

Abstract
C3 glomerulopathy has recently been described as a distinct entity. The underlying mechanism is unregulated activation of the alternate pathway of the complement system. The most common presentation is with an acute nephritic syndrome. The diagnosis is made on immunofluoroscence by the presence of isolated or dominant C3 staining. In this retrospective study, renal biopsy data were collected from 2010 to 2013 patients with C3 glomerulopathy identified and their clinical and biochemical parameters analyzed. Out of 514 biopsies available for analysis, the incidence of C3 glomerulopathy was 1.16% (n = 6). The mean age of the presentation was 26 years and the average estimated glomerular filtration rate was 30.65 ml/min/1.73 m2. The most common histopathological pattern was membranoproliferative glomerulonephritis (n = 4).
Keywords: Alternate pathway, C3 glomerulopathy, membranoproliferative glomerulonephritis, plasma therapy
Introduction
C3 glomerulopathy is recently described condition characterized by the presence of isolated or predominant glomerular C3 deposits.[1,2,3,4,5,6,7] Levy et al.[8] had described a distinct subset of membranoproliferative glomerulonephritis (MPGN) type-I patients with isolated and dominant C3 deposition on immunofluorescence (IF) in 1978, the underlying pathogenic mechanism has only been elucidated in the last decade.[9,10,11,12,13,14,15] C3 glomerulopathy comprises dense deposit disease (DDD), C3GN and complement factor H-related protein 5 (CFHR5) nephropathy.[2,3,4,5,6,7] Studies in the last decade have shown that dysregulation of the alternate pathway of the complement system leads to these disorders.[10,11,12,13,14] Confirmatory tests like electron microscopy (EM) and evaluation of individual complement factor (CF) levels are not easily available, hence it is often difficult to conclusively subclassify the patients into individual disorders.[2,3,4,5,6,7] The long-term outcomes, efficacy of various treatment modalities and transplant outcomes are not yet clear, and need to be elucidated in large epidemiological studies with long follow-up.
Materials and Methods
Retrospective renal biopsy registry data were analyzed from 2010 to 2013. The biopsies were analyzed with hemotoxylin and eosin, Congo red, Masson's trichome, period-acid Schiff, silver methenamine stain and IF. The light microscopic picture was analyzed and classified into various morphological patterns like MPGN, diffuse proliferative GN, isolated mesangial hypercellularity and crescentic GN. The diagnostic criteria was more than 3+ staining with C3 on IF and absence of significant immunoglobulin (Ig) staining or C3 staining two orders of magnitude more than that of IgG or IgM staining.
EM was done in two patients, which were consistent with the findings of C3GN. Demographic, clinical, treatment history and biochemical data, including C3/C4 levels of these patients were analyzed using standard analytical methods.
Results
Among 514 biopsies available for analysis during the above period, six (1.16%) were found to be conforming to C3 glomerulopathy, as defined by the diagnostic criteria mentioned above. Among these six, two samples were further evaluated with EM, which were consistent with the diagnosis. The demographic and clinical profile is given in Table 1.
Table 1.
Demographic and clinical profile of patients with C3GN
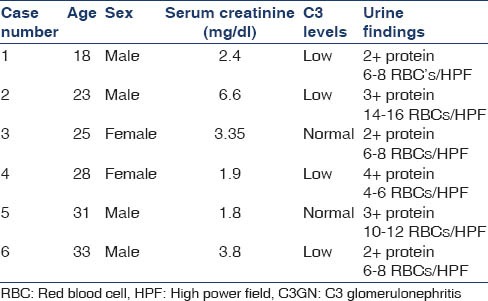
The mean age of the presentation was 26 years, and the average estimated glomerular filtration rate was 30.65 ml/min/1.73 m2 chronic kidney disease (CKD) epidemiology. None of the patients were suspected to be having C3GN prior to the biopsy. All the patients underwent biopsy for evaluation of nephritic syndrome and evaluation of deranged kidney functions. Active urinary sediment was seen in all cases. Three cases presented with acute nephritic syndrome and one had a recent history of upper respiratory tract infection. Other three presented with nephrotic syndrome. Hypertension was seen in four patients. C3 levels were low in four patients. On 1-year follow-up, five patients were available, out of which three had stable renal functions and two had progressed to CKD-5 (Case No: 2 and 6).
