

Abstract
The importance of free radical-induced oxidative damage after traumatic brain injury (TBI) has been well documented. Despite multiple clinical trials with radical-scavenging antioxidants that are neuroprotective in TBI models, none is approved for acute TBI patients. As an alternative antioxidant target, Nrf2 is a transcription factor that activates expression of antioxidant and cytoprotective genes by binding to antioxidant response elements (ARE) within DNA. Previous research has shown that neuronal mitochondria are susceptible to oxidative damage post-TBI, and thus the current study investigates whether Nrf2-ARE activation protects mitochondrial function when activated post-TBI. It was hypothesized that administration of carnosic acid (CA) would reduce oxidative damage biomarkers in brain tissue and also preserve cortical mitochondrial respiratory function post-TBI. A mouse controlled cortical impact (CCI) model was employed with a 1.0mm cortical deformation injury. Administration of CA at 15 minutes post-TBI reduced cortical lipid peroxidation, protein nitration, and cytoskeletal breakdown markers in a dose-dependent manner at 48 hours post-injury. Moreover, CA preserved mitochondrial respiratory function compared to vehicle animals. This was accompanied by decreased oxidative damage to mitochondrial proteins, suggesting the mechanistic connection of the two effects. Lastly, delaying the initial administration of CA up to 8 hours post-TBI was still capable of reducing cytoskeletal breakdown, thereby demonstrating a clinically relevant therapeutic window for this approach. This study demonstrates that pharmacological Nrf2-ARE induction is capable of neuroprotective efficacy when administered after TBI.
Keywords: lipid peroxidation, mitochondria, Nrf2, oxidative damage, traumatic brain injury, 4-hydroxy-2-nonenal
Introduction
It is well established that oxidative stress, and particularly lipid peroxidation (LP), is a deleterious component of many neurodegenerative disorders, which often causes harmful downstream consequences that result in cell death and dysfunction (1-3). More specifically, previous work has demonstrated that free radical mediated LP plays a critical role in the acute pathophysiology of traumatic brain injury (TBI).(1, 4-6) Lipid peroxidation involves free radical-induced oxidation of polyunsaturated fatty acids (e.g., arachidonic, linoleic, and docosahexaenoic acids) in cells and membrane phospholipids at allylic carbons. These peroxidized polyunsaturated fatty acids subsequently undergo phospholipase-mediated hydrolysis and disruption of the membrane phospholipid architecture ensues, leading to eventual loss of proper functioning phospholipid-dependent enzymes, ion channels, and structural proteins. As a consequence of LP-induced membrane damage, peroxidized fatty acids eventually lead to aldehydic breakdown products, including 4-hydroxy-2-nonenal (4-HNE).(7) The aldehyde 4-HNE is highly reactive with many cellular proteins, primarily via Schiff base and Michael adduct reactions with basic (e.g., lysine and histidine) and sulfhydryl (e.g. cysteine) containing amino acids. Such reactions are capable of impairing the function of a variety of cellular proteins, which likely contributes to neurodegenerative processes.(7, 8) Potential sources of post-TBI reactive oxygen species (ROS) that contribute to toxic LP production include iron-dependent Fenton reactions, causing hydroxyl radical (•OH) production and peroxynitrite (PN)-derived free radicals including •OH, nitrogen dioxide (•NO2), and carbonate (•CO3 radicals.(4) Free radical-mediated oxidative damage in acute CNS injury can result in protein modification and mitochondrial dysfunction, largely due to the intrinsic propensity of mitochondria to produce ROS as a byproduct of the electron transport chain function.(2, 9) In fact, previous work by our laboratory (2) and others has demonstrated that one major source of post-injury free radical production is the increased ROS leakage from injured brain mitochondria after injury.(10, 11)
Furthermore, previous work from our laboratory has shown that PN is able to directly inhibit mitochondrial function in the injured brain mitochondria and is associated with elevated 4-HNE.(12, 13) Moreover, direct application of PN to normal mitochondria simulates the effects of in vivo TBI. (12) While LP can directly cause membrane destruction and likely impair mitochondrial function, we recently demonstrated that the LP-derived reactive aldehydes 4-HNE and acrolein themselves can also directly inhibit mitochondrial respiration in vitro in mitochondria isolated from brain and spinal cord. (7) This can most likely be attributed to 4-HNE covalently binding to essential proteins and thereby affecting mitochondrial function.
A major area of investigation in relation to neurodegenerative processes, oxidative stress involves an imbalance in the ratio of harmful reactive oxygen and nitrogen species (ROS/RNS) and protective endogenous antioxidant defense enzymes.(14) An endogenous cytoprotective defense system exists to combat the basal and injury-induced imbalance in ROS/RNS and antioxidant/defense enzymes. This system is primarily under the inducible control of the pleiotropic transcription factor NF-E2-related factor 2 (Nrf2).(14, 15) Nrf2 has been identified as the key mediator of this inducible cytoprotective response via its interaction with the genomic cis-acting enhancer region of defense genes known as the antioxidant response element (ARE).(16, 17) Under normal cellular conditions, Nrf2 is sequestered in the cytoplasm by the repressor protein Keap1.(16) This binding interaction between Nrf2 and Keap1 is thought to facilitate the proteasomal degradation of Nrf2 by recruitment of a Cul3 ubiquitin ligase via the BTB domain of Keap1.(18) However, under conditions of stress (e.g. oxidative stress, ER stress, injury, toxicity, etc.), Nrf2 is released from Keap1 by a proposed hinge-latch mechanism.(19) This release therefore allows for subsequent Nrf2 translocation into the nucleus(20) where it can heterodimerize with small Maf proteins and binds to the ARE of effective cytoprotective genes(14), inducing transcription and consequent production of defense proteins.
In recent years, there have been numerous studies in various different neurodegeneration paradigms that have indicated that manipulation of the Nrf2-ARE pathway can dramatically attenuate multiple pathophysiological processes, including oxidative stress(21), mitochondrial dysfunction(22), and inflammation.(23) Additionally, recent work has demonstrated that this Nrf2-ARE defense response is inducible by a variety of small molecules, as demonstrated in several different in vitro(22, 24) and in vivo(25-27) paradigms. For example, it has recently been shown that the Nrf2-ARE pathway is involved and activated after TBI.(28) Specifically, the promising Nrf2-ARE activator sulforaphane (an isothiocyanate) has been shown to attenuate post-TBI pathophysiology, including blood-brain-barrier dysfunction,(29) edema formation,(30) and cognitive deficits.(31) Another impressive small molecule capable of inducing the Nrf2-ARE response is carnosic acid (CA), which was previously shown to be a more potent and efficacious activator of the ARE in vitro (32) and to be neuroprotective in vivo in a cerebral ischemia paradigm.(32) These protective effects of CA were also demonstrated to be dependent on Nrf2-ARE modulation in vitro.(32) Hence, it could be inferred that CA activates the Nrf2-ARE pathway and hence through downstream mediators is capable of eliciting a robust neuroprotective effect.
