

Abstract
Elevated levels of the transcription factor c-myc are strongly associated with various cancers, and in particular B-cell lymphomas. While many of c-MYC’s functions have been elucidated, its effect on the presentation of antigen (Ag) through the HLA class II pathway has not previously been reported. This is an issue of considerable importance, given the low immunogenicity of many c-MYC-positive tumors. We report here that increased c-MYC expression has a negative effect on the ability of B-cell lymphomas to functionally present Ags/peptides to CD4+ T cells. This defect was associated with alterations in the expression of distinct co-factors as well as interactions of antigenic peptides with class II molecules required for the presentation of class II-peptide complexes and T cell engagement. Using early passage Burkitt’s lymphoma (BL) tumors and transformed cells, we show that compared to B-lymphoblasts, BL cells express decreased levels of the class II editor HLA-DM, lysosomal thiol-reductase GILT, and a 47kDa enolase-like protein. Functional Ag presentation was partially restored in BL cells treated with a c-MYC inhibitor, demonstrating the impact of this oncogene on Ag recognition. This restoration of HLA class II-mediated Ag presentation in early passage BL tumors/cells was linked to enhanced HLA-DM expression and a concurrent decrease in HLA-DO in BL cells. Taken together, these results reveal c-MYC exerts suppressive effects at several critical checkpoints in Ag presentation which contribute to the immunoevasive properties of BL tumors.
Introduction
The c-MYC protein was first identified 30 years ago as a homologue of an avian retroviral oncogene (1). It is a transcription factor encoded by the MYC gene and boasts a target gene network encompassing approximately 15% of all known genes (2–4). The c-MYC protein belongs to the family of basic region helix-loop-helix/leucine zipper transcription factors and its activity is dependent on the formation of heterodimers with MAX, upon which the heterodimers bind to regions of DNA with the CACGTG sequence motif (E-boxes) (5–7). The transcriptional effects of c-myc are thought to be exerted primarily through the recruitment of transcriptional cofactors involved in RNA polymerase II function, as well as the recruitment of histone acetyl transferases, which acetylate lysine residues in histones and cause a more open structure of the chromatin allowing for increased transcription of target genes (8–10). To a lesser extent, c-myc exerts its functions on genes transcribed by RNA polymerases I and III, and may repress transcription through interactions with the Miz-1 transcription factor (11). Overexpression of c-myc also controls genes with a wide array of functions, ranging from cell-cycle progression to differentiation to apoptosis (2, 12).
Transformation of cells by c-MYC protein involves numerous genes (9). Paradoxically, while c-MYC activity induces cell growth and differentiation, it also induces apoptosis. This is achieved through activation of the p53 tumor suppressor and inhibition of cyclin D1 as well as indirect suppression of anti-apoptotic BCL2 and induction of pro-apoptotic BAX and Bim (9, 13, 14). Since its discovery, c-myc has come to be recognized as one of the most commonly activated oncogenes in human cancers and is observed in virtually all malignancies (13, 15). c-MYC protein expression is implicated in the cancer-related deaths of approximately 100,000 people in the United States as well as millions worldwide every year (2, 15, 16). Among malignancies that have a known association with c-myc overexpression, Burkitt Lymphoma (BL) may be the most prominent. Indeed, overexpression of c-myc is a hallmark of BL and activation of c-myc by chromosomal translocation is considered diagnostic for this lymphoid malignancy. In BL, c-myc is translocated to an immunoglobulin locus, resulting in its constitutive expression and cell transformation. The most prominent of these translocations is t(8:14) which is observed in 80% of BL, while t(8:22) and t(8:2) are observed with lesser frequencies (17, 18). These translocations are generated as accidents of antibody affinity maturation during the germinal center reaction through the action of activation-induced cytidine deaminase (AID) (19).
BL is a highly aggressive type of non-Hodgkin’s Lymphoma that occurs most frequently in tropical climates, with a distribution closely following that of holoendemic malaria (20). A sporadic form is observed in other parts of the world and a third form is found associated with HIV. Malaria and EBV, each alone or together, and HIV increase the risk of developing BL, presumably by polyclonal B cell stimulation including activation of AID and by conferring anti-apoptotic functions to cells hit by the c-myc translocation, but the precise contribution remains to be defined (20–23). BL is typically treated effectively with aggressive chemotherapy in young patients, but inferior responses are observed in adults (especially the elderly) and immunodeficient patients (24). Additionally, older and immunodeficient patients are less tolerant of the aggressive chemotherapy required and show increased signs of treatment-associated toxicities. This gap in treatment for these patient groups highlights the need for exploration into improved treatment options which would display lower levels of toxicity. The most ideal treatments would harness the immune system of the individual to target malignant cells. In EBV-positive BL, EBNA-1 is expressed as the only viral protein and it poorly stimulates cytotoxic CD8+ T cells due to its low immunogenicity (25–29). As a result, CD8+ T cell responses to BL are weak and unsustained. While multiple defects in class I antigen presentation and immune escape have been reported (25–29), little is known about disruption of class II presentation by malignant tumors. However, effective tumor immune responses usually involve the stimulation and maintenance of tumor specific CD8+ HLA class I-restricted cytotoxic T cells (CTL) and tumor-specific CD4+ class II-restricted helper T cells (30–32). Several groups have also shown that HLA class II-restricted CD4+ CTL could be generated against BL as well as non-Hodgkins follicular lymphoma (FL) (33–36), suggesting the feasibility of using sufficient tumor specific CD4+ T cells to eliminate B-cell tumors. Most B-cell tumors, including BL, express class II molecules, could provide their own HLA class II Ag presentation and be targets for helper CD4+ T cells. Increased expression of c-MYC imparts reduced immunogenicity to BL cells by antagonizing the NF-κB and interferon pathways (28, 29). In EBV immortalized B-lymphoblastoid cell lines (B-LCL), c-MYC is expressed at low levels and these cells retain a highly immunogenic phenotype with blast formation observed during growth due to expression of LMP1 that activates the canonical as well as the non-canonical NF-κB pathway (37). However, if B-LCLs are transfected to express measurable c-MYC, tumor immunogenicity is greatly diminished and suspension growth is observed, which corresponds to the phenotypes of BL cell lines (29, 38). Clearly, the expression of c-MYC plays an important role in reducing the immunogenicity of BL.
Although c-MYC expression has been described to have multiple effects on malignant cells, the role it plays in HLA class II-mediated Ag presentation and immune recognition has not been reported. Recently, it has been shown that the expression of invariant chain (Ii) regulates the repertoire of tumor peptides presented by class II-positive tumor cells (39). The absence of Ii has also been shown to facilitate CD4+ T cell responses (39, 40). Thus, expression levels of the components of the class II pathway such as Ii, class II-associated invariant chain peptide (CLIP), and HLA-DM/DO molecules in B-cells as well as tumors may modulate immune recognition. Here, studies revealed that cells expressing high levels of c-MYC are poor stimulators of CD4+ T cells. c-MYC expression in these cells was tied to alterations in the expression of HLA-DM, the lysosomal thiol reductase (GILT), and a 47kDa enolase-like protein. We further show that treatment of c-MYC-overexpressing cells with a c-MYC inhibitor led to decreased c-MYC expression, restoration of HLA-DM concurrent with decreased HLA-DO molecules, and partial restoration of class II-mediated Ag presentation. Together these results strongly suggest that c-MYC expression plays a key role in immune evasion via altering HLA class II Ag presentation, and opens the door for novel therapeutics to reverse this deficiency in BL.
Materials and Methods
Cell lines and tumors
Human B-lymphoblastoid cell lines (B-LCL) Frev, Priess and 6.16 were cultured in IMDM (Mediatech, Manassas, VA) supplemented with 10% bovine growth serum (Hyclone, Logan, UT), and 50U/ml penicillin and 50μg/ml streptomycin (Mediatech) (41). The B-LCL EREB2-5 (42) was cultured in RPMI 1640 (Mediatech) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA), 50U/ml penicillin/50μg/ml streptomycin and 2μM estrogen, 1% L-glutamine (Mediatech) and 50μM β-mercaptoethanol (Invitrogen) (43, 44). Human BL (Ramos) and BL-like cell lines (Nalm-6, A1 and P493-6) were maintained in complete RPMI-1640 supplemented with 10% fetal bovine serum, 50U/ml penicillin and 50μg/ml streptomycin, and 1% L-glutamine (41, 45, 46). The wild-type B-LCL, Frev, constitutively expresses HLA-DR4 molecules. Priess cells are homozygous for the expression of HLA-DR4 (DRA*0101, DRB*0401) molecules. The P493-6 cell line was cultured in the presence of 2μM estrogen with or without 5μg/ml tetracycline to control expression of EBNA-2 and c-myc (P493-6.DR4.est and P493-6.DR4.est.tet, respectively) (38). EREB2-5, 6.16, Nalm-6, Ramos, A1, and P493-6 cell lines were retrovirally transduced with a common allele of HLA-DR4 (DRB1*0401) with linked drug selection markers for hygromycin or histidinol resistance (47) to generate EREB2-5.DR4, 6.16.DR4, Nalm-6.DR4, Ramos.DR4, A1.DR4 and P-493-6.DR4 cell lines. 6.16.DR4 cells were further transfected with DMα and DMβ for constitutive expression of HLA-DM molecules to generate 6.16.DR4.DM (41). Frev cells were also transfected with an empty vector or c-myc to generate Frev.vec and Frev.c-myc cell lines.
T cell hybridomas 2.18a and 1.21 recognize Igκ residues 188-203 and 145-159, respectively, and were generated by immunization of DR4-transgenic mice as described previously (44, 48). The T cell hybridoma line 17.9 (generously provided by D. Zaller, Merck Research Laboratories, Rahway, NJ) responds to human serum albumin (HSA) residue 64-76K (49), was cultured in RPMI 1640 with 10% FBS, 50U/ml penicillin/50μg/ml streptomycin and 50μM β-mercaptoethanol (Invitrogen). Institutional approval (HR#17159) for the use of human tissue and tumor samples was obtained. Lymph nodes and blood samples were obtained from two lymphoma patients (TB#2952, TB#7378) through our Hollings Cancer Center Tissue Bank (Medical University of South Carolina, Charleston, USA). TB#2952 and TB#7378 tumors are EBV-positive as analyzed by the Biorepository & Tissue Analysis Shared Resource of the Hollings Cancer Center at MUSC. Blood samples were also obtained from two healthy individuals with written consent (approved protocol HR#17159). Of these (C#16, #C101) and of two tumors (TB#2952, TB#7378), B-cells were isolated using a B-Cell Isolation Kit II (#130-091-151). A portion of B-cells from a healthy individual (#C16) and from a BL tumor (TB#2952) were then infected in vitro with EBV (41), and transfected with HLA-DR4 (DRB1*0401) as described (43). These DR4-expressing early passage cells were used in the functional class II Ag presentation and T cell recognition assays. Cells were cultured in RPMI 1640 with 10% FBS, 50U/ml penicillin/50μg/ml streptomycin and 50μM β-mercaptoethanol (Invitrogen). An explanatory table was also included listing the type and source of the cells and peptide specificity for the T cell hybridomas (Table I).
