAs by using LipofectAMINE 2000 (Invitrogen) and studied 48 hours later.21,33 dsRNA with sequence 5′‐ACUGGAAGCGGAAGAAUAC‐3′ designed to target the open reading frame of bovine ApoER2 (accession number XM_865091) and control dsRNA were purchased from Dharmacon.21 Immunoblot analyses were performed to evaluate effective knockdown of ApoER2 expression.
Primary aortic VSMCs were isolated from C57BL/6 mice by using previously reported procedures with some modifications.30 Briefly, thoracic aortas were dissected from the mice, adventitial tissue was removed, and the aortas were incubated in Hanks' solution containing 1 mg/mL collagenase and 0.5 mg/mL elastase for 30 min at 37°C. The resulting cell suspension was centrifuged at 150g for 5 min and resuspended in 10 mL of DMEM containing 10% FBS. The identity of the isolated cells as VSMCs was confirmed by positive immunostaining with anti–α‐SMA monoclonal antibody (Sigma; clone 1A4). Cells were maintained in DMEM (Sigma‐Aldrich) containing 10% FBS, 100 units/mL penicillin, and 0.1 mg/mL streptomycin, and experiments were done at passages 1 to 4.
Endothelial Cell Proliferation and Migration
For the evaluation of endothelial cell proliferation, BAECs were seeded in 6‐well plates at 1×105 cells/well and grown in EGM2 at 37°C for 24 hours. Cells were then treated with NHIgG (100 μg/mL) or aPLs (100 μg/mL), and cell numbers were counted at 0, 24, and 48 hours. To study endothelial cell migration, BAECs were grown to near‐confluence in 6‐well plates and placed in 1% lipoprotein‐deficient serum (provided by Dr J. Goldstein and Dr M. Brown, University of Texas Southwestern Medical Center) in DMEM for 16 hours, and a defined region of cells was removed by using a single‐edged razor blade.32–33 Cells were treated with vascular endothelial growth factor (VEGF) (50 ng/mL) in the presence or absence of NHIgG or aPLs (100 μg/mL), control IgG (BBG), or β2GPI monoclonal antibody 3F8 (0.3 μg/mL) in DMEM+1% lipoprotein‐deficient serum. In select studies, the cells were also incubated in the absence versus presence of S‐nitroso‐N‐acetyl‐dl‐penicillamine (SNAP, 20 μmol/L) or hydroxyurea (1 mmol/L).35 Twenty‐four hours later, the cells were fixed with 3% paraformaldehyde (Sigma‐Aldrich), permeabilized in 0.2% Triton X‐100 (Bio‐Rad Laboratories), stained with hematoxylin (Fisher Scientific), and viewed under an inverted microscope (Zeiss Axiovert 100 M). The number of cells migrated past a 2000‐μm length of the wound edge was quantified in a minimum of 3 (×40 magnification) fields per well. All endothelial cell culture findings were confirmed in 3 independent experiments.
VSMC Proliferation and Migration
VSMC proliferation was assessed by using previously described methods.36 Briefly, cells were seeded in 96‐well culture plates at 1×104 cells/well and cultured in DMEM containing 0.2% FBS for 48 hours. The cells were incubated with or without PDGF‐BB (30 nmol/L, Invitrogen) in the presence of NHIgG (100 μg/mL) or aPLs (100 μg/mL) for 18 hours at 37°C, followed by incubation with [3H]thymidine (0.2 μCi/well, 28 nmol/L; PerkinElmer) for 6 hours; washed with PBS; and lysed with 0.25 mol/L sodium hydroxide. Radioactivity in cell lysates was quantified with the use of liquid scintillation counting. VSMC migration was evaluated in a Transwell apparatus by using a membrane filter with pore size of 8.0 μm.36 After plating at 2×104 cells/well, cells were treated with or without PDGF‐BB (30 nmol/L) for 4 hours. Nonmigrating cells on the upper surface were scraped gently and removed with PBS wash and wiping with a cotton‐tipped applicator. The filters were fixed in ice‐cold methanol and stained with DAPI, and the number of cells/HPF that had migrated into the membrane was counted microscopically (NIKON Eclipse TE2000‐E, ×40 magnification, 5 fields/chamber). All VSMC culture findings were confirmed in 3 independent experiments.
Statistical Analysis
All data are expressed as mean±SEM. For cell culture experiments, Student t test or 1‐way ANOVA was used to assess differences between 2 groups or among >2 groups, respectively, with Newman–Keuls post‐hoc testing after ANOVA. Findings for the experiments performed in mice were evaluated by using Mann–Whitney tests to assess differences between 2 groups and Kruskal–Wallis analyses followed by Mann–Whitney with Bonferroni correction for comparisons of >2 groups. P<0.05 values were considered significant.
Results
Impact of aPLs on Medial Hypertrophy, Neointima Formation, and Endothelial Repair
To determine if aPLs promote medial hypertrophy and/or neointima formation, we assessed the impact of aPLs on these processes invoked by carotid artery endothelial denudation in mice. Using our previously established methods,30 the carotid arteries of male wild‐type mice were denuded with an epon‐resin intraarterial probe, and the mice were injected with NHIgG or aPLs every 48 hours for 14 days. At the end of the study period, whole neck sections were processed for histological analysis. Compared with noninjured carotid arteries, the endothelial denudation caused medial hypertrophy in both NHIgG‐ and aPL‐treated mice, and aPLs did not enhance the hypertrophy (Figure 1A and 1B). However, endothelial denudation–induced neointima formation was markedly greater in mice injected with aPLs compared with mice given NHIgG (Figure 1A and 1C). Carotid artery lumen area was also reduced in aPL‐treated versus NHIgG‐treated mice (50 904±26 550 μm2 versus 76 727±20 380 μm2, respectively, mean±SEM, n=9 or 10, P<0.05). Thus, aPLs promote neointima formation.
Figure 1.