While previous research has extensively implicated the importance of mitochondria in the pathogenesis of numerous neurodegenerative processes,(11) very little is known with regard to Nrf2-ARE’s potential effects on mitochondrial bioenergetics post injury. Recent work by Greco and colleagues(33) found that a single administration of sulforaphane to naïve animals 40 hours prior to mitochondrial isolation could provide resistance to mitochondrial permeability transition pore formation via Nrf2-ARE mediated defenses. Furthermore, we also recently demonstrated that pre-treating naïve mice with either CA or sulforaphane 48 hours prior to the isolation of cortical mitochondria conferred a robust level of resistance to the mitochondria from subsequent exposure to toxic insults such as 4-HNE, and this effect occurred together with an increase in the mRNA levels of the Nrf2-ARE mediated gene heme oxygenase-1 (HO-1).(34) However, it has not yet been determined if activation of the Nrf2-ARE pathway could provide protection to mitochondria in vivo in the acute post-TBI phase. Thus, the current study investigated whether CA could reduce oxidative damage post-TBI in a dose dependent manner and if CA administration could preserve mitochondrial function post-TBI. It was hypothesized that CA-treated animals would have reduced oxidative damage post-TBI and improved mitochondrial respiratory function as compared to vehicle animals post-injury. It was also hypothesized that even with delayed initial administration of CA to mice, that CA would still be capable of attenuating cytoskeletal breakdown within a clinically relevant therapeutic window.
Materials & Methods
Animals
This study utilized young adult (8 weeks old) male CF-1 mice (Charles River Labs, USA) weighing 28–32 grams at time of surgery. All animals had ad libitum access to food and water and were housed in the Division of Laboratory Animal Resources sector of the University of Kentucky Chandler Medical Center, which is fully accredited by AALAC. All procedures described herein follow protocols approved by the University of Kentucky’s Institutional Animal Care and Use Committee, in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Mouse Model of Controlled Cortical Impact (CCI) TBI
Mice were initially anesthetized in a Plexiglas chamber using 3.0% isoflurane, shaved, weighed, and then placed into a stereotaxic frame (David Kopf, Tujunga, CA, USA). Core body temperature was maintained throughout the surgery process using an underlying heating pad. Throughout the surgical procedure, mice were kept anesthetized by a constant flow of 3.0% isoflurane and oxygen delivered via nose cone. The head was positioned in the horizontal plane with nose bar set at zero. A 2.0cm sagittal incision was made in the scalp and the skin retracted using hemostats to expose the skull. After exposing the skull, a 4.0mm diameter craniotomy was made using a dental bur (SS WHITE, Lakewood, NJ, USA) mounted on a cordless Dremel (Racine, WI, USA) lateral (left) to the sagittal suture, centered between bregma and lambda, while leaving the underlying dura mater intact. Sham-operated (control) mice received anesthesia and all surgical procedures (including craniotomy) but without the controlled cortical impact brain injury. Brain-injured mice received CCI utilizing an electronically controlled pneumatic impacting device (Precision Systems Instrumentation, TBI-0310 Impactor, Fairfax Station, VA, USA) with a 3.0mm diameter, beveled (flat) impactor tip. The impact velocity was held at 3.50 meters per second while the depth of cortical deformation was set at 1.0mm (severe) as described previously.(35) Mortality following this severe CCI brain injury is rare (<5.0%). After injury, the craniotomy was closed by secure placement of a 6.0mm diameter disk of dental acrylic cemented in place with quick bonding liquid cyanoacrylate. The mice were then placed in a Hova-Bator Incubator (model 1583; Randall Burkey Co, Boerne, TX, USA) set at 37°C for at least 30 minutes to prevent post-traumatic hypothermia before returning them to their home cage. Consciousness (i.e. return of right reflex and mobility) was regained within 15 minutes after the surgery. Brain-injured and sham mice typically show no untoward effects after recovering from anesthesia, and resume normal eating, drinking and grooming patterns thereafter. In this study, animals survived from 24 hours (mitochondria assays) to 48 hours (dose response assessments) post-injury depending upon randomized group assignment. Additional details on surgical procedures have been previously published by our laboratory.(35)
Drug Treatments
The pharmacological compound used in this study is CA, which has previously been shown to be a potent activator of the Nrf2-ARE pathway.(32) Carnosic Acid (Sigma, USA) was administered I.P. at 0.3, 1.0, or 3.0 mg/kg in a 10% Ethanol/ 90% PBS vehicle solution. The volumes administered of the compound solution I.P. did not exceed 0.3 ml. Carnosic acid or its vehicle, were administered at 15 minutes post-injury. The doses of CA were chosen based upon previous studies with this compound in a rat acute focal stroke model (32) and our own previously published work in mice.(34)
Chemicals
Sodium pyruvate, malate, rotenone, and carbonyl cyanide p-rifluoromethoxyphenylhydrazone (FCCP), were obtained from Sigma-Aldrich (St. Louis, MO). Oligomycin was obtained from Biomol, USA. 4-hydroxy 2-nonenal (4-HNE) was purchased from EMD Chemicals Inc. (Merck KGaA, Darmstadt, Germany). Chemicals were stored at -20°C as stock solutions. Working solutions for each bioenergetics experiment were always prepared fresh by creating appropriate dilutions in respiration buffer. All materials and reagents for the XF-24 assays were obtained from Seahorse Biosciences (North Billerica, MA, USA).
Isolation of Ficoll-Purified Mitochondria
Cortical brain mitochondria were extracted as previously described.(2) Briefly, mice were decapitated and the brain rapidly removed. Cortical regions were dissected out in an ice-cold Petri dish containing isolation buffer (1mM EGTA, 215mM mannitol, 75mM sucrose, 0.1% BSA, and 20mM HEPES adjusted to a pH of 7.2 with KOH). Brain tissue was homogenized using Potter-Elvehjem homogenizers containing ice-cold isolation buffer. The tissue homogenates were centrifuged twice at 1300g for 3 minutes in an Eppendorf microcentrifuge at 4°C to remove cellular debris and nuclei, and the supernatant was further centrifuged at 13,000g for 10 minutes. The resulting crude mitochondrial pellet was subjected to nitrogen decompression to release synaptic mitochondria, using a nitrogen cell disruption bomb, at 4°C under a pressure of 1200 psi for 10 minutes. After nitrogen disruption, the mitochondrial pellet was resuspended in isolation buffer and layered on top of a discontinuous Ficoll gradient (7.5% and 10%), and centrifuged at 100,000g for 30 minutes. The mitochondrial pellets at the bottom were transferred to microcentrifuge tubes, topped off with isolation buffer without EGTA, and centrifuged at 10,000g for 10 minutes at 4°C to yield a tighter pellet. The final mitochondrial pellet was resuspended in 20-50 microliters of isolation buffer without EGTA to yield a concentration of approximately 10 mg/ml. The final protein concentration was determined using a BCA protein assay kit measuring absorbance at 562nm using a BioTek Synergy HT plate reader (Winooski, VT). These samples were then analyzed for mitochondrial bioenergetics on the Seahorse XF-24 extracellular flux analyzer instrument (Seahorse Bioscience, North Billerica, MA, USA).