Table I.
Cell lines used in this study:
| Cell name(s) | Cell type(s) | HLA-DR4 expression | Reference(s) | |
|---|---|---|---|---|
| Constitutive | Transduced | |||
| Frev | B-lymphoblastoid cell | Yes | No | 41 |
| Priess | B-lymphoblastoid cell | Yes | No | 41 |
| Nalm-6.DR4 | Lymphocytic leukemia cell line | No | Yes | 41, 46 |
| Ramos.DR4 | Burkitt lymphoma cell | No | Yes | 41, 46 |
| 6.16.DR4.DM | B-lymphoblastoid cell line | No | Yes | 41, 46 |
| REEB2-5.DR4 | B-lymphoblast type cell line | No | Yes | 38, 42 |
| A1.DR4 | Burkitt lymphoma type cell line | No | Yes | 38, 42 |
| P493-6.DR4 | Burkitt lymphoma type cell line | No | Yes | 38, 42 |
| TB#2952 | Early passage Burkitt lymphoma | No | Yes | 43 |
| TB#7378 | Early passage Burkitt lymphoma | No | Yes | Unpublished |
| C#16 | Early passage B cells | No | Yes | Unpublished |
| C#101 | Early passage B cells | No | Yes | Unpublished |
| 2.18a | T cell hybridoma against Igκ188-203 | -- | -- | 41, 47 |
| 1.21 | T cell hybridoma against Igκ145-159 | -- | -- | 41, 47 |
| 17.9 | T cell hybridoma against HSA64-76K | -- | -- | 41, 53 |
Antigens and peptides
Human serum albumin (HSA) and human IgG kappa (Igκ) were purchased from Sigma (St. Louis, MO). HSA64-76K (sequence: VKLVNEVTEFAKTK), human Igκ immunodominant κ188-203 (sequence: KHKVYACEVTHQGLSS, referred to as κI), subdominant κ145-159 (sequence: KVQWKVDNALQSGNS, referred to as κII) peptides were produced using Fmoc technology and an Applied Biosystems Synthesizer as described (44, 47, 49–51). The Igκ188-203 and Igκ145-159 peptides were labeled as indicated at the α amino termini by the sequential addition of 2 molecules of Fmoc-6-aminohexanoic acid followed by a single biotin to yield the sequence biotin-aminohexanoic acid-aminohexanoic acid-peptide. Reverse phase HPLC purification and mass spectrometry were used to analyze the peptide and showed a peptide purity > 99%. These DR4-restricted peptides were dissolved in PBS and stored at −20°C until use.
Antigen presentation assays
B-LCL and BL cells were incubated with the whole Ags (Igκ or HSA), or its synthetic epitopes (Igκ188-203, Igκ145-159 or HSA64-76K) for 4h at 37°C in the appropriate cell culture media (43, 44, 51, 52). Cells were then washed and co-cultured with the T cell hybridomas (12.18a and 1.21 cells recognize Igκ188-203 and Igκ145-159 peptides respectively; 17.9 cells recognizes HSA64-76K peptide) for 24h at 37°C. In separate assays, P493-6.DR4 cells were left untreated or treated for 24h with 50μM of the c-MYC inhibitor 10058-F4 (Calbiochem, Billerica, MA). Peptides and Ags were added for the last 4h, and the cells were washed and co-cultured with the appropriate peptide specific T cell hybridomas for 24h. For endogenous Ag presentation assay, cells (Priess EREB2-5.DR4, A1.DR4 and P493-6.DR4) which innately express Igκ were cocultured with the appropriate epitope or peptide specific T cell hybridomas for 24h (47, 51, 53). For all assays, following co-culture with the T cell hybridoma, T cell production of IL-2 was quantitated by ELISA (52, 54). Assays were repeated in triplicate with standard error for triplicate samples within a single experiment being reported.
Western blot analysis
Cell lysates obtained from Frev.vec, Frev.c-myc, EREB2-5.DR4 and A1.DR4 were analyzed by Western blotting as previously described for expression of HLA-DR, Ii and HLA-DM with β-actin (Santa Cruz Biotechnology, Santa Cruz, CA) as a loading control (47, 55-57). Nuclear lysate from Frev.vec, Frev.c-myc, EREB2-5.DR4, A1.DR4, Priess, P493-6.DR4, P493-6.DR4.est.tet, 6.16.DR4.DM, Nalm-6.DR4, Ramos.DR4 were analyzed by Western blotting for expression of c-MYC (Santa Cruz Biotechnology), EBNA-1 and EBNA-2 (gift from Dr. Elisabeth Kremmer, Munich, Germany) with β-actin as the loading control. In separate assays, P493-6.DR4. were left untreated or treated for 24h with 50μM or 100μM of the c-MYC inhibitor 10058-F4. Following treatment, cells were harvested, nuclear lysate obtained, and analyzed by Western blotting for expression of c-MYC with β-actin as the loading control. Densitometry was performed using a ChemiDoc XRS station (Bio-Rad, Hercules, CA) where the protein bands were analyzed using the Quantity One 4.6.3 software (Bio-Rad). Relative protein expression levels were stated as a ratio of specific proteins expressed/β-actin for each sample. Data are representative of at least three separate experiments.
Flow cytometric analysis
Frev, Frev.c-myc, EREB2-5.DR4, A1.DR4, P493-6.DR4, and P493-6.DR4.est/tet cells were stained with antibodies against HLA-DR (L243 antibody) and HLA-DR4 (359-F10 antibody) followed by a secondary FITC conjugate (46, 50, 52). P493-6.DR4 and P493-6.DR4.est/tet cells were also stained with antibodies against CLIP (cer-CLIP antibody), Ii (Pin 1.1 antibody), HLA-DM (MAP1.1-DM antibody) and HLA-DO (Mags.DO5-FITC and isotypes were a gift from L. Denzin, Sloan-Kettering). P493-6.DR4 cells treated with vehicle alone or c-MYC inhibitor 10058-F4 were also stained with antibodies and matched isotype controls for the detection of CLIP and HLA-DM molecules. Primary or early passage B cells and tumors were stained with appropriate antibodies as described above for the detection of HLA-DR, CLIP, HLA-DM and HLA-DO molecules. Samples were then analyzed on FACScan using CellQuest software (BD Biosciences, Mountain View, CA). Background fluorescence was evaluated using irrelevant isotype-matched antibodies (NN4, IN-1) as described (46, 50, 52).
Peptide binding assay
Flow cytometric peptide binding assay was used to determine peptide binding to HLA-DR4 molecules on P493-6.DR4+est/tet (c-myclow) and P493-6.DR4 (c-mychigh) cells using a modified method (58). Briefly, P493-6.DR4 (4×105) cells cultured under c-myc on/off conditions were washed twice with ice-cold PBS containing 1% BSA, and incubated with either vehicle alone or 40μM of biotin-labeled HSA64-76K peptide for overnight separately at 4°C and 37°C. Cells were washed and stained with FITC-conjugated streptavidin (Santa Cruz, sc-2865) for 30min at 4°C, and analyzed by FACScan using CellQuest software (BD Biosciences).
Protein extraction and digestion
Acid eluted surface proteins were obtained from P493-6.DR4 and P493-6.DR4.est.tet cells, run on a non-reducing gel, and stained with Coomassie blue (41). Gel plugs were excised and placed in an eppendorf tube. Each plug was washed with 50mM ammonium bicarbonate and de-stained using 25mM ammonium bicarbonate in 50% acetonitrile. The plugs were dehydrated with 100% acetonitrile, and dried in a speedvac. Each gel plug was covered with Proteomics Grade Trypsin (Sigma) and incubated at 37°C overnight. The supernatant was collected in a clean dry eppendorf tube. Peptides were further extracted with one wash of 25mM ammonium bicarbonate and three washes of 5% formic acid and 50% acetonitrile. The supernatant was collected and pooled after each wash then dried down in a speedvac to ~1μL. Prior to analysis, the samples were reconstituted with 10μl of 0.1% trifluoroacetic acid. Samples were then concentrated with a C18 Ziptip (Millipore) and eluted with 0.1% TFA, 50% acetonitrile, and 7.0mg/mL α-cyano-4-hydroxycinnamic acid directly onto the MALDI target.
MALDI-TOF/TOF mass spectrometry
After protein extraction, digestion and elution, the protein spots were dried completely, and the MALDI target plate was loaded into the Applied Biosystems 4800 Proteomics Analyzer (50). An external calibration was preformed prior to analyzing samples utilizing the manufacturer’s standards and protocols. Samples were analyzed in batch mode using 2000 laser shots per spectrum. First, peptide mass maps were acquired over the m/z range of 800–3500 in reflectron mode with a delayed extraction time optimized for m/z 2000 by averaging 2000 scans to locate peaks of peptide origin. The next batch run performed MS-MS analyses to obtain sequence data on the 20 most abundant peaks from the MS analysis. Upon completion of the batch processing, the data was exported into the GPS Explorer data processing system for interpretation and identification. The MASCOT database-searching algorithm analyzed the data, and summarized the results in report format. Database searches were performed using 2 missed cleavages and one differential modification of methionine oxidation. The top 20 matches were reviewed prior to assigning confident protein identifications.
Statistical analysis
The data are expressed as the mean (±SD) and analyzed using Student’s t-test or one-way ANOVA, with p≤0.05 considered statistically significant. A nonparametric Wilcoxon Rank Sum test was also used when comparing band intensity obtained from densitometric analysis of distinct protein bands detected in western blotting of different cells.
RESULTS
Overexpression of c-MYC imparts a non-immunogenic BL phenotype to B-LCL
Although EBV-positive BL cells express EBV antigens (Ags), these cells are poorly recognized by HLA class I-restricted CD8+ T cells because of diminished viral epitope presentation via the HLA class I pathway. While the Ag specific lysis of tumors is predominantly a function of CD8+ T cells, HLA-class II-restricted CD4+ T cell function is crucial in maintaining sustained immune responses to tumors (59–61). Given the high levels of c-MYC expression associated with BL, the effect of c-MYC overexpression on HLA class II Ag presentation was examined. For these studies, several approaches were used to alter c-MYC levels in human B cell lines to parallel c-MYC expression in BL. The B-LCL Frev was transfected with either an empty vector (Frev.vec) or c-myc (Frev.c-myc). The EREB2-5 cell line was generated from primary human B-cells by co-infecting with an EBNA-2-deficient P3HR1 virus and recombinant EBV encoding an estrogen receptor EBNA-2 fusion protein, to generate a B-LCL whose proliferation is dependent on estrogen (62, 63). These cells were then further transfected for constitutive expression of c-myc to generate the A1 cell line, capable of proliferation in the absence of EBNA-2 (64). Both EREB2-5 and A1 were transduced for expression of a common HLA-DR4 allele. Whole cell lysates from these cells were analyzed by western blotting for expression of c-MYC, EBNA-1 and EBNA-2 proteins (Fig. 1A). Expression of c-MYC was clearly elevated in Frev.c-myc and A1.DR4 cell lines when compared to Frev.vec and EREB2-5.DR4, respectively. For reference, the B-LCL line Priess displays low c-MYC expression. EBNA-1 is expressed at comparable levels in each cell line, while EBNA-2 expression is detected in Frev.vec and EREB2-5.DR4 which carries a mutant form of EBNA-2, and to a much lesser extent in the c-MYC overexpressing cells. The c-MYC overexpressing cells (Frev.c-myc, A1.DR4) also switched from a B-LCL (Frev.vec, EREB2-5.DR4) growth pattern (blast formation) to a BL growth pattern (suspension) (Fig. 1B). Frev.vec, Frev.c-myc, EREB2-5.DR4 and A1.DR4 were then analyzed by flow cytometry for surface expression of HLA class II DR4 (Fig. 1C). Overexpression of c-MYC did not affect expression of surface HLA class II proteins as each cell was found to express comparable levels. The results shown in Fig. 1D indicate that c-MYC overexpressing cells display a reduced capacity to present HSA64-76K peptide to stimulate CD4+ T cells via the class II pathway.