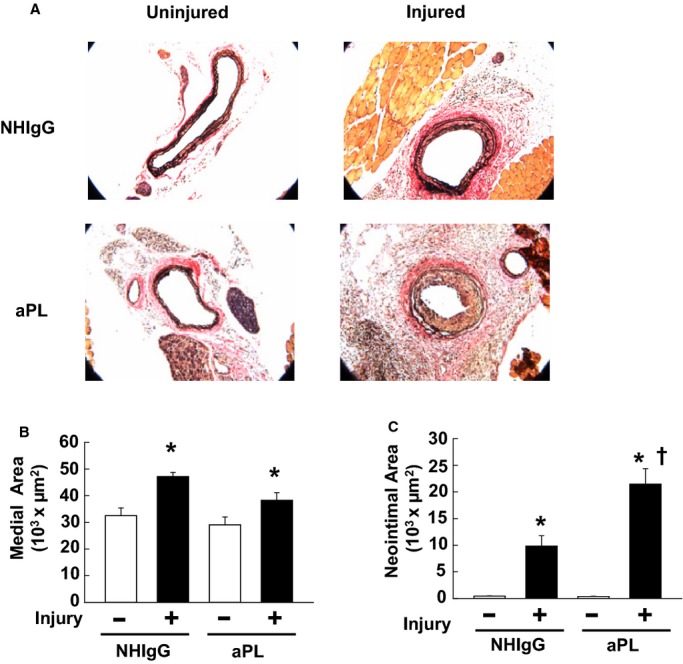
Antiphospholipid antibodies (aPLs) promote carotid artery neointima formation after endothelial denudation in mice. A, Male wild‐type mice were treated with human IgG from healthy nonautoimmune individuals (NHIgG; 100 μg/mouse) or aPLs (100 μg/mouse) 1 day before carotid artery endothelial denudation by using an epon‐resin probe and every other day after denudation for 2 weeks. Medial hypertrophy and neointima formation were then evaluated by histologic analysis. Representative photomicrographs are shown of uninjured (left panels) and injured carotid arteries (right panels). B, Medial area was calculated as the area encircled by the external elastic lamina minus the area encircled by the internal elastic lamina. Values are mean±SEM. For the NHIgG‐treated group (n=10), median was 30 396 (uninjured) and 45 186 (injured). For the aPL‐treated group (n=9), median was 26 861 (uninjured) and 37 477 (injured). C, Neoinimal area was calculated by subtracting the luminal area from the area encircled by the internal elastic lamina. Values are mean±SEM. For the NHIgG‐treated group (n=10), median was 387 (uninjured) and 7974 (injured). For the aPL‐treated group (n=9), median was 288 (uninjured) and 18 226 (injured). *P<0.05 vs uninjured, †P<0.05 vs NHIgG.
Because neointima formation most often is initiated by a loss of endothelial cell monolayer integrity and the severity of neointima development is related to the capacity for endothelial cell monolayer repair,22–26 the effect of aPLs on carotid artery reendothelialization was then tested by using a thermal injury model in which the degree of reendothelialization is readily quantified. The mice received intraperitoneal injections of NHIgG or aPLs 24 hours before injury, on the day of injury, and 24 hours after injury. At 72 hours after injury, the area of remaining denudation was determined by intravascular injection of Evans blue dye and evaluation of its uptake by the carotid artery intimal surface. In a parallel set of studies in mice injected with Evans blue dye later on the day of injury, it was confirmed that the area of initial denudation was similar in mice injected with NHIgG versus those injected with aPLs (data not shown). Compared with mice administered NHIgG, mice treated with aPLs displayed a 6‐fold larger area of remaining denudation at 72 hours after injury (Figure 2A and 2B). Thus, aPLs cause blunting of endothelial repair in vivo in mice.
Figure 2.
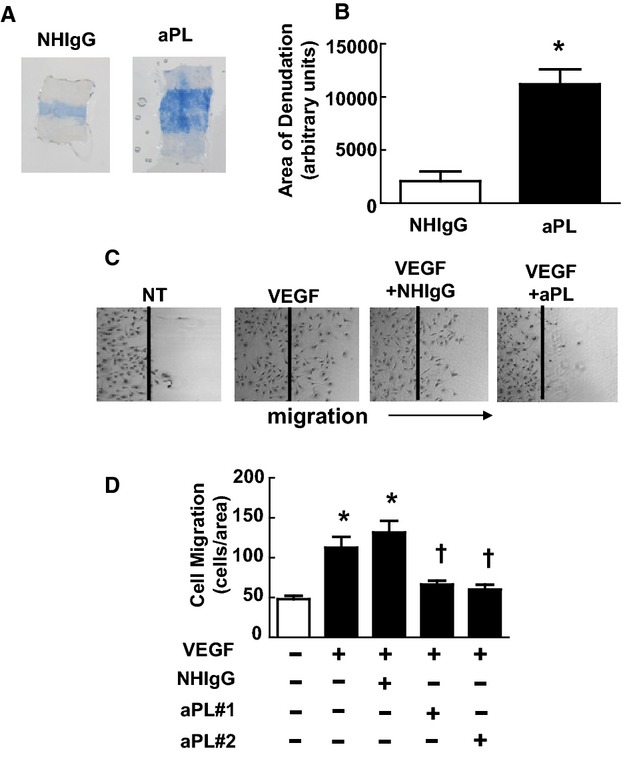
Antiphospholipid antibodies (aPLs) inhibit endothelial repair. A, Male wild‐type mice received human IgG from healthy nonautoimmune individuals (NHIgG) or aPLs 24 hours before carotid artery endothelial denudation, on the day of denudation, and 24 hours after denudation. The remaining area of denudation was evaluated 72 hours later by assessing Evans blue dye uptake. Representative images of the carotid artery intimal surface displaying Evans blue dye incorporation are shown. B, Summary data for the area of remaining denudation is expressed in arbitrary units. Values are mean±SEM. For the NHIgG‐treated group (n=6), median was 1610, and for the aPL‐treated group (n=5), median was 10 710. *P<0.05 vs NHIgG. C, In scratch assays, bovine aortic endothelial cells (BAECs) were wounded and treated with media alone (nontreated [NT]), vascular endothelial growth factor (VEGF) (50 ng/mL), or VEGF in the presence of NHIgG (100 μg/mL) or 1 of 2 different isolates of aPLs (100 μg/mL) for 24 hours, and cell migration was evaluated. Representative images are shown. D, Summary data for migration. Values are mean±SEM, n=8/group, *P<0.05 vs NT, †P<0.05 vs NHIgG.
Endothelial cells and platelets are major cellular targets of aPL action.17,37–38 To determine if there is a direct effect of aPLs on endothelial cells that alters their migratory capacity, scratch assays were performed by using cultured endothelial cells. After monolayer growth to near‐confluence, endothelial cells were removed from a defined region, and cell migration back into the denuded area was assessed over 24 hours under varying conditions. Compared with nontreated cells, cells treated with VEGF predictably displayed enhanced migration, and VEGF‐induced migration was unaffected by the addition of NHIgG (Figure 2C, representative images). In contrast, aPLs completely suppressed VEGF‐induced migration. Summary data are provided in Figure 2D. APL had no discernible effect on endothelial cell proliferation (Figure 3A), and the inhibitory effect of aPLs on endothelial cell migration as assessed in scratch assays was not altered by the presence of the inhibitor of cell proliferation, hydroxyurea (Figure 3B).
Figure 3.
