

Abstract
Background
Kallistatin exerts pleiotropic activities in inhibiting inflammation, apoptosis, and oxidative stress in endothelial cells. Because endothelial progenitor cells (EPCs) play a significant role in vascular repair, we investigated whether kallistatin contributes to vascular regeneration by enhancing EPC migration and function.
Methods and Results
We examined the effect of endogenous kallistatin on circulating EPCs in a rat model of vascular injury and the mechanisms of kallistatin on EPC mobility and function in vitro. In deoxycorticosterone acetate–salt hypertensive rats, we found that kallistatin depletion augmented glomerular endothelial cell loss and diminished circulating EPC number, whereas kallistatin gene delivery increased EPC levels. In cultured EPCs, kallistatin significantly reduced tumor necrosis factor‐α–induced apoptosis and caspase‐3 activity, but kallistatin's effects were blocked by phosphoinositide 3‐kinase inhibitor (LY294002) and nitric oxide (NO) synthase inhibitor (l‐NAME). Kallistatin stimulated the proliferation, migration, adhesion and tube formation of EPCs; however, kallistatin's actions were abolished by LY294002, l‐NAME, endothelial NO synthase–small interfering RNA, constitutively active glycogen synthase kinase‐3β, or vascular endothelial growth factor antibody. Kallistatin also increased Akt, glycogen synthase kinase‐3β, and endothelial NO synthase phosphorylation; endothelial NO synthase, vascular endothelial growth factor, and matrix metalloproteinase‐2 synthesis and activity; and NO and vascular endothelial growth factor levels. Kallistatin's actions on phosphoinositide 3‐kinase–Akt signaling were blocked by LY294002, l‐NAME, and anti–vascular endothelial growth factor antibody.
Conclusions
Endogenous kallistatin plays a novel role in protection against vascular injury in hypertensive rats by promoting the mobility, viability, and vasculogenic capacity of EPCs via enhancing NO and vascular endothelial growth factor levels through activation of phosphoinositide 3‐kinase–Akt signaling. Kallistatin therapy may be a promising approach in the treatment of vascular diseases.
Keywords: angiogenesis, apoptosis, endothelial progenitor cells, kallistatin, nitric oxide
Introduction
Endothelial progenitor cells (EPCs) are a subset of mononuclear cells derived from bone marrow that have the ability to differentiate into mature endothelial cells. As such, EPCs have been identified as an important contributor to endogenous vascular repair by participating in new vessel formation and endothelial regeneration.1 Patients with hypertension, coronary artery disease, chronic renal failure, diabetes, sepsis, and rheumatoid arthritis exhibit decreased numbers of circulating EPCs.1–8 Moreover, EPCs isolated from patients with coronary artery disease and hypertension display an impaired migratory response.2–3 The decline in circulating EPC number can be attributed to increased apoptosis and senescence in addition to reduced growth and migration from bone marrow. Diminished nitric oxide (NO) bioavailability reduces the mobilization of EPCs, thus impairing endogenous vascular repair mechanisms.9–10 However, increased NO levels can be stimulated by a variety of factors, such as statins, red wine intake, and physical exercise, leading to enhanced EPC mobility and functional activity.1,3,11–13 Elevated endothelial NO synthase (eNOS) expression is also linked to improved EPC migratory capacity in vitro.10 Indeed, eNOS was found to be essential to maintain adequate EPC mobilization on stimulation with statins or vascular endothelial growth factor (VEGF).11,14 Pharmacological enhancement of eNOS expression partially reversed the impaired functional activity of EPCs from ischemic cardiomyopathy patients, implicating the critical role of NO for EPC function.12 These findings highly suggest that eNOS‐derived NO serves as a physiological regulator of EPC mobilization and function.
Kallistatin is a plasma protein and tissue kallikrein inhibitor that is widely distributed in tissues relevant to cardiovascular function, including kidneys, heart, and blood vessels.15–19 We have demonstrated that kallistatin exhibits pleiotropic effects by reducing hypertension, inflammation, apoptosis, oxidative stress, angiogenesis, hypertrophy, and fibrosis in animal models and cultured cells.20–27 Kallistatin gene delivery improved cardiac and renal function; decreased oxidative stress, apoptosis, and tissue remodeling; and increased eNOS activation and NO formation in rat models of cardiac ischemia and reperfusion, myocardial infarction, and salt‐induced kidney damage.22–24,26 In cultured endothelial cells, recombinant human kallistatin inhibited vascular inflammation, apoptosis, and oxidative stress via stimulation of eNOS expression and activity as well as NO production.25–26 Collectively, these results indicate that kallistatin is a vascular protective agent by decreasing apoptosis and oxidative stress and promoting NO formation in endothelial cells. Because of the significant role of EPCs in vascular repair, we investigated kallistatin's protective actions against vascular injury by examining its effect on endothelial cell loss and circulating EPC levels using the deoxycorticosterone acetate (DOCA)‐salt hypertensive rat model. We chose this animal model for our studies because these rats exhibit aortic oxidative stress and glomerular capillary loss, both of which are indicative of vascular injury.26,28–29 Furthermore, we elucidated the signaling mechanisms and the pivotal role of NO in mediating kallistatin's regulatory effects on EPC mobility, viability, adhesion, and tube formation in vitro.
Material and Methods
Animal Treatment
All procedures complied with the standard for care and use of animal subjects as stated in the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Resources, National Academy of Sciences, Bethesda, MD).30 The protocols for animal studies were approved by the institutional animal care and use committee of the Medical University of South Carolina. Male Wistar rats (Harlan Sprague Dawley Inc), weighing 200 to 220 g, were anesthetized with an intraperitoneal injection of pentobarbital sodium (50 mg/kg) before undergoing left unilateral nephrectomy. One week after surgery, rats in the sham operation group (n=6) received weekly subcutaneous injections of sesame oil and were provided with tap water. Experimental animals received weekly subcutaneous injections of DOCA (25 mg/kg body weight) suspended in sesame oil and were provided with 1% NaCl drinking water. Two weeks after the initiation of DOCA‐salt treatment, rats were injected intravenously via tail vein with control adenovirus (Ad.Null) or adenovirus containing human kallistatin (Ad.KS) (1×1010 plaque‐forming units, n=6 to 8).26 In another study, DOCA‐salt rats received daily intravenous injection of antibody against rat kallistatin or normal IgG (0.5 mg per rat, n=6 to 8).29 Kidneys and peripheral blood were harvested 2 weeks after gene delivery or 10 days after the start of antibody injection, as previously described.26,29
Flow Cytometry Analysis of Circulating EPCs
Fluorescence‐activated cell analysis was performed to determine the number of CD133+ EPCs in peripheral blood from sham and DOCA‐salt rats. Briefly, peripheral blood was incubated with a fluorescein isothiocyanate (FITC)‐conjugated CD133 antibody (Beckman Coulter, Fullerton, CA). FITC‐labeled anti‐CD45 antibody (BD Biosciences, San Jose, CA) was used for differential gating during flow analysis. FITC‐labeled IgG1a (Beckman Coulter) and phycoerythrin‐labeled IgG2b (Becton Dickinson, Franklin Lakes, NJ) served as the control for color compensation. Analysis was performed with an automated fluorescence‐activated cell counter (Beckman Coulter) in which 1 000 000 events were counted. The absolute cell counts of all measured components per 1 000 000 events in the lymphocyte gate were calculated.31
Immunohistochemical Staining for Capillary Density
Kidneys were fixed in a formaldehyde solution and then dehydrated and embedded. Immunohistochemistry was performed by incubating renal tissue sections (4 μm thick) with a primary antibody against the endothelial cell marker JG‐12 at 4°C overnight. Glomerular capillary density was calculated as the number of capillaries per square millimeter in 10 glomeruli from each rat.26
EPC Culture and Characterization
Peripheral blood from Wistar rats and normal human umbilical cord blood were used as a source of EPCs. Approval for the use of normal human cord blood was obtained by Medical University of South Carolina Human Research (Pro00017277). EPCs were isolated by density gradient centrifugation using Histopaque‐1977 (Sigma, St. Louis, MO) and cultured in EBM‐2 with supplements (EGM2‐MV BulletKit; Clonetics, San Diego, CA), but without hydrocortisone, on human fibronectin‐coated dishes. Colony‐forming units were observed after subsequent outgrowth. Attached cells were obtained after 4 to 7 days of culture and utilized for the in vitro studies. After 3 days, nonadherent cells were removed, and adherent cells were incubated for another 24 hours before the experiments were performed. EPCs were characterized by dual staining for 1,1′‐dioctadecyl‐3,3′,3′‐tetramethylindocarbocyanine, or “Dil,” and acetyl Dil‐labeled low‐density lipoprotein and lectin as well as by expression of CD31 (BD Biosciences) and von Willebrand factor (Sigma) by immunohistochemistry.
