

Abstract
Background
Whether the type of dietary fat could alter cardiometabolic responses to a hypercaloric diet is unknown. In addition, subclinical cardiometabolic consequences of moderate weight gain require further study.
Methods and Results
In a 7‐week, double‐blind, parallel‐group, randomized controlled trial, 39 healthy, lean individuals (mean age of 27±4) consumed muffins (51% of energy [%E] from fat and 44%E refined carbohydrates) providing 750 kcal/day added to their habitual diets. All muffins had identical contents, except for type of fat; sunflower oil rich in polyunsaturated fatty acids (PUFA diet) or palm oil rich in saturated fatty acids (SFA diet). Despite comparable weight gain in the 2 groups, total: high‐density lipoprotein (HDL) cholesterol, low‐density lipoprotein:HDL cholesterol, and apolipoprotein B:AI ratios decreased during the PUFA versus the SFA diet (−0.37±0.59 versus +0.07±0.29, −0.31±0.49 versus +0.05±0.28, and −0.07±0.11 versus +0.01±0.07, P=0.003, P=0.007, and P=0.01 for between‐group differences), whereas no significant differences were observed for other cardiometabolic risk markers. In the whole group (ie, independently of fat type), body weight increased (+2.2%, P<0.001) together with increased plasma proinsulin (+21%, P=0.007), insulin (+17%, P=0.003), proprotein convertase subtilisin/kexin type 9, (+9%, P=0.008) fibroblast growth factor‐21 (+31%, P=0.04), endothelial markers vascular cell adhesion molecule–1, intercellular adhesion molecule–1, and E‐selectin (+9, +5, and +10%, respectively, P<0.01 for all), whereas nonesterified fatty acids decreased (−28%, P=0.001).
Conclusions
Excess energy from PUFA versus SFA reduces atherogenic lipoproteins. Modest weight gain in young individuals induces hyperproinsulinemia and increases biomarkers of endothelial dysfunction, effects that may be partly outweighed by the lipid‐lowering effects of PUFA.
Clinical Trial Registration
URL: http://ClinicalTrials.gov. Unique identifier: NCT01427140.
Keywords: Atherosclerosis, lipids, lipoprotein, nutrition, obesity
Introduction
Central obesity increases the risk of diabetes and cardiovascular disease (CVD). Adult populations tend to gradually increase in weight, explained in part by sedentary lifestyles and high availability of palatable, energy‐dense foods, which facilitates excess energy intake. Moderate weight‐loss improves several metabolic risk factors, but the cardiometabolic consequences of moderate weight gain have been less investigated. The possibility of preventing or reversing cardiovascular disease by dietary interventions has received much interest recently, and the potential anti‐atherogenic effects of unsaturated fats in particular.1 Most dietary interventions, however, have been weight‐loss trials or isocaloric trials, the latter aiming at separating the effect of dietary composition from that of weight change. During isocaloric conditions, polyunsaturated fatty acids (PUFA) reduce low‐density lipoprotein (LDL) cholesterol compared with saturated fatty acids (SFA).2–3 Partial replacement of SFA with PUFA has been associated with moderately reduced risk in cohort studies and controlled trials.4–5 However, it is unknown whether the type of dietary fat plays any role during energy excess and weight gain. Such information would be highly relevant in dietary prevention of obesity‐related disorders, especially considering that a large part of the population is overeating and is in chronic positive energy balance. We recently reported that PUFA causes less liver and visceral fat accumulation than do SFA,6 but the influence on plasma lipoproteins and other CVD risk markers has not been reported. We aimed to study the effects of dietary fat composition during modest weight gain, in a double‐blind, randomized, 2 parallel‐group overfeeding trial using muffins that only differed in the type of fat. We hypothesized that PUFA from sunflower oil could counteract some of the adverse cardiometabolic effects associated with diet‐induced weight gain, in comparison with SFA from palm oil.
Methods
Participants
Fifty‐five healthy volunteers were recruited by local advertising. The details of the study design have been previously described.6 In brief, inclusion criteria were age 20 to 38 years and body mass index 18 to 27 kg/m2, and exclusion criteria were, among others: diabetes; abnormal clinical chemistry results during screening; intolerance to gluten, egg, or milk; medications influencing energy metabolism; omega‐3 supplements; and heavy exercise >3 times/week. All subjects gave written informed consent, and the study was approved by the regional ethical committee.
Study Design
The LIPOGAIN study was a 7‐week, double‐blind, parallel‐group, randomized trial, carried out in Uppsala, Sweden from August through December 2011. The details of the study design and protocol have been previously reported.6
Outcome Measures
The primary outcomes of this study were liver fat content and body composition, measured with magnetic resonance imaging, results that have been previously reported.6 The present study reports on the following parameters: predefined secondary outcome measures of cardiometabolic risk factors including systolic and diastolic blood pressure, fasting blood lipoproteins, lathosterol, proprotein convertase subtilisin/kexin type 9 (PCSK9, a hepatic LDL‐receptor regulator), fibroblast growth factor 21 (FGF21) and nonesterified fatty acids (NEFA); glucose, insulin, homeostasis model assessment of insulin resistance, and proinsulin; markers of endothelial function and coagulation vascular cell adhesion molecule‐1, intercellular adhesion molecule‐1, E‐selectin, endostatin, and von Willebrand factor; and inflammation markers C‐reactive protein, interleukin (IL)‐6, IL‐8, IL‐15, IL‐1 receptor antagonist, tumor necrosis factor α (TNFα), and soluble TNF receptor 2. Samples with C‐reactive protein level ≥10 mg/L were excluded from analyses of inflammation markers, to avoid bias due to acute infections.