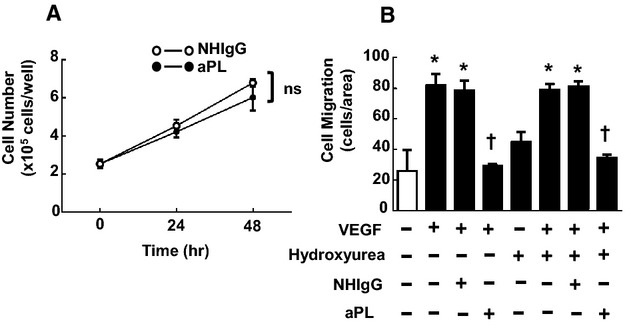
Inhibitory effect of antiphospholipid antibodies (aPLs) on scratch assay for endothelial cell migration does not involve effect on cell proliferation. A, Bovine aortic endothelial cells (BAECs) were seeded in 6‐well plates (1×105 cells/well) and grown for 24 hours. Cells were then incubated for 0, 24, and 48 hours in the presence of human IgG from healthy nonautoimmune individuals (NHIgG) (100 μg/mL) or aPLs (100 μg/mL), and the number of the cells/well was counted. There were no difference between cells treated with NHIgG and aPLs. Values are mean±SEM, n=3/group, ns; not significant. B, In scratch assays, BAECs were wounded and treated with PBS vehicle or hydroxyurea (1 mmol/L), vascular endothelial growth factor (VEGF; \(50 ng/mL), or VEGF in the presence of NHIgG (100 μg/mL) or aPLs (100 μg/mL) for 24 hours, and cell migration was evaluated. Values are mean±SEM, n=3/group, *P<0.05 vs no VEGF, †P<0.05 vs NHIgG.
In addition to the importance of endothelial cell monolayer disruption to neointima development, aberrant VSMC growth and migration can contribute.39 The effect of NHIgG versus aPLs on primary mouse aortic VSMC proliferation was therefore compared. Based on measurement of [3H]‐thymidine incorporation, PDGF‐stimulated VSMC proliferation was similar in NHIgG‐ and aPL‐treated cells (Figure 4A). Using a Transwell apparatus, VSM migration was also evaluated, and PDGF‐stimulated migration was not affected by aPLs (Figure 4B). These collective results indicate that aPLs cause increased neointima formation invoked by endothelial injury, that this is related to marked attenuation of endothelial monolayer repair, and that aPLs have a direct effect on endothelial cells that blunts their capacity for migration.
Figure 4.
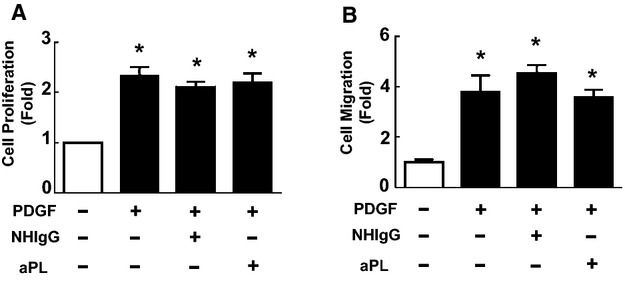
Antiphospholipid antibodies (aPLs) do not promote vascular smooth muscle cell (VSMC) proliferation or migration. A, Proliferation of primary VSMCs isolated from C57BL/6 mice was assessed by quantification of 3H‐thymidine incorporation. Cells were incubated with or without platelet‐derived growth factor (PDGF; 30 nmol/L) for 18 hours in the presence of NHIgG (100 μg/mL) or aPLs (100 μg/mL). Values are mean±SEM, n=3/group, *P<0.05 vs no PDGF. B, VSMCs were incubated with or without PDGF (30 nmol/L) in the presence of NHIgG (100 μg/mL) or aPLs (100 μg/mL) for 24 hours, and cell migration was evaluated. Values are mean±SEM, n=6/group, *P<0.05 vs no PDGF.
Role of β2GPI and ApoER2
The most pathologically relevant aPLs are directed against the phospholipid‐binding protein β2GPI, and aPLs recognizing domain I of β2GPI in particular are highly associated with APS disease severity.17–20 To determine if β2GPI domain I recognition underlies aPL effects on endothelial cell migration, scratch assays were performed testing the effects of a monoclonal antibody to domain I of β2GPI (3F8) versus an IgG control (BBG).21 Mirroring the actions of polyclonal aPLs, 3F8 inhibited endothelial cell migration in response to VEGF, whereas BBG had no effect (Figure 5A).
Figure 5.
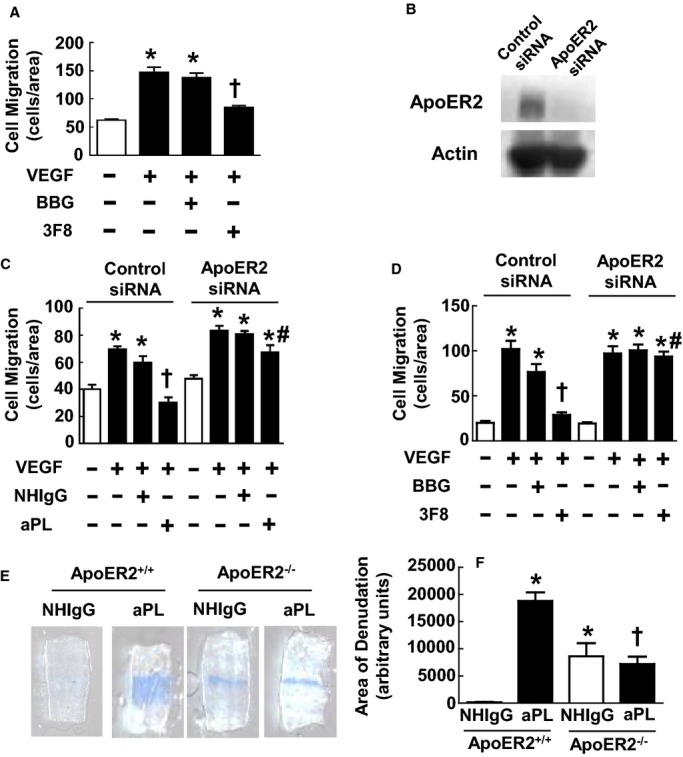
Endothelial cell repair inhibition by antiphospholipid antibodies (aPLs) requires β2‐glycoprotein I (β2GPI) and apolipoprotein E receptor 2 (ApoER2). A, Scratch assays were performed with bovine aortic endothelial cells (BAECs) treated with media alone (basal), vascular endothelial growth factor (VEGF; 50 ng/mL), VEGF+isotype‐matched control IgG (BBG, 0.3 μg/mL), or VEGF+3F8 monoclonal antibody to β2GPI (0.3 μg/mL). Values are mean±SEM, n=8/group, *P<0.05 vs basal, † P<0.05 vs BBG. B, BAECs were transfected with control silent interfering RNA (siRNA) or siRNA targeting ApoER2, and whole‐cell lysates were immunoblotted for ApoER2 and actin. C, Scratch assays were performed 48 hours later with treatments with media alone (basal), VEGF (50 ng/mL), VEGF+human IgG from healthy nonautoimmune individuals (NHIgG; 100 μg/mL), or VEGF+aPLs (100 μg/mL) for 24 hours. Values are mean±SEM, n=4 for no treatment (ApoER2 siRNA) and VEGF, n=6 for no treatment (Control siRNA), VEGF+NHIgG, and VEGF+aPLs*P<0.05 vs basal, †P<0.05 vs NHIgG, #P<0.05 vs Control siRNA. D, Scratch assays were performed 48 hours after cell transfection with control siRNA or siRNA targeting ApoER2, using the treatments described for A. Values are mean±SEM, n=4 for no treatment and VEGF treatment group, and n=8 for VEGF+BBG and VEGF+3F8 treatment group, *P<0.05 vs basal, †P<0.05 vs BBG, #P<0.05 vs Control siRNA. E, Male ApoER2+/+ or ApoER2−/− littermates treated with NHIgG (100 μg/mouse) or aPLs (100 μg/mouse) underwent carotid artery endothelial denudation, and the remaining area of denudation was evaluated 72 hours later. Representative images of the carotid artery intimal surface displaying Evans blue dye incorporation are shown. F, Summary data for the area of remaining denudation at 72 hours are expressed in arbitrary units. Values are mean±SEM. For ApoER2+/+ n=4 (uninjured, median was 161.5) and n=5 (injured, median was 19 087). For ApoER2−/−, n=5 (uninjured, median was 7736) and n=5 (injured, median was 5923). *P<0.05 vs NHIgG in ApoER2+/+, †P<0.05 vs aPLs in ApoER2+/+.