Preparation and Calibration of Seahorse XF-24 Sensor Cartridge Sample Plate
A Seahorse Bioscience XF24 extracellular flux analyzer was used to measure mitochondrial bioenergetics in intact isolated mitochondria as previously described.(34) The XF-24 creates a transient, 7μl chamber in specialized microplates that allows for the determination of oxygen and proton concentrations in real time. The day before the experiment, 1.0 ml of XF Calibrant solution (Seahorse Bioscience) was added to each well of a 24 well dual-analyte sensor cartridge (Seahorse Bioscience). The sensor cartridge was placed back on the 24 well calibration plate and put in a 37°C incubator without CO2 (Seahorse Bioscience, North Billerica, MA, USA) overnight. The day of the experiment, the injection ports on the sensor cartridge were pre-loaded with the appropriate mitochondrial substrates or inhibitors at 10× concentrations. Once the sensor cartridge was loaded with all of the experimental reagents it was placed into the Seahorse XF-24 extracellular flux analyzer for automated calibration. During the sensor calibration, isolated mitochondria were then seeded in 50μl volume of isolation buffer containing 5.0 μg μg of protein (determined by BCA method) per well in XF-24 V7 cell culture microplates. Following the centrifugation of the plates at 2000 rpm for 4 minutes at 4°C, 450μl of respiration buffer (215mM mannitol, 75mM sucrose, 0.1% BSA, 20mM HEPES, 2mM MgCl, 2.5mM KH2PO4 at pH 7.2) at 37°C was gently added to each well for a final volume of 500μl per well at the beginning of the experiment. Plates were immediately placed into the calibrated Seahorse XF-24 extracellular flux analyzer for mitochondrial bioenergetics analysis.
Seahorse XF-24 Assay Protocol for Isolated Mitochondrial Bioenergetics
The following protocol was utilized for the analysis of bioenergetic function in purified mitochondria using the Seahorse Biosciences XF-24 extracellular flux analyzer as previously described.(34) Briefly, pyruvate plus malate plus ADP, oligomycin, FCCP, and rotenone plus succinate were injected sequentially through ports A, B, C, and D, respectively, in the Seahorse Flux Pak cartridges to yield final concentrations of 5.0 mM (pyruvate), 2.5 mM (malate), 1.0 mM (ADP), 1.0 μg/ml (oligomycin), 4.0 μM (FCCP) and 10.0 mM (succinate) plus 100.0 nM (rotenone), respectively.
Western Blot Analysis of Oxidative Damage and Cytoskeletal Breakdown Markers
A Western blotting technique was employed as previously described (34, 35) – with some modifications – to detect 4-HNE adducts in tissue homogenates and mitochondrial pellets. Briefly, at a given time point post-injury, mice received an overdose of sodium pentobarbital (200.0mg/kg IP). The ipsilateral cortex (penumbra tissue and the injured core) and hippocampus were then rapidly dissected out on an ice-chilled stage and immediately transferred to Triton lysis buffer (1.0% Triton, 20.0mM Tris HCL, 150.0mM NaCl, 5.0mM EGTA, 10.0mM EDTA, and 10.0% glycerol) containing protease inhibitors (Complete Mini Protease Inhibitor Cocktail; Roche Diagnostics, Indianapolis, IN, USA). Samples were then sonicated and centrifuged for 30 minutes (13,000rpm at 4°C). The supernatants were collected and the remaining pellet was discarded. In the case of mitochondria pellets, after isolation of the mitochondria the samples were spun down and frozen at -80°C until further analysis. Protein concentrations were determined using a BCA Protein Assay (Pierce; Rockford, IL, USA). An aliquot of each protein sample (15.0μg for 4-HNE and 3-NT; 5.0μg for spectrin) was separated on an SDS–PAGE precast gel (12% Bis-Tris for oxidative damage markers, 3-8% Tris-Acetate for spectrin; w/v acrylamide; Criterion XT, Bio-Rad) using a XT-MES (4-HNE and 3-NT) or XT-Tricine (spectrin) running buffer system and then transferred to nitrocellulose membranes using a semi-dry electro-transferring unit at 15 volts for 30 minutes at room temperature. Preliminary experiments established protein concentration curves in order to ensure that quantified bands were in the linear range. Membranes were incubated for 1 hour at room temperature in 5% milk/TBS blocking solution. The membranes were then incubated overnight at 4°C in blocking solution with 0.5 mM Tween-20 (TBST) containing the appropriate dilution of primary antibody. A mouse monoclonal primary antibody was used for detecting 4-HNE bands (1:100, Japan Institute for Control of Aging, JaICA MHN-100P, Shizouka, Japan). A mouse monoclonal primary antibody was used for detecting 3-NT bands (1:500, Genway #20-321-175259, San Diego, California, USA). A mouse monoclonal primary antibody was also used for detecting alpha spectrin breakdown products (SBDPs), with the 150 kD band representing calpain and caspase-3 generated SBDPs and the 145 kD band being a calpain specific SBDP (1:5000, Affiniti FG6090; Affiniti Biomol, Mamhead Castle, UK). A goat anti-mouse secondary antibody (2 hour incubation at room temperature) conjugated to an infrared dye (1:5000, IRdye800CW, Rockland) was used for detection of the primary labeled bands. Wet membranes were imaged and quantified using Odyssey Infra Red Imaging System (Li-Cor Biosciences, Lincoln, NE, USA). For oxidative damage markers, all bands ranging from 250 kD to 50 kD were quantified for each lane, representing the smear of 4-HNE or 3-NT labeled proteins for each sample. For spectrin, both the 150 kD and 145 kD bands were quantified for each lane, representing spectrin breakdown products for each sample. This was then analyzed as percent of control samples (sham).
Sample Size and Statistical Analysis
The sample sizes for this study were 8-10 per group (dose response analysis) and 9-10 per group (mitochondrial bioenergetics analysis) and 8-10 per group (therapeutic window analysis). Data are presented as group means +/- standard deviation (SD) and were analyzed using GraphPad PRISM version 5.0 (San Diego, CA, USA). Both mitochondrial bioenergetics and immunoblot quantification data were analyzed by appropriately designed ANOVAs followed by Student Newman-Keuls (SNK) post-hoc tests as appropriate. A p value of <0.05 was considered significant for all analyses.