Purification of Recombinant Human Kallistatin
Recombinant human kallistatin was secreted into the serum‐free medium of cultured HEK293T cells, and the cultured medium was concentrated by ammonium sulfate precipitation followed by nickel‐affinity chromatography, as previously described.32–33 The purity of kallistatin was verified by Coomassie blue staining and Western blot using a specific monoclonal antibody.19,33
Detection of EPC Apoptosis
Growth‐arrested EPCs were preincubated with kallistatin (0.2 mol/L) for 30 minutes. EPCs were then treated with tumor necrosis factor‐α (TNF‐α; 10 ng/mL) for an additional 12 hours. To determine the role of the phosphoinositide 3‐kinase (PI3K)‐Akt‐eNOS prosurvival pathway, inhibitors against PI3K (LY294002 [LY], 5 mol/L) and NOS (l‐NAME [Nω‐nitro‐l‐arginine methyl ester], 100 mol/L) were added 30 minutes before kallistatin treatment. EPCs were then incubated in the presence or absence of kallistatin (0.2 mol/L) for 30 minutes, followed by the addition of TNF‐α (10 ng/mL) for 12 hours. EPCs were incubated with Hoechst 33342 (0.4 mg/mL) for 5 minutes and examined under a fluorescence microscope to detect apoptotic nuclei. Caspase‐3 activity was measured by incubating cell extracts with caspase‐3 substrate (10 mol/L).34 The fluorescence of the cleaved substrate was measured by a spectrofluorometer with excitation and emission wavelengths of 380 and 460 nm, respectively.
EPC Proliferation and Migration
Colorimetric assay for 5‐bromo‐2‐deoxyuridine was performed, as described previously.35 Briefly, EPCs were serum deprived, pretreated with LY (5 mol/L) or l‐NAME (100 mol/L) for 30 minutes and then cultured with kallistatin (1 mol/L) for 24 hours prior to the addition of 5‐bromo‐2‐deoxyuridine. A Cell Proliferation ELISA, BrdU (colorimetric) assay (Roche Applied Science, Indianapolis, IN) was performed 12 hours later. For EPC migration, transwell membranes (8 μm; Costar, Corning, NY) were coated on both sides with fibronectin (10 μg/mL) overnight at 4°C. EPCs were detached by trypsinization and resuspended in serum‐free medium (RPMI 1640 containing 0.05% BSA). Serum‐free medium (600 μL) was placed in the lower chambers, and EPCs (120 000) in 100 μL of serum‐free medium were incubated in the upper chamber at 37°C in 5% CO2 for 18 hours. Cells remaining on the upper surface of the filters were mechanically removed, and cells that had migrated to the lower surface were fixed with 4% formaldehyde and counted in 5 fields after staining with 0.1% crystal violet.
EPC Adhesion to Endothelial Cells or Gel Matrix and Tube Formation
EPCs in basal complete medium supplemented with 5% FBS were labeled with Dil (2.5 μg/mL) for 5 minutes at 37°C or 15 minutes at 4°C. Human umbilical vein endothelial cells were pretreated with TNF‐α (1 ng/mL) for 12 hours. Identical cell numbers (1 to 2×105) of EPCs were placed onto culture dishes with and without kallistatin (0.2 mol/L) and were incubated for 3 hours at 37°C. Nonattached cells were gently removed with PBS, and adherent cells were fixed and counted in 10 random fields.5 Moreover, collagen mixture (100 μg/mL; Invitrogen, Grand Island, NY) or fibronectin (100 μg/mL) was coated onto 24‐well plates for 2 hours at 37°C. Wells were blocked with 1% BSA in PBS for 2 hours, and EPCs (1 to 2×105) were added to each well to attach for 1 hour. Adherent cells were stained with 0.1% crystal violet and rinsed with 10% acetic acid to elute the stain from the cells. Attached cells were quantified by analyzing the optical density of the media at a wavelength of 600 nm with a microtiter plate reader.5 For tube formation, phenol‐red free and growth factor‐reduced basement Matrigel (BD Bioscience) was used to coat 24‐well culture plates, which were then incubated for 30 minutes at 37°C to allow solidification. Aliquots of Dil‐labeled EPCs (5×103) were mixed with human umbilical vein endothelial cells (2×104) and seeded onto the Matrigel‐coated plates in the presence or absence of kallistatin (0.2 μmol/L) for 24 hours. The percentages of Dil‐labeled EPCs that had integrated into tube formations were then determined from 5 random high‐power fields for each treatment group.5
Determination of PI3K‐Akt Signaling Pathways on EPC Proliferation and Function
To determine the role of the PI3K‐Akt‐eNOS and Akt–glycogen synthase kinase‐3β (GSK‐3β)–VEGF pathways in EPC proliferation, migration, adhesion, and tube formation, inhibitors against PI3K (LY, 5 mol/L) and NOS (l‐NAME, 100 mol/L) as well as VEGF antibody (10 ng/mL) were added concomitantly with kallistatin. Moreover, cells were transfected with eNOS–small interfering RNA (Dharmacon, Lafayette, CO) or transduced with adenovirus expressing constitutively active GSK‐3β (Ad.GSK‐S9A) overnight prior to assay commencement.
Western Blot Analyses, Gene Expression, and NO Level and NOS Activity Measurements
Western blots of EPC lysates were performed using primary antibodies against phosphorylated and/or total Akt (Ser473), eNOS (Ser1177), GSK‐3β (Ser9) (Cell Signaling, Beverly, MA), and matrix metalloproteinase 2 (MMP‐2; Chemicon, Temecula, CA) overnight at 4°C. Nitrate/nitrite (NOx) production in EPCs was detected in culture medium after 24 hours of incubation with kallistatin (0.2 mol/L), as described previously.25 NOS activity was measured using a colorimetric Nitric Oxide Synthase Assay Kit (Calbiochem, La Jolla, CA). Immunostaining of eNOS was performed, as described previously.25 The expression of forkhead box O1 (FOXO1), Bim, eNOS, VEGF, and MMP‐2 was determined by real‐time polymerase chain reaction (Applied Biosystems, Foster City, CA), as described previously.25–27 Briefly, EPCs were seeded in 6‐well plates (1×105 cells per well). After incubation overnight with EBM‐2 supplemented with 0.1% FBS, kallistatin (0.2 mol/L) was added to the medium, and mRNA was extracted 24 hours later. VEGF protein levels were measured by enzyme‐linked immunosorbent assay (R&D Systems, Minneapolis, MN), and MMP‐2 activity was determined by zymography. The effect of kallistatin on Bim expression induced by H2O2 in EPCs was determined, as described previously.26
Statistical Analysis
Data were analyzed among the groups with the use of 1‐way ANOVA followed by Newman–Keuls multiple comparison test. Unpaired 2‐tailed Student t test was used to locate between‐group differences. Group data were expressed as mean±SEM. Values of all parameters were considered significantly different at a value of P<0.05.