Toll‐like receptors, annexins, and the low‐density lipoprotein receptor family member ApoER2 have all been implicated in aPL actions on target cells, including endothelial cells.21,40–41 Because we and others previously determined that ApoER2 binds directly to the aPL–β2GPI complex adherent to endothelial cells,21,42 a potential requirement for ApoER2 in the antagonism of endothelial cell migration by β2GPI‐recognizing antibody was determined by siRNA knockdown of the receptor. In ApoER2 siRNA–treated cells, ApoER2 protein expression was decreased by 87% (control siRNA, 100.0±12.8% versus ApoER2 siRNA, 12.8±5.12%, n=6/group, mean±SEM, *P<0.05). Representative immunoblots are shown in Figure 5B. Although aPLs or anti‐β2GPI monoclonal antibody 3F8 completely suppressed cell migration in control siRNA‐transfected cells, the impact of aPLs or 3F8 was prevented in cells deficient in ApoER2 (Figure 5C and 5D).
To evaluate whether ApoER2 participates in the attenuation of endothelial repair by aPLs in vivo, carotid artery reendothelialization experiments were performed in ApoER2+/+ versus ApoER2−/− mice administered NHIgG or aPLs. In NHIgG control–treated mice, reendothelialization was reduced in ApoER2−/− versus ApoER2+/+ mice (Figure 5E and 5F). This finding indicates that there is a role for the receptor in endothelial repair under basal conditions, and this was confirmed in nontreated ApoER2+/+ and ApoER2−/− mice (data not shown). Consistent with the observations in Figure 2A and 2B, aPL treatment of ApoER2+/+ mice caused the attenuation of reendothelialization. In contrast, aPL treatment did not further impair reendothelialization in ApoER2−/− mice (Figure 5E and 5F). These cumulative findings indicate that the recognition of domain I of β2GPI by aPLs and ApoER2 are critically involved in the adverse effects of aPLs on endothelial repair.
Role of NO Deficiency
Endothelial migration is governed by both NO‐dependent and NO‐independent processes.33,43–44 Having previously found that aPL actions via ApoER2 antagonize eNOS,21 we determined whether NO deficiency underlies the suppression of endothelial cell migration by aPLs (Figure 6A). Although aPLs inhibited migration stimulated by VEGF, the antagonistic effect of aPLs was fully prevented by the NO donor SNAP. To potentially link β2GPI recognition, NO deficiency, and the attenuation of endothelial cell migration, parallel experiments were performed with the anti‐β2GPI monoclonal antibody (3F8) (Figure 6B). Mirroring the findings with aPLs, SNAP reversed the blunting of VEGF‐stimulated migration by 3F8. To determine if exogenous NO can counteract the negative impact of aPLs on endothelial repair in vivo, the effect of the NO donor molsidomine was evaluated.45 APL‐treated mice were concurrently administered either vehicle control (saline) or molsidomine (5 mg/kg per day), which produces the metabolite 3‐morpholino‐sydnonimine that generates NO.45 Paralleling the findings in cell culture with the NO donor SNAP, despite receiving aPLs, the remaining area of denudation 72 hours after injury was markedly reduced in the molsidomine‐treated mice (Figure 6C and 6D). It was further tested whether the NO donor prevents aPL‐induced neointima formation. Although molsidomine did not affect medial area (Figure 7A and 7B), aPL‐induced neointima formation was reduced in mice given the NO donor (Figure 7A and 7C). The lumen area was also increased in molsidomine‐treated mice (83 650±23 203 μm2, mean±SEM, n=8) compared with mice treated with saline (56 575±19 409 μm2, n=9, *P<0.05). These findings reveal that NO deficiency underlies the adverse impacts of aPLs on endothelial monolayer repair and neointima formation.
Figure 6.
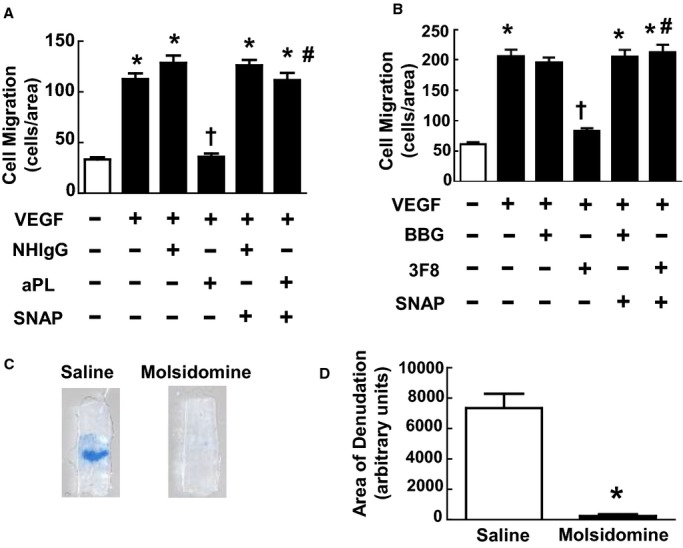
Inhibition of endothelial cell migration and repair by antiphospholipid antibodies (aPLs) entail NO deficiency. A, Scratch assays were performed with bovine aortic endothelial cells (BAECs) treated with media alone (basal), vascular endothelial growth factor (VEGF; 50 ng/mL), VEGF+human IgG from healthy nonautoimmune individuals (NHIgG; 100 μg/mL), VEGF+aPLs (100 μg/mL), VEGF+NHIgG+S‐nitroso‐N‐acetyl‐dl‐penicillamine (SNAP, 20 μmol/L), or VEGF+aPLs+SNAP for 24 hours. Values are mean±SEM, n=8/group, *P<0.05 vs basal, †P<0.05 vs NHIgG, #P<0.05 vs no SNAP. B, Scratch assays were performed with BAEC as described in (A), using the control IgG BBG or anti‐β2GPI antibody 3F8 instead of NHIgG or aPLs, respectively. Values are mean±SEM, n=8/group, *P<0.05 vs basal, †P<0.05 vs BBG, #P<0.05 vs no SNAP. C, Carotid artery reendothelialization was evaluated in aPL‐treated male C57BL/6 mice administered vehicle (saline) or molsidomine (5 mg/kg per day). Representative images of the carotid artery intimal surface displaying Evans blue dye incorporation are shown. D, Summary data for the area of remaining denudation at 72 hours are expressed in arbitrary units. Values are mean±SEM, n=6 for saline‐treated group (median was 7568) and n=7 for molsidomine‐treated group (median was 182). *P<0.05 vs saline.