Results
Carnosic Acid Attenuates Injury-induced Increases in Oxidative Damage and Neuronal Cytoskeletal Degradation in a Dose-Dependent Manner 48 Hours Post-TBI
Oxidative damage measurements of cortical and hippocampal tissue samples from young adult male CF-1 mice treated with 0.3, 1.0, or 3.0 mg/kg CA or vehicle i.p.15 minutes post-injury. At 48 hours post-injury, ipsilateral tissue was collected for Western Blot analysis. We chose this time point because we have previously shown that 48 hours is near the peak of the injury-induced increase in brain tissue LP-related 4-HNE LP (37) and calpain proteolytic cytoskeletal damage (1) in the same mouse CCI model. Quantitative analysis revealed that both 1.0 and 3.0 mg/kg doses of CA significantly attenuated (p < 0.05) 4-HNE in the cortex (see Fig. 1). However, only the 1.0 mg/kg dose of CA significantly attenuated (p < 0.05) 4-HNE levels in the hippocampus, suggesting a U-shaped dose curve (see Fig. 1). In the case of 3-NT (see Fig. 2), both the 1.0 mg/kg and 3.0 mg/kg doses attenuated the injury-induced increases (p < 0.05). Interestingly, the 1.0 mg/kg dose of CA is the same which was previously shown to provide protection to cortical mitochondria from 4-HNE toxicity ex vivo (34). Quantitative analysis of spectrin breakdown products, an indication of cellular cytoskeletal degradation, revealed a significant decrease (p < 0.05) in both the 150 kD and 145 kD breakdown products in the cortex with the 1.0 mg/kg dose. There is a noticeable u-shaped dose response for this endpoint, in which neither the lower or higher doses provided protection but the mid-point 1.0mg/kg dose was effective. One-way ANOVA followed by Student Newman-Keuls post-hoc test. * = p<.05. Error bars represent +/- SD. N = 8-10 per group.
Figure 1.
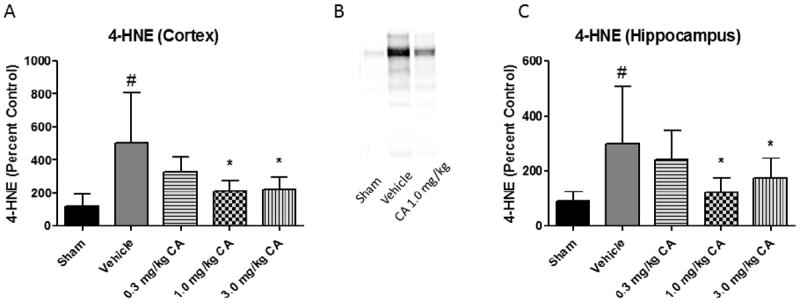
Carnosic Acid Reduces 4-HNE in a Dose Dependent Manner post-TBI. Oxidative damage measurements of cortical and hippocampal tissue samples from young adult male CF-1 mice treated with either 0.3, 1.0, or 3.0 mg/kg carnosic acid (CA) or vehicle 15 minutes post-injury. At 48 hours post-injury, ipsilateral tissue was collected for Western Blot analysis. Quantitative analysis revealed that both 1.0 and 3.0 mg/kg doses of CA significantly attenuated 4-HNE in the cortex. However, only the 1.0 mg/kg dose of CA significantly attenuated 4-HNE levels in the hippocampus. Interestingly, the 1.0 mg/kg dose of CA is the same which was previously shown to provide protection to cortical mitochondria from 4-HNE toxicity ex vivo. One-way ANOVA followed by Student Newman-Keuls post-hoc test. * = p<.05. Error bars represent +/- SD. N = 8-10 per group.
Figure 2.
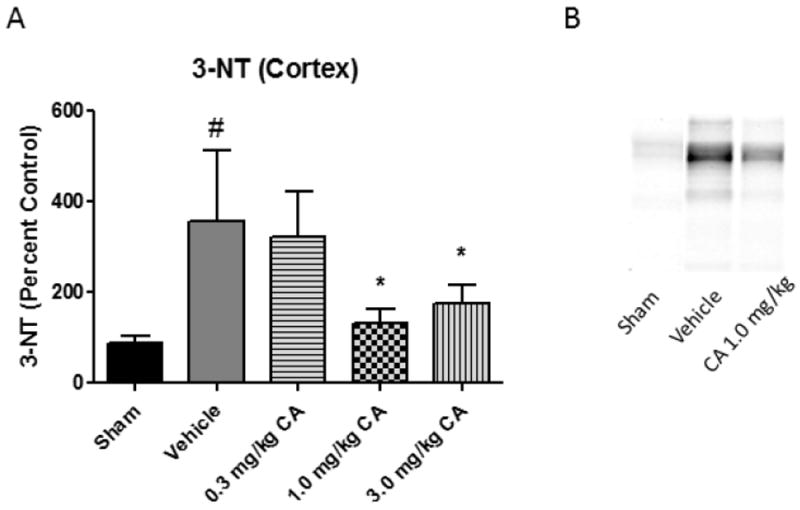
Carnosic Acid Reduces 3-NT in a Dose Dependent Manner post-TBI. Oxidative damage measurements of cortical tissue samples from young adult male CF-1 mice treated with either 0.3, 1.0, or 3.0 mg/kg carnosic acid (CA) or vehicle 15 minutes post-injury. At 48 hours post-injury, ipsilateral tissue was collected for Western Blot analysis. In the case of 3-NT, both the 1.0 mg/kg and 3.0 mg/kg doses attenuated the injury-induced increases in 3-NT. One-way ANOVA followed by Student Newman-Keuls post-hoc test. * = p<.05. Error bars represent +/- SD. N = 8-10 per group.
Carnosic Acid Preserves Mitochondrial Function 24 Hours Post-TBI
Mitochondrial bioenergetics were assessed at 24 hours post-TBI using the Seahorse XF-24 Bioanalyzer for isolated cortical mitochondria from animals treated with either 1.0 mg/kg of carnosic acid or vehicle at 15 minutes post-injury. We chose this time point for the bioenergetic studies based upon our prior studies showing that 24 hours is near the time of maximal injury-induced mitochondrial failure in the mouse CCI model (2). The 1.0 mg/kg dose of carnosic acid significantly increased (p<0.05) mitochondrial respiratory function as compared to vehicle animals for both complex I (ADP Rate) and complex II (Succinate Rate) driven respiration. Moreover, there was no statistical difference between sham animals and CA- treated animals in terms of complex I and complex II respiration, indicating a complete return to sham levels.