Figure 1.

Overexpression of c-MYC imparts a non-immunogenic BL phenotype to B-LCL and diminishes HLA-DR4-mediated CD4+ T cell recognition of BL-type cells. A, The wild-type B-cell line Frev which constitutively expresses HLA-DR4 molecules was either transfected with an empty vector or c-myc by electroporation. EREB2-5 cells are immortalized by EBV expressing a conditional estrogen receptor EBNA2 fusion protein (EREBNA2), and cellular proliferation is dependent on the availability of estrogen. A1 was established by stable transfection of conditionally EBV immortalized EREB2-5 cells with a c-myc/Igκ expression plasmid. The B-LCL-type cell line EREB2-5 and its derivative A1 (BL-type) were retrovirally transduced with HLA-DR4. Frev.vec, Frev.c-myc, EREB2-5.DR4 and A1.DR4 cells were then analyzed by western blotting for c-MYC, EBNA1, and EBNA2 proteins as described in the methods. β-actin was used as a loading control. B, Cells were subjected to microscopy for analysis of morphological characteristics (magnification × 20). C, Cells were stained for surface HLA-DR4 molecules using 359-F10 antibody and matched isotype control (IN-1), and analyzed by a flow cytometer as described. D, For T cell recognition assay, cells were incubated with different concentrations (0, 5, 10, 20μM) of a synthetic HSA64-76K peptide for 4h, washed and cocultured with the peptide-specific T cell hybridoma (17.9) for 24h. T cell production of IL-2 in the culture supernatant was quantitated by ELISA and expressed as pg/ml±SEM. Data are representative of three separate experiments.
Overexpression of c-MYC alters expression levels of HLA class II pathway components
Although c-MYC overexpression did not affect the surface expression of HLA class II proteins, it remained possible that the intracellular components of the class II pathway were affected thus disrupting CD4+ T cell recognition of BL cells. Frev.vec, Frev.c-myc, EREB2-5.DR4 and A1.DR4 whole cell lysates were analyzed by western blotting for expression levels of the class II pathway components HLA-DR, invariant chain (Ii), HLA-DM, and a lysosomal thiol-reductase GILT (Fig. 2A and B). In parallel assays, the expression of cell surface HLA class II proteins was also analyzed (Fig. 2C). Expression levels of both intracellular and cell surface HLA-DR molecules remained unchanged or were minimally altered in the c-MYC low and c-MYC high cells. There was also no significant change observed for Ii protein expression as analyzed by the Wilcoxon Rank Sum test. However, HLA-DM and GILT proteins were significantly decreased in the c-MYC-high cells as compared to c-MYC-low cells (p≤0.0022), suggesting possible implications for c-MYC-induced effects on HLA class II Ag presentation.
Figure 2.

Overexpression of c-MYC minimally influences HLA class II and Ii proteins, but alters other components of the class II pathway. A, B-LCL-type cells (Frev.vec and EREB2-5.DR4) and BL-type cells (Frev.c-myc and A1.DR4) were analyzed by western blotting for the expression of HLA-DR (L243), Ii (Pin 1.1), peptide editor HLA-DM and lysosomal thiol-reductase GILT (Santa Cruz antibodies) proteins. β-actin was used as a loading control. B, Densitometric analysis of protein bands detected in Figure 2A. Data are average density of three independent measurements of a representative band image and expressed as relative density (protein band/actin) ± S.D. Significant differences in relative band intensity were calculated by the Wilcoxon Rank Sum test; *p ≤0.0022. C, Flow cytometric analysis showing percent cell surface class II DR protein expression in BL (c-mychigh) and B-LCL (c-myclow) type cells. Frev.vec, Frev.c-myc, EREB2-5.DR4, and A1.DR4 cells were stained with an antibody against HLA-DR (L243), and a matched isotype (NN4) control, followed by flow cytometric analysis.
Overexpression of c-MYC disrupts Ag processing and presentation via the HLA class II pathway
To examine whether BL cells expressing high levels of c-myc have a diminished capacity for HLA class II-mediated Ag presentation, whole cell lysates were first obtained from the B-LCL 6.16.DR4.DM and two wild-type BL lines Nalm-6 and Ramos, and analyzed by western blotting for expression of c-MYC proteins (Fig. 3A). It was found that the BL lines Nalm-6.DR4 and Ramos.DR4 expressed significantly higher levels of c-MYC than the B-LCL 6.16.DR4.DM. Each cell line was also incubated with either the whole Igκ Ag or synthetic version of κ epitope Igκ188-203 or Igκ145-159, followed by co-culture with the appropriate peptide-specific T cell hybridomas for 24h. Following incubation, the culture supernatant was analyzed by ELISA for IL-2 as a measure of T cell stimulation. Nalm-6.DR4 and Ramos.DR4 displayed a sharply diminished capacity to process and present Igκ-derived epitopes to stimulate CD4+ T cells when compared to 6.16.DR4.DM (Fig. 3B, left panel). Similarly, BL cells failed to functionally present synthetic peptides to activate CD4+ T cells via the HLA class II pathway (Fig. 3B, right panel).
Figure 3.
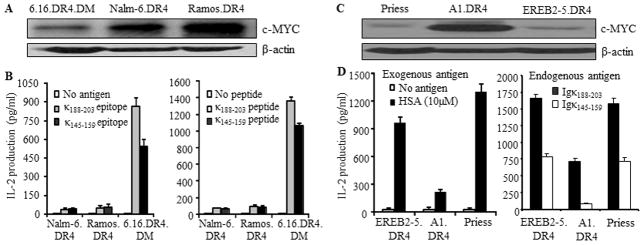
Overexpression of c-MYC disrupts both exogenous and endogenous presentation of Ags to T cells via the HLA class II pathway. A, Western blot analysis of c-MYC protein levels in B-LCL and BL-type cells. HLA-DR4-transfected B-LCL-type cells (6.16.DR4.DM) and BL-type cells (Nalm.DR4, Ramos.DR4) were subjected to western blotting for determining the levels of c-MYC proteins. β-actin was used as a loading control. B, left panel: processing and presentation of natural epitopes from an exogenous Ag (Igκ). B, right panel: presentation of the synthetic forms of Igκ epitopes. Cells (6.16.DR4.DM, Nalm.DR4, and Ramos.DR4) were incubated with either whole Igκ (left panel) or synthetic epitopes (κ188-203 and κ145-159 peptides) (right panel) for 4h. Following incubation, cells were washed and co-cultured with the appropriate epitope or peptide specific T cell hybridomas (2.18a for κ188-203 and 1.21 for κ145-159) for 24h. Supernatants obtained from the co-culture assay were tested by ELISA to determine IL-2 levels as a measure of T-cell stimulation. T cell production of IL-2 was quantitated and expressed as pg/ml. C, Western blot analysis showing c-MYC protein expression in B-LCL (Priess), BL-type (A1.DR4) and B-LCL-type cells (EREB2-5.DR4). D, left panel (exogenous presentation): EREB2-5.DR4, A1.DR4 and Priess cells were incubated with a whole Ag HSA for 4h, followed by coculture with the HSA64-76K peptide specific T cell hybridoma (17.9) for 24h. D, right panel (endogenous presentation): EREB2-5.DR4, A1.DR4 and Priess cells express endogenous Igκ. While EREB2-5.DR4 and A1.DR4 cells are transduced with DR4, Priess cells are homozygous for DR4 alleles. These cells were cocultured with the HLA-DR4-restricted Igκ188-203 and Igκ145-159 peptide specific T cell hybridomas (2.18a for Igκ188-203 and 1.21 for Igκ145-159 respectively) in the absence of exogenous Ag, for 24h. T cell production of IL-2 was quantitated by ELISA and expressed as pg/ml±SEM. Data are representative of three separate experiments.
To investigate whether both exogenous and endogenous routes of class II Ag presentation are disrupted in BL, a number of B-LCL (Priess.DR4, EREB2-5.DR4) and the c-MYC overexpressing counterpart of EREB2-5 cells (A1.DR4, BL-type cells) were analyzed by biochemical and functional assays. It is important to note that these cell lines innately express endogenous Igκ Ag (data not shown). Western blot analysis of whole cell lysates from these cells showed that A1.DR4 cells expressed high levels of c-MYC whereas B-LCL and B-LCL-type cells expressed low levels of c-MYC proteins (Fig. 3C). To further investigate whether BL-type cells expressing high levels of c-myc have a diminished capacity for HLA class II-mediated exogenous Ag presentation, cells (Priess, EREB2-5.DR4 and A1.DR4) were incubated with the whole Ag HSA for 4h (Fig. 3D, left panel). Cells were then washed and cocultured with the HSA64-76K epitope specific T cell hybridoma (17.9) for 24h. The production of IL-2 in the culture supernatant was quantitated by ELISA. Priess and EREB2-5.DR4 cells which express low levels of c-MYC efficiently presented HSA to CD4+ T cells, whereas high c-MYC-expressing A1.DR4 cells failed to optimally present the epitope to the same T cell hybridoma line.
To study the presentation of endogenous Ag, we used two T cell hybridoma lines 2.181 and 1.21 which recognize κ188-203 and κ145-159 peptides respectively. We then cocultured Priess, EREB2-5.DR4 and A1.DR4 cells expressing endogenous Igκ with 2.181 and 1.21 T cell hybridomas followed by analysis of IL-2 production in the culture supernatant (Fig. 3D, right panel). A1.DR4 cells which express high levels of c-MYC failed to optimally present κ188-203 and κ145-159 epitopes to CD4+ T cells. These results strongly suggest that overexpression of c-MYC diminishes both exogenous and endogenous pathways of class II Ag presentation and CD4+ T cell recognition of c-myc overexpressing B-(BL-type) cells.