Dietary Intervention
Participants were instructed to maintain their habitual diet and physical activity level. The PUFA group received muffins high in n‐6 PUFA from sunflower oil containing mainly linoleic acid (18:2n‐6) and the SFA group received muffins high in SFA from palm oil (ie, not palm olein), containing mainly palmitic acid (16:0). The fatty acid profiles of the oils have been described in detail.6 In brief, sunflower oil comprised 65% 18:2n‐6 PUFA, 24% 18:1n‐9, and 6% 16:0, whereas palm oil comprised 9% 18:2n‐6, 37% 18:1n‐9, and 48% 16:0. Both oils were refined, but not hydrogenated. The muffins were otherwise identical in ingredients, taste, and structure; they were baked in large batches under standardized conditions in a metabolic kitchen at Uppsala University, distributed weekly at the clinic, and added to the participants' habitual diets. Each muffin provided 240 kcal of which 51% of energy (%E) was fat, 5%E protein, and 44%E carbohydrate. The amount of muffins was initially 4 per day and increased or decreased by 1 muffin per day depending on the rate of weight gain of the individual, aiming at 3% weight gain in 7 weeks. There was no prespecified period of weight stability during the study. Participants were instructed to abstain from physical exercise or alcohol intake 48 hours before measurements.
Laboratory Analyses
Blood samples were drawn after an overnight fast (12 hours). Blood pressure was measured by an automatic upper arm monitor (Omron M6; OMRON Healthcare Europe, Hoofddorp, the Netherlands). Blood lipids, glucose, insulin, and C‐reactive protein were measured by standard laboratory methods at Uppsala University Hospital. Homeostasis model assessment of insulin resistance was calculated as glucose×insulin/22.5. ELISA was used to determine proinsulin (Mercodia, Uppsala, Sweden), PCSK9 (CycLex, Nagano, Japan), FGF21, IL‐6, IL‐8, IL‐15, IL‐1 receptor antagonist, TNFα, vascular cell adhesion molecule‐1, intercellular adhesion molecule‐1, E‐selectin, endostatin, and TNF receptor 2 (R&D Systems, Minneapolis, MN). Serum unesterified lathosterol was determined by isotope dilution mass spectrometry and corrected for total cholesterol.7 Total NEFA were analyzed using the NEFA FS response kit (Cat. No. 157819910921; DiaSys Diagnostic Systems, Holzheim, Germany), an enzymatic end point method automatically measured on Response 910 (DiaSys Diagnostic Systems) according to the manufacturer's instructions. Von Willebrand factor was measured by ELISA with antisera from Dako (Glostrup, Denmark). The assay was calibrated against Liatest (DiagnosticaStago, Asnieres, France).
Dietary Assessment, Physical Activity, and Compliance
Dietary intake was assessed by 4‐day weighed food records at baseline and at week 7. Dietist XP 3.1 was used for processing of food records. During the same days, participants wore accelerometers (Philips Respironics Actical, Philips Healthcare, DA Best, The Netherlands). Fatty acid composition was measured in plasma cholesterol esters and adipose tissue by gas chromatography.8–9
Statistical Analysis
Power calculations were performed based on the primary outcome liver fat content. Non‐normally distributed variables were log‐transformed, or analyzed with nonparametric tests if normality was not attained as judged by Shapiro–Wilk's test and by visually examining QQ‐plots. Normally distributed variables are given as mean±SD and non‐normally distributed variables as median (interquartile range). Analyses were performed on an intention‐to‐treat basis. Differences in changes between groups were analyzed with a t test or the Mann–Whitney U test. Adjustments for baseline values were not performed. Differences from baseline to follow‐up were analyzed with a paired t test or Wilcoxon signed‐rank test. P‐values <0.05 were considered statistically significant. Data were analyzed with SPSS version 21 and JMP version 10.0.0 software.
Results
Fifty‐five individuals were assessed for eligibility. Of these, n=41 met the inclusion criteria and were randomized. Two individuals left the study before baseline investigations. Of the remaining 39 individuals, all completed the study (Figure 1). The groups were similar at baseline with regard to age and sex, but there were slight imbalances in body weight, body mass index, and waist girth (Table 1). Five individuals were smokers and the vast majority (>90%) were white. It was not feasible to detect potential differences related to sex or race. PUFA and SFA groups consumed on average 3.2 and 3.1 muffins daily, respectively. Dietary intakes before and during the intervention have been described in detail elsewhere6: Total fat intake increased by 5±6%E in both groups, PUFA intake increased by 8±2%E in the PUFA group, and SFA intake increased by 5±3%E in the SFA group, which was also well reflected in both plasma and adipose fatty acids.6 As reported previously in detail,6 the overall diet during the intervention as assessed by repeated 4‐day food records was rather high in fat (≈37%E to 40%E) but more moderate in carbohydrate (≈43%E to 47%E). The added muffins provided ≈5.3%E more linoleic acid, and 4.4%E less palmitic acid, in the PUFA compared with the SFA group. Physical activity did not change or differ between groups, as reported elsewhere.6
Figure 1.
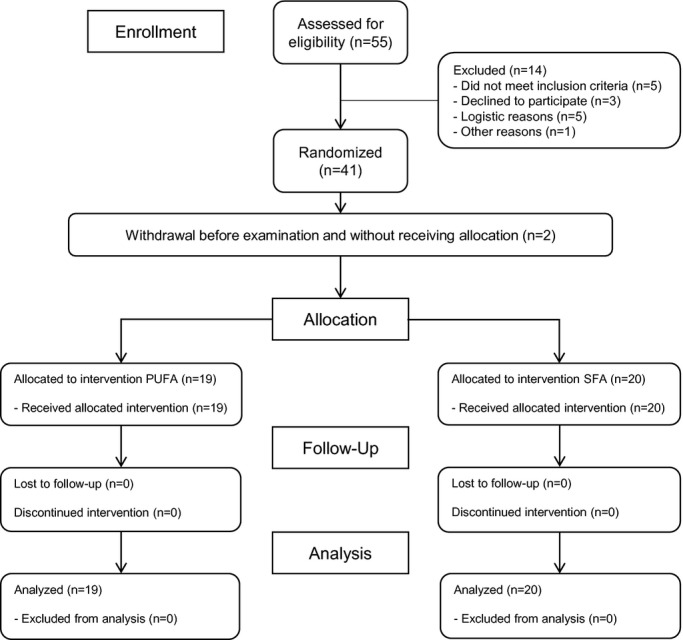
Flow diagram for the LIPOGAIN trial. In the present study, analyses were performed on an intention‐to‐treat basis. Modified with permission from Rosqvist et al.6 PUFA indicates polyunsaturated fatty acids; SFA, saturated fatty acids.