Results
Depletion of Endogenous Kallistatin Augments Endothelial Loss and Reduces Circulating EPC Number in Hypertensive Rats
Endothelial cells in the glomeruli of DOCA‐salt hypertensive rats were identified by immunohistochemical staining with JG‐12 antibody (Figure 1A). Representative images showed that DOCA‐salt rats receiving IgG injection had lower amounts of capillaries in glomeruli compared with the sham group; however, kallistatin depletion by injection with antirat kallistatin antibody further decreased glomerular capillary density. Quantitative analysis indicated that a 44% reduction of glomerular capillary density occurred in the DOCA/IgG group, and depletion of endogenous kallistatin caused a further 25% loss of endothelial cells (Figure 1B). These results indicate that endogenous kallistatin plays a protective role against vascular injury. Moreover, EPCs were isolated from peripheral blood of DOCA‐salt rats and identified by double staining with acetyl Dil‐labeled low‐density lipoprotein and lectin as well as by positive immunostaining for CD31 and von Willebrand factor (Figure 1C). Circulating EPC levels were slightly elevated in the DOCA/Ad.Null group compared with sham rats but with no statistical significance. Kallistatin gene delivery in DOCA‐salt rats markedly induced 6‐ and 4‐fold elevation of circulating EPC levels compared with the sham and DOCA/Ad.Null groups, respectively (Figure 1D). In contrast, depletion of endogenous kallistatin in DOCA‐salt rats further reduced EPC numbers by 75% and 94% compared with the sham and control DOCA/IgG groups, respectively (Figure 1D). These results indicate that endogenous kallistatin modulates both vascular repair and circulating EPC number in hypertensive rats.
Figure 1.
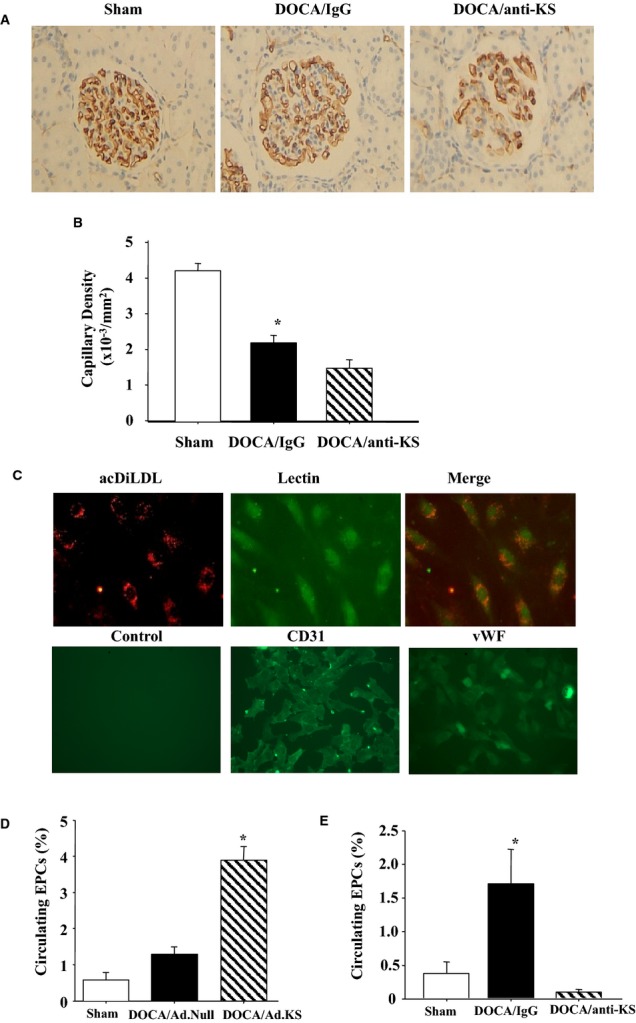
A and B, Representative images of JG‐12 immunostaining in kidney (A) and quantitative analysis of glomerular capillary density after KS antibody administration (B). C, Cultured EPCs are characterized by acDiLDL and lectin staining and by positive immunohistochemical staining for CD31 and vWF. D and E, KS gene delivery increases (D) and KS antibody injection decreases (E) circulating EPC number in DOCA‐salt rats. *P<0.05 vs other groups. Data are expressed as mean±SEM, n=5. acDiLDL indicates acetyl 1,1′‐dioctadecyl‐3,3′,3′‐tetramethylindocarbocyanine–labeled low‐density lipoprotein; Ad.KS, adenovirus containing human KS; Ad.Null, control adenovirus; DOCA, deoxycorticosterone acetate; EPCs, endothelial progenitor cells; KS, kallistatin; vWF, von Willebrand factor.
Kallistatin Inhibits TNF‐α‐induced Apoptosis in Cultured EPCs
Representative images of Hoechst staining showed that kallistatin dramatically inhibited TNF‐α–induced EPC apoptosis; however, PI3K inhibitor (LY) and NOS inhibitor (l‐NAME) blocked kallistatin's protective effect (Figure 2A). Kallistatin alone had no effect on EPC apoptosis. These findings were confirmed by quantitative analysis (Figure 2B). Caspase‐3 activity assay further supported these results (Control: 0.9±0.1; kallistatin: 0.9±0.1; TNF‐α: 2.6±0.2; TNF‐α+kallistatin: 1.1±0.2; TNF‐α+kallistatin/LY: 2.5±0.3; TNF‐α+kallistatin/l‐NAME: 2.6±0.3 pmol/min per mg protein, n=3, P<0.05; Figure 2C). Moreover, kallistatin caused a 2.6‐fold increase in FOXO1 synthesis (n=3, P<0.05; Figure 2D) but a 34% decrease of H2O2‐induced Bim expression (n=3, P<0.05; Figure 2E). These data indicate that kallistatin protects against TNF‐α–induced EPC apoptosis via activation of the PI3K‐Akt‐NOS signaling pathway, leading to increased FOXO1 synthesis and reduced H2O2‐induced Bim expression.
Figure 2.