Inhibition of c-MYC expression partially restores CD4+ T cell recognition of BL-type cells via the HLA class II pathway
To determine the role of c-MYC in diminished class II presentation in BL cells, we employed the cell line P493-6.DR4, which can be reversibly shifted from an LCL-like phenotype (EBNA-2 on, exogenous c-myc off) to a BL-like phenotype (EBNA-2 off, c-myc on) by addition and withdrawal of estrogen plus tetracycline. P493-6 cells expressing DR4 were cultured in the presence (c-myc off) or absence (c-myc on) of estrogen plus tetracycline (P493-6.DR4.est.tet versus P493-6.DR4 cells) (Fig. 4A). When cultured in the presence of estrogen plus tetracycline, c-MYC expression was markedly reduced (Fig. 4A). Cell surface HLA-DR and HLA-DR4 molecules were not significantly altered in P493-6.DR4 vs. P493-6.DR4.est.tet cells regardless of c-MYC expression (Fig. 4B). Cells grown under c-myc on/off conditions were used in T cell assay to determine HLA class II-mediated Ag presentation capability. When cultured in the absence of tetracycline, P493-6.DR4 cells showed a sharply diminished capacity to stimulate T cells via HLA class II as compared to P493-6.DR4.est.tet cells cultured in the presence of estrogen plus tetracycline (Fig 4C). Because P493-6 cells express endogenous kappa antigen, P493-6.DR4 and P493-6.DR4.est.tet cell lines were cocultured with the κ188-203 and κ145-159 peptide specific T cells, followed by the quantitation of T cell production of IL-2 (Fig. 4D, right panel). Likewise, P493-6.DR4 showed a diminished capacity to present endogenous kappa epitopes to stimulate CD4+ T cells via the class II pathway. Taken together, both exogenous and endogenous presentation of class II-restricted peptides, were partially restored by downregulating c-MYC expression and shifting P493-6.DR4 cells towards an LCL-phenotype by the addition of estrogen plus tetracycline.
Figure 4.

Shift from a LCL- to a BL-like phenotype in P493-6 cells did not alter HLA class II protein expression, but decreased CD4+ T cell recognition via both exogenous and endogenous pathway of HLA class II Ag presentation. A, P493-6 cells were reversibly shifted from a LCL- to a BL-like phenotype by withdrawal and re-addition of estrogen (2μM) plus tetracycline (5μg/ml). Western blot analysis of P493-6.DR4 cells for c-MYC protein expression under c-myclow (est/tet) and c-mychigh (no est/tet) culture conditions. β-actin was used as a loading control. B, Flow cytometric analysis of surface HLA-DR (antibody L243) and HLA-DR4 (antibody 359-F10) molecules in c-myclow (B-LCL) and c-mychigh (BL-like) cells. C, Ag presentation assay showing exogenous presentation of HSA Ag/peptide. P493-6.DR4 cells were first cultured under c-mychigh (P493-6.DR4, without est/tet) and c-myclow (P493-6DR4, with est/tet) conditions, and then incubated with the whole HSA or HSA64-76K peptide for 4h, followed by coculture with the HSA64-76K peptide specific T cell hybridoma (17.9) for 24h. D, Ag presentation assay showing endogenous presentation of Igκ. P493-6.DR4 cells grown under c-mychigh (P493-6.DR4, without est/tet) and c-myclow (P493-6DR4, with est/tet) conditions were cocultured with the HLA-DR4-restricted Igκ188-203 and Igκ145-159 peptide specific T cell hybridomas (2.18a for Igκ188-203 and 1.21 for Igκ145-159 respectively) in the absence of exogenous Ag, for 24h. Supernatants obtained from these T cell assays were tested by ELISA to determine IL-2 levels as a measure of T-cell stimulation. T cell production of IL-2 was expressed as mean pg/ml±SEM. E, Peptide binding assay at 4°C. P493-6.DR4 cells grown under c-mychigh (P493-6.DR4, without est/tet) and c-myclow (P493-6DR4, with est/tet) conditions were incubated with vehicle alone or biotin-labeled HSA64-76K peptide for 24h. F, Peptide binding assay at 37°C. Cells (P493-6.DR4) grown under c-mychigh and c-myclow conditions, washed and fixed with 1% paraformaldehyde, and were incubated with biotin-labeled HSA64-76K peptide for 24, followed by staining with FITC-avidin. After washing, cells were analyzed by flow cytometry as described in the methods. Data are representative of three separate experiments.
To investigate whether peptide binding to surface HLA-DR4 molecules was altered in c-myc-high cells, we performed binding assays using biotin-labeled HSA peptide (HSA64-76K) and FITC-labeled avidin, followed by flow cytometric analysis. The binding of DR4-restricted HSA peptide was very low at 4°C (Fig. 4E). But there was a trend that exogenous peptide loading by class II molecules was better in low-c-myc cells as compared to that observed in high-c-myc cells. This difference was more pronounced when the binding assay was performed in fixed cells at 37°C (Fig. 4F). The HSA peptide bound much better to DR4 molecules on low-myc cells as compare to those of high-c-myc cells. These data suggest that the observed differential peptide binding to class II molecules could contribute to reduced peptide presentation and diminished T cell recognition of high-c-myc cells.
To further examine this finding, P493-6.DR4 cells were treated with the c-MYC inhibitor 10058-F4, a small molecule inhibitor which decreases c-MYC expression in tumor cells (65). Whole cell lysates were obtained and analyzed by western blotting for expression of c-MYC protein (Fig. 5A). A dose-dependent decrease in c-MYC expression was consistent with other reports that 10058-F4 treatment results in diminished cellular c-MYC expression. To determine whether inhibition of c-MYC can restore T cell recognition, P493-6.DR4 cells were treated with 50μM 10058-F4 for 24h, followed by incubation with the whole HSA for 4h. Cells were also incubated with the synthetic version of HSA epitope (HSA64-76K) for 4h. After incubation, cells were washed and fixed with 1%paraformaldehyde, and co-cultured with the HSA epitope-specific T cell hybridoma 17.9 for 24h. The culture supernatant was then analyzed by ELISA for IL-2 as a measure of T cell stimulation (Fig. 5B, left panel). It was found that inhibition of c-MYC expression partially restored HLA class II-mediated Ag/peptide presentation and CD4+ T cell recognition of BL-like P493-6 cells.
Figure 5.
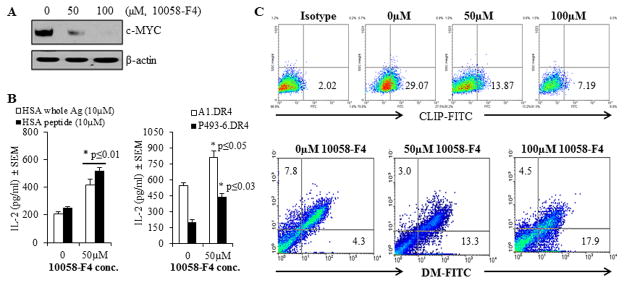
Treatment with the c-MYC inhibitor 10058-F4 partially restored CD4+ T cell recognition of c-MYC overexpressing P493-6 cells via the HLA class II pathway. P493-6 cells were grown in complete RPMI1640 medium in the absence of estrogen and tetracycline (BL-like phenotype). A, Expression of c-MYC proteins in P493-6 cells treated with various concentrations (0, 50 and 100μM) of the c-MYC inhibitor 10058-F4 for 24h. Cells were then washed and subjected to western blotting for analyzing c-MYC protein expression. β-actin was used as a loading control. B, left panel: P493-6 cells were cultured in the presence or absence of c-MYC inhibitor 10058-F4 (50μM) for 24h, followed by the addition of whole HSA or HSA64-76K peptide (10 μM) for the last 4h of incubation. Cells were washed, fixed with 1% paraformaldehyde, and cocultured with the HSA64-76K peptide specific T cell hybridoma (17.9) for 24h. T cell production of IL-2 in the culture supernatant was quantitated by ELISA and expressed as mean pg/ml±SEM. B, right panel: A1.DR4 and P493-6.DR4 cells which express endogenous Igκ were cultured in the presence or absence of c-MYC inhibitor 10058-F4 (50μM) for 24h. Cells were washed, fixed with 1% paraformaldehyde, and cocultured with the HLA-DR4-restricted Igκ188-203 peptide specific T cell hybridoma (2.18a) for 24h. Supernatants obtained were tested by ELISA to determine IL-2 levels as a measure of T-cell proliferation. Data are representative of three separate experiments. *p<0.05. C, Flow cytometric analysis of P493-6 cells treated with the c-MYC inhibitor 10058-F4 for 24h. Cells were first treated with 10058-F4 and stained with an antibody against CLIP (Cer-CLIP antibody) plus a matched isotype control (NN4 antibody) for determining surface CLIP protein expression (upper panel). Cells treated with 10058-F4 were also subjected to intracellular staining with antibodies against HLA-DM and HLA-DO molecules as described in the methods. The lower panel shows the effect of c-MYC inhibitor 10058-F4 on differential expression of HLA-DM and HLA-DO proteins in P493-6 cells as compared to untreated controls.
To determine whether inhibition of c-MYC can also restore the presentation of endogenous Ag, we employed A1.DR4 and P493-6.DR4 cells (both cell lines express high levels of c-MYC) and cocultured with the κ188-203 epitope-specific T cell hybridoma line 2.181 for 24h. After incubation, T cell production of IL-2 production in the culture supernatant was quantitated by ELISA (Fig. 5B, right panel). It was found that inhibition of c-MYC protein by 10058-F4 significantly restored HLA class II-mediated presentation of endogenous Ag Igκ. Taken together, these data suggest that overexpression of c-MYC diminishes both exogenous and endogenous pathways of class II-restricted Ag presentation and CD4+ T cell recognition.
Flow cytometric analysis of P493-6 cells showed that c-MYC inhibitor 10058-F4 dose-dependently reduced cell surface CLIP expression (29% vs. 13% and 29% vs. 7%) (Fig. 5C, upper panel), suggesting that c-MYC affects the number of peptide receptive class II complexes in the cell which may be linked to DM/DO. Further study using intracellular staining of DM/DO molecules confirmed that inhibition of c-MYC elevated DM molecules while down-regulating DO proteins (Fig. 5C, lower panel). These data suggest that the c-MYC inhibitor may partially restore CD4+ T cell recognition by altering DM/DO protein ratio in BL-type cells, and that the disruption of c-MYC expression or function might hold potential in restoring CD4+ T cell recognition of BL.