Figure 7.
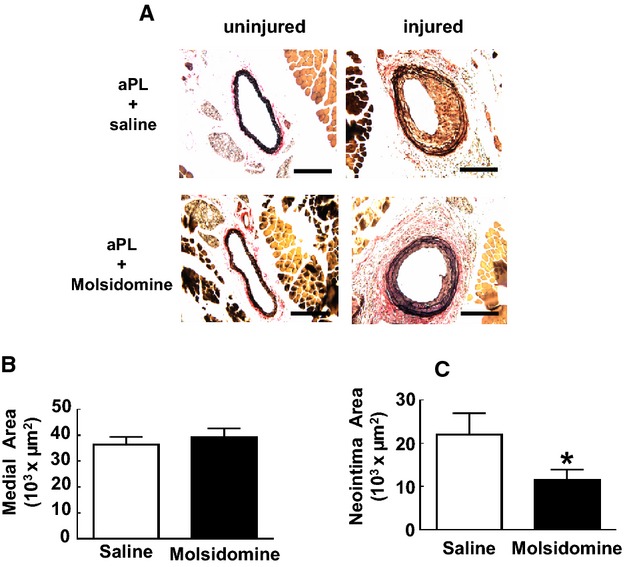
Induction of neointima formation by antiphospholipid antibodies (aPLs) entail NO deficiency. A, Male wild‐type mice were treated with aPLs (100 μg/mouse) 1 day before carotid artery endothelial denudation by using an epon‐resin probe, and every other day after denudation for 2 weeks. Mice were also given daily intraperitoneal injections of vehicle (saline) or molsidomine (5 mg/kg per day). Medial hypertrophy and neointima formation were then evaluated by histologic analysis. Representative photomicrographs are shown of uninjured (left panels) and injured carotid arteries (right panels). B, Medial area was calculated as the area encircled by the external elastic lamina minus the area encircled by the internal elastic lamina. Values are mean±SEM, n=9 for saline‐treated group (median was 33 889) and n=8 for molsidomine‐treated group (median was 39 660). C, Neoinimal area was calculated by subtracting the luminal area from the area encircled by the internal elastic lamina. Values are mean±SEM, n=9 for saline‐treated group (median was 18 917) and n=8 for molsidomine‐treated group (median was 12 661). *P<0.05 vs saline control. IP indicates intraperitoneal.
Discussion
In addition to having greater risk for episodic occurrence of thrombosis, APS patients are predisposed to nonthrombotic vascular disorders. These include exaggerated carotid artery IMT, which is a preclinical indicator of atherosclerosis evident in APS patients independent of other cardiovascular risk factors,4–6,8,46–47 and arterial stenosis in various sites.9,11–15 In the present study, we demonstrate for the first time that aPLs promote neointimal hyperplasia after endothelial injury in mice. We further show that this is related to the attenuation of endothelial monolayer repair and that this involves direct adverse effects of aPLs on endothelial cells. When considered along with our prior discovery that eNOS antagonism by aPLs underlies the thrombotic diathesis characteristic of APS,21 these new observations indicate that direct actions of aPLs on endothelium likely play a major role in both the thrombotic and the nonthrombotic components of vascular disease in individuals with APS.
There are several clinical disorders in which the effects of aPLs on endothelial repair that adversely affect vascular smooth muscle behavior may be applicable. One such disorder is stroke, because APS patients with increased IMT have 3 times greater risk for stroke than do APS patients without IMT.8 A second condition is peripheral vascular disease, which is often a proliferative vasculopathy that leads to stenosis. Patients with APS are 80% more likely to undergo lower extremity vascular surgery than are patients without APS,48 and APS is an independent risk factor for more rapid progression of lower extremity occlusive vascular disease.49 APS patients are also more likely to have premature restenosis of coronary artery bypass grafts.50 Thus, the processes revealed in the current work are relevant not only to preclinical forms of vascular disease such as IMT but also to clinically apparent vascular disorders in APS patients.
Although originally thought to directly recognize anionic phospholipids on the surface of target cells, it has become apparent that pathologically relevant aPLs are directed against the phospholipid‐binding protein β2GPI.17–19 β2GPI is a plasma protein composed of 5 distinct domains (domains I to V), and antibodies against β2GPI increase adhesion molecule expression and the synthesis of cytokines, endothelin‐1, and tissue factor by cultured endothelial cells.20,51 Recent work has further revealed that antibodies specifically recognizing domain I of β2GP are an important pathogenetic component of aPLs.17–20 We previously showed that monoclonal antibodies to domain I of β2GPI cause potent inhibition of eNOS.21 eNOS plays an important role in endothelial repair and in protection against neointimal hyperplasia.43,52 Now we demonstrate in culture that a monoclonal antibody against β2GPI domain I inhibits endothelial cell migration, mimicking the effect of polyclonal human aPLs. In addition, we have implicated ApoER2 in aPL antagonism of cultured endothelial cell migration and in aPL attenuation of endothelial repair in vivo. We and others previously identified ApoER2 as a key transmembrane receptor operative in aPL effects on both endothelial cells and platelets.21,42,53 APL binding to β2GPI induces β2GPI dimerization, which allows the aPL–β2GPI complex to avidly bind to ApoER2.21,53–54 ApoER2 is required for aPL‐induced antagonism of endothelial cell eNOS and the subsequent increase in leukocyte–endothelial cell interaction and thrombus formation.21,42 Using NO donors both in cell culture and in vivo, we now demonstrate that NO deficiency also participates in aPL antagonism of endothelial cell migration and repair. Intricate strategies such as peptide blockade of β2GPI interaction with ApoER2 have been contemplated to combat the clinical sequelae of APS.55–57 However, because we now know that eNOS antagonism underlies both APS‐associated thrombosis21 and APS‐related defects in endothelial monolayer repair contributing to nonthrombotic vascular occlusion, more practical treatments aimed at enhancing NO bioavailability or action can now be considered to combat both the thrombotic and nonthrombotic components of APS‐associated vascular disease.
In addition to resident endothelial cells in the arterial wall, circulating endothelial cells and endothelial progenitor cells may contribute to vascular repair in vivo.58–59 However, these cell populations are not altered in APS patients,60–61 and as such it is unlikely that changes in their function contribute to the attenuation in endothelial repair caused by aPLs in the current study. Leukocytes also modulate vascular repair62–63 and aPL antagonism of eNOS promotes leukocyte–endothelial adhesion,21 and the antibodies also induce tissue factor expression in monocytes,17–18 which can promote neointima formation.64 Thus, there may be processes besides the presently described direct impact of aPLs on endothelial cell migration that may additionally contribute to the exaggerated neointima hyperplasia observed in our study.