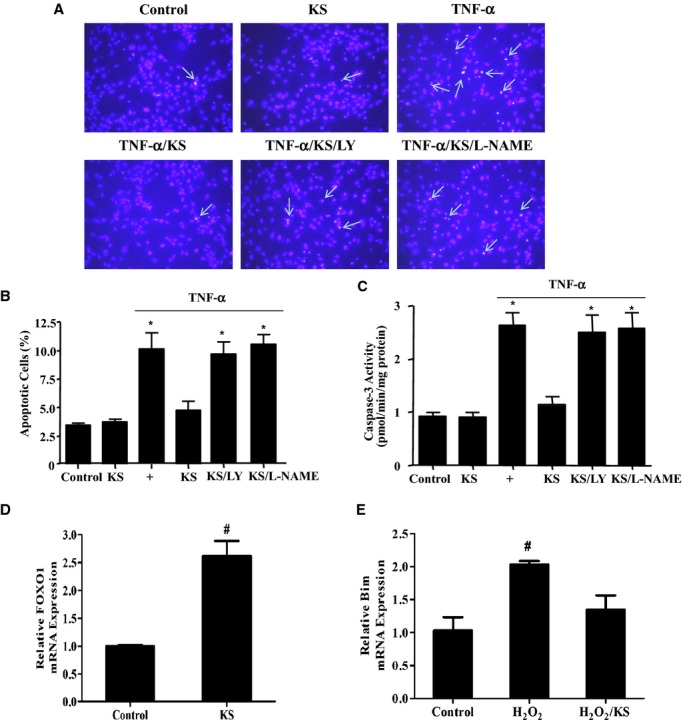
KS inhibits TNF‐α–induced apoptosis in EPCs via phosphoinositide 3‐kinase–Akt–nitric oxide synthase signaling. A, Representative images of apoptotic cells by Hoechst staining. Arrows indicate apoptotic cells. B and C, Apoptosis was verified by quantitative analysis (B) and caspase‐3 activity (C). D and E, KS increases FOXO1 expression (D) but inhibits H2O2‐induced Bim expression (E). *P<0.05 vs control, KS, and TNF‐α/KS; #P<0.05 vs other groups. Data are expressed as mean±SEM, n=3. EPCs indicates endothelial progenitor cells; FOXO1, forkhead box O1; KS, kallistatin; LY, LY294002; TNF‐α, tumor necrosis factor‐α.
Kallistatin Promotes EPC Proliferation, Migration, Adhesion, and Tube Formation Through PI3K‐Akt Signaling Pathways
Kallistatin increased the proliferation of cultured EPCs, as determined by 5‐bromo‐2‐deoxyuridine incorporation, but the effect was blocked by LY and l‐NAME (Figure 3A). Using a modified Boyden chamber method followed by quantitative analysis, kallistatin significantly increased EPC migration; however, the effect was inhibited by LY and l‐NAME (Figure 3B), eNOS–small interfering RNA (Figure 3C), and constitutively active GSK‐3β (Figure 3D). Representative images and quantitative analysis indicated that kallistatin promoted a 2.3‐fold increase in EPC adhesion to endothelial cells (n=3, P<0.05; Figure 4A and 4B) and a 1.7‐fold increase in EPC adhesion to matrix gel (n=3, P<0.05; Figure 4A through 4C). EPC adhesion was mediated by activation of the PI3K‐Akt‐eNOS signaling cascade, as LY and l‐NAME abolished kallistatin's actions (Figure 4A). Moreover, kallistatin stimulated the incorporation of EPCs into endothelial cell–composite tubular structures by 1.8‐fold (n=3, P<0.05; Figure 5A and 5B), as determined by fluorescence and light microscopy showing tube formation with the mixture of EPCs and endothelial cells. Again, LY and l‐NAME significantly inhibited kallistatin's effect. Furthermore, anti‐VEGF antibody and constitutively active GSK‐3β markedly blocked kallistatin‐induced tube formation (Figure 5C and 5D). Consequently, kallistatin promotes EPC migration, adhesion, and tube formation by activating both PI3K‐Akt‐eNOS and Akt‐GSK‐3β‐VEGF signaling pathways.
Figure 3.

KS promotes EPC proliferation and migration via PI3K‐Akt signaling. A through D, KS increases EPC proliferation by BrdU incorporation (A) and migration using modified Boyden chambers (B through D). *P<0.05 vs other groups. Data are expressed as mean±SEM, n=3. Ad.GSK‐S9A indicates adenovirus expressing constitutively active GSK‐3β; Ad.Null, control adenovirus; Brdu, 5‐bromo‐2‐deoxyuridine; eNOS, endothelial nitric oxide synthase; EPCs, endothelial progenitor cells; GSK, glycogen synthase kinase; KS, kallistatin; LY, LY294002; siRNA, small interfering RNA.
Figure 4.
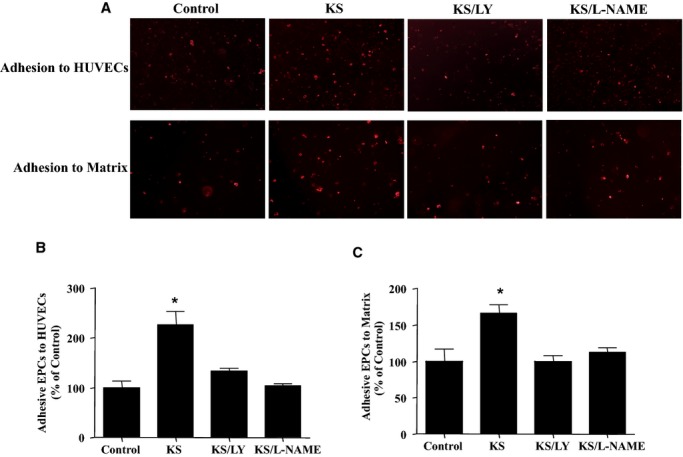
KS promotes EPC adhesion via phosphoinositide 3‐kinase–Akt–nitric oxide synthase signaling. A, Representative images of EPC adhesion to HUVECs (upper panel) and matrix (lower panel). B and C, Quantification of EPC adhesion. *P<0.05 vs other groups. Data are expressed as mean±SEM, n=3. EPCs indicates endothelial progenitor cells; HUVECs, human umbilical vein endothelial cells; KS, kallistatin; LY, LY294002.
Figure 5.
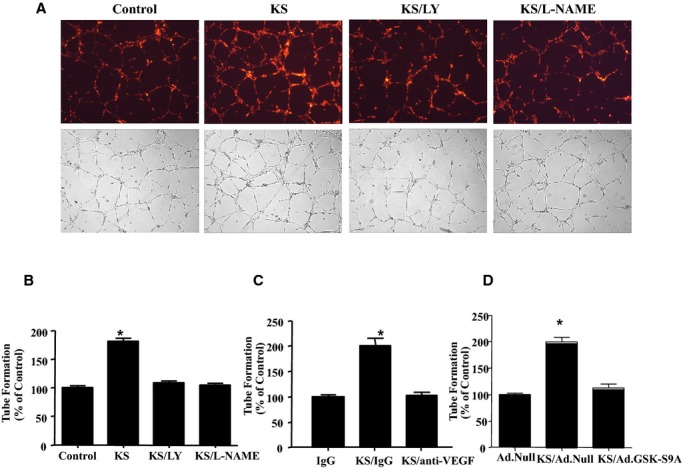
KS promotes EPC tube formation via phosphoinositide 3‐kinase–Akt–nitric oxide synthase signaling. A, Representative images of EPCs incorporating into human umbilical vein endothelial cells, as determined by fluorescent (upper panel) and light (lower panel) microscopy. B through D, Quantification of tube formation. *P<0.01 vs other groups. Data are expressed as mean±SEM, n=3. Ad.GSK‐S9A indicates adenovirus expressing constitutively active GSK‐3β; Ad.Null, control adenovirus; EPCs, endothelial progenitor cells; KS, kallistatin; LY, LY294002; VEGF, vascular endothelial growth factor.
Kallistatin Increases Akt and eNOS Phosphorylation, eNOS Activity and Expression, and NO Production
In cultured EPCs, kallistatin increased the phosphorylation of Akt and eNOS, as determined by Western blot (Figure 6A). In addition, kallistatin stimulated NOS activity by 1.3‐fold (n=3, P<0.05) and expression and NOx levels by 1.7‐fold (n=3, P<0.05) in culture medium (Figure 6B through 6E). Both eNOS phosphorylation and NO secretion were significantly inhibited by LY, indicating that kallistatin‐induced eNOS activity and NO production are mediated by PI3K‐Akt signaling.