The renal biopsy findings are in Table 2.
Table 2.
Renal biopsy findings
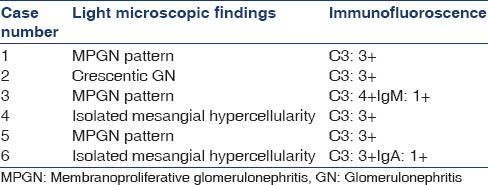
Two cases also underwent EM, which showed sub-endothelial electron-dense deposits.
All patients were treated with angiotensin convertase enzyme inhibitor (ACEI)/angiotensin receptor blockers (ARBs) and other supportive treatment. All patients were treated according to currently accepted guidelines for the treatment of MPGN. Case number 2 was treated with steroids and intravenous cyclophosphamide. Case numbers 1, 3 and 6 were treated with oral steroids; however, they were tapered and stopped after 6 months due to lack of any benefit and also appearance of features of steroid toxicity. Apart from case number 2, none of the patients were treated with cytotoxic drugs.
Discussion
C3 glomerulopathy is an umbrella term, comprising various individual disorders like DDD, C3GN and CFHR5 nephropathy.[1,2,3,4,5,6,7] The putative mechanism in all these disorders has been found to be unregulated activation of the alternate pathway of the complement system.[11,12,13,14,15] Potential abnormalities include genetic deficiencies in regulators of the alternative complement pathway (such as CF H, CFHR1-5, CFI and CD46) and autoantibodies to factor H, factor B and the C3 convertase (known as C3 nephritic factors).[11,12,13,14,15] The net result is unregulated activation of alternate pathway of the complement system. The clinical picture is extremely variable.[16,17,18] DDD typically presents in younger age group and is associated with other extra-renal manifestations like drusen and lipodystrophy.[6,9,19] The prognosis and outcomes after transplant are significantly inferior in patients with DDD.[6,16,20] C3GN has a more variable and indolent course. CFHR5 nephropathy is a familial form of C3 glomerulopathy and is clinically similar to C3GN.[3,4,5,6,7] C3 glomerulopathy is a rare disorder, as confirmed in our study, whose natural history and clinical course is unclear.[6,7,16,17,18]
The morphological picture is heterogeneous,[2,3,4] as seen in our study, and none of the patterns is diagnostic of C3 glomerulopathy. An isolated C3 (i.e. >3+) or C3 dominant (i.e. >2 orders of intensity than any deposit of Ig) pattern on IF is the only feature to suggest an underlying C3 glomerulopathy on routine renal biopsies.[1,2,3,4,5,6] Undoubtedly, EM is essential to further classify these disorders into individual subtypes; however, it is not easily available.[4,5,6,7] The various investigations suggested for evaluation of these disorders include measurement of individual CF levels and screening for mutations of complement regulatory proteins.[5,6,7] These tests are currently available only in research laboratories.
Measurement of C3 and C4 levels is not helpful. As it would be expected from any disease with unregulated alternate pathway activation,[11,12,13,14] C3 levels would be low; however, it can be normal in patients with C3GN[11,12,13,14,15,16,17,18] as seen in one-third of our patients. Hence, it needs to be stressed that normal C3 levels does not rule out C3GN.
It is worthwhile to note that none of the cases discussed above had any evidence of underlying secondary causes such as viral hepatitis, chronic infections or paraproteinemia to explain their MPGN.