Table 1.
Baseline Characteristics
| Whole Sample (n=39) | PUFA Group (n=19) | SFA Group (n=20) | |
|---|---|---|---|
| Age, y | 26.9±4.2 | 26.5±4.6 | 27.4±3.8 |
| Female | 12 (31%) | 6 (32%) | 6 (30%) |
| Current smoker | 5 (13%) | 1 (5%) | 4 (20%) |
| Weight, kg | 65.5±7.6 | 67.7±8.1 | 63.5±6.6 |
| Height, cm | 177.9±8.3 | 177.1±6.8 | 178.7±9.7 |
| BMI, kg/m2 | 20.2 (19.2 to 21.5) | 20.8 (19.5 to 23.3) | 19.7 (18.8 to 20.6) |
| Waist girth, cm | 77.7±5.4 | 79.3±5.5 | 76.1±5.0 |
| Systolic blood pressure, mm Hg | 120.3±9.7 | 120.3±9.6 | 120.4±10.0 |
| Diastolic blood pressure, mm Hg | 73.9±8.1 | 74.2±8.3 | 73.6±8.0 |
Data given as no. (%), mean±SD, or median (interquartile range). BMI indicates body mass index; PUFA, polyunsaturated fatty acids, SFA, saturated fatty acids.
Body weight increased 2.2% or 1.5 kg, with little difference between groups: +1.44±1.1 kg in the PUFA group and +1.48±1.1 kg in the SFA group (P=0.92). Body mass index also increased 2.2% from 20.7 to 21.2 kg/m2 (P<0.001) with no difference between groups (P=0.96). Waist girth increased 1.1% from 77.7 to 78.6 cm (P=0.017) without difference between groups (P=0.85). Body composition data have been previously reported.6 Blood pressure did not change significantly (systolic +0.7 and diastolic +0.2 mm Hg, P>0.22) or differ between groups (P>0.47).
Between‐Group Comparisons
The total:high‐density lipoprotein (HDL) cholesterol, LDL:HDL cholesterol, and apolipoprotein B:AI ratios were lower in the PUFA group compared with the SFA group (Table 2 and Figure 2). There were no significant differences between groups in total, LDL, or HDL cholesterol, apolipoprotein B or AI, triglycerides, PCSK9, lathosterol, or FGF21 (P≥0.10), but non‐HDL cholesterol tended to be lower during the PUFA versus the SFA diet (P=0.06). There were no significant differences between groups for markers of endothelial function (Table 2) or inflammation (P>0.14, not shown).
Table 2.
Lipoproteins, Endothelial Function, and Inflammation Before and After 7 Weeks of Overeating PUFA or SFA
| PUFA Group* Baseline | PUFA Group* Week 7 | PUFA Group* Mean Change | SFA Group Baseline | SFA Group Week 7 | SFA Group Mean Change | P Value* | |
|---|---|---|---|---|---|---|---|
| Total cholesterol, mmol/L | 4.2 (3.9 to 4.6) | 4.2 (3.7 to 4.7) | −0.08±0.42 | 4.1 (3.6 to 5.1) | 4.2 (3.8 to 4.7) | 0.15±0.75 | 0.23 |
| LDL cholesterol, mmol/L | 2.1 (1.7 to 3.1) | 2.0 (1.6 to 2.7) | −0.18±0.33 | 2.3 (2.0 to 2.8) | 2.4 (2.1 to 2.8) | 0.07±0.56 | 0.11 |
| HDL cholesterol, mmol/L | 1.4 (1.0 to 1.5) | 1.4 (1.3 to 1.8) | 0.12±0.18 | 1.4 (1.2 to 1.5) | 1.4 (1.2 to 1.7) | 0.05±0.23 | 0.19 |
| Non‐HDL cholesterol, mmol/L | 2.8 (2.2 to 3.6) | 2.6 (2.2 to 3.2) | −0.21±0.42 | 2.6 (2.3 to 3.6) | 2.7 (2.6 to 3.4) | 0.10±0.58 | 0.06 |
| LDL:HDL cholesterol ratio | 1.62 (1.13 to 2.21) | 1.50 (1.07 to 1.86) | −0.31±0.49 | 1.70 (1.32 to 2.21) | 1.88 (1.48 to 2.25) | 0.05±0.28 | 0.007 |
| Total:HDL cholesterol ratio | 3.15 (2.47 to 3.50) | 2.83 (2.43 to 3.22) | −0.37±0.59 | 3.00 (2.74 to 3.58) | 3.25 (2.73 to 3.61) | 0.07±0.29 | 0.003 |
| Apolipoprotein B, g/L | 0.61 (0.49 to 0.77) | 0.55 (0.48 to 0.72) | −0.05±0.08 | 0.59 (0.55 to 0.74) | 0.62 (0.57 to 0.72) | 0.01±0.11 | 0.12 |
| Apolipoprotein AI, g/L | 1.3 (1.2 to 1.5) | 1.4 (1.2 to 1.6) | 0.06±0.12 | 1.3 (1.2 to 1.4) | 1.3 (1.1 to 1.4) | −0.00±0.17 | 0.28 |
| Apolipoprotein B:AI ratio | 0.47 (0.35 to 0.55) | 0.43 (0.32 to 0.51) | −0.07±0.11 | 0.48 (0.41 to 0.56) | 0.48 (0.40 to 0.61) | 0.01±0.07 | 0.014 |
| Triglycerides, mmol/L | 0.66 (0.54 to 0.77) | 0.65 (0.55 to 0.88) | −0.01±0.26 | 0.64 (0.51 to 0.73) | 0.56 (0.44 to 0.68) | −0.02±0.24 | 0.82 |
| NEFA, mmol/L | 0.6±0.2 | 0.4±0.1 | −0.2±0.2 | 0.5±0.2 | 0.4±0.2 | −0.1±0.2 | 0.17 |