Inhibition of c-MYC down-regulates CLIP expression and reverses the ratio of HLA-DM/HLA-DO molecules in c-MYC overexpressing BL-type P493-6 cells
Having established that treatment with the c-MYC inhibitor 10058-F4 was sufficient to partially restore class II-mediated antigen presentation in P493-6.DR4 cells, we sought to determine the mechanism how c-MYC impairs antigen presentation via the MHC class II pathway. To this end, P493-6.DR4 cells proliferating in the absence of estrogen plus tetracycline (c-myc program) were shifted back to a B-LCL phenotype by addition of estrogen plus tetracycline (38, 63), and then analyzed by flow cytometry for expression of cell surface CLIP and intracellular Ii, as well as DM/DO molecules. While no significant change was observed in Ii expression (93% vs. 94%), cell surface CLIP expression was markedly down-regulated (28% vs. 12%) (Fig. 6A). The shift from a BL-like (P493-6.DR4, A1.DR4) to a B-LCL-phenotype (P493-6.est.tet) led to a sharp increase in HLA-DM molecules (MFI: 194 vs. 801) (Fig. 6B). This increase in HLA-DM molecules was concurrent with a slight decrease in HLA-DO molecules (MFI: 274 vs. 158) and a significant increase (*p<0.001) in the DM/DO protein ratio (Fig. 6C). These data support the notion that the decrease in CD4+ T cell recognition of BL and BL-like P493-6 cells can be linked to alteration of the DM/DO protein ratio and increased surface CLIP expression as a result of overexpression of c-MYC protein.
Figure 6.

Overexpression of c-MYC alters cell surface CLIP by regulating DM/DO ratio in BL/B-LCL type cells. A, DR4 expressing P493-6 cells were cultured under c-myc-on [(P493-6.DR4)(minus estrogen, minus tetracycline)] and c-myc-off conditions [(P493-6.DR4.est.tet)(plus estrogen, plus tetracycline). Cells were then intracellularly stained with antibody against Ii (Pin1.1), followed by addition of FITC-labeled secondary antibody as described. Cells were also stained with cer-CLIP antibody for cell surface CLIP proteins and analyzed by flow cytometry. B, P493-6 cells grown under c-myc-on and c-myc-off conditions were also intracellularly stained with antibodies against HLA-DM/HLA-DO proteins and appropriate isotype controls, followed by flow cytometric analysis. C, Bar graphs showing mean fluorescence intensity ±SEM of HLA-DM and HLA-DO staining in P493-6.DR4 and P493-6.DR4.est.tet cells. Data are representative of at least three separate experiments. *p<0.001. D, Analysis of a 47kDa protein differentially expressed in BL- vs B-LCL-type cells. Acid eluates obtained from BL-type (P493-6.DR4) and B-LCL-type (P493-6.DR4.est.tet) cells were separated and detected on a non-reducing gel as described in the methods. An approximately 47kDa band was excised and analyzed by MALDI-TOF/TOF mass spectrometry. Data shown are representative of at three separate experiments.
Inhibition of c-MYC up-regulates a 47kDa enolase-like acid labile protein and regulates HLA class II-mediated Ag presentation
We have recently shown that a 47kDa enolase-like acid labile protein is highly expressed in B-LCL, and that this protein is nearly absent or expressed at a greatly reduced level in BL cells (41). This enolase-like protein facilitated B-T cell interactions by enhancing HLA class II peptide display to T cells. Studies were conducted to determine if c-MYC impacts the levels of this 47kDa protein. The 47kDa protein band was highly abundant in B-LCL type P493-6.DR4.est/tet cells as compared to BL type P493-6.DR4 cells (Fig. 6D). MALDI-TOF/TOF mass spectrometric analysis confirmed that the 47kDa protein band was an alpha-enolase 1-like molecule (Accession number 4503571; URL: ipi.HUMAN.V3.71). It is important to note that P493-6.DR4 cells express very high levels of c-MYC proteins relative to P493-6.DR4.est/tet cells, and that elevated c-MYC disrupts CD4+ T cell recognition of P493-6.DR4 cells (Fig. 4). In addition, BL-type P493-6.DR4 cells express lower levels of DM proteins as compared to B-LCL-type P493-6.DR4. est/tet cells (Fig. 6A and B). Collectively, these data suggest that the overexpression of c-MYC downregulates HLA-DM as well as the 47kDa enolase-like acid labile protein, leading to disruptions in immune recognition of BL.
High CLIP expression, a decrease in the DM/DO ratio, and impaired MHC class II antigen presentation are also observed in BL tumors ex vivo
To investigate whether high CLIP expression, a decrease in the DM/DO ratio, and impaired Ag presentation via the MHC class II pathway are also observed in primary BL TB#2952 lymphoma was subjected ex vivo to western blotting and flow cytometry, and was compared with an early passage of an EBV transformed B-cell line of a healthy individual (C#16). Western blot analysis showed that TB#2952 expressed higher levels of c-MYC, but equivalent intracellular DRβ proteins as compared with C#16 (Fig. 7A). T cell assay demonstrated that TB#2952 lymphoma which expressed higher levels of c-MYC failed to stimulate CD4+ T cells. Likewise, early passage TB#2952 cells that had been subjected to infection with EBV [TB#2952(+EBV)] had diminished capacity to stimulate CD4+ T cells as compared to EBV infected control cells [C#16(+EBV)] (Fig. 7B), indicating that in vitro EBV infection did not restore the deficiency in Ag presentation observed in BL tumor cells ex vivo.
Figure 7. High CLIP expression, a decrease in the DM/DO ratio, and impaired MHC class II antigen presentation, are also observed in BL tumors ex vivo.
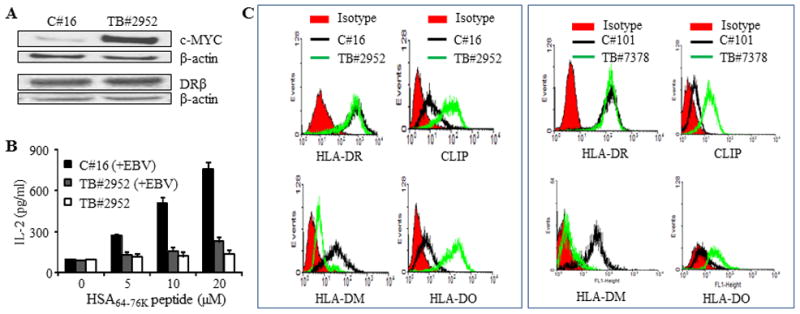
A, Human primary BL (TB#2952) cells were isolated from lymph node samples obtained from lymphoma patients as described in the methods. This primary BL tumor as well as control B-cells from a healthy individual (C#16) were analyzed by western blotting for c-MYC, class II protein expression, and HSA64-76K peptide presentation. In parallel, these B cell tumors and control B-cells were also transduced with DR4β as described. B, HLA-DR4β-transduced early passage TB#2952 tumor cells were either directly tested for HSA64-76K peptide presentation or infected in vitro with EBV [TB#2952(+EBV)] and then used in the T cell assay as described in Figure 1D. Healthy B-cells (C#16) infected in vitro with EBV [C#16(+EBV)] were also tested in the T cell assay for CD4+ T cell recognition, and the data were compared with those of the tumors. Black bars, DR4β-transduced after EBV infection; white bars, DR4β-transduced without EBV infection; grey bars, DR4β-transduced after EBV infection. C, B-cells from healthy individuals (C#16, C#101) and primary BL tumors (TB#2952 and TB#7378) were stained with antibodies against HLA-DR, CLIP and HLA-DM/DO proteins plus appropriate secondary antibodies and matched isotype controls (red) as described in the methods. Cells were then analyzed by flow cytometry.
Flow cytometric analyses of EBV-transformed early passage control C#16 and TB#2952 BL tumor cells showed comparable levels of cell surface HLA-DR molecules, while C#16 expressed higher levels of HLA-DM and lower levels of DO and CLIP proteins (left panels of Fig. 7C). Exactly the same was found with another BL tumor (TB#7378) and control B cell pair of a healthy individual (C#101) (right panels of Fig. 7C). These data further support our findings that the alteration of DM/DO ratio and upregulation of CLIP cause reduced CD4+ T cell recognition of B-cell lymphoma.
DISCUSSION
In the 30 years since its discovery, the c-MYC protein has come to be recognized as one of the most commonly activated oncogenes in human cancers. As a transcription factor that controls up to 15% of all known cellular genes, the effects of c-MYC are wide ranging. Cellular functions and host defenses are frequently controlled by genes within the c-MYC target network including immune regulation (2). In spite of its far-reaching effects, c-MYC protein has never been reported to have an effect on the HLA class II pathway of Ag presentation. We report here for the first time that c-MYC overexpression results in decreased HLA class II-mediated immune recognition of BL.
The HLA class II pathway of Ag presentation is primarily involved in the presentation of exogenous Ags which have been internalized and degraded by antigen presenting cells (APCs). While HLA class I is expressed on every nucleated cell of the body, HLA class II expression is limited to the professional APCs: macrophages, dendritic cells and B-cells (66–69). In the class II pathway, extracellular Ags are internalized and degraded in endolysosomal compartments. Antigenic peptides are generated with the aid of cathepsins and GILT and deficiencies in these proteins lead to the production of peptides which are not optimal for class II-mediated Ag presentation (47, 56, 70, 71). HLA class II is synthesized in the endoplasmic reticulum (ER) lumen bound to Ii which occupies the binding groove to prevent inappropriate peptide binging and to aid in transporting class II through the Golgi and the endolysosomal compartments containing the antigenic peptides (72–74). Here, Ii is degraded by cathepsins leaving class II-associated invariant chain peptide (CLIP) occupying the class II binding groove (66, 68, 73, 75). HLA-DM then mediates the removal of CLIP and the binding of peptide to the class II binding groove (75–79). Removal of CLIP and class II peptide binding is partially modulated by the non-classical HLA-DO which inhibits the activity of HLA-DM. Shifts in the DM/DO ratio may cause an increase in destabilized cell-surface class II-CLIP complexes, thus impairing the HLA class II pathway of Ag presentation (78, 80–83). Most B-cell tumors including BL, express class II molecules, leading to the assumption that these cells should be ready targets for tumor-specific CD4+ T cells. However, we have found that the presentation of nativeAg as well as peptides is influenced by elevated cellular c-MYC. Native Ag requires cellular internalization, processing and epitope loading by class II within endocytic compartments. To better understand this defect in class II presentation in cells with c-mychigh levels, we tested peptide binding to class II molecules at low temperature (4°C) in live cells and at 37°C in paraformaldehyde fixed cells. We found that the peptide binding to class II was reduced in c-mychigh cells as compared to c-myclow cells in each case. Loss of cellular DM can impact binding of exogenous peptides in multiple ways. Our previous reports demonstrated that cellular DM levels can impact peptide presentation as some peptides are endocytosed and bind intracellular class II molecules that may undergo DM editing (49, 57, 84). We similarly showed this was true of peptides which require reduction by the lysosomal-thiol reductase GILT (47, 85). Yet peptide binding studies here suggest that cellular c-MYC levels may also alter the conformation or peptide accessibility of class II molecules. Consistent with this, the conformation of class II molecules is known to be flexible, with loss of DM impacting class II structure and peptide loading (86). Inhibition of c-MYC also resulted in improved CD4+ T cell recognition of BL, suggesting that the alterations in peptide binding to class II molecules along with class II components DM/DO may be regulated by elevated c-MYC in tumor cells. Because the presentation of both exogenous and endogenous Ags were found to be disrupted in BL cells, c-MYC inhibitors could prove important in enhanced immune recognition of B-cell lymphomas.