Figure 6.
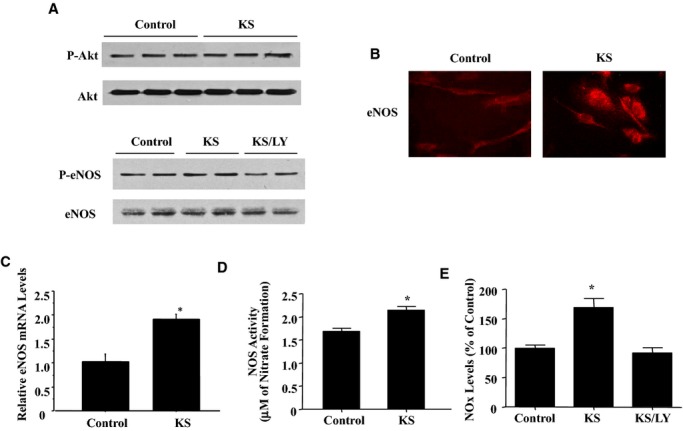
KS activates the phosphoinositide 3‐kinase–Akt–nitric oxide synthase pathway in EPCs. A, Western blots of phosphorylated and total Akt and eNOS. B and C, eNOS expression was determined by immunocytochemistry (B) and real‐time polymerase chain reaction (C). D and E, NOS activity (D) and NOx levels (E). *P<0.01 vs other groups. Data are expressed as mean±SEM, n=3. EPCs indicates endothelial progenitor cells; eNOS, endothelial nitric oxide synthase; KS, kallistatin; LY, LY294002; NOS, nitric oxide synthase; NOx, nitrate/nitrite.
Kallistatin Increases GSK‐3β Phosphorylation and VEGF and MMP‐2 Synthesis via PI3K‐Akt Signaling Pathways
Kallistatin increased GSK‐3β phosphorylation through a PI3K‐Akt signaling event in cultured EPCs as LY blocked kallistatin's effect (Figure 7A). Kallistatin also promoted VEGF secretion into culture medium (Figure 7B) and increased VEGF mRNA levels by 2.6‐fold (n=3, P<0.05; Figure 7C). Likewise, kallistatin upregulated MMP‐2 protein levels and activity (Figure 7D) and elevated mRNA levels by 1.7‐fold (n=3, P<0.05; Figure 7E). Kallistatin's effect on VEGF and MMP‐2 mRNA levels was abolished by LY, l‐NAME, or anti‐VEGF antibody (Figure 7C and 7E). These results indicate that kallistatin triggers the activation of PI3K‐Akt–mediated eNOS‐NO and GSK‐3β–VEGF–MMP‐2 signaling pathways in EPCs.
Figure 7.
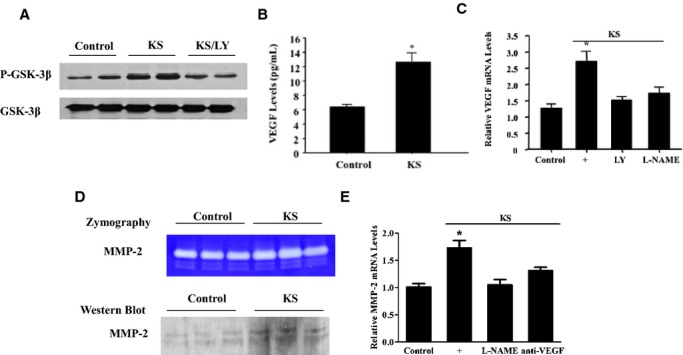
KS increases GSK‐3β phosphorylation as well as VEGF and MMP‐2 levels via phosphoinositide 3‐kinase–Akt pathways in EPCs. A, Western blot of phosphorylated and total GSK‐3β. B and C, Protein (B) and mRNA (C) levels of VEGF. D and E, Zymography and Western blot (D) and mRNA (E) levels of MMP‐2. *P<0.01 vs other groups. Data are expressed as mean±SEM, n=3. EPCs indicates endothelial progenitor cells; GSK‐3β, glycogen synthase kinase‐3β; KS, kallistatin; LY, LY294002; MMP‐2, matrix metalloproteinase‐2; VEGF, vascular endothelial growth factor.
Discussion
This study demonstrates that endogenous kallistatin plays a novel role in protection against vascular injury by increasing circulating EPC levels and promoting the viability, mobility, and function of cultured EPCs. We found that kallistatin depletion decreased circulating EPC levels in DOCA‐salt hypertensive rats, whereas kallistatin gene delivery increased EPC levels. Our previous report showed that kallistatin gene transfer attenuated capillary rarefaction in this animal model.26 Conversely, we further showed in this study that kallistatin antibody injection augmented glomerular capillary loss. Our in vitro studies demonstrated that recombinant kallistatin reduced TNF‐α–induced apoptosis of cultured EPCs, indicating that kallistatin's antiapoptotic actions may be partly responsible for the prevention of glomerular endothelial cell loss. Kallistatin administration also increased the proliferative, migratory, adhesive, and tube‐forming abilities of EPCs by activation of Akt‐eNOS and Akt–GSK‐3β–VEGF signaling pathways. Thus, kallistatin is unique in preventing endothelial injury because it exhibits pleiotropic effects on EPCs by antagonizing TNF‐α–induced apoptosis and directly stimulating the migration and vasculogenic capacity of EPCs by PI3K‐Akt–dependent signaling cascades (Figure 8). EPCs derived from rats and humans displayed similar effects on survival, migration, and functional activity in response to human kallistatin treatment. Consequently, our results strongly implicate human kallistatin as a promising new approach for enhancing endothelial repair in vascular diseases.
Figure 8.
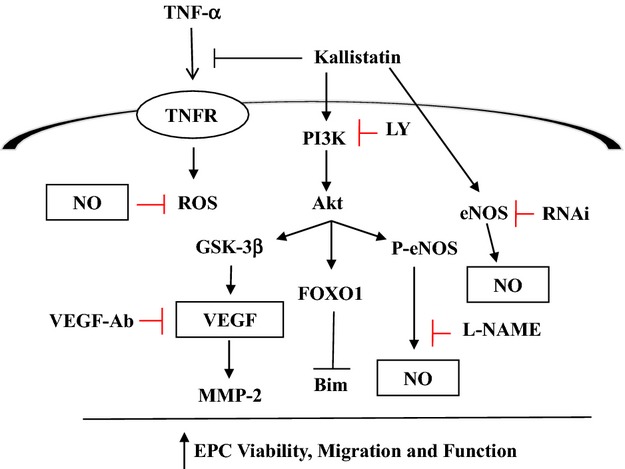
Proposed signaling pathways mediated by KS in promoting EPC viability, migration, adhesion, and tube formation. Ab indicates antibody; eNOS, endothelial nitric oxide synthase; EPCs, endothelial progenitor cells; FOXO1, forkhead box O1; GSK‐3β, glycogen synthase kinase‐3β; LY, LY294002; MMP‐2, matrix metalloproteinase‐2; NO, nitric oxide; PI3K, phosphoinositide 3‐kinase; ROS, reactive oxygen species; TNF‐α, tumor necrosis factor‐α; TNFR, tumor necrosis factor receptor; VEGF, vascular endothelial growth factor.