| PCSK9, ng/mL | 172±35 | 198±34 | 25±33 | 190±52 | 197±46 | 6.8±34 | 0.10 |
| ICAM‐1, pg/L | 136000±24500 | 147000±38700 | 11300±20200 | 135000±19200 | 138000±18300 | 3350±11100 | 0.13 |
| VCAM‐1, pg/L | 329000±62100 | 357000±83500 | 27900±56800 | 344000±50400 | 379000±69200 | 35000±65200 | 0.72 |
| E‐selectin, pg/L | 3971±1461 | 4307±2138 | 337±1031 | 4080±1997 | 4506±1990 | 426±699 | 0.29 |
| Von Willebrand Factor, pg/L | 1.0±0.2 | 1.1±0.2 | 0.06±0.02 | 1.2±0.3 | 1.2±0.3 | −0.01±0.2 | 0.32 |
| Endostatin, pg/L | 40995±7359 | 42636±8249 | 1641±6614 | 40916±6608 | 42441±4908 | 1525±5428 | 0.95 |
| C‐reactive protein, mg/L | 0.5 (0.2 to 0.8) | 0.5 (0.3 to 0.8) | −0.1±2.0 | 0.3 (0.1 to 0.7) | 0.4 (0.3 to 0.8) | 0.09±1.3 | 0.65 |
Data given as mean±SD or median (interquartile range). HDL indicates high‐density lipoprotein; ICAM‐1, intercellular adhesion molecule‐1; LDL, low‐density lipoprotein; NEFA, nonesterified fatty acids; PCSK9, proprotein convertase subtilisin/kexin type 9; PUFA, polyunsaturated fatty acids; SFA, saturated fatty acids; VCAM‐1, vascular cell adhesion molecule‐1.
For apolipoproteins, triglycerides, and C‐reactive protein; PUFA group is n=18 because 1 sample was excluded because the individual was not fasting.
P‐value is for comparison between groups in change from baseline.
Figure 2.
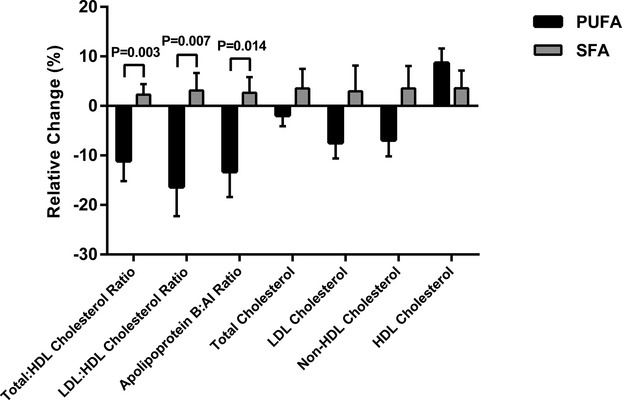
Changes (mean, SEM) in lipoproteins during PUFA and SFA overeating. Relative change denotes mean change/baseline mean. HDL indicates high‐density lipoprotein; LDL, low‐density lipoprotein; PUFA, polyunsaturated fatty acids; SFA, saturated fatty acids.
Whole‐Group Effects
In the 2 groups combined, weight gain was accompanied by a 6% increase in HDL cholesterol (+0.09±0.21 mmol/L, P=0.04), without significant changes in other blood lipids (Figure 3). PCSK9 increased (+9%, +15.8±34.7 ng/mL, P=0.007, Figure 3). There was no change (+2%, +19.4±318 ng/mL, P=0.71) for lathosterol. Total circulating NEFA decreased in the whole study sample during overfeeding (−28%, −0.15±0.24 mmol/L, P<0.001, Figure 3). FGF21 increased (+31%, +34.7±166 ng/mL, P=0.04, Figure 3).
Figure 3.
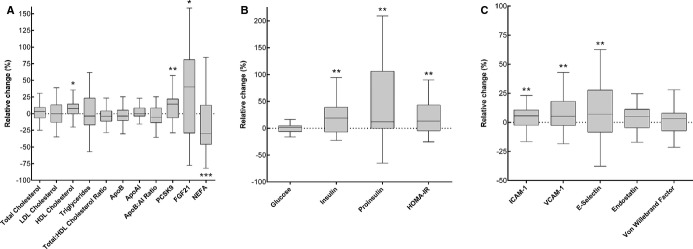
Relative changes during weight gain in the whole study sample (n=39). A, Lipid metabolism. B, Glucose and insulin. C, Endothelial function and coagulation. Calculated as change/baseline value×100. Boxes denote median and IQR, whiskers denote highest/lowest value within 1.5 IQR from upper/lower quartile. *P<0.05, **P<0.01, ***P≤0.001 in paired‐sample t test or Wilcoxon signed‐rank test. ApoB indicates apolipoprotein B; FGF21, fibroblast growth factor‐21; HDL, high‐density lipoprotein; HOMA‐IR, homeostasis model assessment of insulin resistance; ICAM‐1, intercellular adhesion molecule‐1; IQR, ; interquartile range; LDL, low‐density lipoprotein; NEFA, nonesterified fatty acids; PCSK9, proprotein convertase subtilisin/kexin type 9; VCAM‐1, vascular cell adhesion molecule‐1.
There were increases in fasting insulin (mean change +17%, +0.91±1.8 pmol/L, P=0.003), proinsulin (+21%, +0.73±1.6 pmol/L, P=0.007), and homeostasis model assessment of insulin resistance (+18%, +0.20±0.39, P=0.004) (Figure 3). There was no change in fasting plasma glucose (+0.1%, +0.005±0.34 mmol/L, P=0.79).
The endothelial adhesion molecules vascular cell adhesion molecule‐1 (+9%, +31586±60567 pg/L, P=0.002), intercellular adhesion molecule‐1 (+5%, +7231±16459 pg/L, P=0.009), and E‐selectin (+10%, +383±866 pg/L, P=0.009) increased in the whole study sample during weight gain (Figure 3). There were no significant changes for endostatin or von Willebrand factor (P>0.10, Figure 3). For most individuals, little or no change could be detected for inflammation markers (P≥0.10), and thus, these data were not included in the figure.