Along with experiencing greater arterial and venous thrombosis and increased risk of coronary artery disease, myocardial infarction, and cerebrovascular disease,3 female patients with APS have increased complications during pregnancy, including fetal loss, preterm birth, intrauterine growth restriction, preeclampsia, and eclampsia.1,17,65 Although the current work focuses on processes relevant to diseases unrelated to pregnancy, the mechanisms elucidated may also be applicable to the obstetric complications of APS because inhibitory effects of aPLs on primary human endometrial endothelial cell angiogenesis have been reported.66–67 In these studies, there was decreased tube formation in matrigel in culture and blunted in vivo angiogenesis in mice caused by aPLs, as well as aPL suppression of endothelial cell VEGF production. Now, knowing that the blunting of endothelial cell migration by aPLs entails ApoER2 and a decrease in NO bioavailability and that the motility of both systemically derived and endometrial endothelium are tempered by aPLs, the participation of ApoER2 and NO deficiency in preclinical models of obstetric APS can be tested.
Although there is considerable knowledge of the basis for APS‐related thrombosis, how APS affects nonthrombotic vascular disease has been more enigmatic. We now report that aPLs promote neointima formation after endothelial injury in mice, that this is related to the blunting of endothelial repair, and that the attenuation of endothelial repair by aPLs entails NO deficiency mediated by β2GPI and ApoER2. The modulation of endothelial ApoER2 function or NO bioavailability may represent new interventions to prevent the nonthrombotic vascular occlusion and resulting cardiovascular disorders that afflict APS patients.
Acknowledgment
We thank Mohamed Ahmed for technical assistance.
Sources of Funding
This work was supported by NIH HL109604 (Mineo), HL118001 (Shaul and Hui), DK074932 (Hui) and HL20948 (Herz), and by the American Heart Association (12PRE8850012, Ulrich). Additional support was provided by the Crystal Charity Ball Center for Pediatric Critical Care Research (Shaul). We are also indebted to Dr Eugene Frenkel (UTSW) for his assistance recruiting APS patients through support from the Raymond Nasher Cancer Research Fund.
Disclosures
Shen served as a speaker for Janssen and Alexion. The other authors have no conflicting financial interests.
References
- 1.Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med. 2002; 346:752-763. [DOI] [PubMed] [Google Scholar]
- 2.Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, Derksen RH, de Groot PG, Koike T, Meroni PL, Reber G, Shoenfeld Y, Tincani A, Vlachoyiannopoulos PG, Krilis SA. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006; 4:295-306. [DOI] [PubMed] [Google Scholar]
- 3.Soltesz P, Szekanecz Z, Kiss E, Shoenfeld Y. Cardiac manifestations in antiphospholipid syndrome. Autoimmun Rev. 2007; 6:379-386. [DOI] [PubMed] [Google Scholar]
- 4.Ames PR, Antinolfi I, Scenna G, Gaeta G, Margaglione M, Margarita A. Atherosclerosis in thrombotic primary antiphospholipid syndrome. J Thromb Haemost. 2009; 7:537-542. [DOI] [PubMed] [Google Scholar]
- 5.Der H, Kerekes G, Veres K, Szodoray P, Toth J, Lakos G, Szegedi G, Soltesz P. Impaired endothelial function and increased carotid intima‐media thickness in association with elevated von Willebrand antigen level in primary antiphospholipid syndrome. Lupus. 2007; 16:497-503. [DOI] [PubMed] [Google Scholar]
- 6.Ames PR, Margarita A, Sokoll KB, Weston M, Brancaccio V. Premature atherosclerosis in primary antiphospholipid syndrome: preliminary data. Ann Rheum Dis. 2005; 64:315-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ames PR, Margarita A, Delgado AJ, Tommasino C, Iannaccone L, Brancaccio V. Anticardiolipin antibody titre and plasma homocysteine level independently predict intima media thickness of carotid arteries in subjects with idiopathic antiphospholipid antibodies. Lupus. 2002; 11:208-214. [DOI] [PubMed] [Google Scholar]
- 8.Medina G, Casaos D, Jara LJ, Vera‐Lastra O, Fuentes M, Barile L, Salas M. Increased carotid artery intima‐media thickness may be associated with stroke in primary antiphospholipid syndrome. Ann Rheum Dis. 2003; 62:607-610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenthal E, Sangle SR, Taylor P, Khamashta MA, Hughes GR, D'Cruz DP. Treatment of mesenteric angina with prolonged anticoagulation in a patient with antiphospholipid (Hughes) syndrome and coeliac artery stenosis. Ann Rheum Dis. 2006; 65:1398-1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Remondino GI, Mysler E, Pissano MN, Furattini MC, Basta MC, Presas JL, Allievi A. A reversible bilateral renal artery stenosis in association with antiphospholipid syndrome. Lupus. 2000; 9:65-67. [DOI] [PubMed] [Google Scholar]
- 11.Sangle SR, Jan W, Lau IS, Bennett AN, Hughes GR, D'Cruz DP. Coeliac artery stenosis and antiphospholipid (Hughes) syndrome/antiphospholipid anti‐bodies. Clin Exp Rheumatol. 2006; 24:349. [PubMed] [Google Scholar]
- 12.Sangle SR, D'Cruz DP, Jan W, Karim MY, Khamashta MA, Abbs IC, Hughes GR. Renal artery stenosis in the antiphospholipid (Hughes) syndrome and hypertension. Ann Rheum Dis. 2003; 62:999-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sangle SR, D'Cruz DP. Renal artery stenosis: a new facet of the antiphospholipid (Hughes) syndrome. Lupus. 2003; 12:803-804. [DOI] [PubMed] [Google Scholar]
- 14.Sangle SR, D'Cruz DP, Abbs IC, Khamashta MA, Hughes GR. Renal artery stenosis in hypertensive patients with antiphospholipid (Hughes) syndrome: outcome following anticoagulation. Rheumatology (Oxford). 2005; 44:372-377. [DOI] [PubMed] [Google Scholar]
- 15.Peleg H, Bursztyn M, Hiller N, Hershcovici T. Renal artery stenosis with significant proteinuria may be reversed after nephrectomy or revascularization in patients with the antiphospholipid antibody syndrome: a case series and review of the literature. Rheumatol Int. 2012; 32:85-90. [DOI] [PubMed] [Google Scholar]