EPCs are a continuous endogenous source of replenishment for damaged vessels and thus serve to maintain vascular integrity in response to endothelial injury.36–37 Circulating EPC levels, however, are inversely associated with organ dysfunction in patients with hypertension and coronary artery disease, highlighting the role of EPCs in the pathogenesis of cardiovascular diseases.1–3,38 Reduction of the number and function of EPCs limits the capacity for vascular repair. Several factors responsible for suppressed EPC production and number include oxidative stress, apoptosis, senescence, and inflammation.1,39 Our previous studies demonstrated that kallistatin exerts multiple protective effects against dysfunction of the cardiovascular and renal systems by inhibiting oxidative stress, inflammation, and apoptosis and elevating NO levels in experimental animal models and cultured cells.22–27 The PI3K‐Akt‐eNOS signaling pathway has been shown to be essential for EPC survival.11 Kallistatin was found to increase NO levels by stimulating Akt and eNOS phosphorylation in endothelial cells26 and, as shown in the current study, in EPCs. We showed that kallistatin promoted EPC proliferation and abolished TNF‐α–induced apoptosis and caspase activity, but these effects were blocked by PI3K and NOS inhibitors, indicating that activation of the PI3K‐Akt‐eNOS pathway by kallistatin is fundamental to its protective actions in EPC survival. Kallistatin's effect on apoptosis occurred in conjunction with increased FOXO1 synthesis and reduced Bim expression in EPCs, consistent with our findings in endothelial cells.26 Thus, stimulation of the PI3K‐Akt‐eNOS signaling cascade by kallistatin may account, in part, for the increase in circulating EPC number in DOCA‐salt hypertensive rats after kallistatin gene delivery.
The prevention of glomerular capillary loss by kallistatin observed in our previous study26 may be attributed not only to kallistatin's antiapoptotic actions but also to its antioxidant activity in the vasculature. Oxidative stress is a major mediator of vascular injury in cardiovascular pathologies.39–40 We have shown that kallistatin is a significant contributor to vascular homeostasis through its antioxidant actions. Kallistatin gene transfer reduced aortic oxidative stress, whereas kallistatin antibody injection increased it.26,29 Moreover, kallistatin levels in plasma are significantly lower in hypertensive animal models41 and in normotensive rats with oxidative end‐organ damage (Julie Chao, PhD, unpublished data, 2010). Oxidative stress decreases kallistatin expression in cultured endothelial cells,41 leading to insufficient kallistatin levels to maintain healthy vascular homeostasis. Exogenous kallistatin administration, however, prevents organ damage in the heart and kidney by inhibiting apoptosis and oxidative stress and increasing NO bioavailability in several animal models.19,23–26 Furthermore, the current study showed that kallistatin enhanced EPC viability, migration. and functional activity by a PI3K‐Akt‐eNOS–mediated signaling event. These combined findings indicate that kallistatin via NO formation may exert antioxidant actions and contribute to EPC viability, thereby promoting vascular repair and preventing endothelial injury.
Re‐endothelialization is accelerated by the mobilization and function of EPCs through Akt‐dependent eNOS activation and VEGF expression.10–11,10–46 VEGF contributes to neovascularization by augmenting EPC migratory activity, proliferation, and incorporation into endothelial cells.14,47 In this study, we clearly showed that kallistatin promotes EPC migration, adhesion and tube formation by activation of the PI3K‐Akt‐eNOS signaling pathway because PI3K and NOS inhibitors blocked kallistatin's effects. Stimulation of EPC migration and tube formation by kallistatin also involved the Akt–GSK‐3β–VEGF cascade because constitutively active GSK‐3β and anti‐VEGF antibody suppressed the actions of kallistatin. Since VEGF promotes tube formation of EPCs by stimulating MMP‐2 activation,48 it is likely that MMP‐2 plays a role in mediating kallistatin's effect on EPC tube formation. Our results indicate that kallistatin activated Akt–GSK‐3β signaling and increased VEGF and MMP‐2 levels in cultured EPCs. Activation of PI3K‐Akt signaling cascades by VEGF may also account for the increase in EPC migration and tube formation.
EPCs provide a promising approach for endothelial repair by directly contributing to neovascularization and indirectly promoting the function of local endothelial cells in angiogenesis by secretion of angiogenic factors. We showed that EPCs, from either a rat or human source, play an important role in vascular repair by directly contributing to vasculogenesis. Evidently, kallistatin appears to have a dual role in angiogenic signaling. We previously reported that kallistatin inhibits tumor growth and angiogenesis in animal models and in cultured endothelial cells,49–50 and this contrasts with our current findings. In the present study, however, we found that kallistatin exerts proangiogenic activity to promote vascular regeneration by enhancing EPC migration, survival, and neovascularization. Glomerular capillary loss was attenuated by kallistatin administration but was augmented by kallistatin depletion.26,29 Furthermore, we clearly showed that kallistatin directly promoted EPC migration, adhesion, and tube formation through activation of the PI3K‐Akt‐NOS signaling pathway. These combined findings indicate that kallistatin modulates endothelial function by either promoting vascular regeneration or inhibiting angiogenesis, depending on the physiological and pathological conditions.
Kallistatin was first identified in human plasma as a tissue kallikrein‐binding protein15–17; however, numerous studies have shown that kallistatin exerts multiple biological functions unrelated to the tissue kallikrein‐kinin system, such as vasodilation and protection against cardiovascular and renal injury.23–24,26,51 In the current study, we demonstrated that kallistatin reduces vascular injury by enhancing EPC mobility and functional capacity. Likewise, tissue kallikrein‐kinin and kinin B2 receptor signaling have also been observed to promote cardiac neovascularization by enhancing EPC migration and function.52–55 Consequently, kallistatin has a unique protective role in endothelial injury, independent of its interaction with tissue kallikrein.
Sources of Funding
This work was supported by National Institutes of Health grants HL‐118516 and HL‐29397, and C06 RR015455 from the Extramural Research Facilities Program of the National Center for Research Resources.
Disclosures
None.
Acknowledgments
We thank Dr D. Kerjaschki of the University of Vienna for providing JG‐12 antibody and Dr Robin Muise‐Helmericks of the Medical University of South Carolina for providing human umbilical vein endothelial cells.