Discussion
This is the first controlled study in humans that examined the cardiometabolic effects of overfeeding with different types of fat. Even during a hypercaloric state and weight gain, induced by overeating muffins especially high in fat (51%E), but also containing refined carbohydrates (44%E), PUFA in place of SFA improved plasma lipoprotein profile. There were no significant differences between diets in other cardiometabolic risk markers. In the whole group, however, modest weight gain was accompanied by increased markers of endothelial dysfunction and insulin resistance in healthy, normal‐weight individuals. In addition, circulating PCSK9, a suppressor of LDL receptors, increased along with HDL cholesterol and the hepatokine and hormone FGF21. The effects of dietary fat overfeeding on PCSK9 levels, however, require confirmation in future studies using an isocaloric control group.
Previous studies have shown that most long‐chain SFA increase LDL (and HDL) cholesterol, whereas unsaturated fats decrease LDL cholesterol and increase HDL cholesterol to varying extents.2,10 The mechanisms behind such effects have been investigated in isocaloric experimental models, but not in hypercaloric studies in humans. In mice, mechanisms for increases in LDL cholesterol following SFA intake have been demonstrated and include stimulation of the transcription factor sterol regulatory element‐binding protein and co‐activators of the PGC family, which increases the expression of lipogenic genes such as hepatic fatty acid synthase and hydroxymethyl glutaryl coenzyme A reductase.11 On the other hand, PUFA has been demonstrated to bind to peroxisome proliferator‐activated receptor α and thus induce hepatic fatty acid oxidation,12 and suppress sterol regulatory element‐binding protein ‐1,13 which is involved in regulating lipogenesis and could partly explain how PUFA lowers LDL cholesterol.
Overfeeding studies in humans have usually been uncontrolled and of short duration. Studies lasting 4 weeks or more have demonstrated increases in blood pressure,14–15 total14 and LDL cholesterol,15 insulin resistance,16–21 C‐reactive protein,19,21–22 and endothelial dysfunction markers.14,19 Our study indicates that these changes may in part be due to factors other than type of fat such as weight gain itself. The observed increase in proinsulin is a novel finding of interest since proinsulin is an independent predictor of coronary artery disease mortality.23 In line with previous overfeeding studies,21,24 we observed a modest increase in HDL cholesterol. Such increase, however, may not be clinically relevant, since increasing HDL cholesterol using drugs or through genetic variation has not reduced CVD risk.25–26 The reduction in circulating NEFA is in line with previous overfeeding studies17,27 and reflects insulin‐mediated antilipolysis and increased triglyceride storage. We have previously shown in an isocaloric trial that PUFA decreased circulating PCSK9,8 which is another path to LDL cholesterol reduction. PCSK9 increases circulating LDL cholesterol by degradation of the LDL receptor.28 Although increased PCSK9 levels in response to overfeeding is a novel finding, there were no significant differences between diets.
Circulating FGF21 increased during the moderate weight gain, which might have been induced by energy overload rather than excess body fat, since a recent study showed a 3‐fold increase in FGF21 levels during 5 days of intense high‐fat (+50%E) overfeeding.29 The role of elevated FGF21 levels in weight gain is unclear, but may partly reflect a compensatory mechanism to counteract insulin resistance and dyslipidemia.30
Strengths of the study include its double‐blinded, randomized design, absence of dropouts during the study, and good compliance with diets. Limitations include short duration and low power to detect possible differences in some CVD risk factors. Negative results should therefore be interpreted with caution. Also, there was a 2.2% weight gain instead of aimed 3%, and this modest weight gain may have been insufficient to affect these factors in this young and metabolically healthy population. It may thus be inaccurate to generalize to obese and insulin‐resistant populations. Some participants approached or reached the goal of 3% weight gain before week 7 and were thus instructed to decrease the number of daily muffins during the final week(s). In other words, it is likely that some participants were fairly weight stable during the final week and thus might have been in a less anabolic state than the majority of subjects at the follow‐up assessment. Multiple statistical analyses raise the possibility that associations found are due to type‐1 error. It seems biologically plausible, however, to expect similar improvements in lipoproteins during weight gain as in weight stability. The effect size on LDL cholesterol, although not statistically significant, was comparable to what could be expected on the basis of calculations during isocaloric conditions.2 However, the mean difference in change in total:HDL cholesterol ratio (0.44) during overfeeding was more than twice the expected size if these fatty acids had isocalorically replaced one another, and could possibly reflect synergistic effects of weight gain combined with increased intakes of fatty acids and sucrose. Then again, the effect size was not unexpectedly high in comparison to other dietary trials in which test diets were provided and compliance to diets was highly controlled.31 The previously reported decrease in circulating palmitoleic acid (16:1n‐7) during the PUFA diet,6 although not a prespecified study outcome, may also reinforce the conclusion that PUFA seems cardioprotective during positive energy balance, as a recent lipidomics analysis demonstrated that 16:1n‐7 in plasma cholesterol esters was strongly related to CVD.32