Carnosic Acid Reduces Mitochondrial Oxidative Damage 24 Hours Post-TBI
Western blot analysis was performed on the isolated mitochondrial proteins to determine if carnosic acid administration could reduce oxidative damage to mitochondria proteins together with the improved maintenance of bioenergetics. The 1.0 mg/kg dose of CA significantly reduced (p<0.05) levels of both 4-HNE (lipid peroxidation) and 3-NT (protein nitration) markers in cortical mitochondria as compared to vehicle animals. This may serve as a mechanistic explanation for the associated effects on mitochondrial respiration in which CA treatment completely returned oxygen consumption rates to sham levels. Analyzed by one-way ANOVA followed by Student Newman-Keuls post-hoc test. * = p<.05. Error bars represent +/- SD. N = 9-10 per group.
Carnosic Acid Decreases Neuronal Cytoskeletal Degradation with at least an 8 Hour Post-Injury Therapeutic Window
The initial administration of carnosic acid was delayed to either 15 minutes, 1 hour, 4 hours, or 8 hours post-injury for the initial I.P. injection followed by a booster injection at 24 hours post-injury. At 48 hours post-injury, mice were euthanized and the ipsilateral cortical tissue was collected for Western Blot analysis (see methods above) of spectrin breakdown products as an indication of cellular cytoskeletal degradation. The levels of spectrin breakdown products were significantly decreased (p < 0.05) at all delayed time points (15 minutes, 1 hour, 4 hours, and 8 hours) as compared to vehicle for both the 150 kD (caspase and calpain derived) and 145 kD (calpain specific) breakdown products. See Figure 6. Analyzed by one-way ANOVA followed by Student Newman-Keuls post-hoc test. * = p<.05. Error bars represent +/- SD. n = 8-10 per group.
Figure 6.
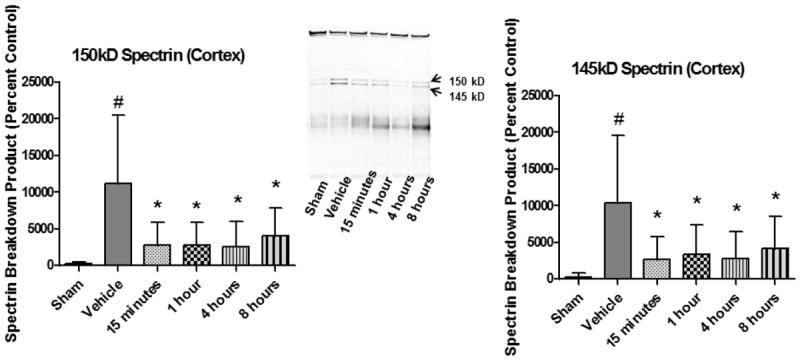
Delayed Initial Administration of Carnosic Acid Provides a Clinically Relevant Therapeutic Window. The initial administration of carnosic acid was delayed to either 15 minutes, 1 hour, 4 hours, or 8 hours post-injury for the initial I.P. injection followed by a booster injection at 24 hours post-injury. At 48 hours post-injury, ipsilateral cortical tissue was collected for Western Blot analysis of spectrin breakdown products as an indication of cellular cytoskeletal degradation. All delayed time points (15 minutes, 1 hour, 4 hours, and 8 hours) were significantly decreased (p < 0.05) as compared to vehicle for both the 150 kD (caspase and calpain derived) and 145 kD (calpain specific) breakdown products. Analyzed by one-way ANOVA followed by Student Newman-Keuls post-hoc test. * = p<.05. Error bars represent +/- SD. n = 8-10 per group.
Discussion
The occurrence of TBI is increasing (3) and represents a major socioeconomic dilemma for developed nations that requires urgency to discover and translate rational therapies for the clinical treatment of this growing epidemic. Over the last few decades, oxidative stress has consistently been identified as one of the key pathological processes underlying the deleterious secondary injury cascade following TBI.(8) Extensive pre-clinical research established the justification underlying clinical trials for three distinct direct antioxidant therapies (e.g. polyethylene glycol conjugated superoxide dismutase, PEG-SOD; tirilazad; and dexanabinol), but unfortunately none have proven effective in rigorous large-scale clinical trials(8) with the possible exception of tirilazad which improved post-TBI survival in patients who had traumatic subarachnoid hemorrhage.(36) Targeting the Nrf2-ARE pathway may provide a pleiotropic approach to attenuate oxidative damage after TBI. The current study provides critical information by demonstrating for the first time the effects of pharmacological Nrf2-ARE activation on protecting mitochondrial function post-TBI. Moreover, the current study is also the first to perform a dose response assessment on a Nrf2-ARE activating drug (e.g. CA) using the rational endpoints of oxidative damage. Although prior studies have investigated compounds that activate the Nrf2-ARE pathway following TBI,(31) none have assessed the dose response profile of such compounds or evaluated the effects on oxidative damage and mitochondrial function or defined what the therapeutic window (extent of post-injury treatment initiation delay) that can still achieve neuroprotection.
Moreover, we recently demonstrated the post-TBI time course of endogenous Nrf2-ARE activation in the absence of Nrf2-ARE activating drugs showing that significant induction which does not occur until 24 hrs post-TBI which is also when significant oxidative damage has occurred.(37) Indeed, the temporal profile of Nrf2-ARE activation closely matched that of oxidative damage post-TBI, suggesting that the posttraumatic turning on of the endogenous antioxidant defenses, in the absence of any pharmacological activator, occurs too slowly to effectively interrupt oxidative damage mechanisms. Thus, any pharmacological activator must augment and speed up Nrf2-ARE related expression so that it antioxidant effects can precede the 24 to 48 hour peak of oxidative damage. Previously, we also demonstrated that administration of CA to mice induces an increase in the critical Nrf2 downstream target heme oxygenase 1. In addition, the CA administration also provided resistance to mitochondria challenged ex vivo by the toxic reactive aldehyde 4-HNE.(34) Moreover, the current study clearly demonstrates that administration of the potent Nrf2-ARE activator CA at an early time point post-TBI (15 minutes post-injury) is capable of attenuating oxidative damage and preserving mitochondrial function thereby extending on our previous ex vivo mechanistic studies with clear evidence of in vivo protection of mitochondria post-TBI.