- 16.de Groot PG, Derksen RH. Pathophysiology of the antiphospholipid syndrome. J Thromb Haemost. 2005; 3:1854-1860. [DOI] [PubMed] [Google Scholar]
- 17.Meroni PL, Borghi MO, Raschi E, Tedesco F. Pathogenesis of antiphospholipid syndrome: understanding the antibodies. Nat Rev Rheumatol. 2011; 7:330-339. [DOI] [PubMed] [Google Scholar]
- 18.Giannakopoulos B, Krilis SA. The pathogenesis of the antiphospholipid syndrome. N Engl J Med. 2013; 368:1033-1044. [DOI] [PubMed] [Google Scholar]
- 19.de Laat B, de Groot PG. Autoantibodies directed against domain I of beta2‐glycoprotein I. Curr Rheumatol Rep. 2011; 13:70-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gushiken FC, Arnett FC, Thiagarajan P. Primary antiphospholipid antibody syndrome with mutations in the phospholipid binding domain of beta(2)‐glycoprotein I. Am J Hematol. 2000; 65:160-165. [DOI] [PubMed] [Google Scholar]
- 21.Ramesh S, Morrell CN, Tarango C, Thomas GD, Yuhanna IS, Girardi G, Herz J, Urbanus RT, de Groot PG, Thorpe PE, Salmon JE, Shaul PW, Mineo C. Antiphospholipid antibodies promote leukocyte‐endothelial cell adhesion and thrombosis in mice by antagonizing eNOS via beta2GPI and apoER2. J Clin Invest. 2011; 121:120-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niimi Y, Azuma H, Hirakawa K. Repeated endothelial removal augments intimal thickening and attenuates EDRF release. Am J Physiol. 1994; 266:H1348-H1356. [DOI] [PubMed] [Google Scholar]
- 23.Cunningham KS, Gotlieb AI. The role of shear stress in the pathogenesis of atherosclerosis. Lab Invest. 2005; 85:9-23. [DOI] [PubMed] [Google Scholar]
- 24.Grote K, Sonnenschein K, Kapopara PR, Hillmer A, Grothusen C, Salguero G, Kotlarz D, Schuett H, Bavendiek U, Schieffer B. Toll‐like receptor 2/6 agonist macrophage‐activating lipopeptide‐2 promotes reendothelialization and inhibits neointima formation after vascular injury. Arterioscler Thromb Vasc Biol. 2013; 33:2097-2104. [DOI] [PubMed] [Google Scholar]
- 25.Kipshidze N, Dangas G, Tsapenko M, Moses J, Leon MB, Kutryk M, Serruys P. Role of the endothelium in modulating neointimal formation: vasculoprotective approaches to attenuate restenosis after percutaneous coronary interventions. J Am Coll Cardiol. 2004; 44:733-739. [DOI] [PubMed] [Google Scholar]
- 26.Ma X, Hibbert B, Dhaliwal B, Seibert T, Chen YX, Zhao X, O'Brien ER. Delayed re‐endothelialization with rapamycin‐coated stents is rescued by the addition of a glycogen synthase kinase‐3beta inhibitor. Cardiovasc Res. 2010; 86:338-345. [DOI] [PubMed] [Google Scholar]
- 27.Jones CI, Barrett NE, Moraes LA, Gibbins JM, Jackson DE. Endogenous inhibitory mechanisms and the regulation of platelet function. Methods Mol Biol. 2012; 788:341-366. [DOI] [PubMed] [Google Scholar]
- 28.Voetsch B, Jin RC, Loscalzo J. Nitric oxide insufficiency and atherothrombosis. Histochem Cell Biol. 2004; 122:353-367. [DOI] [PubMed] [Google Scholar]
- 29.Freedman JE. Oxidative stress and platelets. Arterioscler Thromb Vasc Biol. 2008; 28:s11-s16. [DOI] [PubMed] [Google Scholar]
- 30.Zhu B, Kuhel DG, Witte DP, Hui DY. Apolipoprotein E inhibits neointimal hyperplasia after arterial injury in mice. Am J Pathol. 2000; 157:1839-1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He J, Luster TA, Thorpe PE. Radiation‐enhanced vascular targeting of human lung cancers in mice with a monoclonal antibody that binds anionic phospholipids. Clin Cancer Res. 2007; 13:5211-5218. [DOI] [PubMed] [Google Scholar]
- 32.Schwartz R, Osborne‐Lawrence S, Hahner L, Gibson LL, Gormley AK, Vongpatanasin W, Zhu W, Word RA, Seetharam D, Black S, Samols D, Mineo C, Shaul PW. C‐reactive protein downregulates endothelial NO synthase and attenuates reendothelialization in vivo in mice. Circ Res. 2007; 100:1452-1459. [DOI] [PubMed] [Google Scholar]
- 33.Seetharam D, Mineo C, Gormley AK, Gibson LL, Vongpatanasin W, Chambliss KL, Hahner LD, Cummings ML, Kitchens RL, Marcel YL, Rader DJ, Shaul PW. High‐density lipoprotein promotes endothelial cell migration and reendothelialization via scavenger receptor‐B type I. Circ Res. 2006; 98:63-72. [DOI] [PubMed] [Google Scholar]
- 34.Hui DY. Intimal hyperplasia in murine models. Curr Drug Targets. 2008; 9:251-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gupta SK, Singh JP. Inhibition of endothelial cell proliferation by platelet factor‐4 involves a unique action on S phase progression. J Cell Biol. 1994; 127:1121-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Basford JE, Moore ZW, Zhou L, Herz J, Hui DY. Smooth muscle LDL receptor‐related protein‐1 inactivation reduces vascular reactivity and promotes injury‐induced neointima formation. Arterioscler Thromb Vasc Biol. 2009; 29:1772-1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Urbanus RT, Derksen RH, de Groot PG. Platelets and the antiphospholipid syndrome. Lupus. 2008; 17:888-894. [DOI] [PubMed] [Google Scholar]
- 38.Vega‐Ostertag ME, Pierangeli SS. Mechanisms of aPL‐mediated thrombosis: effects of aPL on endothelium and platelets. Curr Rheumatol Rep. 2007; 9:190-197. [DOI] [PubMed] [Google Scholar]
- 39.Doran AC, Meller N, McNamara CA. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008; 28:812-819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pierangeli SS, Vega‐Ostertag ME, Raschi E, Liu X, Romay‐Penabad Z, De Micheli V, Galli M, Moia M, Tincani A, Borghi MO, Nguyen‐Oghalai T, Meroni PL. Toll‐like receptor and antiphospholipid mediated thrombosis: in vivo studies. Ann Rheum Dis. 2007; 66:1327-1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cockrell E, Espinola RG, McCrae KR. Annexin A2: biology and relevance to the antiphospholipid syndrome. Lupus. 2008; 17:943-951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Romay‐Penabad Z, guilar‐Valenzuela R, Urbanus RT, Derksen RH, Pennings MT, Papalardo E, Shilagard T, Vargas G, Hwang Y, Groot PG, Pierangeli SS. Apolipoprotein E receptor 2 is involved in the thrombotic complications in a murine model of the antiphospholipid syndrome. Blood. 2011; 117:1408-1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kawasaki K, Smith RS, Jr, Hsieh CM, Sun J, Chao J, Liao JK. Activation of the phosphatidylinositol 3‐kinase/protein kinase Akt pathway mediates nitric oxide‐induced endothelial cell migration and angiogenesis. Mol Cell Biol. 2003; 23:5726-5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Michaelis UR, Fleming I. From endothelium‐derived hyperpolarizing factor (EDHF) to angiogenesis: epoxyeicosatrienoic acids (EETs) and cell signaling. Pharmacol Ther. 2006; 111:584-595. [DOI] [PubMed] [Google Scholar]