Our study provides novel knowledge by demonstrating improvements in atherogenic lipoproteins when the excess calories are proportionally high in PUFA. This indicates that fat type not only is of importance in isocaloric diets, but also during energy excess and moderate weight gain. We have recently reported from this study that liver fat and visceral fat accumulation is increased after overfeeding SFA, but prevented by PUFA. The current study extends those findings and demonstrates that mean total:HDL cholesterol, LDL:HDL cholesterol, and apoB:apoAI ratios were 13%, 19%, and 16% lower during overfeeding PUFA compared with SFA, respectively. We believe this is quite a remarkable difference considering the hypercaloric state. However, there were no significant differences between diets with regard to other cardiometabolic risk markers. This is in line with a previous trial using a similar but isocaloric protocol in abdominally obese subjects.8 In that study, there were, in addition to lipid‐lowering effects, also some differences in markers of inflammation and insulin resistance in favor of PUFA, without any adverse effects on oxidative stress.8 Notably, not even during high intakes, and hypercaloric exposure of linoleic acid, there were no adverse changes in any of the inflammation markers. However, the latter is not surprising since the possible pro‐inflammatory fatty acids (ie, 20:3n‐6 and 20:4n‐6) were not increased in plasma after the PUFA diet. Although few studies have specifically tested tropical oils as a source of SFA, current recommendations for CVD prevention are to partially replace SFA with nonhydrogenated PUFA from nontropical oils.33–34
The current reduction in both total:HDL cholesterol ratio and apoB:apoAI ratio may be of clinical relevance. The total:HDL cholesterol ratio was the strongest predictor in the large Prospective Studies Collaboration, about 40% more informative than non‐HDL cholesterol and more than twice as informative as total cholesterol.35 Such data also accord with those of earlier studies in US, Chinese, and North European populations, where total:HDL cholesterol and/or apoB:apoAI ratios showed stronger associations with coronary heart disease risk, compared with other lipids.36–40 As apoB concentrations reflect the number of LDL particles, it can be used as a marker of total atherogenic burden, especially when taking apoAI into account.41
In summary, even modest diet‐induced weight gain resulting mainly from increased fat intake may promote subclinical endothelial dysfunction and insulin resistance in young, healthy, nonobese individuals. In the context of a moderately high‐fat diet, compared with overeating SFA (palmitic acid), PUFA (linoleic acid) instead counteracts some adverse cardiometabolic effects associated with weight gain, mainly by reducing atherogenic lipoproteins. Dietary fat composition thus seems to be important during weight gain, which is a novel finding that could have clinical implications in the long term. Since a large part of the population is in positive energy balance, these results are relevant at the population level, and support the current advice for CVD prevention (ie, partial replacement of SFA with PUFA).
Sources of Funding
This study was mainly funded by the Swedish Research Council (project K2012‐55X‐22081‐01‐3). We also thank Swedish Society of Medicine, Lennanders Foundation, and Uppsala‐Örebro Regional Research Council for support. Rudling received support from the Stockholm and Uppsala City Council (ALF), Swedish Research Council (K2013‐55X‐15075‐10‐3), the Swedish Heart‐Lung and the Diabetes Foundations, the Foundation Leducq, and Cardiovascular Program, Karolinska Institute/Stockholm City Council. The sponsors had no role in the design and conduct or report of the study.
Disclosures
Risérus is the guarantor of the study. Authors have no conflicts of interest to disclose.
Acknowledgments
We thank Siv Tengblad (Uppsala University, Clinical Nutrition and Metabolism) for assessing fatty acids and assistance with baking muffins and Martin Johansson (AarhusKarlshamn Sweden) for kindly donating oils used in the study. The study was performed within Excellence of Diabetes Research in Sweden (EXODIAB).
References
- 1.Estruch R, Ros E, Salas‐Salvadó J, Covas M‐I, Corella D, Arós F, Gómez‐Gracia E, Ruiz‐Gutiérrez V, Fiol M, Lapetra J, Lamuela‐Raventos RM, Serra‐Majem L, Pintó X, Basora J, Muñoz MA, Sorlí JV, Martínez JA, Martínez‐González MA. Primary prevention of cardiovascular disease with a Mediterranean diet. N Engl J Med. 2013; 368:1279-1290. [DOI] [PubMed] [Google Scholar]
- 2.Mensink RP, Zock PL, Kester AD, Katan MB. Effects of dietary fatty acids and carbohydrates on the ratio of serum total to HDL cholesterol and on serum lipids and apolipoproteins: a meta‐analysis of 60 controlled trials. Am J Clin Nutr. 2003; 77:1146-1155. [DOI] [PubMed] [Google Scholar]
- 3.Sanders TA. Fat and fatty acid intake and metabolic effects in the human body. Ann Nutr Metab. 2009; 55:162-172. [DOI] [PubMed] [Google Scholar]