The aforementioned protective effects on mitochondria by administration of CA occur in conjunction with neurochemical evidence of actual neuroprotection in the form of a reduction in calpain-mediated neuronal cytoskeletal degradation as measured by an attenuation of spectrin proteolytic breakdown. It has previously been established that post-traumatic spectrin degradation is produced by both calpain and another cysteine protease caspase 3. The 150 kDa spectrin fragment is produced by both proteases whereas the 145 kDa fragment is calpain-specific.(38). We detected very little of the caspase 3-specific 120 kDa fragment in these or other previous TBI experiments,(1, 39) thus causing us to interpret that the leading source of post-traumatic spectrin degradation is likely due to calpain. Thus, even though Nrf2-ARE activation is predominantly resulting in an attenuation of oxidative damage, it may nevertheless also reduce the increase of this Ca2+-activated protease via preservation of neuronal intracellular Ca2+ homeostatic mechanisms otherwise affected by oxidative damage post-TBI. Our laboratory has previously demonstrated that calpain-mediated spectrin proteolysis peaks at 24 hours after CCI-TBI in mice,(1) and it has been implicated in post-traumatic neurodegeneration.(1, 39) The reduction of calpain-mediated spectrin degradation via predominantly antioxidant mechanisms reported in the current study is similar to the effects previously seen with the direct lipid peroxyl radical scavenger U-83836E.(40)
While these effects of Nrf2-ARE activation on reducing both oxidative damage and cytoskeletal breakdown are impressive, our observation that the neuroprotective therapeutic window is as long as 8 hours suggests that CA’s Nrf2 activating effects may be most relevant for future clinical translation. Similarly, our previous studies in the same mouse controlled cortical impact TBI model as that presently employed with the direct-acting lipid peroxidation inhibitor U-83836E (40) and the mitochondrial permeability transition inhibitor NIM811 (41) also revealed a 12 hour therapeutic window in regards to attenuation of spectrin degradation. Moreover, the extended duration of this window is consistent with our earlier demonstration in the same model that post-TBI mitochondrial failure does not peak until 12 hours, and that subsequently between 12 and 24 hrs, there is a significant increase in spectrin degradation that reaches its post-TBI maximum at 24 hours. In other words, the 12 hour time point represents the time point at which the injured population of mitochondria undergo maximum permeability transition at which time they release their Ca2+ load which results in a secondary wave of calpain-mediated cytoskeletal degradation (1).
In conclusion, it is vital to discover and translate rational therapies for the clinical treatment of TBI as the incidence will continue to rise. More specifically, certain aspects of the secondary injury cascade have been extensively validated, but yet no therapy has been effective in clinical trials.(8) A more pleiotropic approach to antagonize oxidative damage following TBI, such as targeting the Nrf2-ARE pathway, is likely necessary to achieve success. Therefore, the current study provides strong evidence that augmenting the Nrf2-ARE system can be effective at mitigating oxidative damage and the closely linked phenomenon of mitochondria dysfunction post-TBI while still having a clinically relevant therapeutic window for intervention.
Figure 3.
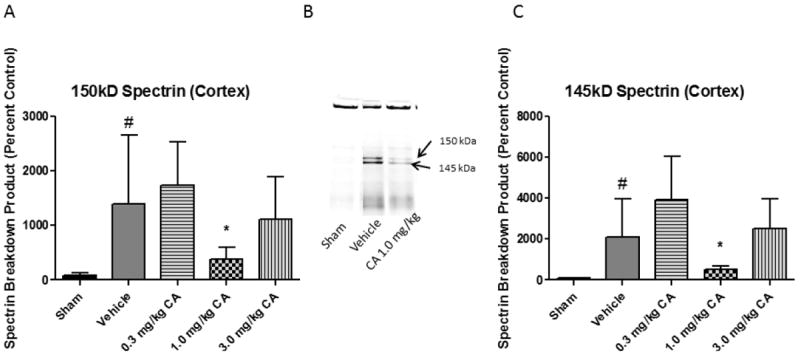
Carnosic Acid Reduces Cytoskeletal Breakdown Following Focal TBI. Cytoskeletal breakdown was assessed in cortical tissue samples from young adult male CF-1 mice treated with either 0.3, 1.0, or 3.0 mg/kg carnosic acid (CA) or vehicle 15 minutes post-injury. At 48 hours post-injury, ipsilateral tissue was collected for Western Blot analysis. In the case of spectrin breakdown products (150 kD and 145 kD bands), both the 1.0 mg/kg and 3.0 mg/kg doses attenuated the injury-induced increases in spectrin breakdown products. This may be due to Nrf2-ARE activation restoring calcium homeostasis indirectly via a reduction in oxidative damage to essential proteins involved in trafficking calcium into and out of the cell. One-way ANOVA followed by Student Newman-Keuls post-hoc test. * = p<.05. Error bars represent +/- SD. N = 8-10 per group.
Figure 4.
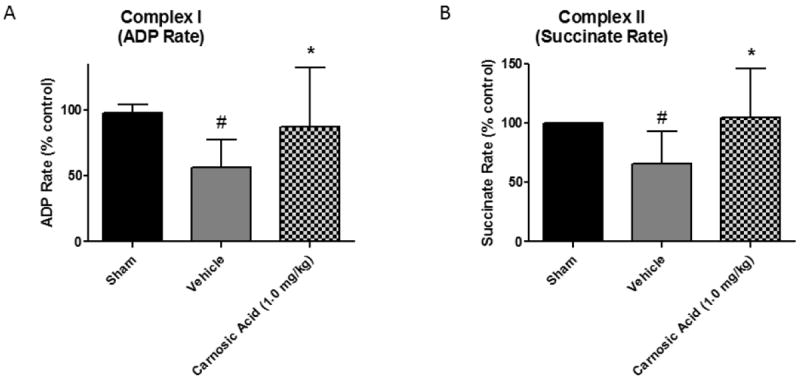
Administration of Carnosic Acid Preserves Mitochondrial Respiratory Function post-TBI. Mitochondrial respiration measurements of cortical mitochondrial samples from young adult male CF-1 mice treated with 1.0 mg/kg carnosic acid (CA) or vehicle at 15 minutes post-injury. The 1.0 mg/kg dose of carnosic acid significantly increased (p<0.05) mitochondrial respiratory function as compared to vehicle animals for both Complex I (ADP Rate) and Complex II (Succinate Rate) driven respiration. Moreover, there was no statistical difference between sham animals and carnosic acid treated animals in terms of Complex I and Complex II respiration, indicating a complete return to sham levels. Analyzed by one-way ANOVA followed by Student Newman-Keuls post-hoc test. * = p<.05. Error bars represent +/- SD. N = 9-10 per group.
Figure 5.