References
- 1.Umemura T, Higashi Y. Endothelial progenitor cells: therapeutic target for cardiovascular diseases. J Pharmacol Sci. 2008; 108:1-6. [DOI] [PubMed] [Google Scholar]
- 2.Vasa M, Fichtlscherer S, Aicher A, Adler K, Urbich C, Martin H, Zeiher AM, Dimmeler S. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res. 2001; 89:E1-E7. [DOI] [PubMed] [Google Scholar]
- 3.Vasa M, Fichtlscherer S, Adler K, Aicher A, Martin H, Zeiher AM, Dimmeler S. Increase in circulating endothelial progenitor cells by statin therapy in patients with stable coronary artery disease. Circulation. 2001; 103:2885-2890. [DOI] [PubMed] [Google Scholar]
- 4.Choi JH, Kim KL, Huh W, Kim B, Byun J, Suh W, Sung J, Jeon ES, Oh HY, Kim DK. Decreased number and impaired angiogenic function of endothelial progenitor cells in patients with chronic renal failure. Arterioscler Thromb Vasc Biol. 2004; 24:1246-1252. [DOI] [PubMed] [Google Scholar]
- 5.Tepper OM, Galiano RD, Capla JM, Kalka C, Gagne PJ, Jacobowitz GR, Levine JP, Gurtner GC. Human endothelial progenitor cells from type ii diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation. 2002; 106:2781-2786. [DOI] [PubMed] [Google Scholar]
- 6.Cribbs SK, Sutcliffe DJ, Taylor WR, Rojas M, Easley KA, Tang L, Brigham KL, Martin GS. Circulating endothelial progenitor cells inversely associate with organ dysfunction in sepsis. Intensive Care Med. 2012; 38:429-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grisar J, Aletaha D, Steiner CW, Kapral T, Steiner S, Seidinger D, Weigel G, Schwarzinger I, Wolozcszuk W, Steiner G, Smolen JS. Depletion of endothelial progenitor cells in the peripheral blood of patients with rheumatoid arthritis. Circulation. 2005; 111:204-211. [DOI] [PubMed] [Google Scholar]
- 8.Ablin JN, Boguslavski V, Aloush V, Elkayam O, Paran D, Caspi D, George J. Effect of anti‐tnfalpha treatment on circulating endothelial progenitor cells (epcs) in rheumatoid arthritis. Life Sci. 2006; 79:2364-2369. [DOI] [PubMed] [Google Scholar]
- 9.Thum T, Fraccarollo D, Schultheiss M, Froese S, Galuppo P, Widder JD, Tsikas D, Ertl G, Bauersachs J. Endothelial nitric oxide synthase uncoupling impairs endothelial progenitor cell mobilization and function in diabetes. Diabetes. 2007; 56:666-674. [DOI] [PubMed] [Google Scholar]
- 10.Aicher A, Heeschen C, Mildner‐Rihm C, Urbich C, Ihling C, Technau‐Ihling K, Zeiher AM, Dimmeler S. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med. 2003; 9:1370-1376. [DOI] [PubMed] [Google Scholar]
- 11.Dimmeler S, Aicher A, Vasa M, Mildner‐Rihm C, Adler K, Tiemann M, Rutten H, Fichtlscherer S, Martin H, Zeiher AM. Hmg‐coa reductase inhibitors (statins) increase endothelial progenitor cells via the pi 3‐kinase/akt pathway. J Clin Invest. 2001; 108:391-397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Landmesser U, Engberding N, Bahlmann FH, Schaefer A, Wiencke A, Heineke A, Spiekermann S, Hilfiker‐Kleiner D, Templin C, Kotlarz D, Mueller M, Fuchs M, Hornig B, Haller H, Drexler H. Statin‐induced improvement of endothelial progenitor cell mobilization, myocardial neovascularization, left ventricular function, and survival after experimental myocardial infarction requires endothelial nitric oxide synthase. Circulation. 2004; 110:1933-1939. [DOI] [PubMed] [Google Scholar]
- 13.Huang PH, Chen YH, Tsai HY, Chen JS, Wu TC, Lin FY, Sata M, Chen JW, Lin SJ. Intake of red wine increases the number and functional capacity of circulating endothelial progenitor cells by enhancing nitric oxide bioavailability. Arterioscler Thromb Vasc Biol. 2010; 30:869-877. [DOI] [PubMed] [Google Scholar]
- 14.Asahara T, Takahashi T, Masuda H, Kalka C, Chen D, Iwaguro H, Inai Y, Silver M, Isner JM. Vegf contributes to postnatal neovascularization by mobilizing bone marrow‐derived endothelial progenitor cells. EMBO J. 1999; 18:3964-3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chao J, Tillman DM, Wang MY, Margolius HS, Chao L. Identification of a new tissue‐kallikrein‐binding protein. Biochem J. 1986; 239:325-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chao J, Chai KX, Chen LM, Xiong W, Chao S, Woodley‐Miller C, Wang LX, Lu HS, Chao L. Tissue kallikrein‐binding protein is a serpin. I. Purification, characterization, and distribution in normotensive and spontaneously hypertensive rats. J Biol Chem. 1990; 265:16394-16401. [PubMed] [Google Scholar]
- 17.Zhou GX, Chao L, Chao J. Kallistatin: a novel human tissue kallikrein inhibitor. Purification, characterization, and reactive center sequence. J Biol Chem. 1992; 267:25873-25880. [PubMed] [Google Scholar]
- 18.Chai KX, Chen LM, Chao J, Chao L. Kallistatin: a novel human serine proteinase inhibitor. Molecular cloning, tissue distribution, and expression in escherichia coli. J Biol Chem. 1993; 268:24498-24505. [PubMed] [Google Scholar]
- 19.Chao J, Schmaier A, Chen LM, Yang Z, Chao L. Kallistatin, a novel human tissue kallikrein inhibitor: levels in body fluids, blood cells, and tissues in health and disease. J Lab Clin Med. 1996; 127:612-620. [DOI] [PubMed] [Google Scholar]
- 20.Chen LM, Chao L, Chao J. Adenovirus‐mediated delivery of human kallistatin gene reduces blood pressure of spontaneously hypertensive rats. Hum Gene Ther. 1997; 8:341-347. [DOI] [PubMed] [Google Scholar]
- 21.Wang CR, Chen SY, Wu CL, Liu MF, Jin YT, Chao L, Chao J. Prophylactic adenovirus‐mediated human kallistatin gene therapy suppresses rat arthritis by inhibiting angiogenesis and inflammation. Arthritis Rheum. 2005; 52:1319-1324. [DOI] [PubMed] [Google Scholar]
- 22.Chao J, Yin H, Yao YY, Shen B, Smith RS, Jr, Chao L. Novel role of kallistatin in protection against myocardial ischemia‐reperfusion injury by preventing apoptosis and inflammation. Hum Gene Ther. 2006; 17:1201-1213. [DOI] [PubMed] [Google Scholar]
- 23.Gao L, Yin H. S. Smith R J, Chao L, Chao J. Role of kallistatin in prevention of cardiac remodeling after chronic myocardial infarction. Lab Invest. 2008; 88:1157-1166. [DOI] [PubMed] [Google Scholar]
- 24.Shen B, Hagiwara M, Yao YY, Chao L, Chao J. Salutary effect of kallistatin in salt‐induced renal injury, inflammation, and fibrosis via antioxidative stress. Hypertension. 2008; 51:1358-1365. [DOI] [PubMed] [Google Scholar]
- 25.Shen B, Smith RS, Jr, Hsu YT, Chao L, Chao J. Kruppel‐like factor 4 is a novel mediator of kallistatin in inhibiting endothelial inflammation via increased endothelial nitric‐oxide synthase expression. J Biol Chem. 2009; 284:35471-35478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen B, Gao L, Hsu YT, Bledsoe G, Hagiwara M, Chao L, Chao J. Kallistatin attenuates endothelial apoptosis through inhibition of oxidative stress and activation of akt‐enos signaling. Am J Physiol Heart Circ Physiol. 2010; 299:H1419-H1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yin H, Gao L, Shen B, Chao L, Chao J. Kallistatin inhibits vascular inflammation by antagonizing tumor necrosis factor‐alpha‐induced nuclear factor kappab activation. Hypertension. 2010; 56:260-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beswick RA, Zhang H, Marable D, Catravas JD, Hill WD, Webb RC. Long‐term antioxidant administration attenuates mineralocorticoid hypertension and renal inflammatory response. Hypertension. 2001; 37:781-786. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, Bledsoe G, Hagiwara M, Shen B, Chao L, Chao J. Depletion of endogenous kallistatin exacerbates renal and cardiovascular oxidative stress, inflammation, and organ remodeling. Am J Physiol Renal Physiol. 2012; 303:F1230-F1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.National Research Council. Guide for the Care and Use of Laboratory Animals. 19967th edWashington, DC: National Academy Press [Google Scholar]