- 4.Astrup A, Dyerberg J, Elwood P, Hermansen K, Hu FB, Jakobsen MU, Kok FJ, Krauss RM, Lecerf JM, Legrand P, Nestel P, Riserus U, Sanders T, Sinclair A, Stender S, Tholstrup T, Willett W. The role of reducing intakes of saturated fat in the prevention of cardiovascular disease: where does the evidence stand in 2010? Am J Clin Nutr. 2011; 93:684-688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mozaffarian D, Micha R, Wallace S. Effects on coronary heart disease of increasing polyunsaturated fat in place of saturated fat: a systematic review and meta‐analysis of randomized controlled trials. PLoS Med. 2010; 7:e1000252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosqvist F, Iggman D, Kullberg J, Cedernaes J, Johansson HE, Larsson A, Johansson L, Ahlstrom H, Arner P, Dahlman I, Riserus U. Overfeeding polyunsaturated and saturated fat causes distinct effects on liver and visceral fat accumulation in humans. Diabetes. 2014; 63:2356-2368. [DOI] [PubMed] [Google Scholar]
- 7.Kempen HJ, Glatz JF, Gevers Leuven JA, van der Voort HA, Katan MB. Serum lathosterol concentration is an indicator of whole‐body cholesterol synthesis in humans. J Lipid Res. 1988; 29:1149-1155. [PubMed] [Google Scholar]
- 8.Bjermo H, Iggman D, Kullberg J, Dahlman I, Johansson L, Persson L, Berglund J, Pulkki K, Basu S, Uusitupa M, Rudling M, Arner P, Cederholm T, Ahlstrom H, Riserus U. Effects of n‐6 PUFAs compared with SFAs on liver fat, lipoproteins, and inflammation in abdominal obesity: a randomized controlled trial. Am J Clin Nutr. 2012; 95:1003-1012. [DOI] [PubMed] [Google Scholar]
- 9.Boberg M, Vessby B, Croon LB. Fatty acid composition of platelets and of plasma lipid esters in relation to platelet function in patients with ischaemic heart disease. Atherosclerosis. 1985; 58:49-63. [DOI] [PubMed] [Google Scholar]
- 10.Sanders TA, Lewis FJ, Goff LM, Chowienczyk PJ. SFAs do not impair endothelial function and arterial stiffness. Am J Clin Nutr. 2013; 98:677-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin J, Yang R, Tarr PT, Wu PH, Handschin C, Li S, Yang W, Pei L, Uldry M, Tontonoz P, Newgard CB, Spiegelman BM. Hyperlipidemic effects of dietary saturated fats mediated through pgc‐1beta coactivation of srebp. Cell. 2005; 120:261-273. [DOI] [PubMed] [Google Scholar]
- 12.Jump DB, Botolin D, Wang Y, Xu J, Christian B, Demeure O. Fatty acid regulation of hepatic gene transcription. J Nutr. 2005; 135:2503-2506. [DOI] [PubMed] [Google Scholar]
- 13.Hannah VC, Ou J, Luong A, Goldstein JL, Brown MS. Unsaturated fatty acids down‐regulate srebp isoforms 1a and 1c by two mechanisms in HEK‐293 cells. J Biol Chem. 2001; 276:4365-4372. [DOI] [PubMed] [Google Scholar]
- 14.Orr JS, Gentile CL, Davy BM, Davy KP. Large artery stiffening with weight gain in humans: role of visceral fat accumulation. Hypertension. 2008; 51:1519-1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Erlingsson S, Herard S, Dahlqvist Leinhard O, Lindstrom T, Lanne T, Borga M, Nystrom FHFast Food Study G. Men develop more intraabdominal obesity and signs of the metabolic syndrome after hyperalimentation than women. Metabolism. 2009; 58:995-1001. [DOI] [PubMed] [Google Scholar]
- 16.Oppert JM, Nadeau A, Tremblay A, Despres JP, Theriault G, Deriaz O, Bouchard C. Plasma glucose, insulin, and glucagon before and after long‐term overfeeding in identical twins. Metabolism. 1995; 44:96-105. [DOI] [PubMed] [Google Scholar]
- 17.Brands M, Swat M, Lammers NM, Sauerwein HP, Endert E, Ackermans MT, Verhoeven AJ, Serlie MJ. Effects of a hypercaloric diet on beta‐cell responsivity in lean healthy men. Clin Endocrinol. 2013; 78:217-225. [DOI] [PubMed] [Google Scholar]
- 18.Erdmann J, Kallabis B, Oppel U, Sypchenko O, Wagenpfeil S, Schusdziarra V. Development of hyperinsulinemia and insulin resistance during the early stage of weight gain. Am J Physiol Endocrinol Metab. 2008; 294:E568-E575. [DOI] [PubMed] [Google Scholar]
- 19.Gupta AK, Johnson WD, Johannsen D, Ravussin E. Cardiovascular risk escalation with caloric excess: a prospective demonstration of the mechanics in healthy adults. Cardiovasc Diabetol. 2013; 12:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samocha‐Bonet D, Campbell LV, Mori TA, Croft KD, Greenfield JR, Turner N, Heilbronn LK. Overfeeding reduces insulin sensitivity and increases oxidative stress, without altering markers of mitochondrial content and function in humans. PLoS One. 2012; 7:e36320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tam CS, Viardot A, Clement K, Tordjman J, Tonks K, Greenfield JR, Campbell LV, Samocha‐Bonet D, Heilbronn LK. Short‐term overfeeding may induce peripheral insulin resistance without altering subcutaneous adipose tissue macrophages in humans. Diabetes. 2010; 59:2164-2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Astrand O, Carlsson M, Nilsson I, Lindstrom T, Borga M, Nystrom FHFast Food Study G. Weight gain by hyperalimentation elevates C‐reactive protein levels but does not affect circulating levels of adiponectin or resistin in healthy subjects. Eur J Endocrinol. 2010; 163:879-885. [DOI] [PubMed] [Google Scholar]
- 23.Zethelius B, Byberg L, Hales CN, Lithell H, Berne C. Proinsulin is an independent predictor of coronary heart disease: report from a 27‐year follow‐up study. Circulation. 2002; 105:2153-2158. [DOI] [PubMed] [Google Scholar]
- 24.Lindstrom T, Kechagias S, Carlsson M, Nystrom FHFast Food Study G. Transient increase in HDL‐cholesterol during weight gain by hyperalimentation in healthy subjects. Obesity (Silver Spring). 2011; 19:812-817. [DOI] [PubMed] [Google Scholar]
- 25.Investigators A‐H. Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes‐Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011; 365:2255-2267. [DOI] [PubMed] [Google Scholar]