- 45.Singh RJ, Hogg N, Joseph J, Konorev E, Kalyanaraman B. The peroxynitrite generator, SIN‐1, becomes a nitric oxide donor in the presence of electron acceptors. Arch Biochem Biophys. 1999; 361:331-339. [DOI] [PubMed] [Google Scholar]
- 46.Charakida M, Besler C, Batuca JR, Sangle S, Marques S, Sousa M, Wang G, Tousoulis D, Delgado AJ, Loukogeorgakis SP, Kworth‐Young C, DCruz D, Luscher T, Landmesser U, Deanfield JE. Vascular abnormalities, paraoxonase activity, and dysfunctional HDL in primary antiphospholipid syndrome. JAMA. 2009; 302:1210-1217. [DOI] [PubMed] [Google Scholar]
- 47.Belizna CC, Richard V, Primard E, Kerleau JM, Cailleux N, Louvel JP, Marie I, Hamidou M, Thuillez C, Levesque H. Early atheroma in primary and secondary antiphospholipid syndrome: an intrinsic finding. Semin Arthritis Rheum. 2008; 37:373-380. [DOI] [PubMed] [Google Scholar]
- 48.Taylor LM, Jr, Chitwood RW, Dalman RL, Sexton G, Goodnight SH, Porter JM. Antiphospholipid antibodies in vascular surgery patients. A cross‐sectional study. Ann Surg. 1994; 220:544-550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lam EY, Taylor LM, Jr, Landry GJ, Porter JM, Moneta GL. Relationship between antiphospholipid antibodies and progression of lower extremity arterial occlusive disease after lower extremity bypass operations. J Vasc Surg. 2001; 33:976-982. [DOI] [PubMed] [Google Scholar]
- 50.Long BR, Leya F. The role of antiphospholipid syndrome in cardiovascular disease. Hematol Oncol Clin North Am. 2008; 22:79-94. [DOI] [PubMed] [Google Scholar]
- 51.Pierangeli SS, Vega‐Ostertag M, Harris EN. Intracellular signaling triggered by antiphospholipid antibodies in platelets and endothelial cells: a pathway to targeted therapies. Thromb Res. 2004; 114:467-476. [DOI] [PubMed] [Google Scholar]
- 52.Moroi M, Zhang L, Yasuda T, Virmani R, Gold HK, Fishman MC, Huang PL. Interaction of genetic deficiency of endothelial nitric oxide, gender, and pregnancy in vascular response to injury in mice. J Clin Invest. 1998; 101:1225-1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Urbanus RT, Pennings MT, Derksen RH, de Groot PG. Platelet activation by dimeric beta2‐glycoprotein I requires signaling via both glycoprotein Ibalpha and apolipoprotein E receptor 2'. J Thromb Haemost. 2008; 6:1405-1412. [DOI] [PubMed] [Google Scholar]
- 54.de Groot PG, Lummel M, Pennings M, Urbanus R, Bas de Laat H, Lenting PJ, Derksen RH. Beta2‐glycoprotein I and LDL‐receptor family members. Thromb Res. 2004; 114:455-459. [DOI] [PubMed] [Google Scholar]
- 55.Ostertag MV, Liu X, Henderson V, Pierangeli SS. A peptide that mimics the Vth region of beta‐2‐glycoprotein I reverses antiphospholipid‐mediated thrombosis in mice. Lupus. 2006; 15:358-365. [DOI] [PubMed] [Google Scholar]
- 56.Ioannou Y, Romay‐Penabad Z, Pericleous C, Giles I, Papalardo E, Vargas G, Shilagard T, Latchman DS, Isenberg DA, Rahman A, Pierangeli S. In vivo inhibition of antiphospholipid antibody‐induced pathogenicity utilizing the antigenic target peptide domain I of beta2‐glycoprotein I: proof of concept. J Thromb Haemost. 2009; 7:833-842. [DOI] [PubMed] [Google Scholar]
- 57.Blank M, Shoenfeld Y, Cabilly S, Heldman Y, Fridkin M, Katchalski‐Katzir E. Prevention of experimental antiphospholipid syndrome and endothelial cell activation by synthetic peptides. Proc Natl Acad Sci USA. 1999; 96:5164-5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Asahara T, Isner JM. Endothelial progenitor cells for vascular regeneration. J Hematother Stem Cell Res. 2002; 11:171-178. [DOI] [PubMed] [Google Scholar]
- 59.Isner JM, Kalka C, Kawamoto A, Asahara T. Bone marrow as a source of endothelial cells for natural and iatrogenic vascular repair. Ann N Y Acad Sci. 2001; 953:75-84. [DOI] [PubMed] [Google Scholar]
- 60.Gresele P, Migliacci R, Vedovati MC, Ruffatti A, Becattini C, Facco M, Guglielmini G, Boscaro E, Mezzasoma AM, Momi S, Pengo V. Patients with primary antiphospholipid antibody syndrome and without associated vascular risk factors present a normal endothelial function. Thromb Res. 2009; 123:444-451. [DOI] [PubMed] [Google Scholar]
- 61.Meroni PL, Borghi MO, Raschi E, Ventura D, Sarzi Puttini PC, Atzeni F, Lonati L, Parati G, Tincani A, Mari D, Tedesco F, Inflammatory response, the endothelium. Thromb Res. 2004; 114::329-334. [DOI] [PubMed] [Google Scholar]
- 62.Saxena A, Rauch U, Berg KE, Andersson L, Hollender L, Carlsson AM, Gomez MF, Hultgardh‐Nilsson A, Nilsson J, Bjorkbacka H. The vascular repair process after injury of the carotid artery is regulated by IL‐1RI and MyD88 signalling. Cardiovasc Res. 2011; 91:350-357. [DOI] [PubMed] [Google Scholar]
- 63.Simon DI. Inflammation and vascular injury: basic discovery to drug development. Circ J. 2012; 76:1811-1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Breitenstein A, Camici GG, Tanner FC. Tissue factor: beyond coagulation in the cardiovascular system. Clin Sci (Lond). 2010; 118:159-172. [DOI] [PubMed] [Google Scholar]
- 65.Ruiz‐Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet. 2010; 376:1498-1509. [DOI] [PubMed] [Google Scholar]
- 66.Di Simone N, D'Ippolito S, Marana R, Di NF, Castellani R, Pierangeli SS, Chen P, Tersigni C, Scambia G, Meroni PL. Antiphospholipid antibodies affect human endometrial angiogenesis: protective effect of a synthetic peptide (TIFI) mimicking the phospholipid binding site of beta glycoprotein I. Am J Reprod Immunol. 2013; 70:299-308. [DOI] [PubMed] [Google Scholar]
- 67.Di Simone N, Di NF, D'Ippolito S, Castellani R, Tersigni C, Caruso A, Meroni P, Marana R. Antiphospholipid antibodies affect human endometrial angiogenesis. Biol Reprod. 2010; 83:212-219. [DOI] [PubMed] [Google Scholar]