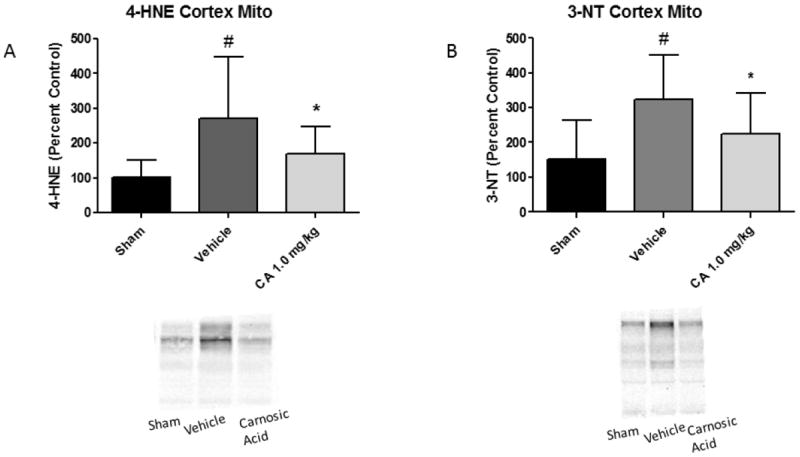
Administration of Carnosic Acid Reduces Oxidative Damage to Cortical Mitochondria post-TBI. Oxidative damage measurements of cortical mitochondrial samples from young adult male CF-1 mice treated with 1.0 mg/kg carnosic acid (CA) or vehicle at 15 minutes post-injury. Western blot analysis was performed on the isolated mitochondrial proteins to determine if carnosic acid administration could reduce oxidative damage to mitochondria proteins. The 1.0 mg/kg dose of carnosic acid significantly reduced (p<0.05) levels of both 4-HNE (lipid peroxidation) and 3-NT (protein nitration) markers in cortical mitochondria as compared to vehicle animals. This may serve as a mechanistic explanation for the associated effects on mitochondrial respiration in which carnosic acid treatment completely returned oxygen consumption rates to sham levels. Analyzed by one-way ANOVA followed by Student Newman-Keuls post-hoc test. * = p<.05. Error bars represent +/- SD. N = 9-10 per group.
Acknowledgments
This work was supported by grants NIH-NIDA 1T32 DA022738, NIH-NINDS 1T32 NS077889, NIH-NINDS 2P30 NS051220-01, and funds from the Kentucky Spinal Cord & Head Injury Research Trust.
Footnotes
Disclosure/Conflict of Interest
No competing financial interests exist.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Deng Y, Thompson BM, Gao X, Hall ED. Temporal relationship of peroxynitrite-induced oxidative damage, calpain-mediated cytoskeletal degradation and neurodegeneration after traumatic brain injury. Exp Neurol. 2007 May;205(1):154–65. doi: 10.1016/j.expneurol.2007.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singh IN, Sullivan PG, Deng Y, Mbye LH, Hall ED. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective therapy. J Cereb Blood Flow Metab. 2006 Nov;26(11):1407–18. doi: 10.1038/sj.jcbfm.9600297. [DOI] [PubMed] [Google Scholar]
- 3.Thurman DJ, Alverson C, Dunn KA, Guerrero J, Sniezek JE. Traumatic brain injury in the United States: A public health perspective. J Head Trauma Rehabil. 1999 Dec;14(6):602–15. doi: 10.1097/00001199-199912000-00009. [DOI] [PubMed] [Google Scholar]
- 4.Smith SL, Andrus PK, Zhang JR, Hall ED. Direct measurement of hydroxyl radicals, lipid peroxidation, and blood-brain barrier disruption following unilateral cortical impact head injury in the rat. J Neurotrauma. 1994 Aug;11(4):393–404. doi: 10.1089/neu.1994.11.393. [DOI] [PubMed] [Google Scholar]
- 5.Ansari MA, Roberts KN, Scheff SW. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radic Biol Med. 2008 Aug 15;45(4):443–52. doi: 10.1016/j.freeradbiomed.2008.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kontos HA, Wei EP. Superoxide production in experimental brain injury. J Neurosurg. 1986 May;64(5):803–7. doi: 10.3171/jns.1986.64.5.0803. [DOI] [PubMed] [Google Scholar]
- 7.Vaishnav RA, Singh IN, Miller DM, Hall ED. Lipid peroxidation-derived reactive aldehydes directly and differentially impair spinal cord and brain mitochondrial function. J Neurotrauma. 2010 Jul;27(7):1311–20. doi: 10.1089/neu.2009.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bains M, Hall ED. Antioxidant therapies in traumatic brain and spinal cord injury. Biochim Biophys Acta. 2012 May;1822(5):675–84. doi: 10.1016/j.bbadis.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pandya JD, Pauly JR, Nukala VN, Sebastian AH, Day KM, Korde AS, et al. Post-Injury Administration of Mitochondrial Uncouplers Increases Tissue Sparing and Improves Behavioral Outcome following Traumatic Brain Injury in Rodents. J Neurotrauma. 2007 May;24(5):798–811. doi: 10.1089/neu.2006.3673. [DOI] [PubMed] [Google Scholar]
- 10.Azbill RD, Mu X, Bruce-Keller AJ, Mattson MP, Springer JE. Impaired mitochondrial function, oxidative stress and altered antioxidant enzyme activities following traumatic spinal cord injury. Brain Res. 1997 Aug 15;765(2):283–90. doi: 10.1016/s0006-8993(97)00573-8. [DOI] [PubMed] [Google Scholar]
- 11.Sullivan PG, Thompson MB, Scheff SW. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp Neurol. 1999 Nov;160(1):226–34. doi: 10.1006/exnr.1999.7197. [DOI] [PubMed] [Google Scholar]
- 12.Singh IN, Sullivan PG, Hall ED. Peroxynitrite-mediated oxidative damage to brain mitochondria: Protective effects of peroxynitrite scavengers. J Neurosci Res. 2007 Aug 1;85(10):2216–23. doi: 10.1002/jnr.21360. [DOI] [PubMed] [Google Scholar]
- 13.Deng-Bryant Y, Singh IN, Carrico KM, Hall ED. Neuroprotective effects of tempol, a catalytic scavenger of peroxynitrite-derived free radicals, in a mouse traumatic brain injury model. J Cereb Blood Flow Metab. 2008 Jun;28(6):1114–26. doi: 10.1038/jcbfm.2008.10. [DOI] [PubMed] [Google Scholar]
- 14.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 15.Chan JY, Han XL, Kan YW. Isolation of cDNA encoding the human NF-E2 protein. Proc Natl Acad Sci U S A. 1993 Dec 1;90(23):11366–70. doi: 10.1073/pnas.90.23.11366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999 Jan 1;13(1):76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci U S A. 1994 Oct 11;91(21):9926–30. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004 Aug;24(16):7130–9. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tong KI, Kobayashi A, Katsuoka F, Yamamoto M. Two-site substrate recognition model for the Keap1-Nrf2 system: a hinge and latch mechanism. Biol Chem. 2006 Oct-Nov;387(10-11):1311–20. doi: 10.1515/BC.2006.164. [DOI] [PubMed] [Google Scholar]
- 20.Jain AK, Bloom DA, Jaiswal AK. Nuclear import and export signals in control of Nrf2. J Biol Chem. 2005 Aug 12;280(32):29158–68. doi: 10.1074/jbc.M502083200. [DOI] [PubMed] [Google Scholar]