- 26.Voight BF, Peloso GM, Orho‐Melander M, Frikke‐Schmidt R, Barbalic M, Jensen MK, Hindy G, Holm H, Ding EL, Johnson T, Schunkert H, Samani NJ, Clarke R, Hopewell JC, Thompson JF, Li M, Thorleifsson G, Newton‐Cheh C, Musunuru K, Pirruccello JP, Saleheen D, Chen L, Stewart A, Schillert A, Thorsteinsdottir U, Thorgeirsson G, Anand S, Engert JC, Morgan T, Spertus J, Stoll M, Berger K, Martinelli N, Girelli D, McKeown PP, Patterson CC, Epstein SE, Devaney J, Burnett MS, Mooser V, Ripatti S, Surakka I, Nieminen MS, Sinisalo J, Lokki ML, Perola M, Havulinna A, de Faire U, Gigante B, Ingelsson E, Zeller T, Wild P, de Bakker PI, Klungel OH, Maitland‐van der Zee AH, Peters BJ, de Boer A, Grobbee DE, Kamphuisen PW, Deneer VH, Elbers CC, Onland‐Moret NC, Hofker MH, Wijmenga C, Verschuren WM, Boer JM, van der Schouw YT, Rasheed A, Frossard P, Demissie S, Willer C, Do R, Ordovas JM, Abecasis GR, Boehnke M, Mohlke KL, Daly MJ, Guiducci C, Burtt NP, Surti A, Gonzalez E, Purcell S, Gabriel S, Marrugat J, Peden J, Erdmann J, Diemert P, Willenborg C, Konig IR, Fischer M, Hengstenberg C, Ziegler A, Buysschaert I, Lambrechts D, Van de Werf F, Fox KA, El Mokhtari NE, Rubin D, Schrezenmeir J, Schreiber S, Schafer A, Danesh J, Blankenberg S, Roberts R, McPherson R, Watkins H, Hall AS, Overvad K, Rimm E, Boerwinkle E, Tybjaerg‐Hansen A, Cupples LA, Reilly MP, Melander O, Mannucci PM, Ardissino D, Siscovick D, Elosua R, Stefansson K, O'Donnell CJ, Salomaa V, Rader DJ, Peltonen L, Schwartz SM, Altshuler D, Kathiresan S. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012; 380:572-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meugnier E, Bossu C, Oliel M, Jeanne S, Michaut A, Sothier M, Brozek J, Rome S, Laville M, Vidal H. Changes in gene expression in skeletal muscle in response to fat overfeeding in lean men. Obesity (Silver Spring). 2007; 15:2583-2594. [DOI] [PubMed] [Google Scholar]
- 28.Park SW, Moon YA, Horton JD. Post‐transcriptional regulation of low density lipoprotein receptor protein by proprotein convertase subtilisin/kexin type 9a in mouse liver. J Biol Chem. 2004; 279:50630-50638. [DOI] [PubMed] [Google Scholar]
- 29.Vienberg SG, Brons C, Nilsson E, Astrup A, Vaag A, Andersen B. Impact of short‐term high‐fat feeding and insulin‐stimulated FGF21 levels in subjects with low birth weight and controls. Eur J Endocrinol. 2012; 167:49-57. [DOI] [PubMed] [Google Scholar]
- 30.Woo YC, Xu A, Wang Y, Lam KS. Fibroblast growth factor 21 as an emerging metabolic regulator: clinical perspectives. Clin Endocrinol. 2013; 78:489-496. [DOI] [PubMed] [Google Scholar]
- 31.Iggman D, Gustafsson IB, Berglund L, Vessby B, Marckmann P, Riserus U. Replacing dairy fat with rapeseed oil causes rapid improvement of hyperlipidaemia: a randomized controlled study. J Intern Med. 2011; 270:356-364. [DOI] [PubMed] [Google Scholar]
- 32.Stegemann C, Pechlaner R, Willeit P, Langley SR, Mangino M, Mayr U, Menni C, Moayyeri A, Santer P, Rungger G, Spector TD, Willeit J, Kiechl S, Mayr M. Lipidomics profiling and risk of cardiovascular disease in the prospective population‐based Bruneck study. Circulation. 2014; 129:1821-1831. [DOI] [PubMed] [Google Scholar]
- 33.Mozaffarian D, Appel LJ, Van Horn L. Components of a cardioprotective diet: new insights. Circulation. 2011; 123:2870-2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eckel RH, Jakicic JM, Ard JD, de Jesus JM, Houston Miller N, Hubbard VS, Lee IM, Lichtenstein AH, Loria CM, Millen BE, Nonas CA, Sacks FM, Smith SC, Jr, Svetkey LP, Wadden TA, Yanovski SZ, Kendall KA, Morgan LC, Trisolini MG, Velasco G, Wnek J, Anderson JL, Halperin JL, Albert NM, Bozkurt B, Brindis RG, Curtis LH, DeMets D, Hochman JS, Kovacs RJ, Ohman EM, Pressler SJ, Sellke FW, Shen WK, Smith SC, Jr, Tomaselli GF. 2013 AHA/ACC guideline on lifestyle management to reduce cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014; 129:S76-S99. [DOI] [PubMed] [Google Scholar]
- 35.Prospective Studies C. Lewington S, Whitlock G, Clarke R, Sherliker P, Emberson J, Halsey J, Qizilbash N, Peto R, Collins R. Blood cholesterol and vascular mortality by age, sex, and blood pressure: a meta‐analysis of individual data from 61 prospective studies with 55,000 vascular deaths. Lancet. 2007; 370:1829-1839. [DOI] [PubMed] [Google Scholar]
- 36.Lamarche B, Despres JP, Moorjani S, Cantin B, Dagenais GR, Lupien PJ. Triglycerides and HDL‐cholesterol as risk factors for ischemic heart disease: results from the Quebec cardiovascular study. Atherosclerosis. 1996; 119:235-245. [DOI] [PubMed] [Google Scholar]
- 37.van Lennep JE, Westerveld HT, van Lennep HW, Zwinderman AH, Erkelens DW, van der Wall EE. Apolipoprotein concentrations during treatment and recurrent coronary artery disease events. Arterioscler Thromb Vasc Biol. 2000; 20:2408-2413. [DOI] [PubMed] [Google Scholar]
- 38.Walldius G, Jungner I, Holme I, Aastveit AH, Kolar W, Steiner E. High apolipoprotein B, low apolipoprotein A‐I, and improvement in the prediction of fatal myocardial infarction (AMORIS study): a prospective study. Lancet. 2001; 358:2026-2033. [DOI] [PubMed] [Google Scholar]
- 39.Chien KL, Hsu HC, Su TC, Chen MF, Lee YT, Hu FB. Apolipoprotein B and non‐high density lipoprotein cholesterol and the risk of coronary heart disease in Chinese. J Lipid Res. 2007; 48:2499-2505. [DOI] [PubMed] [Google Scholar]
- 40.Gotto AM, Jr, Whitney E, Stein EA, Shapiro DR, Clearfield M, Weis S, Jou JY, Langendorfer A, Beere PA, Watson DJ, Downs JR, de Cani JS. Relation between baseline and on‐treatment lipid parameters and first acute major coronary events in the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS). Circulation. 2000; 101:477-484. [DOI] [PubMed] [Google Scholar]
- 41.Sacks FM. The apolipoprotein story. Atheroscler Suppl. 2006; 7:23-27. [DOI] [PubMed] [Google Scholar]