Treatment generally consists of supportive measures like ACEI/ARBs. Role of steroids and other immunosuppressive drugs is unclear. It is suggested that immunosuppressive drugs be avoided in these patients due to the increased risk of infection associated with these agents.[16,17,18,21,22] Infections in these patients with underlying abnormalities of innate immunity may cause complement activation and trigger inflammation, which could exacerbate nephritis.[9,16,17,21] Drugs such as steroids, cytotoxic drugs and rituximab may be useful in patients with proven antibodies against complement regulatory proteins. These drugs along with steroids may also have a role in patients with crescentic GN.[9,21]
Plasma therapy may be useful in patients with regulatory factor deficiency or with circulating C3 nephritic factor (known to cause unregulated activation of alternate pathway). Plasma infusion may replace the deficient factor. Plasma exchange helps by removing C3 nephritic factor. The frequency and duration of plasma therapy is unclear. If there is adequate clinical response, it needs to be continued indefinitely.[9,16,17,18,21]
In view of the recent advances made in understanding of the underlying pathophysiological mechanisms involved in these diseases, it seems logical to attempt therapeutic inhibition of complement C3 or C5.
Eculizumab is a monoclonal antibody that prevents C5 activation and has been used in many patients with C3 glomerulopathy with mixed results. The major limiting factor regarding the use of Eculizumab is its prohibitive cost. Eculizumab has not been tried in any large controlled studies, hence there are no recommendations regarding the duration and frequency of treatment. If there is an adequate response, it needs to be continued indefinitely.[20,21,22]
Overall, the therapeutic options are limited and beyond supportive treatment there is nothing much to offer these patient at present.
Transplantation is the best long-term option in patients who progress to CKD-ESRD. However, the risk of disease recurrence and graft loss is considerable. There are no studies, which have evaluated the long-term outcomes among renal transplant recipients. It could be deduced that the high rate of recurrence among MPGN patients could actually be occurring in the subset of patients who had underlying C3GN; however, this is conjectural. Combined liver and kidney transplant is advisable in patients with CF mutations.[6,9,20,22]
Conclusion
C3 glomerulopathy generally affects young adults. The clinical features are mild in most of them; however, the long-term outcomes are not yet known. The light microscopic picture can be variable. Specific treatment is not available. Plasma therapy is indicated in patients with progressive disease and documented CF deficiencies. Close follow-up and supportive treatment with ACEI or ARBs is advised in all patients.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Pickering M, Cook HT. Complement and glomerular disease: New insights. Curr Opin Nephrol Hypertens. 2011;20:271–7. doi: 10.1097/MNH.0b013e328345848b. [DOI] [PubMed] [Google Scholar]
- 2.Bomback AS, Appel GB. Pathogenesis of the C3 glomerulopathies and reclassification of MPGN. Nat Rev Nephrol. 2012;8:634–42. doi: 10.1038/nrneph.2012.213. [DOI] [PubMed] [Google Scholar]
- 3.Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis: Pathogenetic heterogeneity and proposal for a new classification. Semin Nephrol. 2011;31:341–8. doi: 10.1016/j.semnephrol.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 4.Fakhouri F, Frémeaux-Bacchi V, Noël LH, Cook HT, Pickering MC. C3 glomerulopathy: A new classification. Nat Rev Nephrol. 2010;6:494–9. doi: 10.1038/nrneph.2010.85. [DOI] [PubMed] [Google Scholar]
- 5.Hou J, Markowitz GS, Bomback AS, Appel GB, Herlitz LC, Barry Stokes M, et al. Toward a working definition of C3 glomerulopathy by immunofluorescence. Kidney Int. 2014;85:450–6. doi: 10.1038/ki.2013.340. [DOI] [PubMed] [Google Scholar]