- 21.Johnson JA, Johnson DA, Kraft AD, Calkins MJ, Jakel RJ, Vargas MR, et al. The Nrf2-ARE pathway: an indicator and modulator of oxidative stress in neurodegeneration. Ann N Y Acad Sci. 2008 Dec;1147:61–9. doi: 10.1196/annals.1427.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Calkins MJ, Jakel RJ, Johnson DA, Chan K, Kan YW, Johnson JA. Protection from mitochondrial complex II inhibition in vitro and in vivo by Nrf2-mediated transcription. Proc Natl Acad Sci U S A. 2005 Jan 4;102(1):244–9. doi: 10.1073/pnas.0408487101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rojo AI, Innamorato NG, Martin-Moreno AM, De Ceballos ML, Yamamoto M, Cuadrado A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia. 2010 Apr;58(5):588–98. doi: 10.1002/glia.20947. [DOI] [PubMed] [Google Scholar]
- 24.Li J, Johnson D, Calkins M, Wright L, Svendsen C, Johnson J. Stabilization of Nrf2 by tBHQ confers protection against oxidative stress-induced cell death in human neural stem cells. Toxicol Sci. 2005 Feb;83(2):313–28. doi: 10.1093/toxsci/kfi027. [DOI] [PubMed] [Google Scholar]
- 25.Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci. 2008 Dec 10;28(50):13574–81. doi: 10.1523/JNEUROSCI.4099-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao X, Sun G, Zhang J, Strong R, Dash PK, Kan YW, et al. Transcription factor Nrf2 protects the brain from damage produced by intracerebral hemorrhage. Stroke. 2007 Dec;38(12):3280–6. doi: 10.1161/STROKEAHA.107.486506. [DOI] [PubMed] [Google Scholar]
- 27.Son TG, Camandola S, Arumugam TV, Cutler RG, Telljohann RS, Mughal MR, et al. Plumbagin, a novel Nrf2/ARE activator, protects against cerebral ischemia. J Neurochem. 2010 Mar;112(5):1316–26. doi: 10.1111/j.1471-4159.2009.06552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jin W, Wang H, Yan W, Zhu L, Hu Z, Ding Y, et al. Role of Nrf2 in protection against traumatic brain injury in mice. J Neurotrauma. 2009 Jan;26(1):131–9. doi: 10.1089/neu.2008.0655. [DOI] [PubMed] [Google Scholar]
- 29.Zhao J, Moore AN, Redell JB, Dash PK. Enhancing expression of Nrf2-driven genes protects the blood brain barrier after brain injury. J Neurosci. 2007 Sep 19;27(38):10240–8. doi: 10.1523/JNEUROSCI.1683-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao J, Moore AN, Clifton GL, Dash PK. Sulforaphane enhances aquaporin-4 expression and decreases cerebral edema following traumatic brain injury. J Neurosci Res. 2005 Nov 15;82(4):499–506. doi: 10.1002/jnr.20649. [DOI] [PubMed] [Google Scholar]
- 31.Dash PK, Zhao J, Orsi SA, Zhang M, Moore AN. Sulforaphane improves cognitive function administered following traumatic brain injury. Neurosci Lett. 2009 Aug 28;460(2):103–7. doi: 10.1016/j.neulet.2009.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Satoh T, Kosaka K, Itoh K, Kobayashi A, Yamamoto M, Shimojo Y, et al. Carnosic acid, a catechol-type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S-alkylation of targeted cysteines on Keap1. J Neurochem. 2008 Feb;104(4):1116–31. doi: 10.1111/j.1471-4159.2007.05039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greco T, Fiskum G. Brain mitochondria from rats treated with sulforaphane are resistant to redox-regulated permeability transition. J Bioenerg Biomembr. 2010 Dec;42(6):491–7. doi: 10.1007/s10863-010-9312-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller DM, Singh IN, Wang JA, Hall ED. Administration of the Nrf2-ARE activators sulforaphane and carnosic acid attenuates 4-hydroxy-2-nonenal-induced mitochondrial dysfunction ex vivo. Free Radic Biol Med. 2013 Apr;57:1–9. doi: 10.1016/j.freeradbiomed.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hall ED, Wang JA, Miller DM. Relationship of nitric oxide synthase induction to peroxynitrite-mediated oxidative damage during the first week after experimental traumatic brain injury. Exp Neurol. 2012 Dec;238(2):176–82. doi: 10.1016/j.expneurol.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marshall LF, Maas AI, Marshall SB, Bricolo A, Fearnside M, Iannotti F, et al. A multicenter trial on the efficacy of using tirilazad mesylate in cases of head injury. J Neurosurg. 1998 Oct;89(4):519–25. doi: 10.3171/jns.1998.89.4.0519. [DOI] [PubMed] [Google Scholar]
- 37.Miller DM, Wang JA, Buchanan AK, Hall ED. Temporal and Spatial Dynamics of Nrf2-ARE Mediated Gene Targets in Cortex and Hippocampus Following Controlled Cortical Impact Traumatic Brain Injury in Mice. J Neurotrauma. 2014 Mar 14; doi: 10.1089/neu.2013.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang KK. Calpain and caspase: can you tell the difference? Trends Neurosci. 2000 Jan;23(1):20–6. doi: 10.1016/s0166-2236(99)01479-4. [DOI] [PubMed] [Google Scholar]
- 39.Thompson SN, Gibson TR, Thompson BM, Deng Y, Hall ED. Relationship of calpain-mediated proteolysis to the expression of axonal and synaptic plasticity markers following traumatic brain injury in mice. Exp Neurol. 2006 Sep;201(1):253–65. doi: 10.1016/j.expneurol.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 40.Mustafa AG, Wang JA, Carrico KM, Hall ED. Pharmacological inhibition of lipid peroxidation attenuates calpain-mediated cytoskeletal degradation after traumatic brain injury. J Neurochem. 2011 May;117(3):579–88. doi: 10.1111/j.1471-4159.2011.07228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mbye LH, Singh IN, Carrico KM, Saatman KE, Hall ED. Comparative neuroprotective effects of cyclosporin A and NIM811, a nonimmunosuppressive cyclosporin A analog, following traumatic brain injury. J Cereb Blood Flow Metab. 2009 Jan;29(1):87–97. doi: 10.1038/jcbfm.2008.93. Epub 2008 Aug 20. [DOI] [PMC free article] [PubMed] [Google Scholar]