We have shown here that overexpression of c-MYC leads to decreased class II-mediated immunogenicity in BL, by regulating the expression of key components of the HLA class II pathway. Elevation of c-MYC was found to cause a shift from the immunogenic B-LCL phenotype (growth with blast formation) to the non-immunogenic BL phenotype (growth as suspension cells) as reported previously (38). This shift in phenotype correlated with a decrease in HLA class II-mediated immunogenicity for several different peptide Ags. The cause of the c-MYC-associated decrease in immunogenicity does not seem to be linked to expression of class II proteins as both c-mychigh and c-myclow cells expressed comparable cell surface class II levels. Rather as shown here, c-MYC overexpression influences cellular expression of DM, DO and GILT, critical components which modulate the efficiency of class II Ag presentation. GILT reduces disulfide bonds in Ags which allows proteins to be unfolded and further processed by acidic proteases (56, 70, 71, 87). The importance of GILT for the class II pathway has been demonstrated in GILT−/− mice which are defective in their ability to process Ag, although they express normal levels of class II (47, 88, 89). It is plausible that the decreased immunogenicity observed in our c-mychigh cell lines is at least partially attributable to downregulation of the critical endolysosomal reductase GILT.
Secondly, we show that overexpression of c-MYC leads to decreased expression of the class II pathway component HLA-DM. As discussed above, HLA-DM mediates the removal of CLIP from the peptide binding groove as well as the binding of antigenic peptides. The functional importance of HLA-DM to class II-mediated Ag presentation was first recognized in B-LCL with mutations in DM which displayed impaired class II-peptide complex formation and defects in exogenous Ag presentation (68, 76, 80, 90). In keeping with this finding, we also demonstrate that expression of cell surface CLIP is increased in c-mychigh cells. Given that one function of HLA-DM is to mediate the release of CLIP from the class II peptide binding groove, it is expected that in conditions of decreased HLA-DM expression, CLIP would remain bound to class II proteins and be expressed on the cell surface. Having demonstrated that overexpression of c-MYC was correlated with decreased class II-mediated Ag presentation and that this effect may be attributable to downregulation of GILT and HLA-DM, we next sought to determine if inhibition of c-MYC restored Ag presentation. Cells treated with the small molecule c-MYC inhibitor 10058-F4, partially restored class II-Ag presentation. When B-cells were treated with 10058-F4, cellular c-MYC levels dropped while a shift in the intracellular DM/DO ratio was observed. Treatment of B-cells with 10058-F4 also resulted in decreased CLIP binding to class II molecules, a result consistent with DM editing of CLIP and the formation of increased numbers of peptide-receptive class II molecules available to present exogenous and endogenous antigenic peptides. Further analyses with primary tumors confirmed that elevated DO and CLIP molecules disrupt CD4+ T cell recognition of BL.
It should be noted that inhibition of c-MYC was not sufficient to fully restore class II-mediated Ag presentation. This is in keeping with our previously published (91) and current findings that a 47kDa α-enolase 1-like acid labile protein is nearly absent or expressed at a greatly reduced level in BL, and this deficiency may attenuate class II-mediated functional Ag presentation. The expression of α-enolase molecules has been shown to downregulate c-MYC (92, 93); thus, the 47kDa α-enolase 1-like molecule may regulate c-myc as well as components of the HLA class II pathway to promote immune recognition of BL.
Acknowledgments
We thank Elisabeth Kremmer for a gift of the anti-EBNA-2 antibody. We also thank Ms. Abigail W. Lauer (Public Health Sciences at MUSC) for statistical analysis.
Abbreviations used in this article
- BL
Burkitt lymphoma
- EBV
Epstein-Barr Virus
- B-LCL
B-lymphoblastoid cell line
- HLA
human leukocyte antigen
- Ii
invariant chain
- CLIP
class II-associated invariant chain peptide
- DM
HLA-DM
- DO
HLA-DO
- GILT
γ-IFN-inducible lysosomal thiol reductase
- κ
kappa
Footnotes
This work was supported by grants from the National Institutes of Health (R01 CA129560 and R01 CA129560-S1 to AH, and RO1AI079065 to JSB). The research presented in this article was also supported in part by the Tissue Biorepository and Flow Cytometry Shared Resource as part of the Hollings Cancer Center at the Medical University of South Carolina which is funded by a Cancer Center Support Grant P30 CA138313.
Authorship and Conflict of Interest
Contribution: A.H. conceived and designed the experiments, interpreted data and wrote the manuscript; J.M.G., C.M., J.F., S.A., Am. H., and A.H. performed the research, analyzed and interpreted data, performed statistical analysis, and wrote the manuscript; B.K., G.W.B., and J.B. provided vital new reagents, interpreted data, and wrote the manuscript. The authors have no financial conflict of interest.
References
- 1.Vennstrom B, Sheiness D, Zabielski J, Bishop JM. Isolation and characterization of c-myc, a cellular homolog of the oncogene (v-myc) of avian myelocytomatosis virus strain 29. J Virol. 1982;42:773–779. doi: 10.1128/jvi.42.3.773-779.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dang CV, O’Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Semin Cancer Biol. 2006;16:253–264. doi: 10.1016/j.semcancer.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 3.Lin Z, Yin Q, Flemington E. Identification of a negative regulatory element in the Epstein-Barr virus Zta transactivation domain that is regulated by the cell cycle control factors c-Myc and E2F1. J Virol. 2004;78:11962–11971. doi: 10.1128/JVI.78.21.11962-11971.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allday MJ. How does Epstein-Barr virus (EBV) complement the activation of Myc in the pathogenesis of Burkitt’s lymphoma? Semin Cancer Biol. 2009;19:366–376. doi: 10.1016/j.semcancer.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Slack GW, Gascoyne RD. MYC and aggressive B-cell lymphomas. Adv Anat Pathol. 2011;18:219–228. doi: 10.1097/PAP.0b013e3182169948. [DOI] [PubMed] [Google Scholar]
- 6.Larsson LG, Henriksson MA. The Yin and Yang functions of the Myc oncoprotein in cancer development and as targets for therapy. Exp Cell Res. 2010;316:1429–1437. doi: 10.1016/j.yexcr.2010.03.025. [DOI] [PubMed] [Google Scholar]
- 7.Grandori C, Cowley SM, James LP, Eisenman RN. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu Rev Cell Dev Biol. 2000;16:653–699. doi: 10.1146/annurev.cellbio.16.1.653. [DOI] [PubMed] [Google Scholar]
- 8.Knoepfler PS, Zhang XY, Cheng PF, Gafken PR, McMahon SB, Eisenman RN. Myc influences global chromatin structure. EMBO J. 2006;25:2723–2734. doi: 10.1038/sj.emboj.7601152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer. 2008;8:976–990. doi: 10.1038/nrc2231. [DOI] [PubMed] [Google Scholar]
- 10.Arabi A, Wu S, Ridderstrale K, Bierhoff H, Shiue C, Fatyol K, Fahlen S, Hydbring P, Soderberg O, Grummt I, Larsson LG, Wright AP. c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat Cell Biol. 2005;7:303–310. doi: 10.1038/ncb1225. [DOI] [PubMed] [Google Scholar]
- 11.Eilers M, Eisenman RN. Myc’s broad reach. Genes Dev. 2008;22:2755–2766. doi: 10.1101/gad.1712408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoffman B, Liebermann DA. Apoptotic signaling by c-MYC. Oncogene. 2008;27:6462–6472. doi: 10.1038/onc.2008.312. [DOI] [PubMed] [Google Scholar]
- 13.Wang C, Lisanti MP, Liao DJ. Reviewing once more the c-myc and Ras collaboration: converging at the cyclin D1-CDK4 complex and challenging basic concepts of cancer biology. Cell Cycle. 2011;10:57–67. doi: 10.4161/cc.10.1.14449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Egle A, Harris AW, Bouillet P, Cory S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:6164–6169. doi: 10.1073/pnas.0401471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene. 1999;18:3004–3016. doi: 10.1038/sj.onc.1202746. [DOI] [PubMed] [Google Scholar]
- 16.Dang CV, Resar LM, Emison E, Kim S, Li Q, Prescott JE, Wonsey D, Zeller K. Function of the c-Myc oncogenic transcription factor. Exp Cell Res. 1999;253:63–77. doi: 10.1006/excr.1999.4686. [DOI] [PubMed] [Google Scholar]
- 17.Boxer LM, Dang CV. Translocations involving c-myc and c-myc function. Oncogene. 2001;20:5595–5610. doi: 10.1038/sj.onc.1204595. [DOI] [PubMed] [Google Scholar]
- 18.Gerbitz A, Mautner J, Geltinger C, Hortnagel K, Christoph B, Asenbauer H, Klobeck G, Polack A, Bornkamm GW. Deregulation of the proto-oncogene c-myc through t(8;22) translocation in Burkitt’s lymphoma. Oncogene. 1999;18:1745–1753. doi: 10.1038/sj.onc.1202468. [DOI] [PubMed] [Google Scholar]
- 19.Robbiani DF, Nussenzweig MC. Chromosome translocation, B cell lymphoma, and activation-induced cytidine deaminase. Annual review of pathology. 2013;8:79–103. doi: 10.1146/annurev-pathol-020712-164004. [DOI] [PubMed] [Google Scholar]
- 20.Mutalima N, Molyneux E, Jaffe H, Kamiza S, Borgstein E, Mkandawire N, Liomba G, Batumba M, Lagos D, Gratrix F, Boshoff C, Casabonne D, Carpenter LM, Newton R. Associations between Burkitt lymphoma among children in Malawi and infection with HIV, EBV and malaria: results from a case-control study. PLoS One. 2008;3:e2505. doi: 10.1371/journal.pone.0002505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rasti N, Falk KI, Donati D, Gyan BA, Goka BQ, Troye-Blomberg M, Akanmori BD, Kurtzhals JA, Dodoo D, Consolini R, Linde A, Wahlgren M, Bejarano MT. Circulating epstein-barr virus in children living in malaria-endemic areas. Scand J Immunol. 2005;61:461–465. doi: 10.1111/j.1365-3083.2005.01589.x. [DOI] [PubMed] [Google Scholar]