- 6.Barbour TD, Pickering MC, Terence Cook H. Dense deposit disease and C3 glomerulopathy. Semin Nephrol. 2013;33:493–507. doi: 10.1016/j.semnephrol.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pickering MC, D’Agati VD, Nester CM, Smith RJ, Haas M, Appel GB, et al. C3 glomerulopathy: Consensus report. Kidney Int. 2013;84:1079–89. doi: 10.1038/ki.2013.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levy M, Gubler MC, Sich M, Beziau A, Habib R. Immunopathology of membranoproliferative glomerulonephritis with subendothelial deposits (Type I MPGN) Clin Immunol Immunopathol. 1978;10:477–92. doi: 10.1016/0090-1229(78)90160-5. [DOI] [PubMed] [Google Scholar]
- 9.Barbour TD, Pickering MC, Cook HT. Recent insights into C3 glomerulopathy. Nephrol Dial Transplant. 2013;28:1685–93. doi: 10.1093/ndt/gfs430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Servais A, Frémeaux-Bacchi V, Lequintrec M, Salomon R, Blouin J, Knebelmann B, et al. Primary glomerulonephritis with isolated C3 deposits: A new entity which shares common genetic risk factors with haemolytic uraemic syndrome. J Med Genet. 2007;44:193. doi: 10.1136/jmg.2006.045328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sethi S, Fervenza FC, Zhang Y, Nasr SH, Leung N, Vrana J, et al. Proliferative glomerulonephritis secondary to dysfunction of the alternative pathway of complement. Clin J Am Soc Nephrol. 2011;6:1009–17. doi: 10.2215/CJN.07110810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lachmann PJ. The amplification loop of the complement pathways. Adv Immunol. 2009;104:115–49. doi: 10.1016/S0065-2776(08)04004-2. [DOI] [PubMed] [Google Scholar]
- 13.Servais A, Noël LH, Roumenina LT, Le Quintrec M, Ngo S, Dragon-Durey MA, et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. 2012;82:454–64. doi: 10.1038/ki.2012.63. [DOI] [PubMed] [Google Scholar]
- 14.Leroy V, Fremeaux-Bacchi V, Peuchmaur M, Baudouin V, Deschênes G, Macher MA, et al. Membranoproliferative glomerulonephritis with C3NeF and genetic complement dysregulation. Pediatr Nephrol. 2011;26:419–24. doi: 10.1007/s00467-010-1734-4. [DOI] [PubMed] [Google Scholar]
- 15.Hawfield A, Iskandar SS, Smith RJ. Alternative pathway dysfunction in kidney disease: A case report and review of dense deposit disease and C3 glomerulopathy. Am J Kidney Dis. 2013;61:828–31. doi: 10.1053/j.ajkd.2012.11.045. [DOI] [PubMed] [Google Scholar]
- 16.Sethi S, Fervenza FC, Zhang Y, Zand L, Vrana JA, Nasr SH, et al. C3 glomerulonephritis: Clinicopathological findings, complement abnormalities, glomerular proteomic profile, treatment, and follow-up. Kidney Int. 2012;82:465–73. doi: 10.1038/ki.2012.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Medjeral-Thomas NR, O’Shaughnessy MM, O’Regan JA, Traynor C, Flanagan M, Wong L, et al. C3 glomerulopathy: Clinicopathologic features and predictors of outcome. Clin J Am Soc Nephrol. 2014;9:46–53. doi: 10.2215/CJN.04700513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walker PD, Ferrario F, Joh K, Bonsib SM. Dense deposit disease is not a membranoproliferative glomerulonephritis. Mod Pathol. 2007;20:605–16. doi: 10.1038/modpathol.3800773. [DOI] [PubMed] [Google Scholar]
- 19.Darouich S, Goucha R, Jaafoura MH, Zekri S, Kheder A, Ben Maiz H. Membranoproliferative glomerulonephritis with isolated C3 deposits: Case report and literature review. Ultrastruct Pathol. 2011;35:42–6. doi: 10.3109/01913123.2010.532902. [DOI] [PubMed] [Google Scholar]
- 20.McCaughan JA, O’Rourke DM, Courtney AE. Recurrent dense deposit disease after renal transplantation: An emerging role for complementary therapies. Am J Transplant. 2012;12:1046–51. doi: 10.1111/j.1600-6143.2011.03923.x. [DOI] [PubMed] [Google Scholar]
- 21.Nester CM, Smith RJ. Treatment options for C3 glomerulopathy. Curr Opin Nephrol Hypertens. 2013;22:231–7. doi: 10.1097/MNH.0b013e32835da24c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vivarelli M, Pasini A, Emma F. Eculizumab for the treatment of dense-deposit disease. N Engl J Med. 2012;366:1163–5. doi: 10.1056/NEJMc1111953. [DOI] [PubMed] [Google Scholar]