- 31.Chan YH, Lam TH, Lau KK, Yiu KH, Siu CW, Li SW, Chan HT, Tam S, Lau CP, Tse HF. Dietary intake of phytoestrogen is associated with increased circulating endothelial progenitor cells in patients with cardiovascular disease. Eur J Cardiovasc Prev Rehabil. 2011; 18:360-368. [DOI] [PubMed] [Google Scholar]
- 32.Chen VC, Chao L, Chao J. Reactive‐site specificity of human kallistatin toward tissue kallikrein probed by site‐directed mutagenesis. Biochim Biophys Acta. 2000; 1479:237-246. [DOI] [PubMed] [Google Scholar]
- 33.Li P, Bledsoe G, Yang ZR, Fan H, Chao L, Chao J. Human kallistatin administration reduces organ injury and improves survival in a mouse model of polymicrobial sepsis. Immunology. 2014; 142:216-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chao J, Yin H, Gao L, Hagiwara M, Shen B, Yang ZR, Chao L. Tissue kallikrein elicits cardioprotection by direct kinin b2 receptor activation independent of kinin formation. Hypertension. 2008; 52:715-720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J, Yang Z, Li P, Bledsoe G, Chao L, Chao J. Kallistatin antagonizes wnt/beta‐catenin signaling and cancer cell motility via binding to low‐density lipoprotein receptor‐related protein 6. Mol Cell Biochem. 2013; 379:295-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mikirova NA, Jackson JA, Hunninghake R, Kenyon J, Chan KW, Swindlehurst CA, Minev B, Patel AN, Murphy MP, Smith L, Alexandrescu DT, Ichim TE, Riordan NH. Circulating endothelial progenitor cells: a new approach to anti‐aging medicine? J Transl Med. 2009; 7:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Besler C, Doerries C, Giannotti G, Luscher TF, Landmesser U. Pharmacological approaches to improve endothelial repair mechanisms. Expert Rev Cardiovasc Ther. 2008; 6:1071-1082. [DOI] [PubMed] [Google Scholar]
- 38.Werner N, Kosiol S, Schiegl T, Ahlers P, Walenta K, Link A, Bohm M, Nickenig G. Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med. 2005; 353:999-1007. [DOI] [PubMed] [Google Scholar]
- 39.Fiorito C, Rienzo M, Crimi E, Rossiello R, Balestrieri ML, Casamassimi A, Muto F, Grimaldi V, Giovane A, Farzati B, Mancini FP, Napoli C. Antioxidants increase number of progenitor endothelial cells through multiple gene expression pathways. Free Radic Res. 2008; 42:754-762. [DOI] [PubMed] [Google Scholar]
- 40.Touyz RM. Reactive oxygen species in vascular biology: role in arterial hypertension. Expert Rev Cardiovasc Ther. 2003; 1:91-106. [DOI] [PubMed] [Google Scholar]
- 41.Shen B, Chao L, Chao J. Pivotal role of jnk‐dependent foxo1 activation in downregulation of kallistatin expression by oxidative stress. Am J Physiol Heart Circ Physiol. 2010; 298:H1048-H1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walter DH, Rittig K, Bahlmann FH, Kirchmair R, Silver M, Murayama T, Nishimura H, Losordo DW, Asahara T, Isner JM. Statin therapy accelerates reendothelialization: a novel effect involving mobilization and incorporation of bone marrow‐derived endothelial progenitor cells. Circulation. 2002; 105:3017-3024. [DOI] [PubMed] [Google Scholar]
- 43.Heeschen C, Aicher A, Lehmann R, Fichtlscherer S, Vasa M, Urbich C, Mildner‐Rihm C, Martin H, Zeiher AM, Dimmeler S. Erythropoietin is a potent physiologic stimulus for endothelial progenitor cell mobilization. Blood. 2003; 102:1340-1346. [DOI] [PubMed] [Google Scholar]
- 44.Urao N, Okigaki M, Yamada H, Aadachi Y, Matsuno K, Matsui A, Matsunaga S, Tateishi K, Nomura T, Takahashi T, Tatsumi T, Matsubara H. Erythropoietin‐mobilized endothelial progenitors enhance reendothelialization via akt‐endothelial nitric oxide synthase activation and prevent neointimal hyperplasia. Circ Res. 2006; 98:1405-1413. [DOI] [PubMed] [Google Scholar]
- 45.Nakano M, Satoh K, Fukumoto Y, Ito Y, Kagaya Y, Ishii N, Sugamura K, Shimokawa H. Important role of erythropoietin receptor to promote vegf expression and angiogenesis in peripheral ischemia in mice. Circ Res. 2007; 100:662-669. [DOI] [PubMed] [Google Scholar]
- 46.Santhanam AV, d'Uscio LV, Peterson TE, Katusic ZS. Activation of endothelial nitric oxide synthase is critical for erythropoietin‐induced mobilization of progenitor cells. Peptides. 2008; 29:1451-1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iwaguro H, Yamaguchi J, Kalka C, Murasawa S, Masuda H, Hayashi S, Silver M, Li T, Isner JM, Asahara T. Endothelial progenitor cell vascular endothelial growth factor gene transfer for vascular regeneration. Circulation. 2002; 105:732-738. [DOI] [PubMed] [Google Scholar]
- 48.Wu Y, Dai J, Schmuckler NG, Bakdash N, Yoder MC, Overall CM, Colman RW. Cleaved high molecular weight kininogen inhibits tube formation of endothelial progenitor cells via suppression of matrix metalloproteinase 2. J Thromb Haemost. 2010; 8:185-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miao RQ, Agata J, Chao L, Chao J. Kallistatin is a new inhibitor of angiogenesis and tumor growth. Blood. 2002; 100:3245-3252. [DOI] [PubMed] [Google Scholar]
- 50.Miao RQ, Chen V, Chao L, Chao J. Structural elements of kallistatin required for inhibition of angiogenesis. Am J Physiol Cell Physiol. 2003; 284:C1604-C1613. [DOI] [PubMed] [Google Scholar]
- 51.Chao J, Stallone JN, Liang YM, Chen LM, Wang DZ, Chao L. Kallistatin is a potent new vasodilator. J Clin Invest. 1997; 100:11-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krankel N, Katare RG, Siragusa M, Barcelos LS, Campagnolo P, Mangialardi G, Fortunato O, Spinetti G, Tran N, Zacharowski K, Wojakowski W, Mroz I, Herman A, Manning Fox JE, MacDonald PE, Schanstra JP, Bascands JL, Ascione R, Angelini G, Emanueli C, Madeddu P. Role of kinin b2 receptor signaling in the recruitment of circulating progenitor cells with neovascularization potential. Circ Res. 2008; 103:1335-1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krankel N, Kuschnerus K, Muller M, Speer T, Mocharla P, Madeddu P, Bader M, Luscher TF, Landmesser U. Novel insights into the critical role of bradykinin and the kinin b2 receptor for vascular recruitment of circulating endothelial repair‐promoting mononuclear cell subsets: alterations in patients with coronary disease. Circulation. 2013; 127:594-603. [DOI] [PubMed] [Google Scholar]
- 54.Yao Y, Sheng Z, Li Y, Fu C, Ma G, Liu N, Chao J, Chao L. Tissue kallikrein‐modified human endothelial progenitor cell implantation improves cardiac function via enhanced activation of akt and increased angiogenesis. Lab Invest. 2013; 93:577-591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yao Y, Sheng Z, Li Y, Yan F, Fu C, Ma G, Liu N, Chao J, Chao L. Tissue kallikrein promotes cardiac neovascularization by enhancing endothelial progenitor cell functional capacity. Hum Gene Ther. 2012; 23:859-870. [DOI] [PMC free article] [PubMed] [Google Scholar]