- 22.Njie R, Bell AI, Jia H, Croom-Carter D, Chaganti S, Hislop AD, Whittle H, Rickinson AB. The effects of acute malaria on Epstein-Barr virus (EBV) load and EBV-specific T cell immunity in Gambian children. J Infect Dis. 2009;199:31–38. doi: 10.1086/594373. [DOI] [PubMed] [Google Scholar]
- 23.Piriou E, Kimmel R, Chelimo K, Middeldorp JM, Odada PS, Ploutz-Snyder R, Moormann AM, Rochford R. Serological evidence for long-term Epstein-Barr virus reactivation in children living in a holoendemic malaria region of Kenya. J Med Virol. 2009;81:1088–1093. doi: 10.1002/jmv.21485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aldoss IT, Weisenburger DD, Fu K, Chan WC, Vose JM, Bierman PJ, Bociek RG, Armitage JO. Adult Burkitt lymphoma: advances in diagnosis and treatment. Oncology (Williston Park) 2008;22:1508–1517. [PubMed] [Google Scholar]
- 25.Sharipo A, Imreh M, Leonchiks A, Imreh S, Masucci MG. A minimal glycine-alanine repeat prevents the interaction of ubiquitinated I kappaB alpha with the proteasome: a new mechanism for selective inhibition of proteolysis. Nat Med. 1998;4:939–944. doi: 10.1038/nm0898-939. [DOI] [PubMed] [Google Scholar]
- 26.Fogg MH, Wirth LJ, Posner M, Wang F. Decreased EBNA-1-specific CD8+ T cells in patients with Epstein-Barr virus-associated nasopharyngeal carcinoma. Proc Natl Acad Sci U S A. 2009;106:3318–3323. doi: 10.1073/pnas.0813320106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee SP, Brooks JM, Al-Jarrah H, Thomas WA, Haigh TA, Taylor GS, Humme S, Schepers A, Hammerschmidt W, Yates JL, Rickinson AB, Blake NW. CD8 T cell recognition of endogenously expressed epstein-barr virus nuclear antigen 1. The Journal of experimental medicine. 2004;199:1409–1420. doi: 10.1084/jem.20040121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Piriou E, van Dort K, Nanlohy NM, van Oers MH, Miedema F, van Baarle D. Loss of EBNA1-specific memory CD4+ and CD8+ T cells in HIV-infected patients progressing to AIDS-related non-Hodgkin lymphoma. Blood. 2005;106:3166–3174. doi: 10.1182/blood-2005-01-0432. [DOI] [PubMed] [Google Scholar]
- 29.Schlee M, Schuhmacher M, Holzel M, Laux G, Bornkamm GW. c-MYC impairs immunogenicity of human B cells. Advances in cancer research. 2007;97:167–188. doi: 10.1016/S0065-230X(06)97007-9. [DOI] [PubMed] [Google Scholar]
- 30.Aubert RD, Kamphorst AO, Sarkar S, Vezys V, Ha SJ, Barber DL, Ye L, Sharpe AH, Freeman GJ, Ahmed R. Antigen-specific CD4 T-cell help rescues exhausted CD8 T cells during chronic viral infection. Proc Natl Acad Sci U S A. 2011;108:21182–21187. doi: 10.1073/pnas.1118450109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fu T, Voo KS, Wang RF. Critical role of EBNA1-specific CD4+ T cells in the control of mouse Burkitt lymphoma in vivo. J Clin Invest. 2004;114:542–550. doi: 10.1172/JCI22053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matloubian M, Concepcion RJ, Ahmed R. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J Virol. 1994;68:8056–8063. doi: 10.1128/jvi.68.12.8056-8063.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paludan C, Bickham K, Nikiforow S, Tsang ML, Goodman K, Hanekom WA, Fonteneau JF, Stevanovic S, Munz C. Epstein-Barr nuclear antigen 1-specific CD4(+) Th1 cells kill Burkitt’s lymphoma cells. Journal of immunology. 2002;169:1593–1603. doi: 10.4049/jimmunol.169.3.1593. [DOI] [PubMed] [Google Scholar]
- 34.Khanna R, Burrows SR, Steigerwald-Mullen PM, Moss DJ, Kurilla MG, Cooper L. Targeting Epstein-Barr virus nuclear antigen 1 (EBNA1) through the class II pathway restores immune recognition by EBNA1-specific cytotoxic T lymphocytes: evidence for HLA-DM-independent processing. Int Immunol. 1997;9:1537–1543. doi: 10.1093/intimm/9.10.1537. [DOI] [PubMed] [Google Scholar]
- 35.Khanna R, Burrows SR, Thomson SA, Moss DJ, Cresswell P, Poulsen LM, Cooper L. Class I processing-defective Burkitt’s lymphoma cells are recognized efficiently by CD4+ EBV-specific CTLs. Journal of immunology. 1997;158:3619–3625. [PubMed] [Google Scholar]
- 36.Long HM, Zuo J, Leese AM, Gudgeon NH, Jia H, Taylor GS, Rickinson AB. CD4+ T-cell clones recognizing human lymphoma-associated antigens: generation by in vitro stimulation with autologous Epstein-Barr virus-transformed B cells. Blood. 2009;114:807–815. doi: 10.1182/blood-2008-12-194043. [DOI] [PubMed] [Google Scholar]
- 37.Saito N, Courtois G, Chiba A, Yamamoto N, Nitta T, Hironaka N, Rowe M, Yamamoto N, Yamaoka S. Two carboxyl-terminal activation regions of Epstein-Barr virus latent membrane protein 1 activate NF-kappaB through distinct signaling pathways in fibroblast cell lines. The Journal of biological chemistry. 2003;278:46565–46575. doi: 10.1074/jbc.M302549200. [DOI] [PubMed] [Google Scholar]
- 38.Staege MS, Lee SP, Frisan T, Mautner J, Scholz S, Pajic A, Rickinson AB, Masucci MG, Polack A, Bornkamm GW. MYC overexpression imposes a nonimmunogenic phenotype on Epstein-Barr virus-infected B cells. Proc Natl Acad Sci U S A. 2002;99:4550–4555. doi: 10.1073/pnas.072495599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chornoguz O, Gapeev A, O’Neill MC, Ostrand-Rosenberg S. Major histocompatibility complex class II+ invariant chain negative breast cancer cells present unique peptides that activate tumor-specific T cells from breast cancer patients. Molecular & cellular proteomics: MCP. 2012;11:1457–1467. doi: 10.1074/mcp.M112.019232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thompson JA, Srivastava MK, Bosch JJ, Clements VK, Ksander BR, Ostrand-Rosenberg S. The absence of invariant chain in MHC II cancer vaccines enhances the activation of tumor-reactive type 1 CD4+ T lymphocytes. Cancer immunology, immunotherapy: CII. 2008;57:389–398. doi: 10.1007/s00262-007-0381-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.God JM, Zhao D, Cameron CA, Amria S, Bethard JR, Haque A. Disruption of HLA class II antigen presentation in Burkitt lymphoma: implication of a 47,000 MW acid labile protein in CD4+ T-cell recognition. Immunology. 2014;142:492–505. doi: 10.1111/imm.12281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kempkes B, Spitkovsky D, Jansen-Durr P, Ellwart JW, Kremmer E, Delecluse HJ, Rottenberger C, Bornkamm GW, Hammerschmidt W. B-cell proliferation and induction of early G1-regulating proteins by Epstein-Barr virus mutants conditional for EBNA2. EMBO J. 1995;14:88–96. doi: 10.1002/j.1460-2075.1995.tb06978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Amria S, Cameron C, Stuart R, Haque A. Defects in HLA class II antigen presentation in B-cell lymphomas. Leuk Lymphoma. 2008;49:353–355. doi: 10.1080/10428190701814305. [DOI] [PubMed] [Google Scholar]
- 44.Balkan W, Martinez AF, Fernandez I, Rodriguez MA, Pang M, Troen BR. Identification of NFAT binding sites that mediate stimulation of cathepsin K promoter activity by RANK ligand. Gene. 2009;446:90–98. doi: 10.1016/j.gene.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 45.Polack A, Hortnagel K, Pajic A, Christoph B, Baier B, Falk M, Mautner J, Geltinger C, Bornkamm GW, Kempkes B. c-myc activation renders proliferation of Epstein-Barr virus (EBV)-transformed cells independent of EBV nuclear antigen 2 and latent membrane protein 1. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:10411–10416. doi: 10.1073/pnas.93.19.10411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Radwan FF, Zhang L, Hossain A, Doonan BP, God JM, Haque A. Mechanisms regulating enhanced human leukocyte antigen class II-mediated CD4 + T cell recognition of human B-cell lymphoma by resveratrol. Leuk Lymphoma. 2012;53:305–314. doi: 10.3109/10428194.2011.615423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haque MA, Li P, Jackson SK, Zarour HM, Hawes JW, Phan UT, Maric M, Cresswell P, Blum JS. Absence of gamma-interferon-inducible lysosomal thiol reductase in melanomas disrupts T cell recognition of select immunodominant epitopes. The Journal of experimental medicine. 2002;195:1267–1277. doi: 10.1084/jem.20011853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma C, Whiteley PE, Cameron PM, Freed DC, Pressey A, Chen SL, Garni-Wagner B, Fang C, Zaller DM, Wicker LS, Blum JS. Role of APC in the selection of immunodominant T cell epitopes. J Immunol. 1999;163:6413–6423. [PubMed] [Google Scholar]
- 49.Pathak SS, Blum JS. Endocytic recycling is required for the presentation of an exogenous peptide via MHC class II molecules. Traffic. 2000;1:561–569. doi: 10.1034/j.1600-0854.2000.010706.x. [DOI] [PubMed] [Google Scholar]
- 50.Hossain A, God JM, Radwan FF, Amria S, Zhao D, Bethard JR, Haque A. HLA class II defects in Burkitt lymphoma: bryostatin-1-induced 17 kDa protein restores CD4+ T-cell recognition. Clin Dev Immunol. 2011;2011:780839. doi: 10.1155/2011/780839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li P, Haque MA, Blum JS. Role of disulfide bonds in regulating antigen processing and epitope selection. J Immunol. 2002;169:2444–2450. doi: 10.4049/jimmunol.169.5.2444. [DOI] [PubMed] [Google Scholar]
- 52.Zhao D, Amria S, Hossain A, Sundaram K, Komlosi P, Nagarkatti M, Haque A. Enhancement of HLA class II-restricted CD4+ T cell recognition of human melanoma cells following treatment with bryostatin-1. Cell Immunol. 2011;271:392–400. doi: 10.1016/j.cellimm.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hossain A, Radwan FF, Doonan BP, God JM, Zhang L, Bell PD, Haque A. A possible cross-talk between autophagy and apoptosis in generating an immune response in melanoma. Apoptosis: an international journal on programmed cell death. 2012;17:1066–1078. doi: 10.1007/s10495-012-0745-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Younger AR, Amria S, Jeffrey WA, Mahdy AE, Goldstein OG, Norris JS, Haque A. HLA class II antigen presentation by prostate cancer cells. Prostate Cancer Prostatic Dis. 2008;11:334–341. doi: 10.1038/sj.pcan.4501021. [DOI] [PubMed] [Google Scholar]
- 55.Haque A, Das A, Hajiaghamohseni LM, Younger A, Banik NL, Ray SK. Induction of apoptosis and immune response by all-trans retinoic acid plus interferon-gamma in human malignant glioblastoma T98G and U87MG cells. Cancer Immunol Immunother. 2007;56:615–625. doi: 10.1007/s00262-006-0219-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goldstein OG, Hajiaghamohseni LM, Amria S, Sundaram K, Reddy SV, Haque A. Gamma-IFN-inducible-lysosomal thiol reductase modulates acidic proteases and HLA class II antigen processing in melanoma. Cancer Immunol Immunother. 2008;57:1461–1470. doi: 10.1007/s00262-008-0483-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Amria S, Hajiaghamohseni LM, Harbeson C, Zhao D, Goldstein O, Blum JS, Haque A. HLA-DM negatively regulates HLA-DR4-restricted collagen pathogenic peptide presentation and T cell recognition. Eur J Immunol. 2008;38:1961–1970. doi: 10.1002/eji.200738100. [DOI] [PubMed] [Google Scholar]
- 58.Rothbard JB, Busch R, Howland K, Bal V, Fenton C, Taylor WR, Lamb JR. Structural analysis of a peptide--HLA class II complex: identification of critical interactions for its formation and recognition by T cell receptor. International immunology. 1989;1:479–486. doi: 10.1093/intimm/1.5.479. [DOI] [PubMed] [Google Scholar]
- 59.Zhang S, Zhang H, Zhao J. The role of CD4 T cell help for CD8 CTL activation. Biochem Biophys Res Commun. 2009;384:405–408. doi: 10.1016/j.bbrc.2009.04.134. [DOI] [PubMed] [Google Scholar]
- 60.Castellino F, Germain RN. Cooperation between CD4+ and CD8+ T cells: when, where, and how. Annu Rev Immunol. 2006;24:519–540. doi: 10.1146/annurev.immunol.23.021704.115825. [DOI] [PubMed] [Google Scholar]
- 61.Khanolkar A, Badovinac VP, Harty JT. CD8 T cell memory development: CD4 T cell help is appreciated. Immunol Res. 2007;39:94–104. doi: 10.1007/s12026-007-0081-4. [DOI] [PubMed] [Google Scholar]
- 62.Pajic A, Staege MS, Dudziak D, Schuhmacher M, Spitkovsky D, Eissner G, Brielmeier M, Polack A, Bornkamm GW. Antagonistic effects of c-myc and Epstein-Barr virus latent genes on the phenotype of human B cells. Int J Cancer. 2001;93:810–816. doi: 10.1002/ijc.1404. [DOI] [PubMed] [Google Scholar]
- 63.Pajic A, Polack A, Staege MS, Spitkovsky D, Baier B, Bornkamm GW, Laux G. Elevated expression of c-myc in lymphoblastoid cells does not support an Epstein-Barr virus latency III-to-I switch. J Gen Virol. 2001;82:3051–3055. doi: 10.1099/0022-1317-82-12-3051. [DOI] [PubMed] [Google Scholar]
- 64.Schlee M, Holzel M, Bernard S, Mailhammer R, Schuhmacher M, Reschke J, Eick D, Marinkovic D, Wirth T, Rosenwald A, Staudt LM, Eilers M, Baran-Marszak F, Fagard R, Feuillard J, Laux G, Bornkamm GW. C-myc activation impairs the NF-kappaB and the interferon response: implications for the pathogenesis of Burkitt’s lymphoma. International journal of cancer. Journal international du cancer. 2007;120:1387–1395. doi: 10.1002/ijc.22372. [DOI] [PubMed] [Google Scholar]
- 65.Huang MJ, Cheng YC, Liu CR, Lin S, Liu HE. A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle arrest, apoptosis, and myeloid differentiation of human acute myeloid leukemia. Exp Hematol. 2006;34:1480–1489. doi: 10.1016/j.exphem.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 66.Rocha N, Neefjes J. MHC class II molecules on the move for successful antigen presentation. EMBO J. 2008;27:1–5. doi: 10.1038/sj.emboj.7601945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Haque A, Blum JS. New insights in antigen processing and epitope selection: development of novel immunotherapeutic strategies for cancer, autoimmunity and infectious diseases. J Biol Regul Homeost Agents. 2005;19:93–104. [PubMed] [Google Scholar]
- 68.Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol. 2013;31:443–473. doi: 10.1146/annurev-immunol-032712-095910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gras S, Burrows SR, Turner SJ, Sewell AK, McCluskey J, Rossjohn J. A structural voyage toward an understanding of the MHC-I-restricted immune response: lessons learned and much to be learned. Immunol Rev. 2012;250:61–81. doi: 10.1111/j.1600-065X.2012.01159.x. [DOI] [PubMed] [Google Scholar]
- 70.West LC, Cresswell P. Expanding roles for GILT in immunity. Curr Opin Immunol. 2013;25:103–108. doi: 10.1016/j.coi.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Norton DL, Haque A. Insights into the Role of GILT in HLA Class II Antigen Processing and Presentation by Melanoma. J Oncol. 2009;2009:142959. doi: 10.1155/2009/142959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stern LJ, Potolicchio I, Santambrogio L. MHC class II compartment subtypes: structure and function. Curr Opin Immunol. 2006;18:64–69. doi: 10.1016/j.coi.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 73.Landsverk OJ, Bakke O, Gregers TF. MHC II and the endocytic pathway: regulation by invariant chain. Scand J Immunol. 2009;70:184–193. doi: 10.1111/j.1365-3083.2009.02301.x. [DOI] [PubMed] [Google Scholar]
- 74.Chen X, Jensen PE. MHC class II antigen presentation and immunological abnormalities due to deficiency of MHC class II and its associated genes. Exp Mol Pathol. 2008;85:40–44. doi: 10.1016/j.yexmp.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Painter CA, Stern LJ. Conformational variation in structures of classical and non-classical MHCII proteins and functional implications. Immunol Rev. 2012;250:144–157. doi: 10.1111/imr.12003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Denzin LK, Fallas JL, Prendes M, Yi W. Right place, right time, right peptide: DO keeps DM focused. Immunol Rev. 2005;207:279–292. doi: 10.1111/j.0105-2896.2005.00302.x. [DOI] [PubMed] [Google Scholar]
- 77.Martin R, Olivares M, Perez M, Xaus J, Torre C, Fernandez L, Rodriguez JM. Identification and evaluation of the probiotic potential of lactobacilli isolated from canine milk. Veterinary journal. 2010;185:193–198. doi: 10.1016/j.tvjl.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 78.Busch R, Rinderknecht CH, Roh S, Lee AW, Harding JJ, Burster T, Hornell TM, Mellins ED. Achieving stability through editing and chaperoning: regulation of MHC class II peptide binding and expression. Immunol Rev. 2005;207:242–260. doi: 10.1111/j.0105-2896.2005.00306.x. [DOI] [PubMed] [Google Scholar]
- 79.Morris P, Shaman J, Attaya M, Amaya M, Goodman S, Bergman C, Monaco JJ, Mellins E. An essential role for HLA-DM in antigen presentation by class II major histocompatibility molecules. Nature. 1994;368:551–554. doi: 10.1038/368551a0. [DOI] [PubMed] [Google Scholar]
- 80.Guce AI, Mortimer SE, Yoon T, Painter CA, Jiang W, Mellins ED, Stern LJ. HLA-DO acts as a substrate mimic to inhibit HLA-DM by a competitive mechanism. Nat Struct Mol Biol. 2013;20:90–98. doi: 10.1038/nsmb.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen X, Jensen PE. The expression of HLA-DO (H2-O) in B lymphocytes. Immunol Res. 2004;29:19–28. doi: 10.1385/IR:29:1-3:019. [DOI] [PubMed] [Google Scholar]
- 82.Xiu F, Cote MH, Bourgeois-Daigneault MC, Brunet A, Gauvreau ME, Shaw A, Thibodeau J. Cutting edge: HLA-DO impairs the incorporation of HLA-DM into exosomes. J Immunol. 2011;187:1547–1551. doi: 10.4049/jimmunol.1100199. [DOI] [PubMed] [Google Scholar]
- 83.Glazier KS, Hake SB, Tobin HM, Chadburn A, Schattner EJ, Denzin LK. Germinal center B cells regulate their capability to present antigen by modulation of HLA-DO. J Exp Med. 2002;195:1063–1069. doi: 10.1084/jem.20012059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pathak SS, Lich JD, Blum JS. Cutting edge: editing of recycling class II:peptide complexes by HLA-DM. Journal of immunology. 2001;167:632–635. doi: 10.4049/jimmunol.167.2.632. [DOI] [PubMed] [Google Scholar]
- 85.Haque MA, Hawes JW, Blum JS. Cysteinylation of MHC class II ligands: peptide endocytosis and reduction within APC influences T cell recognition. Journal of immunology. 2001;166:4543–4551. doi: 10.4049/jimmunol.166.7.4543. [DOI] [PubMed] [Google Scholar]
- 86.Yin L, Trenh P, Guce A, Wieczorek M, Lange S, Sticht J, Jiang W, Bylsma M, Mellins ED, Freund C, Stern LJ. Susceptibility to HLA-DM protein is determined by a dynamic conformation of major histocompatibility complex class II molecule bound with peptide. The Journal of biological chemistry. 2014;289:23449–23464. doi: 10.1074/jbc.M114.585539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Phipps-Yonas H, Semik V, Hastings KT. GILT expression in B cells diminishes cathepsin S steady-state protein expression and activity. Eur J Immunol. 2013;43:65–74. doi: 10.1002/eji.201242379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Maric M, Arunachalam B, Phan UT, Dong C, Garrett WS, Cannon KS, Alfonso C, Karlsson L, Flavell RA, Cresswell P. Defective antigen processing in GILT-free mice. Science. 2001;294:1361–1365. doi: 10.1126/science.1065500. [DOI] [PubMed] [Google Scholar]
- 89.O’Donnell PW, Haque A, Klemsz MJ, Kaplan MH, Blum JS. Cutting edge: induction of the antigen-processing enzyme IFN-gamma-inducible lysosomal thiol reductase in melanoma cells Is STAT1-dependent but CIITA-independent. Journal of immunology. 2004;173:731–735. doi: 10.4049/jimmunol.173.2.731. [DOI] [PubMed] [Google Scholar]
- 90.Denzin LK, Cresswell P. Sibling rivalry: competition between MHC class II family members inhibits immunity. Nat Struct Mol Biol. 2013;20:7–10. doi: 10.1038/nsmb.2484. [DOI] [PubMed] [Google Scholar]
- 91.God JM, Zhao D, Cameron CA, Amria S, Bethard JR, Haque A. Disruption of HLA class II antigen presentation in Burkitt lymphoma: implication of a 47 000 MW acid labile protein in CD4(+) T-cell recognition. Immunology. 2014;142:492–505. doi: 10.1111/imm.12281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Subramanian A, Miller DM. Structural analysis of alpha-enolase. Mapping the functional domains involved in down-regulation of the c-myc protooncogene. The Journal of biological chemistry. 2000;275:5958–5965. doi: 10.1074/jbc.275.8.5958. [DOI] [PubMed] [Google Scholar]
- 93.Sedoris KC, Thomas SD, Miller DM. c-myc promoter binding protein regulates the cellular response to an altered glucose concentration. Biochemistry. 2007;46:8659–8668. doi: 10.1021/bi7003558. [DOI] [PubMed] [Google Scholar]